DOI:
10.1039/D0RA00531B
(Paper)
RSC Adv., 2020,
10, 10519-10525
Porous SnO2 nanostructure with a high specific surface area for improved electrochemical performance†
Received
18th January 2020
, Accepted 27th February 2020
First published on 11th March 2020
Abstract
Tin oxide (SnO2) has been attractive as an alternative to carbon-based anode materials because of its fairly high theoretical capacity during cycling. However, SnO2 has critical drawbacks, such as poor cycle stability caused by a large volumetric variation during the alloying/de-alloying reaction and low capacity at a high current density due to its low electrical conductivity. In this study, we synthesized a porous SnO2 nanostructure (n-SnO2) that has a high specific surface area as an anode active material using the Adams fusion method. From the Brunauer–Emmett–Teller analysis and transmission electron microscopy, the as-prepared SnO2 sample was found to have a mesoporous structure with a fairly high surface area of 122 m2 g−1 consisting of highly-crystalline nanoparticles with an average particle size of 5.5 nm. Compared to a commercial SnO2, n-SnO2 showed significantly improved electrochemical performance because of its increased specific surface area and short Li+ ion pathway. Furthermore, during 50 cycles at a high current density of 800 mA g−1, n-SnO2 exhibited a high initial capacity of 1024 mA h g−1 and enhanced retention of 53.6% compared to c-SnO2 (496 mA h g−1 and 23.5%).
1. Introduction
Li-ion batteries (LIBs) are electrochemical power sources that are mostly used in portable electronic devices, electric vehicles, and energy storage systems. The LIBs are secondary electrochemical cells, which can repeatedly perform the charging and discharging process using the Li+ ion behaviour between the anode and cathode.1–5 During discharging, a spontaneous oxidation reaction occurs at the anode, producing electrical energy from chemical energy with the reduction reaction at the cathode.6 In contrast, during charging, a spontaneous oxidation reaction occurs at the cathode, storing electrical energy with the reduction reaction at the anode.7 Among these components in the LIBs, the anode is an electrode at which a spontaneous electrochemical oxidation of Li+ ions occurs during discharging.8
In particular, anode materials for high-performance LIBs need to have high capacity, high power density, long cycle life, and excellent stability.9 Among the known anode candidates, tin(IV) oxide (SnO2) exhibits both conversion (eqn (1)) and alloying/de-alloying (eqn (2)) reactions during cycling as well as a fairly high theoretical capacity of 711–781 mA h g−1.10,11 However, SnO2 has critical drawbacks, such as poor cycle stability caused by a large volumetric variation during the alloying/de-alloying reaction and low initial capacity due to its low electrical conductivity.12,13 Thus, to overcome these drawbacks of SnO2 anode, various research efforts using alloy structures, nanostructure control, and doping, have been carried out.14–21 The doped SnO2 anodes with Sb and F, despite having improved electrical conductivity, showed a limitation to the relief of volumetric expansion. Furthermore, the composite structure of SnO2 with electronic conductive materials exhibited a decreased capacity due to a reduced amount of SnO2 as an active material.
| | |
SnO2 + 4Li+ → Sn + 2Li2O
| (1) |
| | |
Sn + xLi+ → LixSn (0 ≤ x ≤ 4.4)
| (2) |
Typically, porous metal oxides with high specific surface areas have been prepared using the Adams fusion method due to their various merits such as facile preparation procedure, low reaction temperature, short reaction time, and high yield.22–26 Thus, in this study, the SnO2 sample prepared using the Adams fusion method, with high specific surface areas and particular pore structures showed improved LIB performance compared to a commercial SnO2 because of the enhanced stability caused by relieved volumetric expansion during cycling.
| | |
SnCl2 + NaNO3 → NaCl + Sn(NO3)2
| (3) |
| | |
Sn(NO3)2 → SnO2 + NO2
| (4) |
2. Experimental
2.1 Synthesis of porous SnO2 nanostructures
SnO2 nanostructures were prepared using the Adams fusion method with different weight ratios of SnCl2·2H2O to NaNO3. 2.8 g of SnCl2·2H2O as a Sn source was dispersed with 5.0 g NaNO3 in 100 mL de-ionized (DI) water using ultra-sonication. The dispersed solutions were evaporated at 80 °C until the solvent was completely evaporated. The dried samples were heated in a 500 °C box furnace for 2 h and then washed with DI water several times. Finally, the resulting powders were obtained using a freeze dryer.
2.2 Materials characterization
The crystal structure of the as-prepared samples was characterized using an X-ray diffractometer (XRD, Bruker, D2 Phase system) operating at 40 kV and 100 mA with an X-ray source of Cu Kα (λ = 0.15418 nm) and a Ni filter. The morphology of the samples was confirmed using a field-emission scanning electron spectroscope (FE-SEM, ZEISS, Gemini 300). The particle size and crystal structure of the samples were characterized using a Cs-corrected transmission electron microscope (CS-TEM, JEM-ARM200F, JEOL Ltd, Japan). The specific surface area and pore structure of the samples preheated under a pressure of 100 mm Hg at 473 K for 6 h were characterized using a nitrogen adsorption/desorption analyzer (Micromeritics ASAP 2020).
2.3 Cell assembly and electrochemical characterization
To characterize the electrochemical properties of the samples, coin-type half cells with the samples as anodes were prepared in an Ar-filled glove box. The slurry was prepared using a paste mixer with the as-prepared powder samples as anode active materials (70 wt%), Ketjen black as a conducting agent (Wellcos, 10 wt%), and polyvinylidene fluoride (10 wt% PVDF, Alfa Aesar, 20 wt%) as a binder. The as-prepared slurry was coated on Cu foil (Hohsen Corporation) using a doctor blade method and then dried in a 110 °C convection oven for 24 h. The half-cell was assembled with an active material-coated electrode, 1.1 M LiPF6 as an electrolyte, polyethylene (Wellcos) as a separator, and lithium metal as a counter electrode. Charge–discharge curves of the samples were obtained using a multichannel battery tester (WBCS3000L, Wonatech Co.) in the potential range of 0–3 V vs. Li/Li+ with various current densities of 100, 300, 500, 800 and 1600 mA g−1. Cyclic voltammograms (CVs) of the samples were measured at a scan rate of 0.02 mV s−1 in the potential range of 0–3 V vs. Li/Li+. Galvanostatic intermittent titration technique (GITT) measurement was performed at a current density of 100 mA g−1 for 10 min intervals, with rest periods of 60 min, in the potential range of 0–3 V Li/Li+.
