DOI:
10.1039/D0RA00830C
(Paper)
RSC Adv., 2020,
10, 9657-9662
Synthesis and fluorescence properties of butadiyne-linked linear and cyclic carbazole oligomers†
Received
28th January 2020
, Accepted 27th February 2020
First published on 6th March 2020
Abstract
Butadiyne-linked linear and cyclic carbazole oligomers were successfully synthesized. The intensity of the emission band in the 0–0 band of the highly planar macrocyclics decreased compared to that of the 0–1 band. Contrary to this, for larger macrocycles having reduced planarity, the intensity of the emission of the 0–0 band increased as in the cases of the linear compounds. This suggests that the emission color of the π-conjugated molecule can be controlled not only by the difference between the cyclic and chain structures but also by the control of the planarity, and is expected to be a new principle for molecular design in the development of fluorescent materials.
Introduction
The development of fluorescent molecules is attracting attention from the viewpoint of application to organic EL, bioimaging, two-photon fluorescent materials, and so on. In order to make a molecule that emits UV light emit visible light, it is necessary to expand the π-conjugation system to reduce the HOMO–LUMO energy difference, whereas the principle of molecular design for controlling the emission colour is required. Carbazole has a planar structure in which two benzene rings are bridged by a nitrogen atom, and exhibit strong UV emission. Several carbazole oligomers and polymers have been reported so far.1–11 Since the bond angle between the 3- and 6-positions of carbazole is 90°, the oligomerization from these positions is expected to construct a stable cyclic tetramer. So, ethynyl-linked1,6,8 and directly linked5,11 cyclic carbazole tetramers have been synthesized. However, there have been no reports on butadiyne-linked cyclic tetramer or short oligomers connected at the 3,6-positions except for trimers and tetramers linked at the 1,8-positions.9 It has been reported that two-photon absorption efficiency can be greatly improved by using the butadiyne linkage to connect π-conjugated systems between porphyrins.12,13 Further, macrocyclic compounds composed of self-assembled porphyrins linked with fluorene14 or carbazole15 using the ethynylene linkage resulted in two-photon absorption enhancement. In this study, we report synthesis of butadiyne-linked linear and cyclic carbazole oligomers connected at the 3,6-positions, and their absorption and photoluminescence properties. In addition, attention has recently been focused on the emission properties of π-conjugated cyclic compounds,16–31 and this study may also be interesting in this respect. The structures of synthesized compounds monomer 1, linear trimer 3L, linear tetramer 4L, cyclic trimer 3R, cyclic tetramer 4R, cyclic pentamer 5R, and cyclic hexamer 6R are presented in Scheme 1.
