
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of novel N-(6-methanesulfonyl-benzothiazol-2-yl)-3-(4-substituted-piperazin-1-yl)-propionamides with cholinesterase inhibition, anti-β-amyloid aggregation, neuroprotection and cognition enhancing properties for the therapy of Alzheimer's disease†
Chandra Bhushan Mishra‡
a,
Shruti Shalini‡a,
Siddharth Gusaina,
Amresh Prakash b,
Jyoti Kumaria,
Shikha Kumaria,
Anita Kumari Yadava,
Andrew M. Lynnc and
Manisha Tiwari*a
b,
Jyoti Kumaria,
Shikha Kumaria,
Anita Kumari Yadava,
Andrew M. Lynnc and
Manisha Tiwari*a
aDr. B. R. Ambedkar Centre for Biomedical Research, University of Delhi, New Delhi 110007, India. E-mail: mtiwari07@gmail.com
bAmity Institute of Integrative Sciences and Health (AIISH), Amity University Haryana, Amity Education Valley, Gurgaon-122413, India
cSchool of Computational & Integrative Sciences, Jawaharlal Nehru University, New Delhi 110067, India
First published on 5th May 2020
Abstract
A novel series of benzothiazole–piperazine hybrids were rationally designed, synthesized, and evaluated as multifunctional ligands against Alzheimer's disease (AD). The synthesized hybrid molecules illustrated modest to strong inhibition of acetylcholinesterase (AChE) and Aβ1-42 aggregation. Compound 12 emerged as the most potent hybrid molecule exhibiting balanced functions with effective, uncompetitive and selective inhibition against AChE (IC50 = 2.31 μM), good copper chelation, Aβ1-42 aggregation inhibition (53.30%) and disaggregation activities. Confocal laser scanning microscopy and TEM analysis also validate the Aβ fibril inhibition ability of this compound. Furthermore, this compound has also shown low toxicity and is capable of impeding loss of cell viability elicited by H2O2 neurotoxicity in SHSY-5Y cells. Notably, compound 12 significantly improved cognition and spatial memory against scopolamine-induced memory deficit in a mouse model. Hence, our results corroborate the multifunctional nature of novel hybrid molecule 12 against AD and it may be a suitable lead for further development as an effective therapeutic agent for therapy in the future.
1. Introduction
Alzheimer's disease (AD) is a predominant source of irreversible dementia, resulting in more than 75% of the dementia cases worldwide. It is known to be a multifactorial neurodegenerative disorder designated by progressive loss of memory and other cognitive functions. The main risk factor of AD is aging; however the mechanism underlying the foundation of AD due to aging is yet to be firmly elucidated. It is clinically marked by the progression from episodic memory problems to a slow global decline of cognitive function. Patients in end-stage AD become bedridden and are highly dependent on custodial care, with an average life span of 9 years after diagnosis.1 AD leaves an enormous emotional and financial burden on patients, their families and society.There are approximately 44 million people affected by Alzheimer's disease, and it is expected to increase three times by 2050; these perturbing numbers show that Alzheimer's disease (AD) remains a serious socio-economical problem.2 AD is estimated to have cost the world $604 billion in 2010 alone.3 These costs are staggering, particularly in light of worldwide increase in the number of AD cases. AD remains the most prevalent unmet medical need because of its chronicity, cost, severity, and lack of mechanism based treatment.4
Extensive research from several years has still not been able to establish the exact molecular-mechanistic aspects of AD. Clinically, AD is indicated by widespread neuronal cell death in the brain, which corresponds to deposition of abundant fibrillar plaques, primarily comprising the beta-amyloid (Aβ) peptide.5,6 The identified pathological hallmarks of AD are senile plaques of amyloid beta protein, intracellular neurofibrillary tangles (NFTs), and neuronal degeneration. Accumulation of Aβ peptides encourages conformational changes that lead to further non-covalent polymerization into a heterogeneous range of oligomeric class that finally forms amyloid fibrils.7 Vulnerability of brain to oxidative stress is due to its high demand for oxygen, abundant lipid content and relative scarcity of antioxidant enzymes.8–10 Consequences of neuronal loss because of oxidation include upregulation of proinflammatory cytokines and irreparable DNA damage.11,12 Metal dyshomeostasis in the body, especially in the brain, can be a critical mechanism for neurodegenerative processes, including oxidative stress generation or amyloid plaques aggregation.13
Despite the fact that Alzheimer's disease (AD) was first described over 100 years ago, disease-modifying treatment has not yet been developed for this disease. There is no complete cure for AD, but some medicines can ameliorate its clinical symptoms by having an impact on cognitive functions, delaying functional decline and improving behavioural changes.14,15 Still, their therapeutic interest is limited owing to their side effects and to the fact that they only treat symptoms. There are only four drugs in the market for the treatment of AD, three cholinesterase inhibitors (Donepezil, Rivastigmine, Galantamine) and an N-methyl-D-aspartate (NMDA) receptor AD antagonist (Memantine) thereby limiting its treatment possibilities. No new drugs have been approved for the treatment of AD since 2003.16,17 Decades of research in AD pathophysiology has led to target identification for new drug development. Nowadays, multi targeted directed ligands (MTDL) approach of AD therapy is widely used, where ChE inhibition is joined with additional biological properties.18 Drugs with neuroprotective and disease-modifying properties are of greater research interest. Multi-target-directed ligands (MTDL) strategy has been used to develop a variety of hybrid compounds having capability to act simultaneously in diverse biological targets. The complex pathophysiology of AD may necessitate combination or multi drug targeted ligand therapy as disease modifying strategy.19 MTDLs approach involves molecular hybridization technique in which two or more pharmacophore of bioactive molecule are incorporated into single drug molecule. This approach reduces the side effects and lead to a better pharmacokinetic profile of the MTDL compared to two or more separate individual drug representing respective single pharmacophore.20 This approach of discovery of drug candidate which is capable of targeting multiple factors involved in AD complex pathogenesis and having multiple properties like antioxidant, neuroprotective, metal chelation, anti-inflammatory, anti-Aβ aggregation and cholinesterase and secretase inhibitory activities, would greatly facilitate in improving therapeutic strategies.21
In this context, we have previously developed various multifunctional synthetic molecules against AD such as donepezil derivatives, indandione derivatives and diallyl disulphide derivatives.22–24 In the present work, we have rationally designed novel N-(6-methanesulfonyl-benzothiazol-2-yl)-3-(4-substituted-piperazin-1-yl)-propionamides as potential multifunctional agents for AD, which consists of benzothiazole moiety as well as piperazine moiety.
Benzothiazole are compounds with fused benzene and thiazole moiety containing nitrogen and sulphur, which has fascinated interest in central nervous system (CNS)-related drug discovery. Various studies have reported that benzothiazole derivatives possess a broad spectrum of biological activities including AChE inhibitory action, Aβ-binding ability and neuroprotective property.25,26 For example, benzothiazole containing Aβ binding agents thioflavin-T and PIB are widely used in detecting amyloid fibrils in vivo as well in vitro.27 Additionally, a well-known neuroprotective drug riluzole containing benzothiazole core has shown very potent neuroprotective effect in various in vitro as well as in vivo models and is widely used clinically for therapy of amyotrophic lateral sclerosis.28 Moreover, benzothiazole derivative sabeluzole (NMDA antagonist), has also been reported to impede the clinical progression of AD.29 Conversely, piperazine is a flexible ring system consisting of a six-member ring with two nitrogen atoms at 1, 4 positions. Piperazines are reported for various therapeutic uses and specially, this ring system has been widely used for CNS drug discovery.30–32 Studies have supported that piperazine derivatives show a very high binding affinity to amyloid, capability to cross blood brain barrier and in addition they also provide rapid elimination of non-specific signals from the brain.33 Moreover, piperazines containing neuroprotective agents VK-28 as well as SP-04 are well studied as effective neuroprotective agents in various neurodegenerative models34.
A lot of efforts have been made by several research groups to modify donepezil pharmacophore (standard AChE inhibitor) by various approaches such as bioisosteric replacement of pharmacophoric elements, hybridization of two pharmacophore, and linker optimization. Indeed, some of the modified leads have shown potential multifunctional activities for the treatment of AD.35 It is well studied that basic piperidine moiety of standard AChE inhibitor donepezil actively participates in interaction with the AChE active site and this basic moiety is considered as an essential pharmacophoric element to produce inhibitory action.36
Therefore, by considering these evidences benzothiazole–piperazine hybrid molecules have been designed to develop effective multifunctional agents against AD. In due course, benzothiazole and piperazine pharmacophore have been hybridized by using a flexible propanamide linker. We have made bioisosteric replacement of the piperidine ring of donepezil with piperazine ring and it was thought that this bioisosteric replacement may provide better pharmacological action against AD. Additionally, it is also assumed that the flexible propanamide linker will provide enough flexibility to hybrid pharmacophore for adequate adjustment at CAS as well as the PAS binding site of AChE active site. Thus, this drug designing strategy may confer a series of benzothiazole–piperazine hybrid molecules as potential multifunctional agents that might display polypharmacological action all in concert, such as AChE inhibition, Aβ disaggregation, and neuroprotection (Fig. 1). Synthesized compounds have been evaluated for their acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibitory action, Aβ disaggregation activity, metal chelation potency and neuroprotective capability. Additionally, molecular docking and molecular dynamic (MD) simulation have also been carried out to explore molecular level interaction of these molecules with AChE and Aβ peptide. Moreover, in vivo efficacy of the most active compound was illustrated in the scopolamine induced mouse model of dementia.