3. Results and discussion
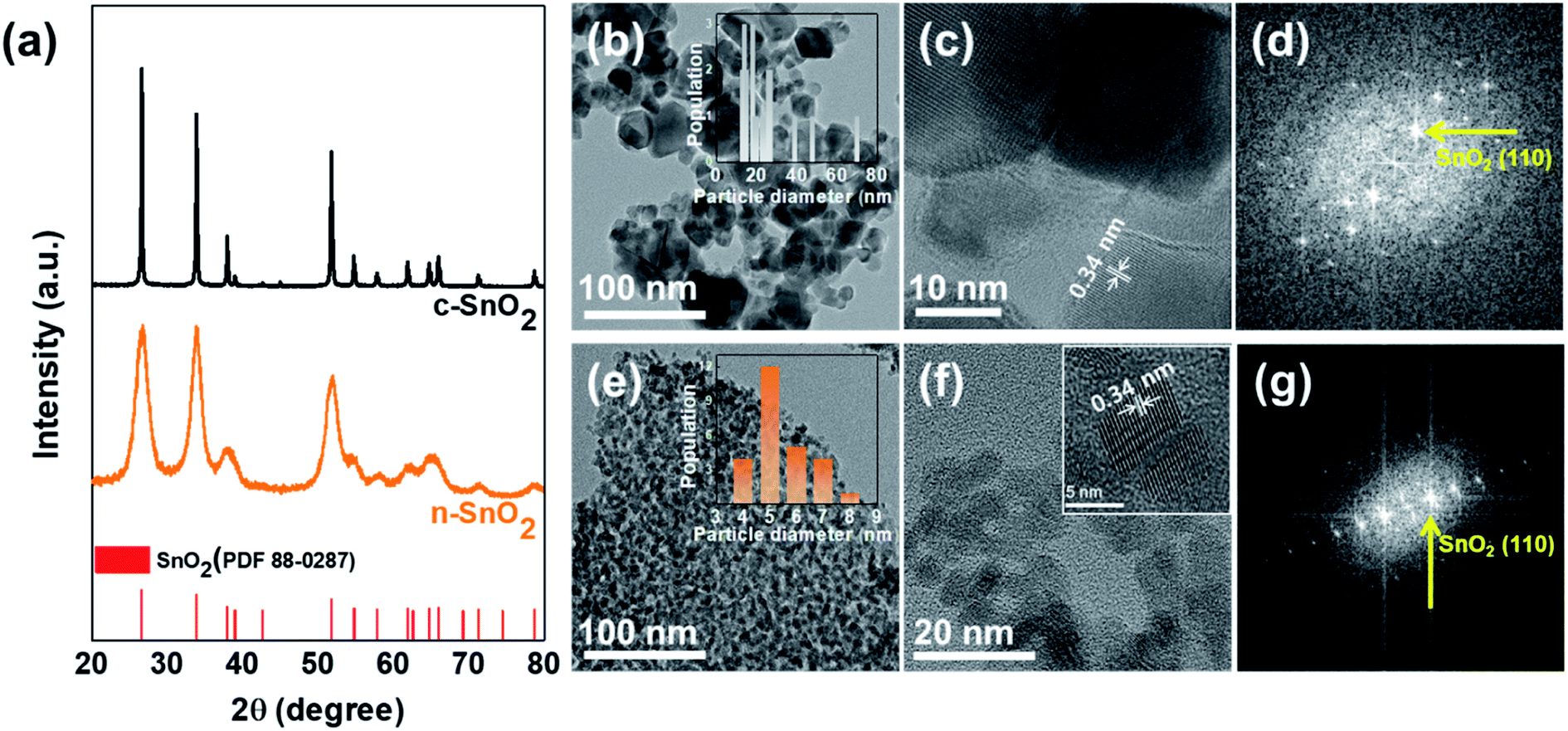
Fig. 1(a) shows the XRD patterns of the c-SnO2 and n-SnO2 samples prepared using the Adams fusion method. The XRD characteristic peaks, corresponding to the (110), (101), and (200) planes of the tetragonal SnO2 structure, appeared at 26.6°, 33.9°, and 38.0° respectively. All of the samples exhibited a tetragonal structure with a = b = 4.74 Å, c = 3.19 Å (P42/mn (136) group, PDF 88-0287).27 However, the c-SnO2 showed sharp XRD peaks, while the as-prepared n-SnO2 showed relatively broad XRD peaks. Compared to the c-SnO2, the broad XRD peaks for the as-prepared SnO2 samples imply a nanostructure formed through the Adams fusion method.28 The particle size and structure of the samples were characterized using a TEM analysis (Fig. 1(b–g)). The n-SnO2 showed an average particle size of ∼5.5 nm with a fairly narrow size distribution, whereas the c-SnO2 showed a relatively broad size distribution and average particle size of 24 nm. Furthermore, as compared to the c-SnO2, the n-SnO2 synthesized using the Adams fusion method was found to be significantly porous without serious aggregation. From the high-resolution TEM image and fast Fourier transform (FFT) pattern, the (110) planes in the c-SnO2 and n-SnO2 were observed. In particular, compared to the c-SnO2, the selective area electron diffraction (SAED) pattern of the n-SnO2 demonstrates a nano-sized SnO2 with a high crystalline structure (Fig. S1†).