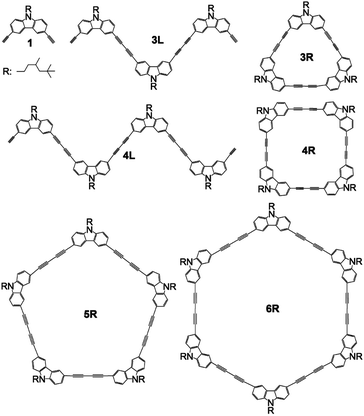 |
| | Scheme 1 Structures of 1, 3L, 4L, 3R, 4R, 5R, and 6R. | |
Results and discussion
Synthesis
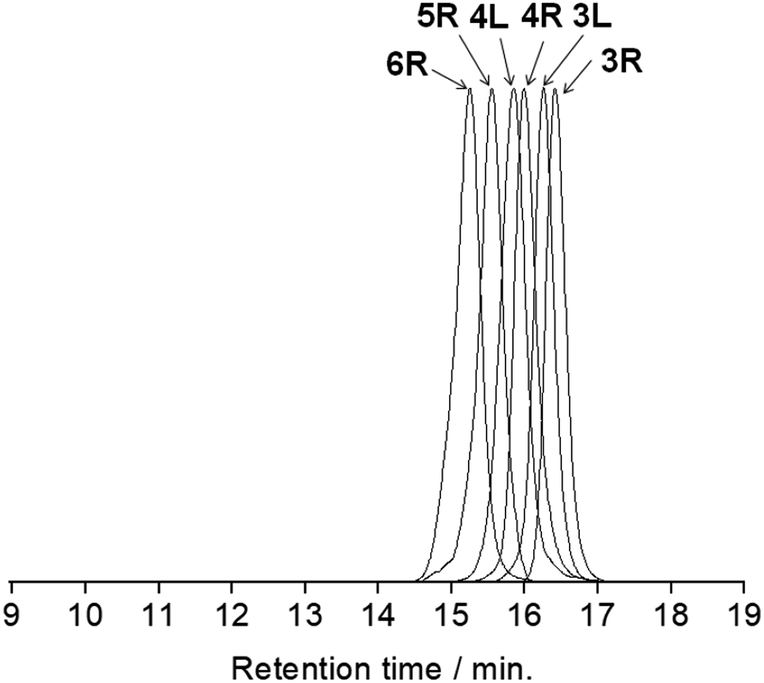
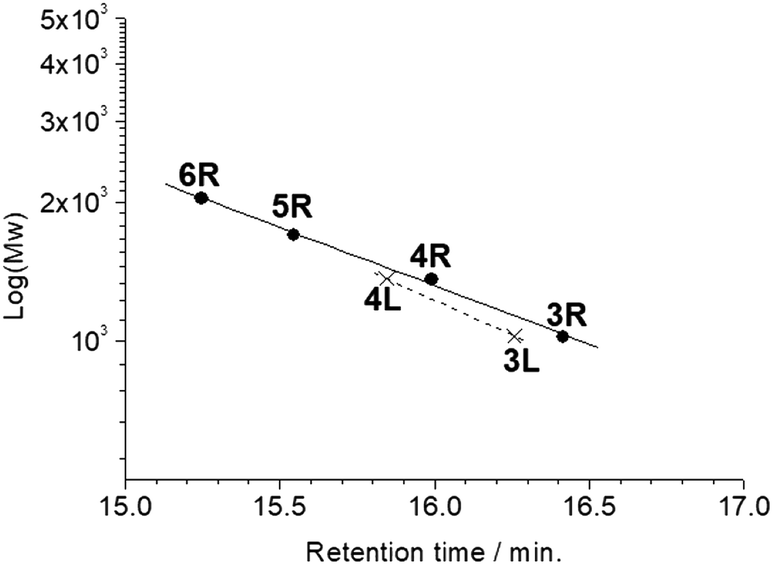
All the butadiyne-linked oligomers were synthesized using Pd(PPh3)2Cl2 under an oxygen atmosphere in THF/triethylamine.32 3R, 4R, and 5R were prepared at 60 °C for 26 hours. In the analytical GPC (gel permeation chromatography) data using chroloform as an eluent after the reaction (ESI†), main three peaks corresponding to 3R and 5R as well as 4R were observed. Each peak was isolated using preparative GPC with the yields of 6% for 3R, 11% for 4R, and 6% for 5R, respectively. In order to suppress cyclization, synthesis of 3L and 4L was performed by reducing the amount of Pd(PPh3)2Cl2 and lowering the reaction temperature to 50 °C as compared to the synthesis of the cyclic compounds. Each compound was isolated using preparative GPC with the yields of 2% for 3L and 4% for 4L, respectively. In 1H NMR spectra of 3L and 4L, a signal of the terminal alkyne, which disappeared in the cyclic compounds 3R and 4R, was observed at 3.09–3.10 ppm as two protons (ESI†). Further, in the MALDI-TOF MASS measurements, the molecular ion peaks of 3L and 4L were two hydrogens larger than those of the corresponding cyclic molecules 3R and 4R (ESI†). The cyclic hexamer 6R could be synthesized from linear trimer 3L with the formation of 3R, and separated by preparative GPC with 20% yield. Fig. 1 and 2 show the analytical GPC chart containing all isolated compounds and logarithm plots of molecular weight vs. retention time, respectively. As seen in Fig. 2, 6R fits on the GPC calibration curve drawn from 3R, 4R, and 5R instead of 3L and 4L. In the NMR spectra of 6R, the terminal alkyne signal was not observed, and one set of carbazole was observed as seen in 3R and 4R. These data indicate the formation of 6R.
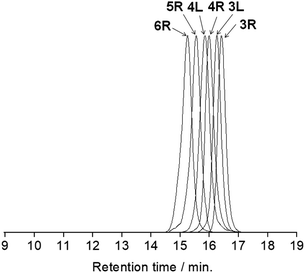 |
| | Fig. 1 The analytical GPC chart containing all isolated compounds. Chroloform was used as an eluent. | |
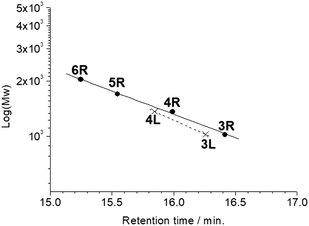 |
| | Fig. 2 Logarithm plots of molecular weight vs. retention time. | |
Optical properties
The absorption and fluorescence spectra of synthesized compounds were measured in chloroform at room temperature. Comparing the linear compounds 3L and 4L, both the absorption and fluorescence of 4L shifted slightly longer than those of 3L due to the expansion of the π-conjugation, and the shapes of the spectra were similar (Fig. 3). 3L and 4L showed light blue emission as shown in Fig. 4.