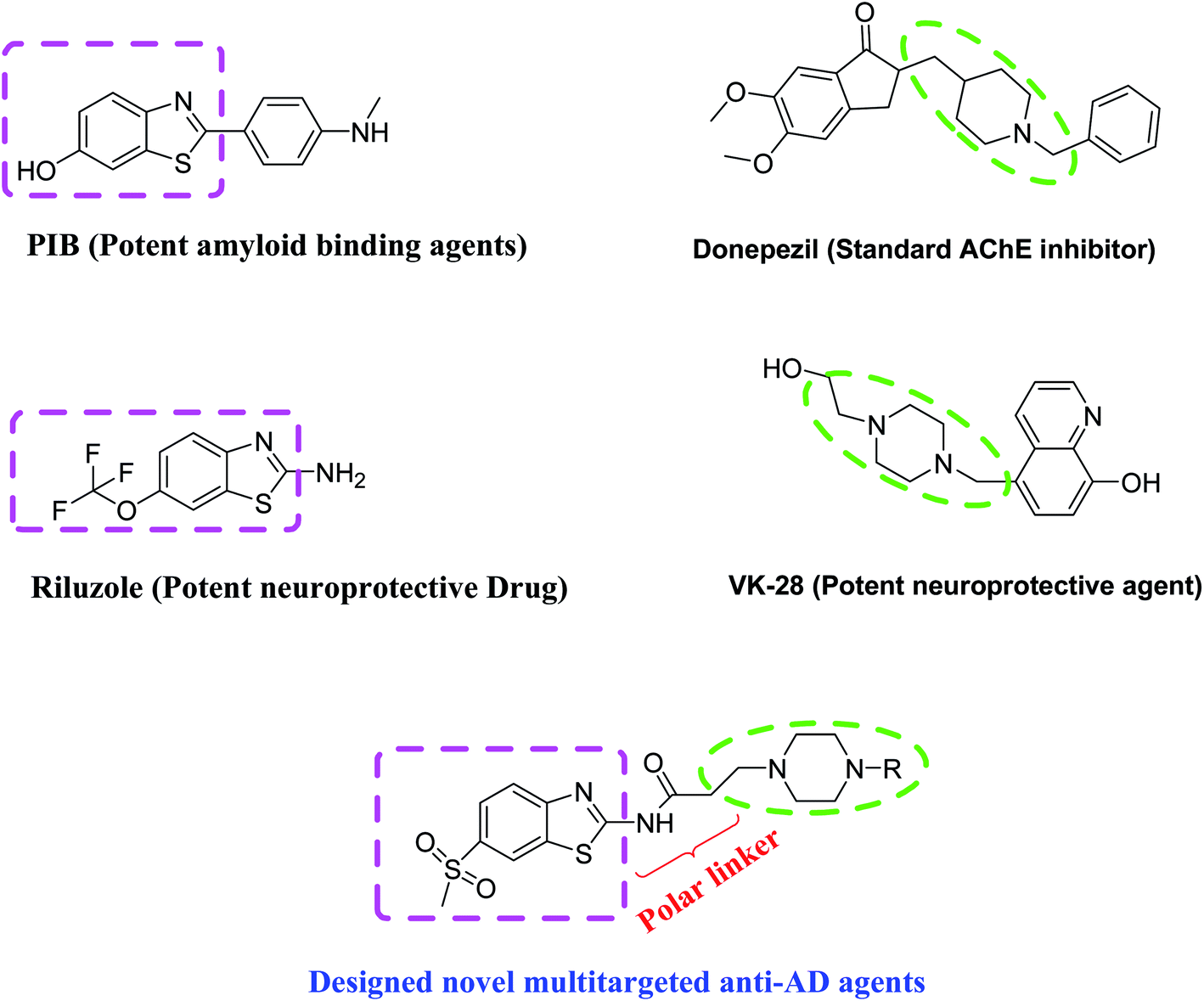 | ||
| Fig. 1 Designing strategy of multifunctional benzothiazole–piperazine hybrids. | ||
2. Results and discussion
2.1. Synthesis
| Compound number | R-group | Compound number | R-group |
|---|---|---|---|
| 4 (85%) | 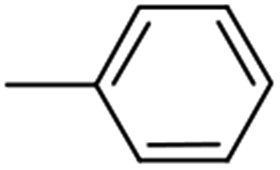 |
11 (88%) |  |
| 5 (88%) | 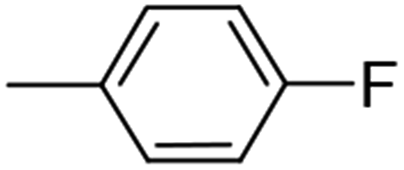 |
12 (74%) | 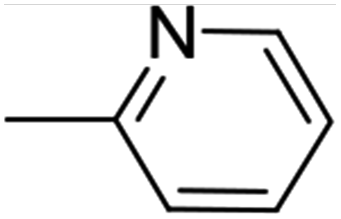 |
| 6 (75%) | 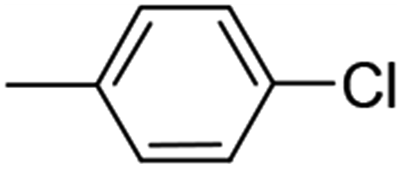 |
13 (81%) | 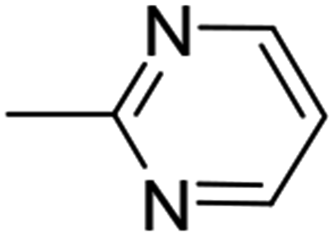 |
| 7 (82%) | 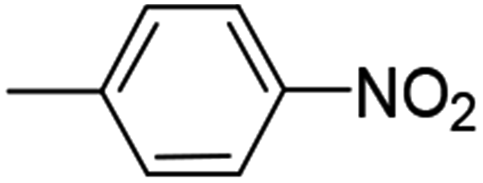 |
14 (69%) | 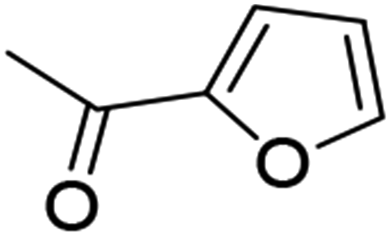 |
| 8 (84%) | 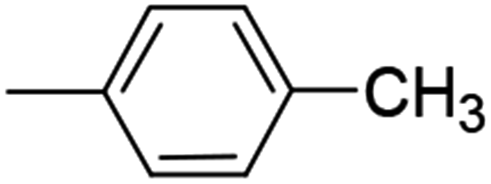 |
15 (86%) | 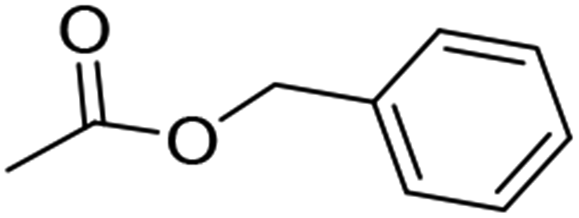 |
| 9 (79%) | 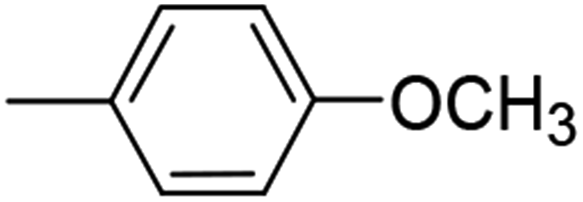 |
16 (75%) |  |
| 10 (76%) | 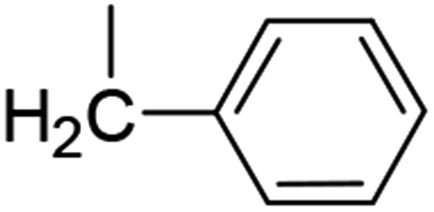 |
17 (71%) |  |
 | ||
| Scheme 1 Reagents and conditions: (A) KSCN, Br2, glacial acetic acid, 0–5 °C, 6 h; (B) 3-Chloropropionyl chloride, TEA, 1,4-dioxane, reflux, 8 h; (C) Substituted piperazines, NaHCO3, NaI, dried acetonitrile, reflux, 8–12 h. | ||
2.2. In vitro studies
| Compounds | IC50 ± SD (μM) AChEa | IC50 ± SD (μM) BuChEb | Self Aβ1-42 aggregation inhibitionc (%) |
|---|---|---|---|
| a Half maximal inhibitory concentration of AChE from electric eel (means ± SD of three experiments).b Half maximal inhibitory concentration of BuChE from equine serum (means ± SD of three experiments).c Self-induced Aβ1-42 aggregation inhibition (50 μM) by tested inhibitors at 50 μM by thioflavin-T based fluorescence method (means ± SD of three experiments).d na-no activity. | |||
| 4 | 24.14 ± 0.721 | >100 | 54.29502 ± 0.265 |
| 5 | 18.31 ± 0.777 | >100 | 55.53029 ± 0.134 |
| 6 | 13.05 ± 0.813 | >100 | 51.74112 ± 0.289 |
| 7 | 11.25 ± 1.803 | >100 | 54.34428 ± 0.427 |
| 8 | 20.18 ± 1.435 | >100 | 54.43522 ± 0.159 |
| 9 | 14.45 ± 1.032 | >100 | 52.31708 ± 0.114 |
| 10 | 7.834 ± 0.810 | >100 | 53.40836 ± 0.322 |
| 11 | 4.548 ± 0.984 | >100 | 52.22614 ± 0.513 |
| 12 | 2.319 ± 0.410 | >100 | 53.30605 ± 0.541 |
| 13 | 10.87 ± 0.657 | >100 | 53.6736 ± 0.228 |
| 14 | 14.59 ± 0.438 | >100 | 54.48827 ± 0.885 |