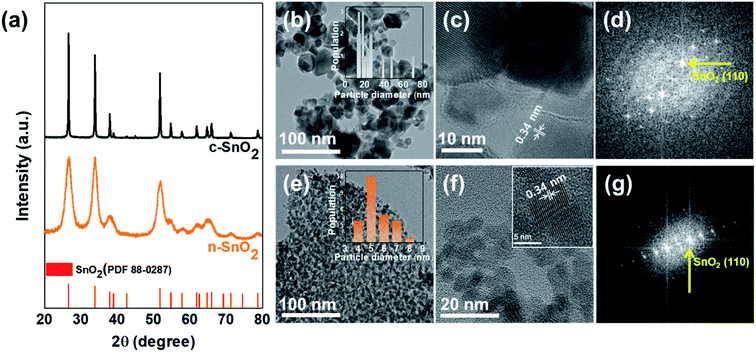 |
| | Fig. 1 (a) XRD patterns of c-SnO2 and n-SnO2. The Cs-TEM and HRTEM images, and Fast Fourier Transform (FFT) patterns of (b–d) c-SnO2 and (e–g) n-SnO2. | |
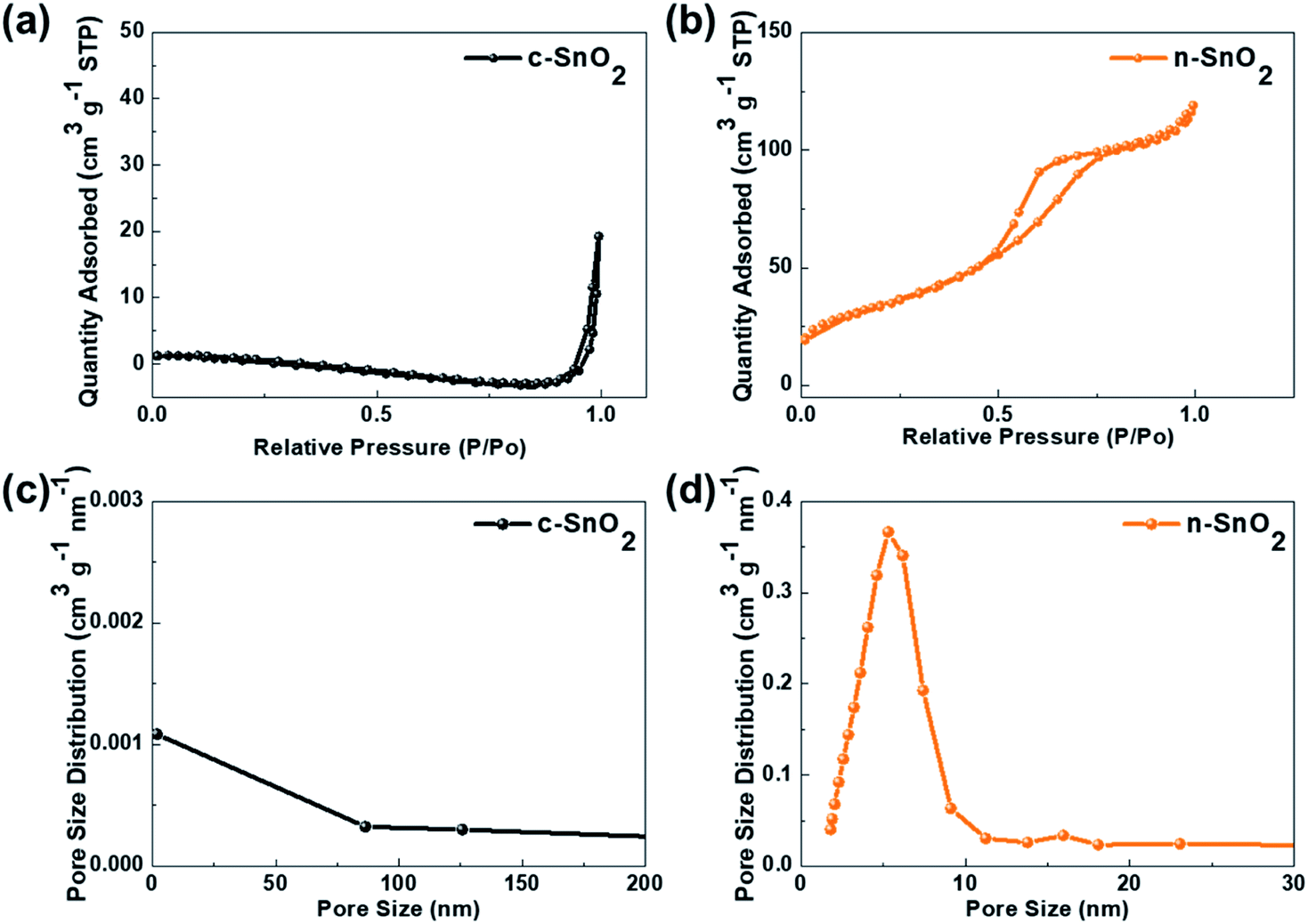
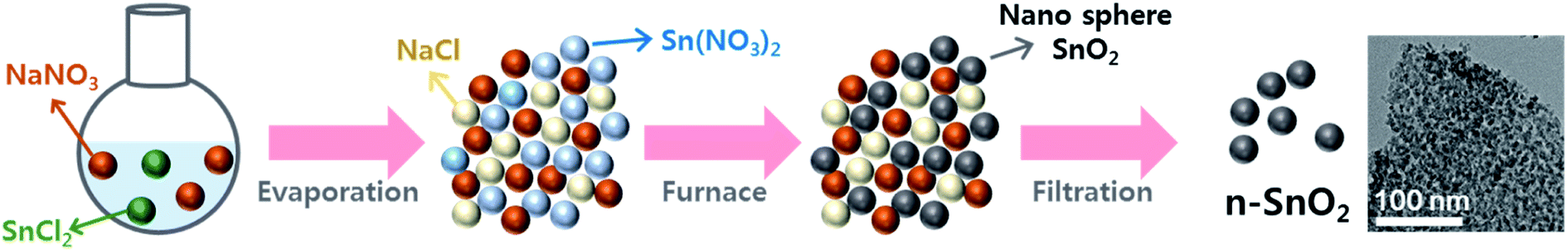
Fig. 2 shows the nitrogen adsorption–desorption isotherms of c-SnO2 and n-SnO2 samples prepared using the Adams fusion method. Compared to the c-SnO2, the as-prepared n-SnO2 sample exhibited type IV curves, demonstrating that the SnO2 sample synthesized using the Adams fusion method has a mesoporous structure.29 The specific surface areas of c-SnO2 and n-SnO2 were 1.6 and 122 m2 g−1, respectively. In particular, in contrast to c-SnO2, n-SnO2 showed a distinct pore size distribution. During the synthesis of SnO2 using the Adams fusion method, the generated NO2 gas and NaCl solid phase can result in a porous SnO2 nanostructure. The n-SnO2 has representative and minor pore sizes of 5.3 and 16.0 nm, respectively. Fig. S2† shows the SEM images of c-SnO2 and n-SnO2 prepared using the Adams fusion method. The c-SnO2 exhibited an irregular shape with agglomeration at a micron size. On the other hand, the n-SnO2 samples prepared using the Adams fusion method showed a porous structure with free volumes. Consequently, the SnO2 sample prepared using facile synthesis in the absence of complex additives or templates exhibited significantly high specific surface areas, compared to the SnO2 samples (58.6, 12.6–102.4, and 37.7–53.6 m2 g−1) prepared using microwave-hydrothermal, sol–gel, and co-precipitation methods, respectively.30–32 In the preparation using the Adams fusion method (Fig. 3), followed by the process in eqn (3) in an evaporator, a gas phase of NO2 can be generated during the formation of a solid phase of SnO2 in a furnace (eqn (4)). The generated NO2 gas may induce the formation of pores, resulting in a porous SnO2 structure. In addition, NaCl as a by-product formed within the sample was removed by washing with DI water, forming a porous SnO2 structure with free spaces in the powder.