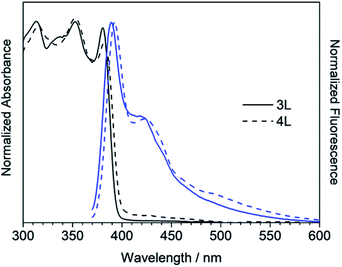 |
| | Fig. 3 UV/Vis absorption (black) and fluorescence (blue) spectra of 3L and 4L in chloroform. | |
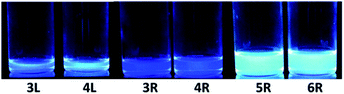 |
| | Fig. 4 Emission colour excited at 365 nm. | |
Secondly, cyclic compounds 3R and 4R were examined (Fig. 5). Compared with 3L and 4L, the absorption intensity around at 370 to 390 nm became weak, and these split into two peaks probably due to changes in vibration mode, but not clearly at this time. In the fluorescence spectra, emission from the 0–0 band near 400 nm was also weak. These results indicate that the cyclization changed the spectral shape for trimer and tetramer. The fluorescence spectra did not depend on concentration and solvent (acetone, THF, toluene, and hexane). A similar result was reported for cyclic and linear thiophene oligomers, where the intensity of the 0–0 band of the highly planar cyclic compound was lower than that of the linear structure.26 3R and 4R emitted blue light as presented in Fig. 4 due to the shift of the maximum emission band to around 440 nm.
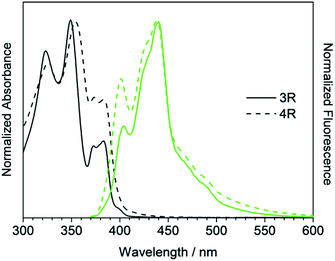 |
| | Fig. 5 UV/Vis absorption (black) and fluorescence (green) spectra of 3R and 4R. | |
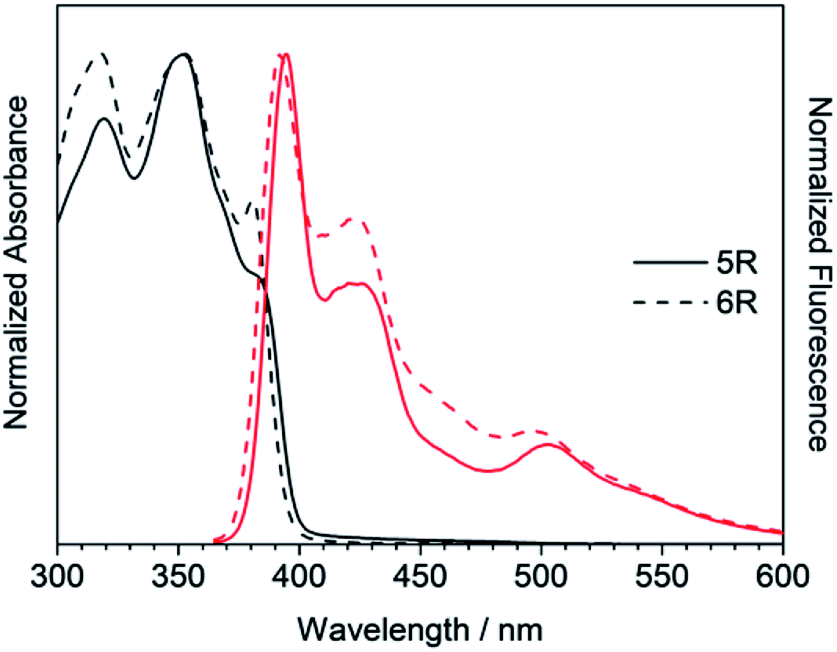
Next, the larger macrocycles 5R and 6R were measured. As shown in Fig. 6, although these are cyclic molecules, the shapes of the fluorescence spectra are different from those of 3R and 4R. The emission intensity from the 0–0 band around 400 nm increased similar to the linear molecules 3L and 4L, and a band around 500 nm, which was observed as small shoulder in other compounds, appeared clearly. In the absorption spectra, the absorption intensities at 380 to 390 nm were intermediate between linear and cyclic compounds for trimers and tetramers, and the splitting disappeared like linear molecules. These data indicate that the increase in the ring size increased the flexibility and lowered the planarity. These results are also consistent with the reversal of the relationship between the 0–0 and 0–1 band intensities obtained for the larger cyclic oligothiophene.26 For the emission band around 500 nm, although clear information cannot be obtained from the absorption spectra, it may be a change in the vibration mode due to the increase in carbazole unit and flexibility. In the case of the ethynylene-linked cyclic tetramer in THF solution,6 the S0–S1 absorption around at 375 nm was relatively weak, whereas the maximum fluorescence probably emitted from 0–0 band was observed at around 410 nm contrary to this study. On the other hand, the directly linked cyclic oligomer (mixture of 4-mer to 12-mer) exhibited higher emission intensity from the 0–1 band than the 0–0 band.5 The butadiyne-linked carbazole tetramer connected at the 1,8-positions also showed stronger emission in the 0–1 band, but in trimer, the 0–0 band was stronger.9 As seen in Fig. 4, the emission colour of 5R and 6R became light blue near white due to the relatively strong emission at around 500 nm.