| 15 | 19.48 ± 0.452 | >100 | 52.44591 ± 1.203 |
| 16 | 5.244 ± 0.690 | >100 | 45.3109 ± 0.241 |
| 17 | 26.43 ± 0.551 | >100 | 53.40836 ± 0.805 |
| Rivastigmine | 4.45 ± 0.37 | 1.32 ± 0.09 | n.ad |
| Galantamine | 3.12 ± 0.44 | 3.12 ± 0.44 | n.ad |
| Tacrine | 0.13 ± 0.21 | 0.054 ± 0.85 | n.ad |
| Donepezil | 0.049 ± 0.05 | 8.71 ± 1.36 | n.ad |
| Curcumin | n.ad | n.ad | 50.23 ± 1.201 |
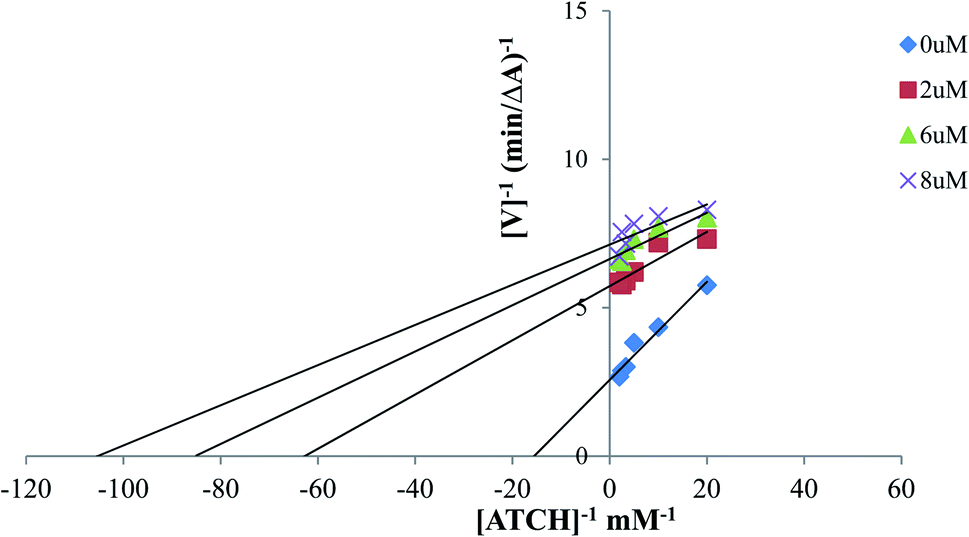 | ||
| Fig. 2 Lineweaver–Burk reciprocal plot of AChE initial velocity with increasing substrate concentrations showing kinetic study of AChE inhibition mechanism with compound 12. | ||
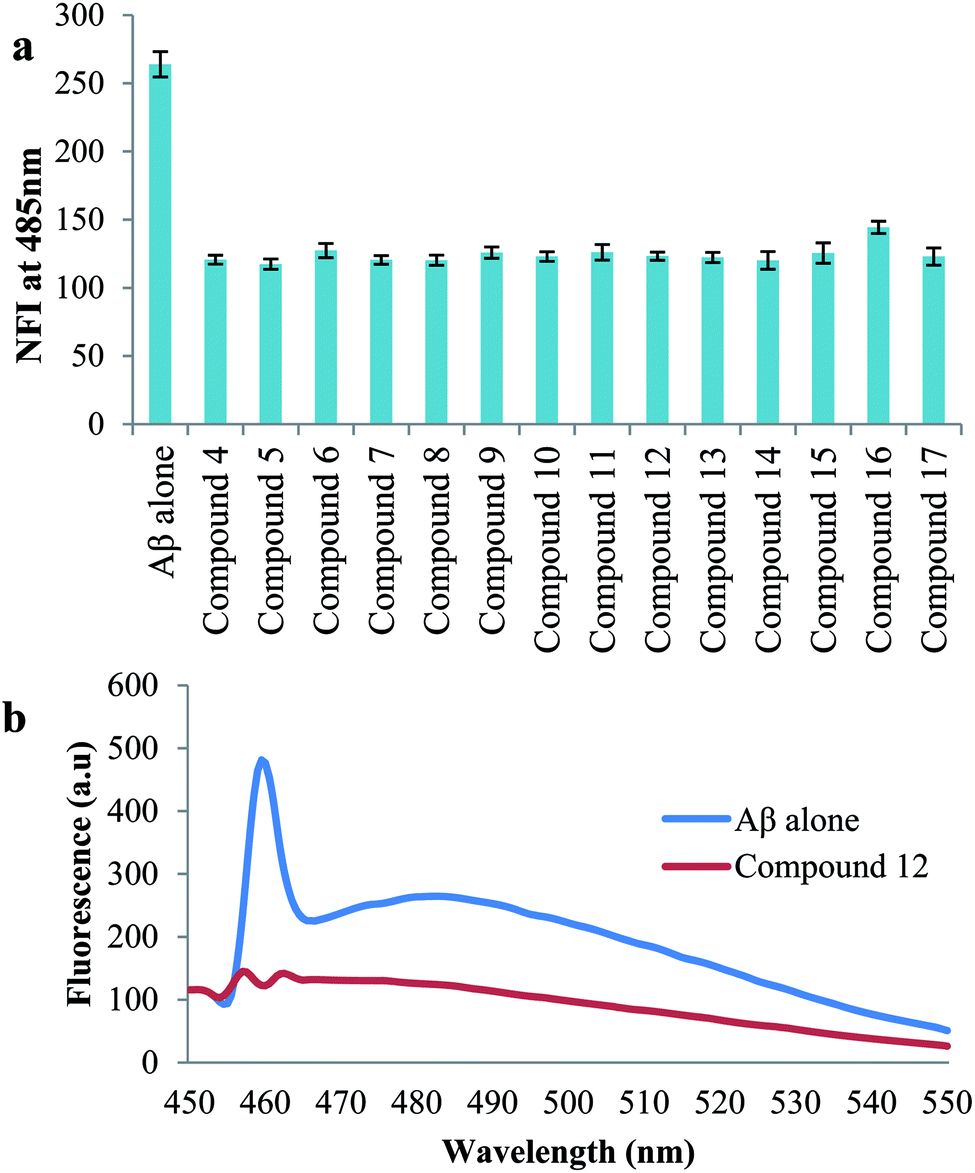 | ||
| Fig. 3 (a) Inhibition of Aβ42 aggregates studied by thioflavin (ThT) assay. Aβ42 (50 μM) was incubated alone and independently with compounds and their influence on Aβ aggregation was quantified by measuring ThT fluorescence intensity, represented as normalized fluorescence intensity (NFI) at 485 nm for a given time point (48 h); (b) ThT emission fluorescence spectra (range 450–550 nm) of Aβ1-42 alone in blue and in the presence of compound 12 (50 μM) in red. | ||
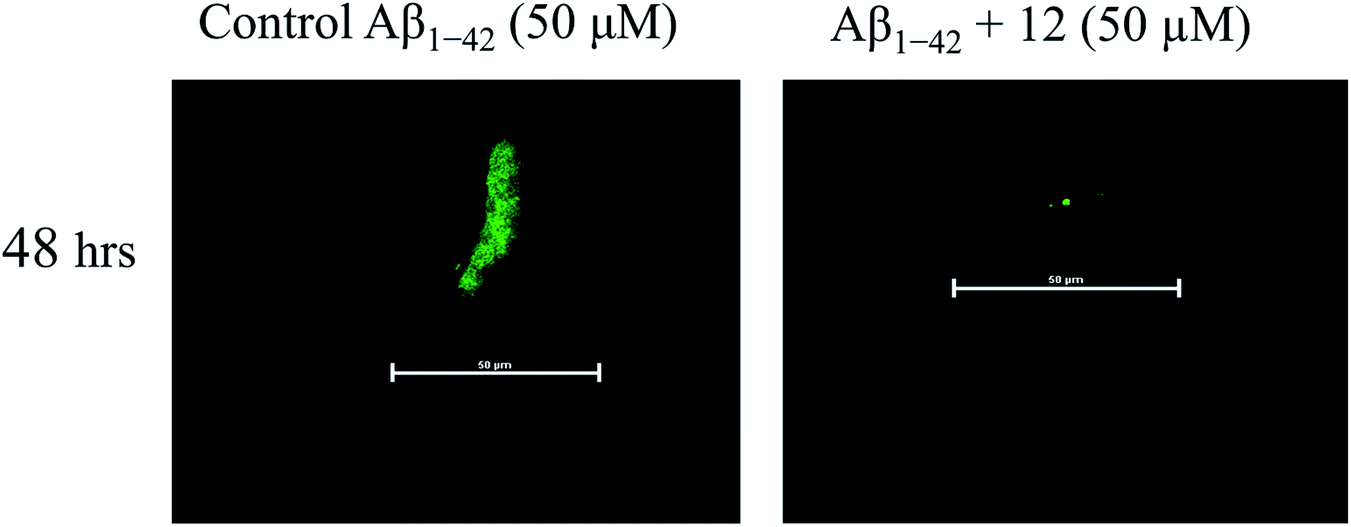 | ||
| Fig. 4 Assessment of the effect of compound 12 on anti-aggregation property of amyloid beta peptide (Aβ1-42). Confocal laser scanning microscopic analysis of the deposited amyloid beta aggregates as assessed in 48 h. The fluorescence intensity of confocal microscopic image represents a vision of Aβ1-42 amyloid aggregates, with representative Aβ1-42 peptide (control) co-incubated with the presence of compound 12. | ||
 | ||
| Fig. 5 TEM analysis of Aβ1-42 aggregation in the presence of compound 12: (a) Aβ1-42 alone (50 μM), incubated at 37 °C for 48 h. (b) Aβ1-42 (50 μM) and curcumin (50 μM) were incubated at 37 °C for 48 h. (c) Aβ1-42 (50 μM) and 12 (50 μM) were incubated at 37 °C for 48 h. | ||
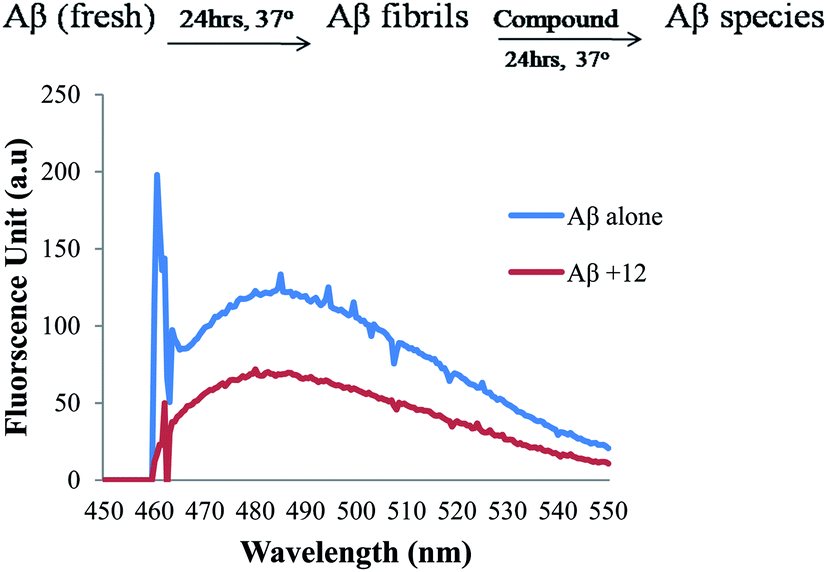 | ||
| Fig. 6 ThT emission fluorescence spectra (range 450–550 nm) of Aβ1-42 alone in blue and in the presence of compound 12 (50 μM) in red for disaggregation studies. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 12–Cu(II) complex.
:1, 12–Cu(II) complex.