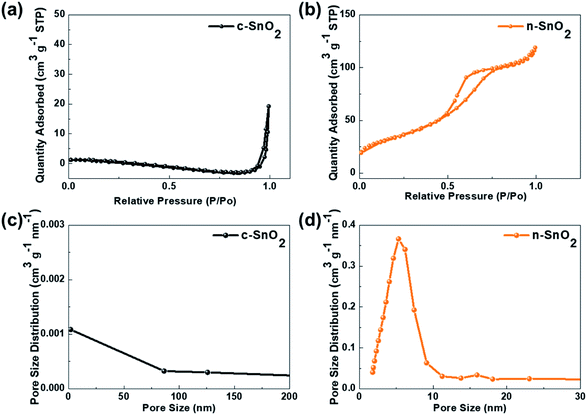 |
| | Fig. 2 N2 adsorption/desorption isotherms of (a) c-SnO2 and (b) n-SnO2. Pore size distribution of (c) c-SnO2 and (d) n-SnO2. | |
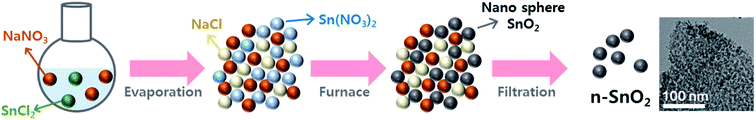 |
| | Fig. 3 Schematic illustration of formation mechanism of the n-SnO2 using the Adams fusion method. | |
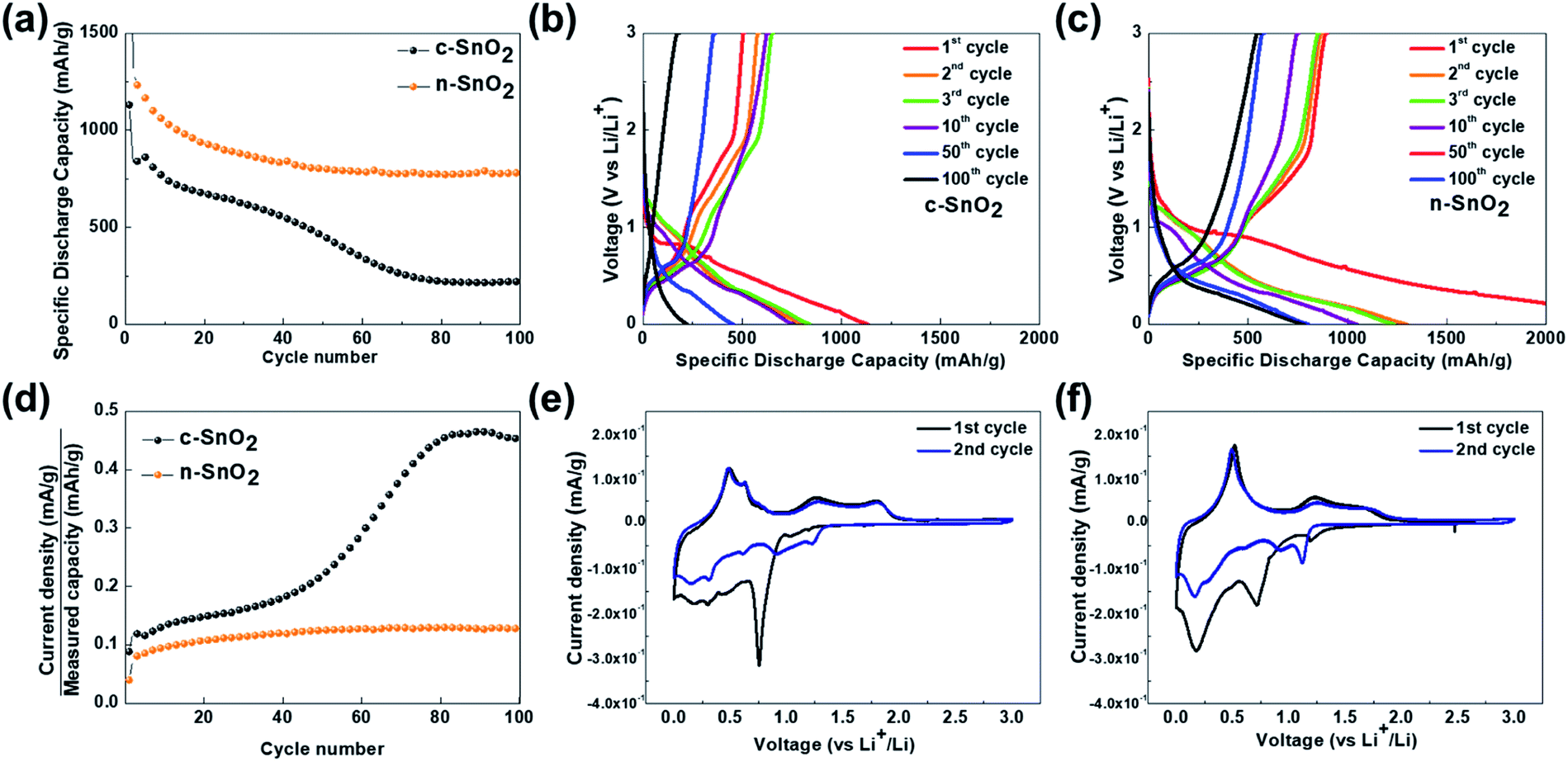
Fig. 4(a–d) show the charge–discharge characteristic curves of the samples as anodes measured at a current density of 100 A g−1 in a potential range of 0–3 V vs. Li/Li+ for 100 cycles. The initial discharge capacities of c-SnO2 and n-SnO2 were 839 and 1233 mA h g−1, respectively. In particular, the high initial capacity of n-SnO2 might result from the superior activity of the nano-sized active material with short Li+ ion diffusion pathway. The c-SnO2 exhibited significantly decreased capacity with increasing cycle numbers and a retention of 26% after 100 cycles. On the other hand, n-SnO2 maintained the discharge capacities during cycling and showed a retention of 63%. The improved cycling performance of the n-SnO2 anode can result when pulverization and aggregation of the active material are prevented due to the relieved volumetric variation in the porous structure consisting of nano-sized particles. Fig. 4(d) shows the plots of C rate versus cycle number of the samples for 100 cycles. In this study, the C-rate can be defined as an applied current density/the measured capacity during cycling. Thus, during cycling, the C-rate can be increased due to the deteriorated performance of an active material.33 n the 1st cycle, the samples exhibited almost the same C rates measured at the same current density because of active participation of all the phases in the samples. However, with increasing cycling number, the increased C rate implies a decreased actual amount of the electrochemically participated active material caused by the deteriorated electrochemical performance of the active material. Compared to the initial cycles, c-SnO2 showed a considerable increment in the C rate of ∼275%, whereas n-SnO2 showed a relatively slight increase in the C rate of ∼15%. During the cycling, n-SnO2 exhibited a relatively low C rate, i.e., the improved LIB performance, preventing the pulverization and crack caused by the volumetric variation of the electrode during cycling. Fig. 4(e) and (f) show the CVs of c-SnO2 and n-SnO2 