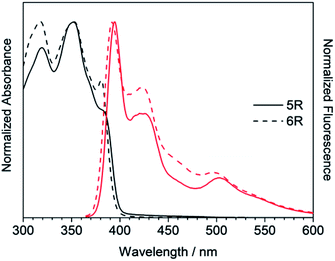 |
| | Fig. 6 UV/Vis absorption (black) and fluorescence (red) spectra of 5R and 6R. | |
Absorption and fluorescence data are summarized in Table 1. Increasing the number of units to 5 and 6 reduced the quantum yield. In 4R, since the bond angle is 90° between carbazole 3- and 6-positions, the steric distortion is the smallest among the cyclic compounds giving the relatively large quantum yield value.
Table 1 Photophysical data of carbazole oligomers
| Compound |
Absorption/nm |
Fluorescencea/nm |
Quantum yield |
| The excitation wavelengths for fluorescence measurements were selected to the absorption peak around 350 nm. |
| 3L |
381 |
389, 419 |
0.08 |
| 3R |
373, 383 |
404, 440 |
0.08 |
| 4L |
382 |
392, 423 |
0.17 |
| 4R |
373, 383 |
401, 438 |
0.18 |
| 5R |
380–385 (shoulder) |
394, 429, 503 |
0.15 |
| 6R |
381 |
392, 424, 497 |
0.11 |
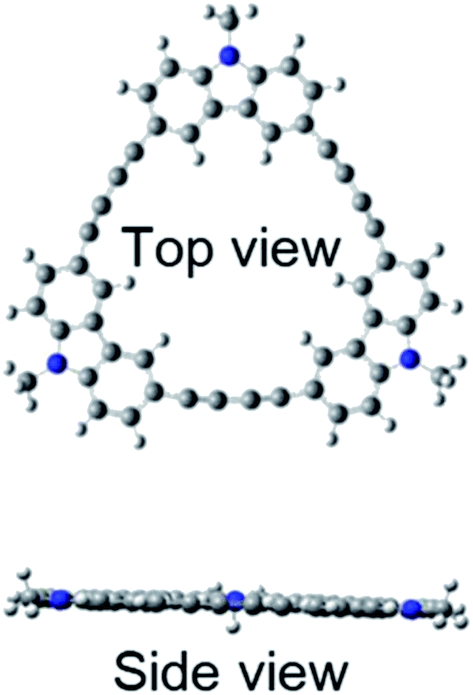
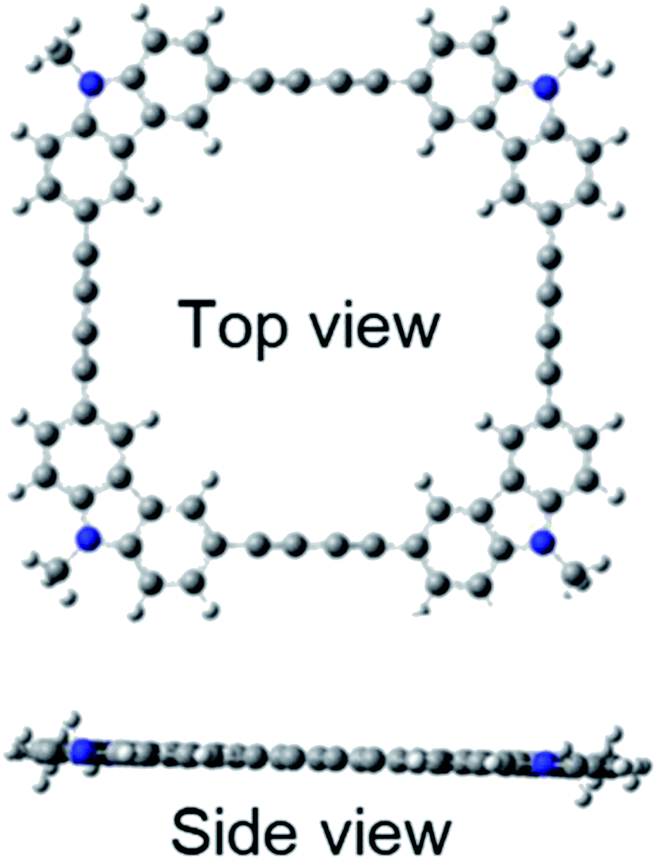
Quantum chemical calculations using Gaussian 09 were performed to further investigate these absorption and fluorescence properties. Geometry optimization in the ground state was calculated using DFT with CAM-B3LYP and 6-31G(d). Chloroform was used as a solvent, and CPCM was employed as a solvent model. To increase the calculation speed, the trimethylhexyl group at the N-position was converted to a methyl group. Fig. S16 and S17 in ESI† show the optimized structures of 3L and 4L. Since 3L and 4L have a chain structure, the rotation barrier of the triple bond is relatively low, so that they are flexible and do not have rigid planarity. In the solution, various conformations other than the optimized structure may be taken due to the flexibility. On the other hand, 3R and 4R have complete planar structures, as demonstrated in Fig. 7 and 8. However, even in the cyclic structure, the planar geometry cannot be maintained in 5R and 6R due to increased flexibility (ESI†). Again, for 5R and 6R, various conformations other than optimized structures can be adopted in solution. According to the theory,25 the ratio of 0–0 and 0–1 bands responds to the product of the exciton coherence number and the conformation factor, where the model shows that the 0–0 band is highly sensitive to chain bending. This may explain the small 0–0 bands observed in 3R and 4R without chain bending.