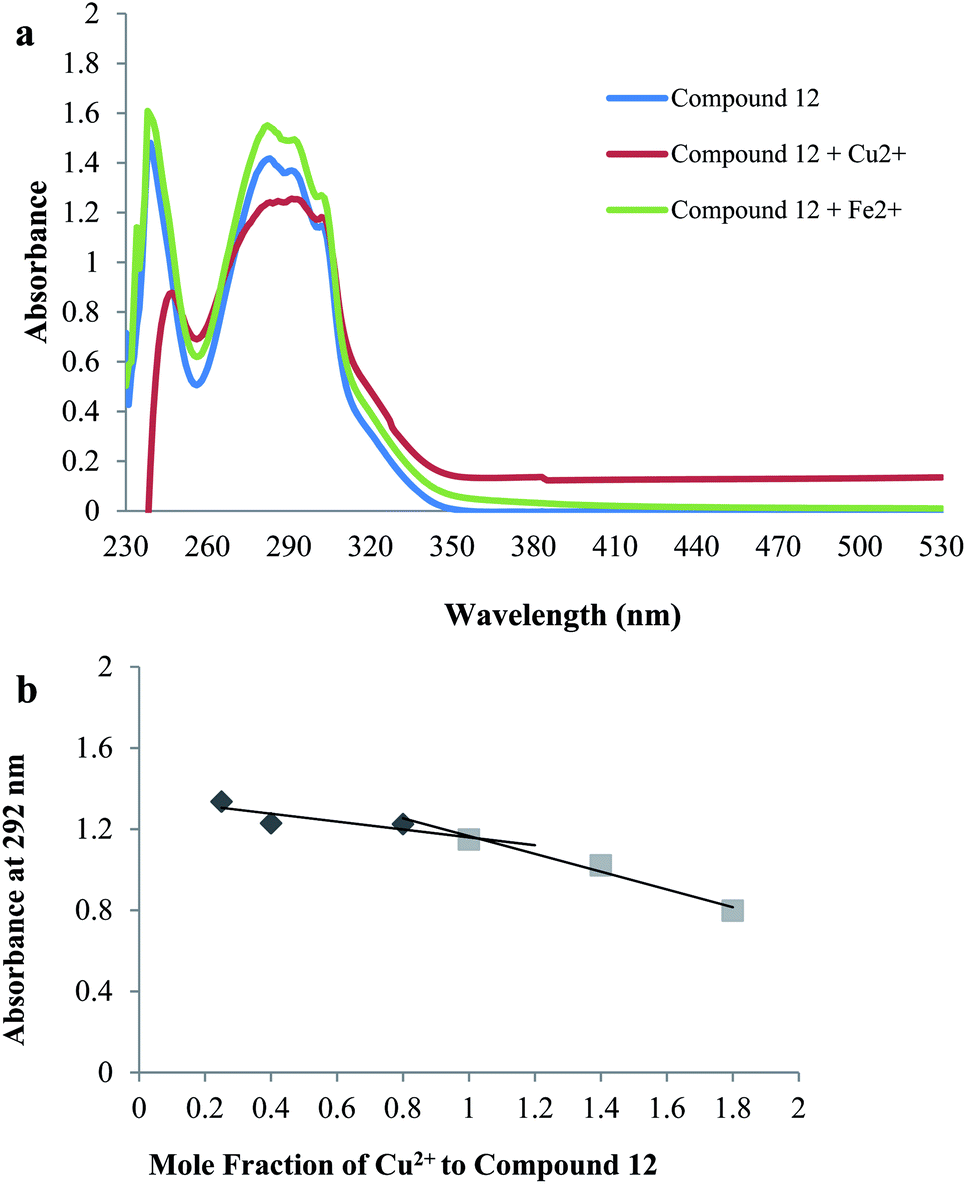 | ||
| Fig. 7 Metal chelation study: (a) UV absorbance spectrum of compound 12 (60 μM) alone in blue or in the presence of CuSO4 (60 μM) in red, FeSO4 (60 μM) in green in buffer (20 mM HEPES, 150 mM NaCl, pH 7.4); (b) determination of the stoichiometry of complex Cu2+–12 by Job's method. | ||
 | ||
| Fig. 8 Proposed binding mode of compound 12 in the active site of AChE (PDB: 1EVE). The black dashed lines represent hydrogen bonds. | ||
Assessment of Fig. 9 to understand the binding mode of compound 12 with Aβ1-42 shows that compound 12 interacts well with Aβ1-42. Compound 12 is transversely fitted along the major helix, and Lys28 is involved in H-bond interaction with a methylsulfonyl group of compound 12. In addition, propanamide and pyridine piperazine fragments display hydrophobic interactions with residues Lys16, Leu17, Val18, Phe20, Ala21, Asp23, Val24, ser26, Ile31, Leu34 and Met35 resulting in stabilized compound 12–Aβ1-42 complex very nicely. These results indicate that the hydrophobic interactions and the hydrogen bonding interaction play crucial roles in the effective inhibition of Aβ1-42 aggregation.
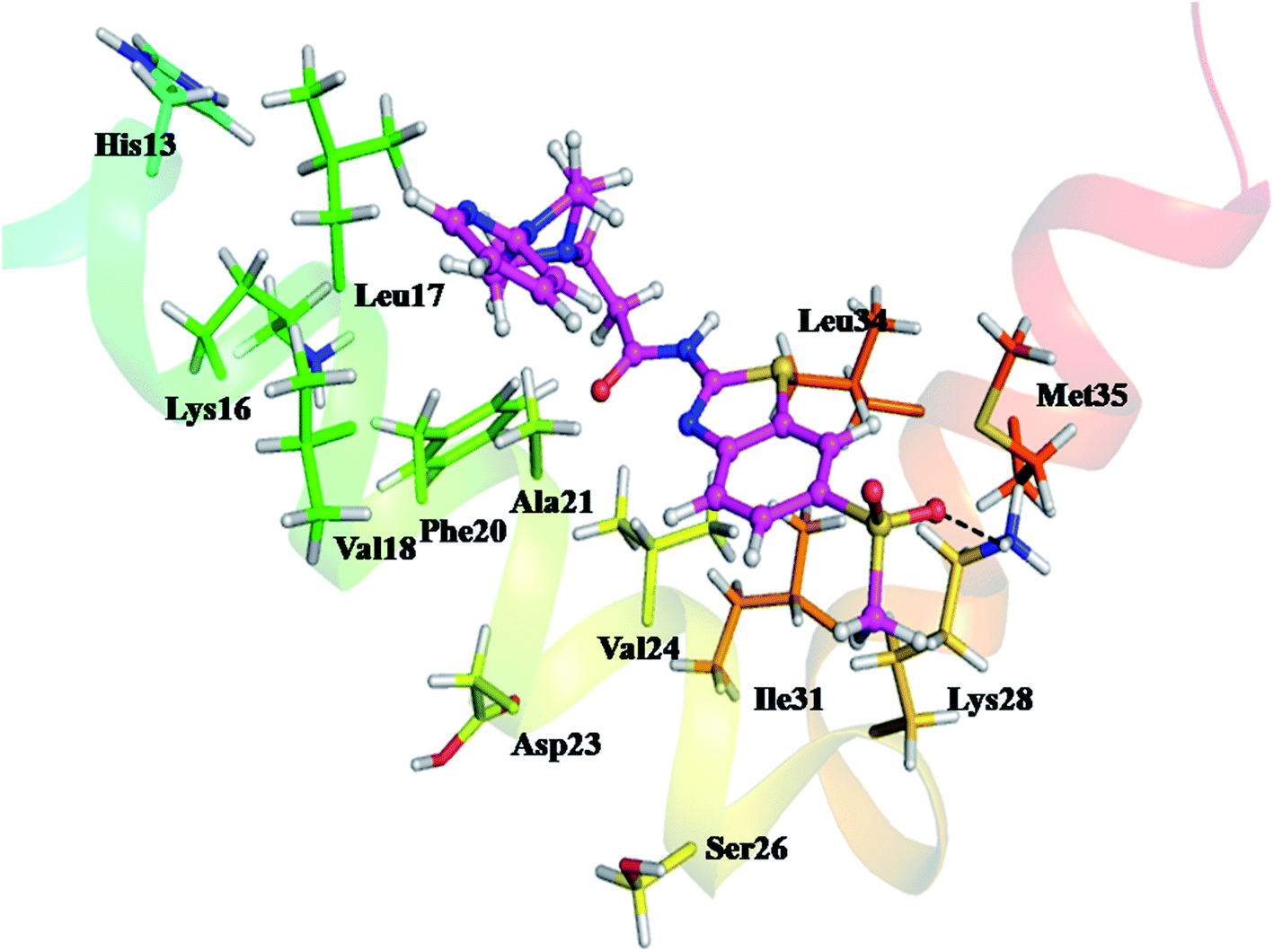 | ||
| Fig. 9 Proposed binding mode of compound 12 in the active site of Aβ1-42 (PDB: 1IYT). The blue dashed lines represent hydrogen bonds. | ||
 | ||
| Fig. 10 Molecular dynamic simulation studies with AChE (a) The equilibrium average of Cα RMSD of AChE (black), AChE–12 docked complex (red) in water at 300 K; (b) time evolution plot of radius of gyration (Rg) plot of AChE (black), AChE–12 complex (red) in water at 300 K; (c) time evolution plot of solvent accessible surface area (SASA) plot of AChE (black), AChE–12 complex (red) in water at 300 K; (d) root mean square fluctuation (RMSF) plot of AChE (black), AChE–12 complex (red) in water at 300 K; (e) structural snapshot of AChE–12 complex at the time interval of 10 ns, during the simulation in water at 300 K. | ||
Another effective parameter, radius of gyration (Rg) is applied to investigate the conformational stability and integrity of protein and protein–ligand complex46 (Fig. 10b). The Rg plot of all Cα-atoms shows that the structural composition of AChE is retained in water, having Rg value 2.30 ± 0.02 nm. However, the structural integrity of AChE–12 complex remains stable around Rg value 2.27 ± 0.02 nm. This slight decrease in Rg value indicates the stable molecular interaction of protein–ligand complex.
To determine the structural stability of AChE and AChE–12 complex we also monitored the solvent accessible surface area (SASA) as shown in Fig. 10c. We observed that the structure of both, AChE and AChE–12 complex having almost similar average SASA value ∼310 nm2. This result suggested that the ligand is nicely fitted at the active site of AChE, thus, the conformational dynamics analyses RMSD, Rg and SASA results were consistent, indicating the stable interaction of AChE–12 complex.