samples, respectively, as anodes measured at a scan rate of 0.02 mV s−1 in a potential range of 0–3 V vs. Li/Li+. In the 1st cycle measured during a anodic scan, i.e. discharge process, the samples exhibited anodic characteristic peaks at ∼1.2 and ∼0.8 V, associated with the electrochemical reduction of SnO2 to SnO and SnO to Sn, respectively, with the formation of solid electrolyte interphase (SEI) (eqn (5)–(7)).34 In addition, the anodic peaks of the samples corresponding to the alloying reaction of Li+ ions and Sn appeared between 0.5 and 0.1 V (eqn (8)).35 In the 1st cycles measured during a cathodic scan, i.e. charge process, the samples exhibited the cathodic characteristic peaks at ∼0.56 and ∼1.25 V, respectively, associated with the electrochemical oxidation of LixSny, i.e., de-alloying process of LixSny (eqn (5), (6) and (8)).36,37 Furthermore, in the 2nd scan, the samples showed the SEI layer formation at 0.5–1.0 V with alloying/de-alloying and conversion reactions. In particular, compared to c-SnO2, the n-SnO2 sample exhibited relatively distinct electrochemical reactions, which correspond to the conversion and alloying/de-alloying reactions. The more evident alloying/de-alloying and conversion reactions for the n-SnO2 sample during cycling might be attributed to sufficient electrochemical active sites and short diffusion length in a porous nanostructure.
| | |
SnO2 + 2Li+ + 2e− ↔ SnO + Li2O
| (5) |
| | |
SnO + 2Li+ + 2e− ↔ Sn + Li2O
| (6) |
| | |
SnO2 + 4Li+ + 2e− ↔ Sn + 2Li2O
| (7) |
| | |
Sn + xLi+ + xe− ↔ LixSn (0 ≤ x ≤ 4.4)
| (8) |
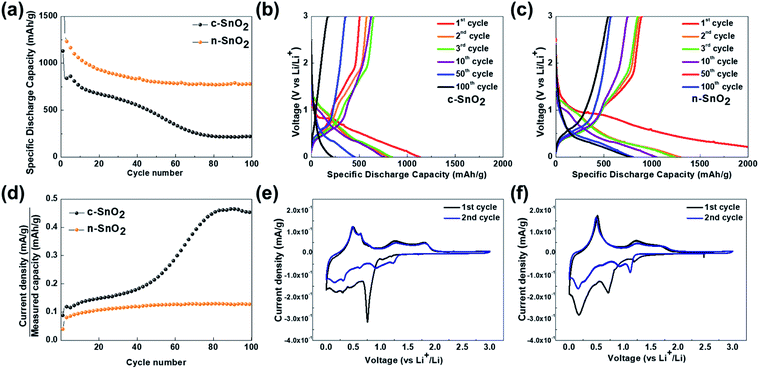 |
| | Fig. 4 (a) Cycling performance of the anode samples measured at a current density of 100 mA g−1 in a potential range of 0–3 V vs. Li/Li+ for 100 cycles. Charge–discharge curves of (b) c-SnO2 and (c) n-SnO2 measured at a current density of 100 mA g−1 for 100 cycles. (d) Plots of C rate vs. cycle number of the samples. Cyclic voltammograms (CVs) of (e) c-SnO2 and (f) n-SnO2 measured at a scan rate of 0.02 mV s−1 in a potential range of 0–3 V vs. Li/Li+. | |
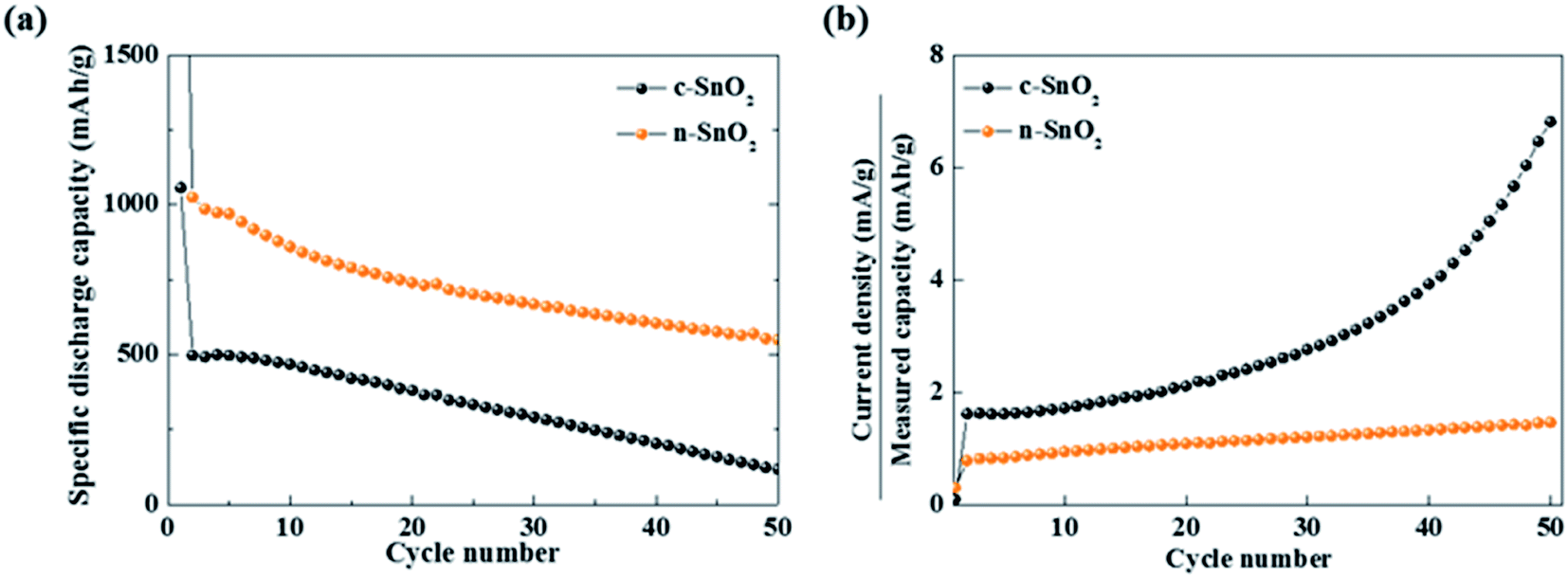