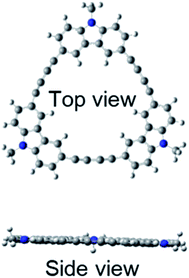 |
| | Fig. 7 Optimized structure of 3R. | |
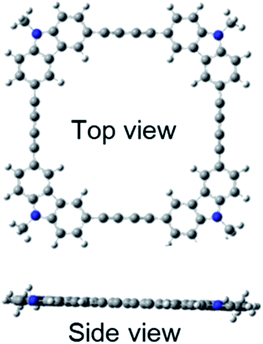 |
| | Fig. 8 Optimized structure of 4R. | |
Next, in order to investigate the effect of the molecular structure on the electronic transition, the excited state was calculated by TD-DFT. The S0–S1 transitions for 3R and 4R were calculated as forbidden with the oscillator strength of zero due to the cancellation of the transition dipole (ESI†). In contrast, the S0–S1 transitions in the chain origomers 3L and 4L, and large rings 5R and 6R were allowed because flexibility and low planarity eliminate the transition dipole cancellation. These results correspond to the fact that the absorption bands at around 380 nm for 3R and 4R were weaker than other oligomers. Further, the HOMO–LUMO absorption intensity of 3R was about half compared to that of 4R, suggesting that 4R was more flexible than 3R due to increased number of carbazoles and triple bonds. From the results at the calculation level used in this study, further quantitative discussion seems to be difficult between experimental and calculated values for wavelength and oscillator strength. For comparison, the calculated oscillator strengths were plotted vs. wavelength together with measured spectra (ESI†). Still, the question remains that the fluorescence quantum yields of 3R and 4R are not lower compared with 3L and 4L. Further, from the experimental results including 1H NMR, where no significant ring current effect was observed in 3R and 4R, it cannot be judged whether the cyclic conjugate is stronger than linear. More detailed theoretical calculations including vibrations, and time-resolved measurements will be needed to clear these.
Experimental
General procedures
Analytical GPC measurements were performed using an Agilent 1100 HPLC system with a JAIGEL 3HA column. Preparative GPC separations were made using a JAI LC-908 recycling HPLC system with two JAIGEL 3H columns in series. 1H NMR spectra were recorded on Bruker AV III 500 HD spectrometer. UV-vis spectra were measured using a JASCO V-630 spectrometer. Fluorescence measurements were performed on a Hitachi F-2500 spectrophotometer. MALDI-TOF mass spectra were obtained with a Bruker Autoflex with dithranol as a matrix. 3,6-Diiodo-9-(3,5,5-trimethyl)hexylcarbazole was prepared according to the literature.15
3,6-Bis(trimethylsilylethynyl)-9-(3,5,5-trimethyl)hexylcarbazole. Under a nitrogen atmosphere, after dissolving 3,6-diiodo-9-(3,5,5-trimethyl)hexylcarbazole (0.25 g, 0.46 mmol), Pd(PPh3)2Cl2 (0.1 g, 0.17 mmol), CuI (0.01 g, 0.34 mmol), and triethylamine (15 ml) in dry THF (75 ml), TMS-acetylene (0.2 ml, 1.42 mmol) was added to the solution and stirred at 40 °C for 6.5 hours. Saturated aqueous ammonium chloride solution was added, and the mixture was extracted with chloroform. Purification by silica gel column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chloroform = 3:1) gave a viscous yellow liquid to yield 0.21 g (0.43 mmol, 91%). 1H NMR (500 MHz, CDCl3): δ = 8.19 (d, 2H), 7.56–7.58 (dd, 2H), 7.26–7.28 (d, 2H), 4.22–4.29 (m, 2H), 1.76–1.84 (m, 1H), 1.59–1.65 (m, 2H), 1.14–1.30 (dd, 3H), 1.07–1.08 (d, 2H), 0.87 (s, 9H), 0.30 (s, 18H) ppm.