To investigate conformational fluctuations, and the effect of local dynamics on the structural complex of AChE–12, and residues involved in molecular interaction, we calculated the average fluctuation of each amino acids of AChE. The RMSF plot of all Cα-atoms is shown in Fig. 10d. The RMSF results clearly indicate that the active site residues which were involved in interaction with compound 12 having lower average fluctuation. Furthermore, the almost complete overlapping RMSF plots of AChE and AChE–12 complex signifies no conformational changes in native structure of protein upon the binding of ligand. Thus, to confirm the results we also measured the evolution plot of secondary structure (ESI Fig. S1 and S2†) and the structural snapshots of AChE–12 (Fig. 10e) at time interval of 10 ns. Both analyses show consistent results with RMSF which again confirm the favourable interaction of 12 at active site of AChE.
The conformational stability of Aβ1-42–12 complex was measured using structural order parameters, RMSD, Rg and SASA. Fig. 11a shows that RMSD plot of Aβ1-42 remains stable around 0.5 nm in water at 300 K. The consecutive drifts in trajectory, during the initial 0–20 ns represents the flexible nature of Aβ1-42, however, it attains equilibrium at ∼20 ns and the stable structure can be seen for 20–100 ns of simulation. However, the plot shows lower RMSD value upon binding with 12, during initial 0–25 ns of simulation. The trajectory of Aβ1-42–12 complex finds stable at 25–40 ns, followed by continuous increase in RMSD ∼40–55 ns. With the drift of ∼0.5 nm which suggested the conformational flipping, it attains stable equilibrium at ∼55–95 ns and during the last 5 ns, it dropped down to the conformational state observed for ligand unbound Aβ1-42.
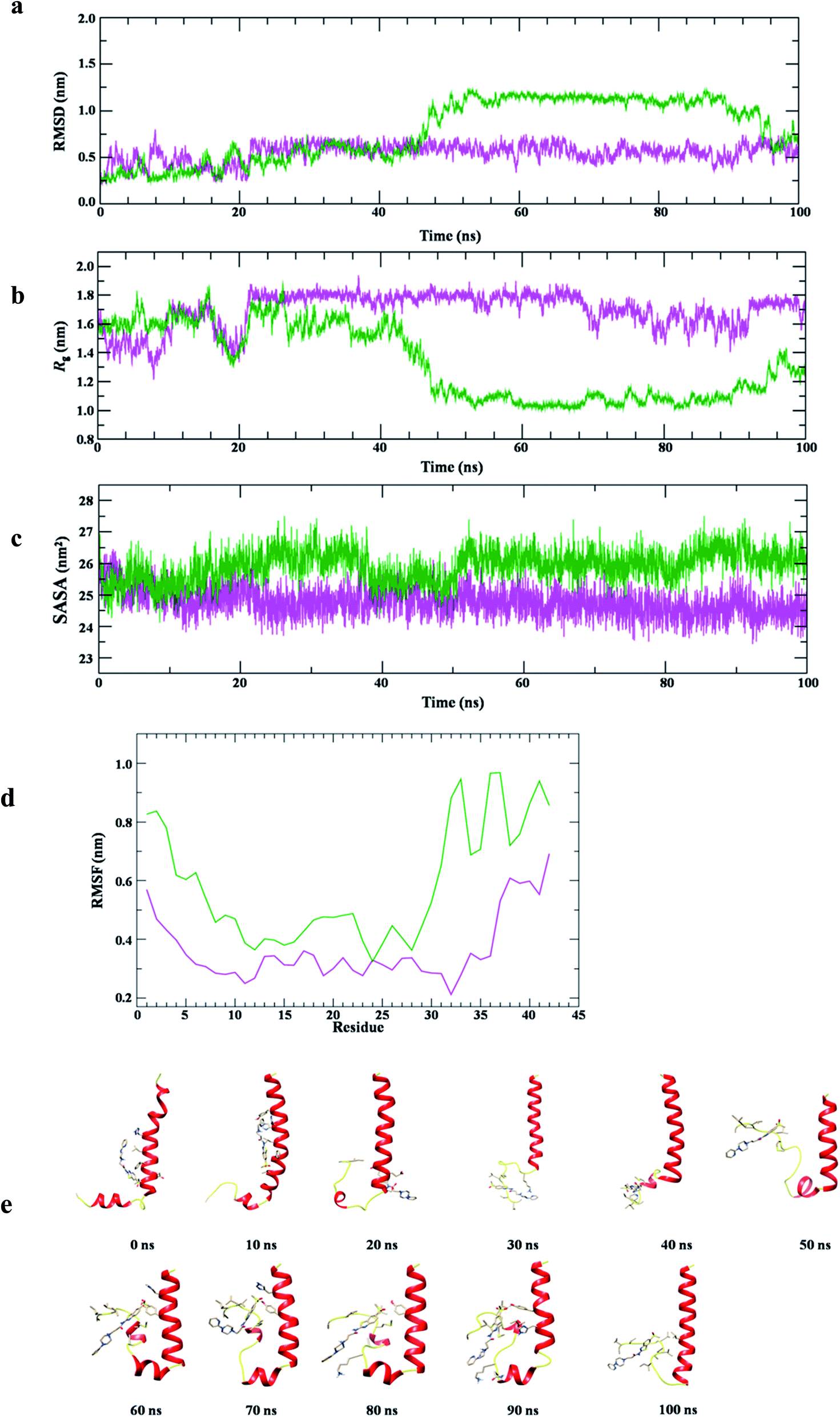 | ||
| Fig. 11 Molecular dynamic simulation with Aβ1-42; (a) the equilibrium average of Cα RMSD of Aβ1-42 (magenta), Aβ1-42–12 docked complex (green) in water at 300 K; (b) time evolution plot of radius of gyration (Rg) plot of Aβ1-42 (magenta), Aβ1-42–12 docked complex (green) in water at 300 K; (c) time evolution plot of solvent accessible surface area (SASA) plot of Aβ1-42 (magenta), Aβ1-42–12 docked complex (green) in water at 300 K; (d) root mean square fluctuation (RMSF) plot of Aβ1-42 (magenta), Aβ1-42–12 docked complex (green) in water at 300 K; (e) structural snapshot of Aβ1-42–12 complex at the time interval of 10 ns, during the simulation in water at 300 K. | ||
The comparative structural stability and integrity of Aβ1-42 and Aβ1-42–12 complex examined by Rg plot (Fig. 11b) shows initially flexible Rg trajectory for Aβ1-42 which attains equilibrium ∼20 ns and structural dynamics remains stable with average Rg value 1.8 ± 0.02 nm for the period of 20–100 ns. Differently, the Rg plot of Aβ1-42–12 complex shows initial fluctuation around Rg value 1.8 nm which is dropped down to 1.3 nm at ∼20 ns, thereafter, with the drift of 0.3 the trajectory remains fluctuating around Rg value 1.6 which can be observed during 24–40 ns. We find subsequent decrease in Rg value ∼48 ns and the stable conformation of protein–ligand complex with Rg value 1.1 nm is observed during the period of 50–100 ns of simulation. Thus, the Rg analysis also anticipated the two major conformational states of Aβ1-42–12 complex. However, the SASA plot (Fig. 11c) of Aβ1-42 remains stable ∼26 nm2, whereas, we find slightly lower SASA value (∼25 nm2) as compared protein alone in water. This observation suggested the stable association of 12 with Aβ1-42. We also examine RMSF plot (Fig. 11d) to understand the contribution of active site residues on conformational stability of Aβ1-42–12 complex. RMSF plot shows that the residues belonging to Aβ1-42 having average fluctuation ≤0.4 nm, whereas, the average fluctuation of Aβ1-42 is slightly increases upon binding with 12. The secondary structure analysis shows that the helical structure of Aβ1-42 remains consistent with crystal structure, however, we observed the propensity to form turn like structure for the residues belonging to N-and C-terminal of Aβ (ESI Fig. S3 and S4†). The binding of 12 disrupted the salt bridge between the Asp23–Lys28 which prevent the self-aggregation of Aβ1-42, thus, we observed turn like structure at C-terminal of Aβ1-42–12 complex, however, the helical structure of Aβ at N-terminal remains consistent with crystal structure. The structural snapshot of Aβ1-42–12 complex at the time interval of 10 ns (Fig. 11e) provide the clear evidence that binding interaction of 12 is largely localized at the C-terminal and the bulky side chain of 12 create structural hindrance for salt bridge associated, thus prevent the protein aggregation the pathological state of Alzheimer disease.47
To further explore the dynamic properties of the investigated structures in our simulations, the essential dynamics (ED) analysis on the Cα atoms was performed.48 ED actually reflects the overall conformational space of protein and protein–12 complex during simulations (Fig. 12A).The projection of trajectories onto the first two principal components (PC1, PC2) shows the motion of investigated system: AChE and AChE–12 complex in phase space and is illustrated in Fig. 12A.
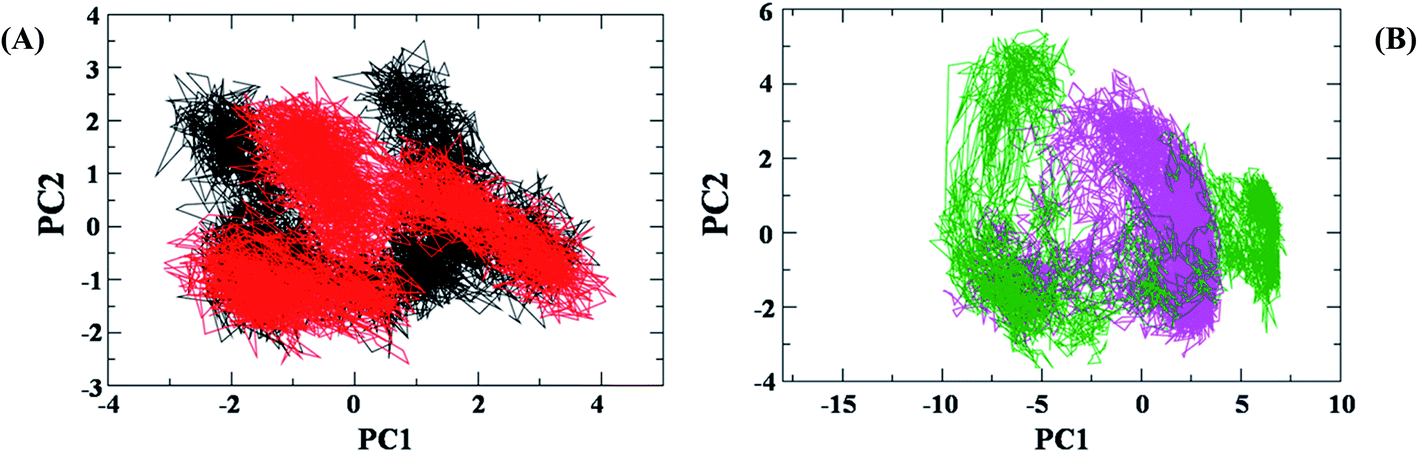 | ||
| Fig. 12 Principal component (PC) analysis of protein and protein–ligand complex: (A) the collective motion of AChE (black), AChE–12 complex (red) using projections of MD trajectories on two eigenvectors corresponding to the first two PCs. (B) Aβ1-42 (green) and Aβ1-42–12 docked complex (magenta). | ||
In the ED analysis shows, both AChE and AChE–12 complex show similar phase space behaviour, however, the ligand 12 decreases the flexibility in the AChE protein which is suggesting the normal function of the protein upon complex formation with 12.