Characteristic curves of specific discharge capacity vs. cycle number were obtained at a relatively high current density of 800 mA g−1 for 50 cycles (Fig. 5(a)). The c-SnO2 showed an initial capacity of 496 mA h g−1 and retention of 23.5% after 50 cycles. The n-SnO2 showed a relatively high initial capacity of 1024 mA h g−1 and retention of 53.6% after 50 cycles. The improved high rate performance of the porous nanoparticle SnO2 anode samples can be attributed to increased reaction sites and decreased Li+ ion diffusion length caused by the porous nanostructure with a high specific surface area. Furthermore, as reported in the literature, the porous nanostructure materials for the LIBs exhibited significantly enhanced electrochemical properties due to relieved volumetric expansion during cycling.38,39 The increased C rate under a high constant current density implies the deteriorated electrochemical performance of the electrode during fast cycling. As shown in Fig. 5(b), among the anode samples, c-SnO2 exhibited the highest C-rate variations. Compared to the initial cycles, c-SnO2 showed a considerable C rate increment of ∼323%, whereas n-SnO2 showed a relatively slight C rate increase of ∼85%. During the rapid cycling, the improved cycling performance of the n-SnO2 can result from the relieved volumetric expansion caused by a porous structure of the n-SnO2 consisting of nano-sized particles.
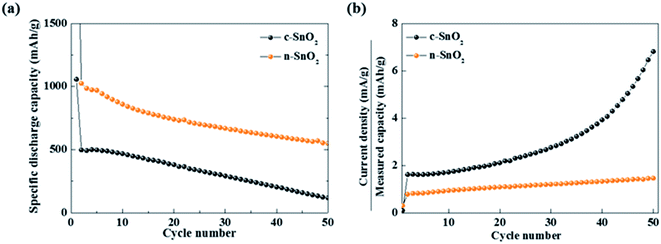 |
| | Fig. 5 (a) Specific discharge capacities measured at a current density of 800 mA g−1 for 50 cycles. (b) Plots of C rate vs. cycle number of the anode samples. | |
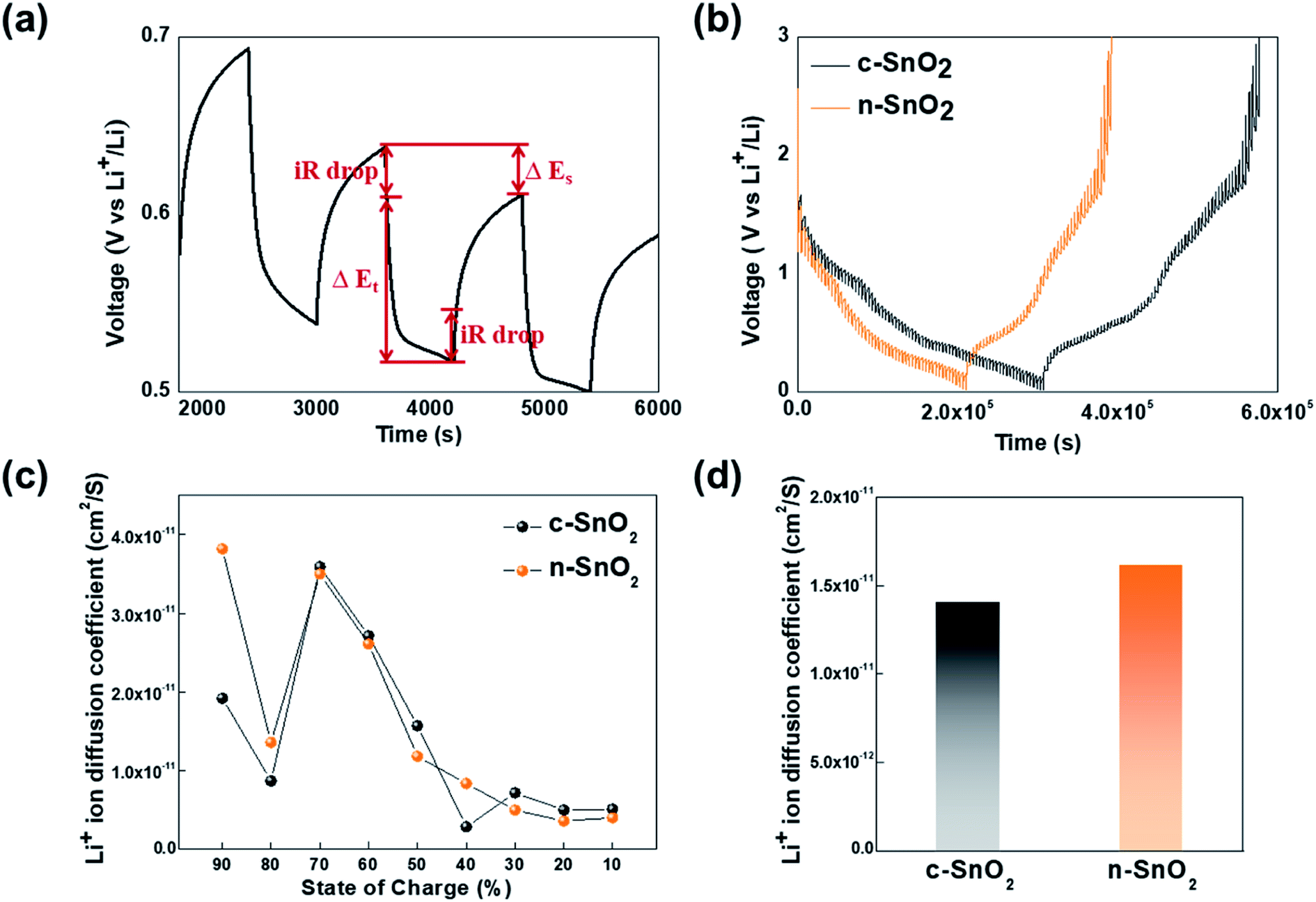
To investigate the Li+ ion path length in the SnO2 anode samples, the Li+ ion diffusion coefficient was measured using the GITT analysis, followed by one cycle for an initial stabilization, at a current density of 100 mA g−1 for 10 min intervals, with rest periods of 60 min, in the potential range of 0–3 V Li/Li+ (Fig. 6). The Li+ ion diffusion coefficients of the samples were calculated using eqn (9).
| |
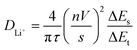 | (9) |
where
τ is the duration of the current pulse (10 min),
n is the number of moles (c-SnO
2; 6.97 × 10
−6 and n-SnO
2; 5.11 × 10
−6 mol),
V is the molar volume of the electrode (c-SnO
2; 76.204 and n-SnO
2; 103.915 cm
3 mol
−1),
s is the electrode/electrolyte contact area (1.32 cm
2), Δ
Es is the steady-state voltage change, and Δ
Et is the voltage change during the constant current pulse. The Li
+ ion diffusion coefficients of the samples with the state of charge were evaluated (
Fig. 6(c)). Compared to the c-SnO
2, the n-SnO
2 exhibited an improved diffusion coefficient of Li