:chloroform = 3:1) gave a viscous yellow liquid to yield 0.21 g (0.43 mmol, 91%). 1H NMR (500 MHz, CDCl3): δ = 8.19 (d, 2H), 7.56–7.58 (dd, 2H), 7.26–7.28 (d, 2H), 4.22–4.29 (m, 2H), 1.76–1.84 (m, 1H), 1.59–1.65 (m, 2H), 1.14–1.30 (dd, 3H), 1.07–1.08 (d, 2H), 0.87 (s, 9H), 0.30 (s, 18H) ppm.
3,6-Diethynyl-9-(3,5,5-trimethyl)hexylcarbazole 1. After dissolving 3,6-bis(trimethylsilylethynyl)-9-(3,5,5-trimethyl)hexylcarbazole (0.21 g, 0.44 mol) in CHCl3 (100 ml), TBAF (0.4 ml, 1.14 mmol) was added, and the mixture was stirred at room temperature for 3 hours. Silica gel column chromatography was performed to give 1 in 87% yield. 1H NMR (500 MHz, CDCl3): δ = 8.21 (d, 2H), 7.59–7.61 (dd, 2H), 7.30–7.32 (d, 2H), 4.22–4.29 (m, 2H), 3.08 (s, 2H), 1.79–1.87 (m, 1H), 1.59–1.67 (m, 2H), 1.15–1.31 (dd, 3H), 1.08–1.09 (d, 2H), 0.87 (s, 9H) ppm.
3R, 4R, and 5R32. Under an oxygen atmosphere, 1 (70 mg, 0.20 mmol), Pd(PPh3)2Cl2 (140 mg, 0.16 mmol), and triethylamine (15 ml) were dissolved in dry THF (70 ml), and the mixture was stirred at 60 °C. After stirring for 23 h, water was added and extracted with chloroform. The solution was washed with saturated aqueous NH4Cl and water. The crude was purified by preparative GPC (column; JAIGEL 3H, eluent; chloroform) to give 3R (6%), 4R (11%), and 5R (6%). 3R: 1H NMR (500 MHz, CDCl3): δ = 8.44 (d, 6H), 7.56–7.58 (dd, 6H), 7.32–7.34 (d, 6H), 4.24–4.32 (m, 6H),1.83–1.90 (m, 3H), 1.63–1.71 (m, 6H), 1.17–1.32 (s, 9H), 1.11–1.12 (d, 6H), 0.89 (s, 27H) ppm; MALDI-TOF Mass calcd for C75H75N3: 1017.60, found: 1018.01 [M + H]+; UV/Vis (CHCl3) λmax/nm (ε/mol−1 dm3 cm−1) = 323 (76600), 349 (90800), 373 (33000), 383 (35300). 4R: 1H NMR (500 MHz, CDCl3): δ = 8.33 (d, 8H), 7.66–7.68 (dd, 8H), 7.34–7.36 (d, 8H), 4.24–4.32 (m, 8H), 1.82–1.89 (m, 4H), 1.63–1.67 (m, 8H), 1.18–1.33 (m, 12H), 1.11–1.12 (d, 8H), 0.89 (s, 36H) ppm; MALDI-TOF Mass calcd for C100H100N4: 1356.79; found: 1357.33 [M + H]+; UV/Vis (CHCl3) λmax/nm (ε/mol−1 dm3 cm−1) = 326 (75200), 354 (95000), 373 (58300), 383 (57300). 5R: 1H NMR (500 MHz, CDCl3): δ = 8.28 (d, 10H), 7.68–7.70 (dd, 10H), 7.34–7.36 (d, 10H), 4.24–4.33 (m, 10H), 1.83–1.88 (m, 5H), 1.63–1.70 (m, 10H), 1.17–1.33 (m, 15H), 1.11–1.12 (d, 10H), 0.89 (s, 45H) ppm; MALDI-TOF Mass calcd for C125H125N5: 1695.99; found: 1696.71 [M + H]+; UV/Vis (CHCl3) λmax/nm (ε/mol−1 dm3 cm−1) = 319 (86900), 352 (100000), 380 (55500).