Differently, the ED analysis of Aβ1-42 and Aβ1-42–12 complex shows that the protein–12 complex has not explored the same conformational space as the native protein, Aβ1-42 (Fig. 12B). Instead, it is restricted to small excursions slightly away from its initial conformation. In this Fig. 12B, we can see irregular stretches or clusters of line in the ED plot of Aβ. Two features are very apparent from these plots. Firstly, the clusters are well defined in Aβ1-42–12 complex structure than the protein Aβ1-42 alone. Secondly, the Aβ–12 complex covers a smaller region on the plot particularly along PC1 plane compared to that covered by native Aβ1-42. It is also clear from all the plots that the flexibility of Aβ1-42 decreases upon binding to 12, suggesting that 12 can efficiently prevent the self-aggregation of Aβ1-42, the pathological state of Alzheimer disease.49,50 Our observation thus corroborates with the idea of higher flexibility of Aβ1-42 at 300 K.
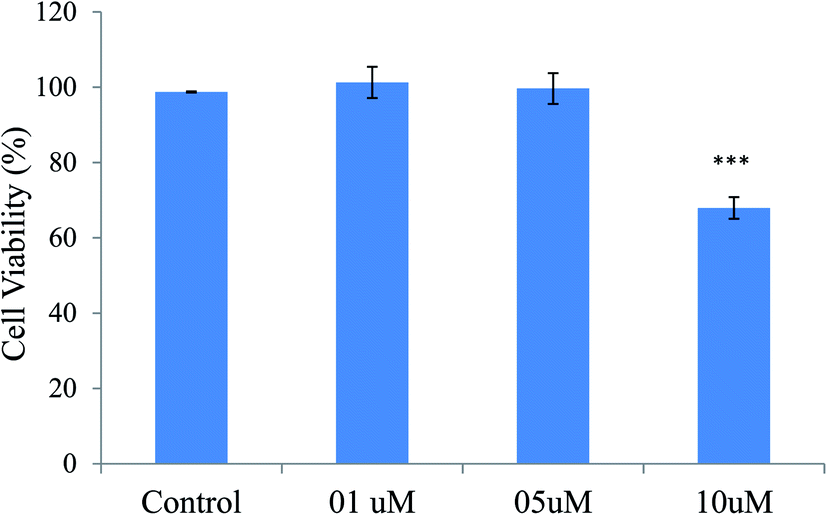 | ||
| Fig. 13 Effects of various concentrations of compound 12 on cell viability in SHSY5Y cells after treatment for 24 h. Cell viability was measured by MTT assay. Data were shown as mean ± SD of three independent experiments and each included triplicate sets. ***p < 0.001 vs. control, statistical analysis was performed. | ||
 | ||
| Fig. 14 A: Protective effects of compound 12 on H2O2-induced cell death in SHSY5Y cells. Cells were incubated with different concentration of 12 for 3 h and then treated with 200 μM H2O2 for 24 h. Cell viability was measured by MTT assay. Data were shown as mean ± SD of three independent experiments and each included triplicate sets. ###p < 0.001 compared to control, ***p < 0.001 compared to H2O2 treated cells; B: phase-contrast micrographs showing morphological changes in H2O2-induced neurotoxicity and neuroprotection of compound 12 in SH-SY5Y cells. (a) Cells without treatment showed healthy shapes. (b) H2O2 alone (200 μM) induced neurotoxicity. (c) Co-treatment of compound 12 (5 μM) for 24 h with H2O2 (200 μM) and showed neuroprotection. | ||
2.3. In vivo studies
The clinical manifestation of dementia is the impairment of learning as well as memory and is considered as a main symptom of AD. Scopolamine is a known muscarinic receptor antagonist that interferes with transmission of acetylcholine in the central nervous system51 inducing dysregulation in cholinergic signaling pathway causing severe damage in learning, and short term maintenance of spatial memory tasks. Therefore, anti-dementia and nootropic effect compound 12 was evaluated against scopolamine-induced mice model of dementia.For the study of learning and memory in rodents which relies on the intrinsic tendency of mice to stay in closed spaces, EPM model is used widely.52 Therefore, compound 12 was evaluated for anti-amnesic activities in this model. During the training trial, our result showed no significant difference in the transfer latencies among the treatment groups in EPM i.e. all the mice of the different groups came into the closed arm of the maze within stipulated time period. Nonetheless, in the scopolamine treated group (3 mg kg−1), the transfer latency time increased significantly when compared to the control group, which indicated the memory impairment during the retention trial (Table 2). In comparison, there were significant reduction in the transfer latency in the donepezil (5 mg kg−1) treated group that showcased its potential in reversing memory impairment induced by scopolamine.

Similarly, animals treated with compound 12 at 10 mg kg−1 and 20 mg kg−1 significantly reversed the effects of scopolamine. Compound 12 at both the doses showed significant reductions of transfer latencies (Table 2), which indicates its anti-dementia and nootropic potential against scopolamine induced memory deficit.
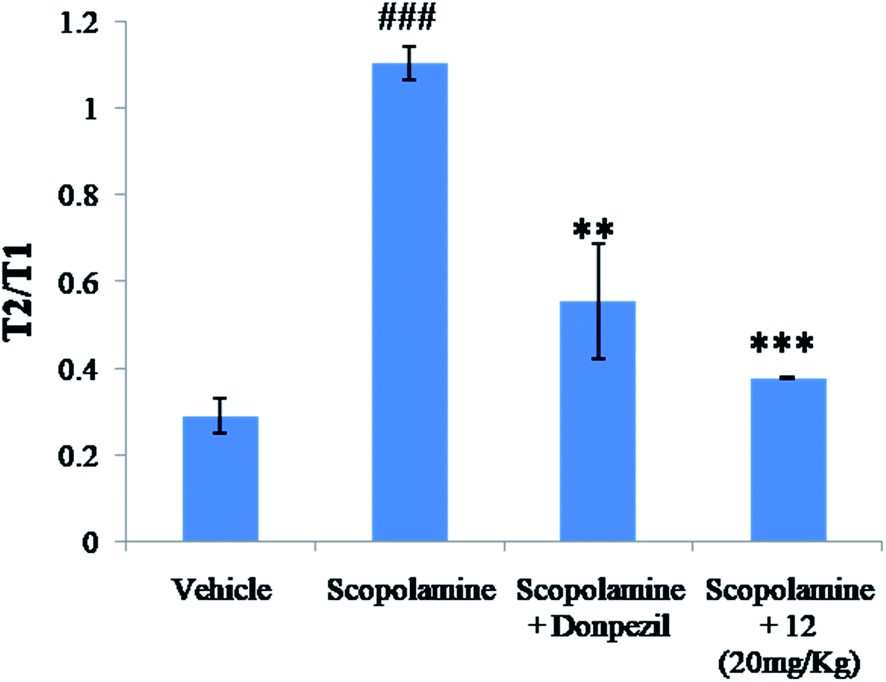 | ||
| Fig. 15 Treatment effect of test compound 12 at 20 mg kg−1 on the T2/T1 (second interaction trial/first interaction trial ratio) in social recognition test in scopolamine induced model. Data is expressed as mean SEM of 6 animals. ###p < 0.001 compared to vehicle, ***p < 0.001 and **p < 0.01 compared to scopolamine treated groups. | ||
3. Conclusion
In conclusion, a series of multifunctional benzothiazole–piperazine hybrids (4–17) were developed to target multifaceted nature of Alzheimer's disease. The synthesized derivatives potentially and selectively inhibited AChE over BuChE with IC50 in micromolar range. Furthermore, the derivatives inhibited Aβ1-42 aggregation efficiently. The results obtained from numerous in vitro and in vivo experiments established the multimodal mechanism of action of most active compound 12. In molecular docking studies, compound 12 adequately interacts with AChE and Aβ peptide. Further, we performed MD simulation for compound 12 which reveals its proficient binding with AChE and Aβ and steady interaction with the vital active site residual during 100 ns simulation. Compound 12 was non-cytotoxic to neuronal cells SHSY-5Y. This compound could efficiently reduce death induced by H2O2 treatment in the SHSY-5Y cells. It potentially attenuated scopolamine-induced cognitive impairments in mice model in the EPM and social recognition behavioral assays. Therefore, these properties together proved the multifunctional nature of 12, and its ability to alleviate multifaceted nature of AD. Furthermore, there is an enormous possibility for further development of this best lead molecule. Efforts in enhancing the efficiency of compound 12 through various chemical modifications have already been instigated by this research group.4. Experimental methods
4.1. General
All the reagents and dried organic solvents were obtained from Sigma Aldrich (St. Louis, MO, USA.), TCI Chemicals (Tokyo, Japan), and Merck (Darmstadt, Germany). 1H and 13C NMR spectra were acquired with a Jeol NMR spectrophotometer USA at 400 and 100 MHz, respectively. Chemical shifts are shown in parts per million (ppm) proportional to tetramethylsilane (TMS). The coupling constants (J) are presented in hertz (Hz), and the splitting patterns are designated by using abbreviations: s (singlet), d (doublet), t (triplet), m (multiplet), brs (broad singlet), and dd (double doublet). LC/MS data were recorded with an Agilent 6310 Ion trap LC/MS system and elemental analysis (C, H and N) was carried out on Elementar analysensysteme. Melting points were taken in open capillaries by using KSPII melting point apparatus (KRUSS, Germany). Purity of all target compounds were analysed by using Shimadzu reverse phase HPLC system (Kyoto, Japan) coupled with a photodiode array detector (PDA) and C-18 column. Acetonitrile and methanol (80:20) were used as a mobile phase, keeping flow rate 1 ml min−1. All target compounds displayed >97% purity.