+ ion with the state of charge (SOC). In particular, significantly higher diffusion coefficients of the n-SnO
2 in the initial ranges of the SOC (90% and 80%) might be attributed to short Li
+ ion diffusion length and large reaction sites provided by the porous structure of the n-SnO
2 consisting of nano-sized particles. However, from the SOC 70%, the samples showed fairly similar trend. Thus, it can be speculated that the porous nanostructure of the n-SnO
2 might significantly affect the shorten Li
+ ion diffusion length in the initial discharging.
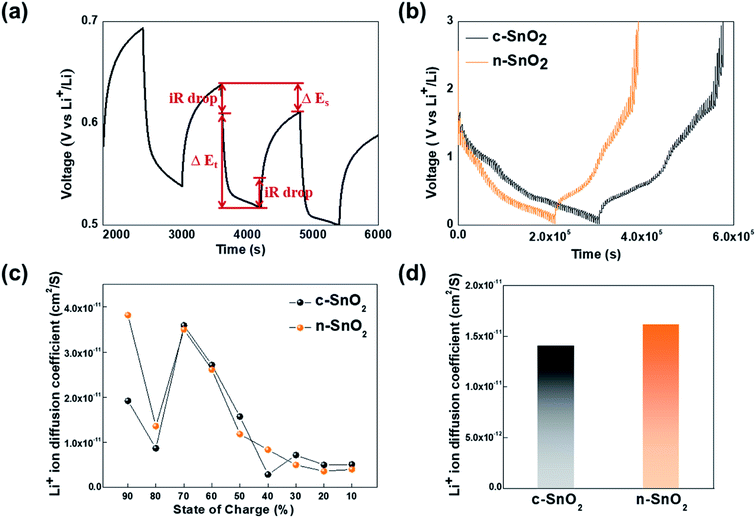 |
| | Fig. 6 (a) A single titration profile of the sample in the GITT during discharging. (b) Plots of discharge potential of the samples measured for three discharge cycles in a potential range of 0–3 V vs. Li/Li+. (c) Plots of diffusion coefficient of Li+ ion vs. the state of charge and (d) comparison of the average diffusion coefficient for the anode samples. | |
Furthermore, the average Li+ ion diffusion coefficients of c-SnO2 and n-SnO2 for the SOC from 90% to 10% were determined to be 1.41 × 10−11 and 1.62 × 10−11 cm2 S−1, respectively (Fig. 6(d)). Thus, the improved Li+ ion diffusivity of n-SnO2 during the cycling can result from increased reaction sites and decreased Li+ ion pathway caused by the porous nanostructure with a high specific surface area of the n-SnO2 synthesized using the Adams fusion method.
4. Conclusions
In this study, a porous SnO2 nanostructure (n-SnO2) with a high specific surface area as an anode for the LIBs was synthesized using the Adams fusion method. The as-prepared n-SnO2 anode was found to have a mesoporous structure consisting of high-crystalline nanoparticles. Compared to a commercial SnO2, n-SnO2 showed significantly improved LIB performance, i.e., high initial capacity, improved rate performance, and high retention due to increased number of electrochemical active sites, reduced Li+ ion diffusion length, and relieved volumetric expansion.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Technology Development Program to Solve Climate Changes and the Hydrogen Energy Innovation Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT (NRF-2017M1A2A2086648 and no. 2019M3E6A1104186).
Notes and references
- Z. Zhang, H. Zhao, Z. Du, X. Chang, L. Zhao, X. Du, Z. Li, Y. Teng, J. Fang and K. Świerczek, ACS Appl. Mater. Interfaces, 2017, 35880–35887 CrossRef CAS PubMed.