3L and 4L. Under an oxygen atmosphere, 1 (100 mg, 0.29 mmol), Pd(PPh3)2Cl2 (100 mg, 0.11 mmol), and triethylamine (20 ml) were dissolved in dry THF (70 ml), and the mixture was stirred at 50 °C. After stirring for 53 h, water was added and extracted with chloroform. The solution was washed with saturated aqueous NH4Cl and water. The crude was purified by preparative GPC (column; JAIGEL 3H, eluent; chloroform) to give 3L (2%) and 4L (4%). 3L: 1H NMR (500 MHz, CDCl3): δ = 8.28–8.29 (dd, 4H), 8.24 (d, 2H), 7.66–7.69 (m, 4H), 7.61–7.63 (dd, 2H), 7.32–7.36 (m, 6H), 4.25–4.33 (m, 6H), 3.09 (s, 2H), 1.81–1.89 (m, 3H), 1.63–1.69 (m, 6H), 1.16–1.32 (m, 9H), 1.11–1.12 (d, 6H), 0.88 (s, 27H) ppm; MALDI-TOF Mass calcd for C75H77N3: 1019.61; found: 1020.03 [M + H]+; UV/Vis (CHCl3) λmax/nm (ε/mol−1 dm3 cm−1) = 313 (70300), 353 (70200), 381 (68000). 4L: 1H NMR (500 MHz, CDCl3): δ = 8.24–8.33 (m, 8H), 7.61–7.70 (m, 8H), 7.32–7.36 (m, 8H), 4.24–4.32 (m, 8H), 3.10 (s, 2H), 1.82–1.89 (m, 4H), 1.63–1.67 (m, 8H), 1.16–1.33 (m, 12H), 1.10–1.12 (m, 8H), 0.89 (s, 36H) ppm; MALDI-TOF Mass calcd for C100H102N4: 1358.81; found: 1359.34 [M + H]+; UV/Vis (CHCl3) λmax/nm (ε/mol−1 dm3 cm−1) = 315 (78600), 353 (81600), 382 (71600).
6R. Under an oxygen atmosphere, 3R (8 mg, 7.63 μmol), Pd(PPh3)2Cl2 (8 mg, 8.8 μmol), and triethylamine (2 ml) were dissolved in dry THF (10 ml), and the mixture was stirred at 60 °C. After stirring for 21 h, water was added and extracted with chloroform. The solution was washed with saturated aqueous NH4Cl and water. The crude was purified by preparative GPC (column; JAIGEL 3H, eluent; chloroform) to give 6R (20%). 1H NMR (500 MHz, CDCl3): δ = 8.29 (d, 12H), 7.65–7.67 (dd, 12H), 7.33–7.35 (d, 12H), 4.22–4.32 (m, 12H), 1.81–1.89 (m, 6H), 1.62–1.70 (m, 12H), 1.16–1.32 (m, 18H), 1.10–1.11 (d, 12H), 0.88 (s, 54H) ppm; MALDI-TOF Mass calcd for C150H150N6: 2035.19; found: 2036.29 [M + H]+; UV/Vis (CHCl3) λmax/nm (ε/mol−1 dm3 cm−1) = 317 (90400), 353 (90100), 381 (63200).
Summary
In summary, we succeeded in synthesizing butadiyne-linked linear and cyclic carbazole oligomers. Although the cyclic tetramer 4R having a stable structure was expected to be the main product, 3L, 4L, 3R, and 5R could also successfully be isolated. Furthermore, a cyclic hexamer 6R could be synthesized from 3L. As pointed out in the literatures,25,26 the intensity of the emission band in the 0–0 band of the highly planar macrocyclics 3R and 4R decreased compared to that of the 0–1 band. On the other hand, in the macrocycles 5R and 6R having reduced planarity, the intensity of the emission band of the 0–0 band increased as in the cases of the linear compounds 3L and 4L. As a result, by controlling the oligomer structure, different emission colors, light blue (3L and 4L), blue (3R and 4R), and pale blue close to white (5R and 6R) could be expressed. This indicates that the emission color of the π-conjugated molecule can be controlled not only by the difference between the cyclic and chain structures but also by the control of the planarity, and is expected to be a new principle for molecular design in the development of fluorescent materials. Further, these compounds will be evaluated as two-photon fluorescent materials expected to be applied to 3D displays and other applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grant-in-Aids for Scientific Research (C) (no. 18K05258) from Ministry of Education, Culture, Sports, Science and Technology, Japan (Monbu Kagakusho). The MALDI-TOF measurement was carried out at Instrumental Analysis Center at Yokohama National University.
Notes and references
- W. Zhang and J. S. Moore, J. Am. Chem. Soc., 2004, 126, 12796 CrossRef CAS PubMed.
- Y. Zhang, T. Wada and H. Sasabe, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2041–2047 CrossRef CAS.
- S. Maruyama, Y. Zhang, T. Wada and H. Sasabe, J. Chem. Soc., Perkin Trans. 1, 1999, 41–46, 10.1039/A807158F.
- S. Maruyama, H. Suzuki, X.-t. Tao, T. Wada, H. Sasabe, S. Miyata and T. Kamata, Phys. Chem. Chem. Phys., 2000, 2, 3565–3569 RSC.
- J. Ostrauskaite and P. Strohriegl, Macromol. Chem. Phys., 2003, 204, 1713–1718 CrossRef CAS.
- K. Balakrishnan, A. Datar, W. Zhang, X. Yang, T. Naddo, J. Huang, J. Zuo, M. Yen, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2006, 128, 6576–6577 CrossRef CAS PubMed.
- B. Schmaltz, A. Rouhanipour, H. J. Räder, W. Pisula and K. Müllen, Angew. Chem., Int. Ed., 2009, 48, 720–724 CrossRef CAS PubMed.