4.2. Synthesis of 6-methanesulfonyl-benzothiazol-2-yl amine (1)
4-Methylsulfony aniline (10 millimole) and potassium thiocyanate (40 millimole) were stirred in glacial acetic acid at room temperature (RT) for 1 h. After that, bromine (10 millimole in 10 ml glacial acetic acid) was added dropwise by keeping temperature 0–5 °C. After addition of bromine, reaction was continued for 6 h at RT. Salt appeared in reaction mixture was filtered and dissolved in water. Salt solution was treated with ammonium hydroxide to give benzothiazole-2-amine in good yield.4.3. Synthesis of 3-chloro-N-(6-methanesulfonyl-benzothiazol-2-yl)-propionamide (3)
An equimolar amount of benzothiazole amine 1 (10 millimole), 3-chloropropionyl chloride (10 millimole) and TEA (catalytic amount) were refluxed in 1,4-dioxane. Reaction mixture was poured in chilled water and obtained precipitate was filtered to give key intermediate 3-chloro-N-(6-methanesulfonyl-benzothiazol-2-yl)-propionamide (2).4.4. Synthesis of N-(6-methanesulfonyl-benzothiazol-2-yl)-3-(4-substituted-piperazin-1-yl)-propionamide (4–17)
An equimolar ratio of key intermediate 3 and substituted piperazines were refluxed in acetonitrile with catalytic amount of NaHCO3 and NaI for 8–14 h. Reaction mixture was diluted with water and crude product was extracted with ethyl acetate, dried with anhydrous sodium sulphate. Evaporation of ethyl acetate under reduced pressure was provided crude products. Crude products were purified with column chromatography using chloroform and methanol (90:5) to give pure target compounds 4–17 in very good yield.
4.5. Inhibition of AChE and BuChE by synthesized derivatives
To determine the compound's ability to inhibit AChE and BuChE revised Ellman methods were performed. Concisely, stock solution of test compounds (10 mM) were prepared in 100% DMSO and diluted using autoclaved water to attain a final concentration range between (0.01–100) μM. Six different range of drug concentration was used to perform the assay. Enzyme dilutions and substrate dilutions were prepared according to the protocol. This assay was performed as reported by our lab previously.22,544.6. Kinetic characterization of AChE inhibition by compound 12
To study the kinetics of AChE inhibition, three concentrations of compound 12 (2 μM, 6 μM and 8 μM) were selected. A reciprocal plot of 1/velocity versus 1/substrate was plotted at low substrate concentrations by using modified Ellman's method.544.7. Inhibition of self-mediated Aβ1-42 aggregation assay
Marketed amyloid beta peptides were obtained and dissolved in hexafluoroisopropanol (HFIP) at 8 mg ml−1. The HFIP was evaporated and then Aβ1-42 stocks were prepared in DMSO, finally diluted in reaction buffer (50 mM phosphate buffer (pH 7.4)). For the inhibition of self-mediated Aβ1-42 aggregation experiment, ThT assay was performed as reported by our research group.244.8. Disaggregation of self-induced Aβ1-42 aggregation
For the study of disaggregation of self-induced Aβ1-42 aggregation, peptide (100 μM) was incubated at 37 °C for 48 h to form amyloid beta fibrils and further the compounds were added and incubated again at 37 °C for 24 h. ThT method was used to quantify amyloid fibril formation.544.9. Inhibition of self-mediated Aβ1-42 aggregation by ThT assay using confocal microscopy
Peptide (10 μl) and compounds (10 μl) were incubated for 48 h at 37 °C. 5 μl from incubation mixture was diluted with 10 μM of ThT (1:2) i.e., 10 μl of ThT in glycine–NaOH and placed on glass slide. Fluorescence signal of samples were analyzed at 488 nm by confocal laser scanning microscope.
4.10. Inhibition of self-mediated Aβ1-42 aggregation using TEM
Solution of Aβ stock was diluted with 50 mM phosphate buffer (pH 7.4), and further incubated in the presence and absence of compound 12 at 37 °C for 48 h. The final concentrations of Aβ peptide and 12 were 50 μM, respectively. Aliquots (10 μl) of the samples were put on carbon-coated copper grid. Each grid was negatively stained with 0.5% phosphotungstate (PTA) solution for 1 min at RT. Specimen was further transferred for imaging with transmission electron microscopy (JEOL JEM-1400).554.11. Metal chelation studies
The ability of compounds to chelate metal ions was performed by UV-spectroscopy. Compound 12 (60 μM) was dissolved in HEPES buffer with and without the metal ions. UV absorption spectra were recorded with wavelength ranging from 200 to 600 nm. The stoichiometry of the complex 12–Cu2+ was determined by Job's method. The tested compound and copper ion were incubated at different molar ratios and UV absorption was plotted.554.12. Molecular docking stdies
Atomic coordinates of AChE (PDB ID: 1EVE) and Aβ1-42 (PDB ID: 1YT) were taken from the Protein Data Bank (www.rcsb.org). Autodock 4.2 with standard protocol was used to dock the synthesized compound 12 with AChE and Aβ1-42.16 Protein–ligands interactions were assessed by applying Lamarckian genetic algorithm (LGA).The most encouraging free binding energy and docking orientations lying within the range of 2.0 Å in root-mean square deviation (RMSD) tolerance were used to cluster the molecule and ranked accordingly. PyMOL was employed to visualize molecular interactions. All calculations were carried out on workstation machines running Linux x86 as operating systems.564.13. Molecular dynamic (MD) simulation studies
MD simulations were carried out with GROMACS 4.6.5 suite of programs, using charm force-field and spc216 water molecules. The trajectory files were analyzed using GROMACS utilities: g_gyrate, g_rmsd and g_rmsf to obtain the radius of gyration (Rg), root mean square deviation (RMSD) and root mean square fluctuation (RMSF). The coordinates of AChE and Aβ1-42 were minimized with steepest descent, up to a tolerance of 100 kJ mol−1 to remove all bad contacts. Two independent MD simulations were carried out with standard protocol in water for AChE and AChE–12 complex, respectively at 300 K to evaluate the structural changes, binding affinity and stability in course of simulation. Similarly, another set of MD simulations were performed for Aβ1-42 and Aβ1-42–12complex. The SHAKE algorithm was used to restrain the covalent bonds containing hydrogen atoms at their equilibrium distances and periodic boundary conditions were applied. Particle Mesh Ewald (PME) method was employed for computing the long-range electrostatic interactions. A 1.4 nm cut-off for van der Waals interactions and a 1.0 nm cut-off for Coulombs interaction were adopted in the simulation, respectively. Analytical graphs were plotted using Xmgrace, VMD and PyMol were used for structure and molecular interaction analysis.22,534.14. Cytotoxicity of synthetic compounds on SHSY-5Y cell line
To study the cytotoxicity of the compound 12 on neuronal cells, MTT assay was performed. 1 × 104 cells per well were seeded in 96-well plates. The medium was removed after 24 h and compound 12 at different concentrations (5 μM, 10 μM and 20 μM) for 24 h at 37 °C were added. After 24 h, MTT (5 mg ml−1 stock) solution (20 μl) per well were added at 37 °C for 4 h. MTT was removed thereafter and formazon crystals were then dissolved in DMSO. The absorbance at 570 nm was recorded. Results have been shown as the mean ± SD of three independent experiments.224.15. Neuroprotection against H2O2 induced neurotoxicity
To study the neuroprotective effects of the compound against hydrogen peroxide (H2O2) induced neurotoxicity, MTT assay was performed. 2 × 104 per well SH-SY5Y cells were seeded in 96-well plates. Serum free medium was added after 24 h of incubation, containing different concentrations of the compounds 12 followed by 200 μM H2O2 after an interval of 3 hours at 37 °C for 24 h. Cells without treatment were used as healthy controls and cells treated with H2O2 as control group were used and the results were evaluated as percentage of control. Photomicrographs were also captured using camera attached to the microscope (Olympus, Japan) after 24 h of treatment to assess morphological changes in SH-SY5Y cells.244.16. Animals
Swiss albino male mice (25–30 g, 2–3 weeks; n = 6 each group) was procured from animal house facility of Dr. B. R. Ambedkar Center for Biomedical Research, University of Delhi. Delhi, India. Polypropylene cages were used for housing the animals and optimum conditions (temperature 25 ± 2 °C, 12 h light/12 h dark cycle, 50 ± 5% relative humidity and free access to food and water ad libitum) were maintained under the standard laboratory environmental conditions. Thus, all animal procedures were performed in accordance with the guidelines for care and use of laboratory animals of Institutional Animal Ethics Committee (IAEC), Dr. B.R. Ambedkar Center for Biomedical Research, Delhi University and approved by the animal ethics Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India.4.17. Drugs and experimental design
Scopolamine hydrobromide was procured from Sigma-Aldrich (MO, USA).Scopolamine was dissolved in saline and given intraperitoneally (i.p.) in a dose of 3 mg kg−1. Five groups with n = 6 were designed and mice were randomly allocated in each group. Animals in group I were given scopolamine 3 mg kg−1 b. wt per day, i.p51,52. Group II animals were treated together with scopolamine 3 mg kg−1 b. wt per day, i.p. and standard drug Donepezil (5 mg kg−1 b. wt per day; i.p). In groups III & IV, scopolamine followed by treatment with compound 12 (10 and 20 mg kg−1 b. wt per day, i.p.) were given. Animals in group V were used as vehicle controls and they were administered saline (daily, i.p). For 7 consecutive days, all the drugs including scopolamine were given daily. Behavioural studies were carried out, at the end of treatment schedules. Elevated plus maze (EPM) task and social recognition test were performed for each animal in each group.4.18. Elevated plus maze test (EPM)
There are two open arms (50 cm × 10 cm) and two closed arms (50 cm × 10 cm × 30 cm) in the elevated plus maze. There is a central platform elevated to a height of 50 cm from the floor. The experiments were performed in the light phase. At one edge of the arm, we placed the mice facing the opposite side and recorded the time taken by animals to enter the closed arm which was documented as transfer of latency (TL).524.19. Social recognition test
The behavioural assay was performed according to the protocol given by Timmermann et al.57 The adult mice and the juvenile (8–12 g) were kept together to make acquainted to themselves, for 1 h in the test room. Thereafter, the adult mice were brought back to their respective cages. The first interactive trial (T1) of the adult mice with the juvenile mice began after 30 min of habituation period. The adult mice were placed with the juvenile and the interaction time (T1) was recorded and observed simultaneously for 5 min. This was subsequently repeated for all the groups. The second trial began after 120 min in the same test cage pursued by recording the time in seconds of adult mice interacting with the juvenile mice. The obtained results were given in the form of recognition ratios (T2/T1). Index of improved short term recognition memory is the decrease in T2:T1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Author C. B. M would like to acknowledge the financial support from DHR for Young Scientist fellowship. Author S. S is thankful to Ministry of AYUSH for the financial support (JRF). M. T wishes to acknowledge University of Delhi, UGC-SAP and DST-PURSE for providing research funds to carry out the study. The authors gratefully acknowledge animal facility of ACBR and NMR facility of USIC and TEM facility of SAIF, AIIMS.References
- M. Citron, Nat. Neurosci., 2002, 5, 1055–1057 CrossRef CAS PubMed.