- A. Mishra, A. Mehta, S. Basu, S. J. Malode, N. P. Shetti, S. S. Shukla, M. N. Nadagouda and T. M. Aminabhavi, Mater. Sci. Energy Technol., 2018, 1, 182–187 Search PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- W. Yuan, Y. Zhang, L. Cheng, H. Wu, L. Zheng and D. Zhao, J. Mater. Chem. A, 2016, 4, 8932–8951 RSC.
- M.-C. Kim, S.-J. Kim, S.-B. Han, D.-H. Kwak, E.-T. Hwang, D.-M. Kim, G.-H. Lee, H.-S. Choe and K.-W. Park, J. Mater. Chem. A, 2015, 3, 23003 RSC.
- S. Goriparti, E. Miele, F. D. Angelis, E. D. Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- E. Kamali Heidari, A. Kamyabi-Gol, M. H. Sohi and A. Ataie, J. Ultrafine Grained Nanostruct. Mater., 2018, 51(1), 1–12 Search PubMed.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 5 CrossRef.
- S. Choi, M.-C. Kim, S.-H. Moon, J.-E. Lee, Y.-K. Shin, E.-S. Kim and K.-W. Park, J. Ind. Eng. Chem., 2018, 64, 344–351 CrossRef CAS.
- Y. Zhang, H. Zhang, J. Zhang, J. Wang and Z. Li, RSC Adv., 2015, 5, 106258 RSC.
- Y. Cheng, A. Nie, L.-Y. Gan, Q. Zhang and U. Schwingenschlögl, J. Mater. Chem. A, 2015, 3, 19483 RSC.
- Y. Wang and T. Chen, Electrochim. Acta, 2009, 53, 3510–3515 CrossRef.
- H. Xu, L. Shi, Z. Wang, J. Liu, J. Zhu, Y. Zhao, M. Zhang and S. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 27486–27493 CrossRef CAS PubMed.
- J. Santos-Peña, T. Brousse, L. Sánchez, J. Morales and D. M. Schleich, J. Power Sources, 2001, 97–98, 232–234 CrossRef.
- F. D. Wu, M. Wu and Y. Wang, Electrochem. Commun., 2011, 13, 433–436 CrossRef CAS.
- H. Wang, Y. Wu, Y. Bai, W. Zhou, Y. An, J. Li and L. Guo, J. Mater. Chem., 2011, 21, 10189 RSC.
- D. Su, C. Wang, H. Ahn and G. Wang, Phys. Chem. Chem. Phys., 2013, 15, 12543 RSC.
- D. Pham-Cong, J. S. Park, J. H. Kim, J. Kim, P. V. Braun, J. H. Choi, S. J. Kim, S. Y. Jeong and C. R. Cho, Carbon, 2017, 111, 28–37 CrossRef CAS.
- J.-S. Park, Y. J. Oh, J. H. Kim and Y. C. Kang, Mater. Charact., 2020, 161, 110099 CrossRef CAS.
- H. Du, Z. Liu, K. Huang, M. Yu, H. Sun and B. Geng, J. Power Sources, 2019, 441, 227172 CrossRef CAS.
- Q. Tian, Y. Chen, F. Chen, W. Zhang, J. Chen and L. Yang, J. Alloys Compd., 2019, 809, 151793 CrossRef CAS.
- R. Adams and R. L. Shriner, J. Am. Chem. Soc., 1923, 45(9), 2171–2179 CrossRef CAS.
- Y. Zhang, C. Wang, Z. Mao and N. Wang, Mater. Lett., 2007, 61, 1205–1209 CrossRef CAS.
- C. Felix, T. Maiyalagan, S. Pasupathi, B. Bladergroen and V. Linkov, Micro Nanosyst., 2012, 4, 186–191 CrossRef CAS.
- E. Oakton, D. Lebedev, A. Fedorov, F. Krumeich, J. Tillier, O. Sereda, T. J. Schmidt and C. Copéret, New J. Chem., 2016, 40, 1834–1838 RSC.
- C. Felix, B. J. Bladergroen, V. Linkov, B. G. Pollet and S. Pasupathi, Catalysts, 2019, 9, 318 CrossRef.
- J. Fan, E. Menéndez, M. Guerrero, A. Quintana, E. Weschke, E. Pellicer and J. Sort, Nanomaterials, 2017, 7, 348 CrossRef PubMed.
- A. Kar, S. Sain, S. Kundu, A. Bhattacharyya, S. K. Pradhan and A. Patra, ChemPhysChem, 2015, 16, 1017–1025 CrossRef CAS PubMed.
- P.-Y. Chang, K. Bindumadhavan and R.-A. Doong, Nanomaterials, 2015, 5, 2348–2358 CrossRef CAS PubMed.
- C. Gu, W. Guan, J.-J. Shim, Z. Fang and J. Huang, CrystEngComm, 2017, 19, 708–715 RSC.
- A. K. Haridas, C. S. Sharma, N. Y. Hebalkar and T. N. Rao, Mater. Today Energy, 2017, 4, 14–24 CrossRef.
- D. Narsimulu, E. S. Srinadhu and N. Satyanarayana, Materialia, 2018, 4, 276–281 CrossRef.
- H. Kim, M.-C. Kim, S. Choi, S.-H. Moon, Y.-S. Kim and K.-W. Park, Nanoscale, 2019, 11, 17415 RSC.
- J. Y. Cheong, C. Kim, J. S. Jang and I.-D. Kim, RSC Adv., 2016, 6, 2920 RSC.
- B. Huang, X. Li, Y. Pei, S. Li, X. Cao, R. C. Massé and G. Cao, Small, 2016, 12, 1945–1955 CrossRef CAS PubMed.
- O. Cevher and H. Akbulut, Acta Phys. Pol., A, 2017, 131, 204–206 CrossRef CAS.
- N. Wu, W. Du, X. Gao, L. Zhao, G. Liu, X. Liu, H. Wu and Y.-B. He, Nanoscale, 2018, 10, 11460 RSC.
- Y. Jiang, T. Yuan, W. Sun and M. Yan, ACS Appl. Mater. Interfaces, 2012, 4, 6216–6220 CrossRef CAS PubMed.
- H.-S. Chen and R. V. Kumar, RSC Adv., 2012, 2, 2294–2301 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00531b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*