- A. D. Finke, D. E. Gross, A. Han and J. S. Moore, J. Am. Chem. Soc., 2011, 133, 14063–14070 CrossRef CAS PubMed.
- C. Maeda, T. Yoneda, N. Aratani, M.-C. Yoon, J. M. Lim, D. Kim, N. Yoshioka and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 5691–5694 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- L. S. Coumont and J. G. C. Veinot, Tetrahedron Lett., 2015, 56, 5595–5598 CrossRef CAS.
- K. Ogawa, A. Ohashi, Y. Kobuke, K. Kamada and K. Ohta, J. Am. Chem. Soc., 2003, 125, 13356–13357 CrossRef CAS PubMed.
- M. Drobizhev, Y. Stepanenko, Y. Dzenis, A. Karotki, A. Rebane, P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 2004, 126, 15352–15353 CrossRef CAS.
- J. E. Raymond, A. Bhaskar, T. Goodson, N. Makiuchi, K. Ogawa and Y. Kobuke, J. Am. Chem. Soc., 2008, 130, 17212–17213 CrossRef CAS PubMed.
- O. Varnavski, J. E. Raymond, Z. S. Yoon, T. Yotsutuji, K. Ogawa, Y. Kobuke and T. Goodson, J. Phys. Chem. C, 2014, 118, 28474–28481 CrossRef CAS.
- J. Kromer, I. I. Rios-Carreras, G. Fuhrmann, C. Musch, M. Wunderlin, T. Debaerdemaeker, E. Mena-Osteritz and P. Bauerle, Angew. Chem., Int. Ed. Engl., 2000, 39, 3481–3486 CrossRef CAS PubMed.
- M. Mayor and C. Didschies, Angew. Chem., Int. Ed. Engl., 2003, 42, 3176–3179 CrossRef CAS PubMed.
- B. M. Wong, J. Phys. Chem. C, 2009, 113, 21921–21927 CrossRef CAS PubMed.
- F. Zhang, G. Gotz, H. D. Winkler, C. A. Schalley and P. Bauerle, Angew. Chem., Int. Ed. Engl., 2009, 48, 6632–6635 CrossRef CAS PubMed.
- S. Yamago, Y. Watanabe and T. Iwamoto, Angew. Chem., Int. Ed., 2010, 49, 757–759 CrossRef CAS PubMed.
- J. E. Donehue, O. P. Varnavski, R. Cemborski, M. Iyoda and T. Goodson, J. Am. Chem. Soc., 2011, 133, 4819–4828 CrossRef CAS PubMed.
- T. Iwamoto, Y. Watanabe, Y. Sakamoto, T. Suzuki and S. Yamago, J. Am. Chem. Soc., 2011, 133, 8354–8361 CrossRef CAS PubMed.
- T. J. Sisto, M. R. Golder, E. S. Hirst and R. Jasti, J. Am. Chem. Soc., 2011, 133, 15800–15802 CrossRef CAS PubMed.
- T. Nishihara, Y. Segawa, K. Itami and Y. Kanemitsu, J. Phys. Chem. Lett., 2012, 3, 3125–3128 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, J. Phys. Chem. B, 2014, 118, 8352–8363 CrossRef CAS PubMed.
- P. Kim, K. H. Park, W. Kim, T. Tamachi, M. Iyoda and D. Kim, J. Phys. Chem. Lett., 2015, 6, 451–456 CrossRef CAS PubMed.
- C.-K. Yong, P. Parkinson, D. V. Kondratuk, W.-H. Chen, A. Stannard, A. Summerfield, J. K. Sprafke, M. C. O'Sullivan, P. H. Beton, H. L. Anderson and L. M. Herz, Chem. Sci., 2015, 6, 181–189 RSC.
- F. Chen, Y. S. Hong, S. Shimizu, D. Kim, T. Tanaka and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 10639–10642 CrossRef CAS PubMed.
- Y. Nagata, S. Kato, Y. Miyake and H. Shinokubo, Org. Lett., 2017, 19, 2718–2721 CrossRef CAS PubMed.
- F. Chen, T. Tanaka, T. Mori and A. Osuka, Chem.–Eur. J., 2018, 24, 7489–7497 CrossRef CAS PubMed.
- Y. Matsuo, F. Chen, K. Kise, T. Tanaka and A. Osuka, Chem. Sci., 2019, 10, 11006–11012 RSC.
- R. W. Wagner, T. E. Johnson, F. Li and J. S. Lindsey, J. Org. Chem., 1995, 60, 5266–5273 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data for GPC, NMR, mass, and quantum chemical calculations. See DOI: 10.1039/d0ra00830c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Shohei Tanaka and
Kyosuke Shimura
*,
Shohei Tanaka and
Kyosuke Shimura