- B. Klimova, P. Maresova and K. Kuca, Curr. Alzheimer Res., 2016, 13, 1249–1258 CrossRef CAS PubMed.
- P. Marešová, H. Mohelská, J. Dolejš and K. Kuča, Curr. Alzheimer Res., 2015, 12, 903–911 CrossRef PubMed.
- H. C. Beerens, C. Sutcliffe, A. Renom-Guiteras, M. E. Soto, R. Suhonen, A. Zabalegui, C. Bökberg, K. Saks and J. P. Hamers, J. Am. Med. Dir. Assoc., 2014, 15, 54–61 CrossRef PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- T. P. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- S. Ayala, P. Genevaux, C. Hureau and P. Faller, ACS Chem. Neurosci., 2019, 10, 3366–3374 CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- G. Nesi, S. Sestito, M. Digiacomo and S. Rapposelli, Curr. Top. Med. Chem., 2017, 17(27), 3062–3079 Search PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- G. Caruso, S. F. Spampinato, V. Cardaci, F. Caraci, M. A. Sortino and S. Merlo, Curr. Pharm. Des., 2019, 25(45), 4771–4781 CrossRef CAS PubMed.
- K. A. Jellinger, J. Cell. Mol. Med., 2010, 14, 457–487 CrossRef CAS PubMed.
- A. I. Bush, J. Alzheimer's Dis., 2013, 33, S277–S281 Search PubMed.
- M. L. Seux, J. Rotrou and A. S. Rigaud, PSN, 2008, 6, 82–90 CrossRef.
- K. Blennow, M. J. de Leon and H. Zetterberg, Lancet, 2006, 368, 387–403 CrossRef CAS.
- K. G. Yiannopoulou and S. G. Papageorgiou, Ther. Adv. Neurol. Disord., 2013, 6, 19–33 CrossRef CAS PubMed.
- R. Anand, K. D. Gill and A. A. Mahdi, Neuropharmacology, 2014, 76, 27–50 CrossRef CAS PubMed.
- R. Ali, I. A. Sheikha, N. R. Jabirb and M. A. Kamal, Am. J. Neuroprot. Neuroregener., 2012, 4, 136–144 CrossRef.
- S. Sestito, S. Wang and Q. Chen, et al., Eur. J. Med. Chem., 2019, 174, 216–225 CrossRef CAS PubMed.
- M. Maspero, D. Volpato and D. Cirillo, et al., Bioorg. Chem., 2020, 96, 103633 CrossRef CAS PubMed.
- F. J. Pérez-Areales, A. L. Turcu and M. Barniol-Xicota, et al., Eur. J. Med. Chem., 2019, 180, 613–626 CrossRef PubMed.
- C. B. Mishra, S. Kumari, A. Manral, A. Prakash, V. Saini, A. M Lynn and M. Tiwari, Eur. J. Med. Chem., 2017, 125, 736–750 CrossRef CAS PubMed.
- A. Manral, V. Saini, P. Meena and M. Tiwari, Bioorg. Med. Chem., 2015, 23, 6389–6403 CrossRef CAS PubMed.
- C. B. Mishra, A. Manral, S. Kumari, V. Saini and M. Tiwari, Bioorg. Med. Chem., 2016, 24, 3829–3841 CrossRef CAS PubMed.
- J. M. Song, A. M. Di Battista, Y. M. Sung, J. M. Ahn, R. S. Turner, J. Yang, D. T. Pak, H. K. Lee and H. S. Hoe, Exp. Neurol., 2014, 252, 105–113 CrossRef CAS PubMed.
- R. Ali and N. Siddiqui, J. Chem., 2013, 1–12 Search PubMed.
- C. A. Mathis, N. S. Mason, B. J. Lopresti and W. E. Klunk, Semin. Nucl. Med., 2012, 42, 423–432 CrossRef PubMed.
- C. B Mishra, S. Kumari and M. Tiwari, Eur. J. Med. Chem., 2015, 6, 1–34 CrossRef PubMed.
- N. Guzior, A. Wieckowska, D. Panek and B. Malawska, Curr. Med. Chem., 2015, 22(3), 373–404 CrossRef CAS PubMed.
- C. B Mishra, S. Kumari, A. Angeli, S. M. Monti, M. Buonanno, M. Tiwari and C. T. Supuran, J. Med. Chem., 2017, 60, 2456–2469 CrossRef PubMed.
- C. B. Mishra, S. Kumari, A. Angeli, S. Bua, M. Tiwari and C. T. Supuran, J. Med. Chem., 2018, 61, 3151–3165 CrossRef CAS PubMed.
- S. Kumari, C. B. Mishra and M. Tiwari, Pharmacol. Rep., 2016, 68, 250–258 CrossRef CAS PubMed.
- D. Özkay Ümide, C. Ö. Devrim, Y. Ozkay and Y. Ozturk, Pharmacol. Rep., 2012, 64, 834–847 CrossRef.
- S. Stayte and B. Vissel, Front. Neurosci., 2014, 8, 113 Search PubMed.
- A. K. Rathi, R. Syed, H. S. Shin and R. V. Patel, Expert Opin. Ther. Pat., 2016, 26(7), 777–797 CrossRef CAS PubMed.
- L. Peauger, R. Azzouz, V. Gembus, M. L. Ţînţaş, J. S. O. Sopková-de, P. Bohn, C. Papamicaël and V. Levacher, J. Med. Chem., 2017, 60, 5909–5926 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres Jr and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- D. R. Liston, et al., Pharmacology of selective acetylcholinesterase inhibitors: implications for use in Alzheimer's disease, Eur. J. Pharmacol., 2004, 486, 9–17 CrossRef CAS PubMed.
- Li Luo, et al., Design, synthesis and evaluation of phthalide alkyl tertiary amine derivatives as promising acetylcholinesterase inhibitors with high potency and selectivity against Alzheimer's disease, Bioorg. Med. Chem., 2020, 115400 CrossRef CAS PubMed.
- A. Kumar, A. Singh and Ekavali, Pharmacol. Rep., 2015, 67(2), 195–203, DOI:10.1016/j.pharep.2014.09.004.
- H. Aoyama and T. Doura, Bioorg. Med. Chem. Lett., 2020, 30, 126888 CrossRef PubMed.
- G. A. de Souza, et al., J. Enzyme Inhib. Med. Chem., 2019, 34, 631–637 CrossRef PubMed.
- N. A. Vyas, S. B. Singh, A. S. Kumbhar, D. S. Ranade, G. R. Walke, P. P. Kulkarni, V. Jani, U. B. Sonavane, R. R. Joshi and S. Rapole, Inorg. Chem., 2018, 57, 7524–7535 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- H. Du, X. Liu, J. Xie and F. Ma, ACS Chem. Neurosci., 2019, 10, 2397–2407 CrossRef CAS PubMed.
- O. Crescenzi, S. Tomaselli, R. Guerrini, S. Salvadori, A. M. D'Ursi, P. A. Temussi and D. Picone, Eur. J. Biochem., 2002, 269, 5642–5648 CrossRef CAS PubMed.
- S. Vilasi, R. Carrotta, C. Ricci, G. C. Rappa, F. Librizzi, V. Martorana and M. R. Mangione, ACS Chem. Neurosci., 2019, 10(8), 3565–3574 CrossRef CAS PubMed.
- A. Prakash, G. Dixit, N. K. Meena, R. Singh, P. Vishwakarma, S. Mishra and A. M. Lynn, J. Biomol. Struct. Dyn., 2018, 36(9), 2391–2406 CrossRef CAS PubMed.
- L. Vugmeyster, M. A. Clark, I. B. Falconer, D. Ostrovsky, D. Gantz, W. Qiang and G. L. Hoatson, J. Biol. Chem., 2016, 291(35), 18484–18495 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- A. Blokland, Scopolamine Rev., 2005, 1, 1–76 Search PubMed.
- A. C. Sharma and S. K. Kulkarni, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 1992, 16, 117–125 CrossRef CAS PubMed.
- C. B. Mishra, S. Kumari, A. Prakash, R. Yadav, A. K. Tiwari, P. Pandey and M. Tiwari, Eur. J. Med. Chem., 2018, 151, 520–532 CrossRef CAS PubMed.
- T. Umar, S. Shalini, M. K. Raza, S. Gusain, J. Kumar, P. Seth, M. Tiwari and N. Hoda, Eur. J. Med. Chem., 2019, 175, 2–19 CrossRef CAS PubMed.
- T. Umar, S. Shalini, M. K. Raza, S. Gusain, J. Kumar, W. Ahmed, M. Tiwari and N. Hoda, MedChemComm, 2018, 9, 1891–1904 RSC.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng., Des. Sel., 1995, 8(2), 127–134 CrossRef CAS PubMed.
- D. B. Timmermann, K. Sandager-Nielsen, T. Dyhring, M. Smith, A. M. Jacobsen, E. Ø. Nielsen, M. Grunnet, J. K. Christensen, D. Peters, K. Kohlhaas, G. M. Olsen and P. K. Ahring, Br. J. Pharmacol., 2012, 167, 164–182 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: In silico study data, 1H and 13C spectra of all synthesized compounds have been kept as supplementary data. See DOI: 10.1039/d0ra00663g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |