
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface enhanced Raman spectroscopic studies on the adsorption behaviour of nitric oxide on a Ru covered Au nanoparticle film
Ming Geac,
Qian Wua,
Lu Yina,
Minmin Xuab,
Yaxian Yuan*a,
Qinghua Guoa and
Jianlin Yao *ab
*ab
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: jlyao@suda.edu.cn; yuanyaxian@suda.edu.cn
bCollaborative Innovation Center for New-type Urbanization and Social Governance of Jiangsu Province, Soochow University, Suzhou 215123, China
cCollege of Chemistry and Chemical Engineering, Nantong University, Nantong 226001, China
First published on 26th March 2020
Abstract
Nitric oxide (NO) is very interesting because of its effects on air pollution and especially biological systems. The adsorption behavior of NO molecules has fundamental importance with great technical challenges due to complex processes and species identification. Herein, the NO adsorption behavior on a Ru surface has been investigated using well-designed surface enhanced Raman spectroscopy (SERS) substrates. A Au nanoparticle monolayer film on ITO was employed as the electrode and Ru layers were electrochemically deposited. The internal SERS effect from the Au nanoparticles with high sensitivity and the metallic surfaces of Ru with practical application were integrated into a composite Au/Ru substrate. The molecular adsorption and dissociation of NO were observed simultaneously by SERS. A competitive relationship between adsorption and dissociation was observed at higher NO pressure, and the 3-fold and 2-fold bridge and top adsorption configurations appeared on the surface and were associated with different νNO vibrational frequencies. The results indicated that 3-fold bridge sites are preferred for dissociation over other structures. The dissociation of NO produced adsorbed atomic nitrogen and oxygen species to form Ru–N and Ru–O bonds, respectively. The dissociation process, especially for linear NO, was site dependent and blocked at higher pressure or coverage. Due to the change in adsorption energy and coverage, a conversion of the adsorption configuration from bridge to top was observed in the initial stage of NO adsorption, and this was followed by a mixture of bridge and top configurations of NO and dissociated species. A two-step dissociation mechanism and the steps of NO adsorption were proposed. The present study suggested that the SERS technique with appropriate attractive metal overlayers provided a significant and possibly even a valuable approach to explore adsorption behavior and kinetics at gas–solid interfaces.
Introduction
Nitric oxide (NO) is a naturally produced free radical in the human body. The misregulation of NO always leads to various diseases, such as inflammation,1 neurological disorders2 and cancer.3 Moreover, NO is one of the most common air pollutants released in the combustion of fossil fuels, which causes harm to both human health and the environment.4,5 From a medical point of view as well as an industrial perspective, the detection and determination of NO in a rapid, highly sensitive way is an important and significant challenge.For many years, various techniques have been developed for the detection of NO, such as fluorescence,6,7 chemiluminescence,8 electron spin resonance spectrophotometry,9 colorimetry10 and electrochemical methods.11,12 Among these methods, fluorescence and electrochemical techniques have become powerful tools for the investigation of this radical species. The fluorescence approach allows in situ detection, but it is susceptible to photobleaching and phototoxicity.13 Electrochemical detection is a feasible method for measuring NO owing to the small electrode size, non-destructive mode of analysis, and minimal or no reagent requirements. For instance, semiconducting metal oxides are the most widely used gas sensors due to the remarkable change in their electrical conductivity in gaseous atmosphere.14 However, these techniques have limited applicability in complex media, and exhibit long reaction times, and low sensitivity and selectivity. In contrast, high sensitivity and selective detection make surface enhanced Raman spectroscopy (SERS) widely used in surface science, sensing, and in situ monitoring of chemical reactions.15–18 SERS-based sensors allow the continuous, precise, and real-time detection of NO. So far, the SERS-based measurement of NO in living cells has been achieved through integrating SERS-active nanostructures with Raman reporter molecules. After the reaction between the reporter and NO, the changes in the spectral features allow us to recognize NO.19,20 These achievements demonstrated that SERS could be developed as one of the powerful tools for the real-time and in situ monitoring of NO.
The Group VIII metals are of particular interest for catalytic oxidation reactions in the gas phase. For example, Ru based catalysts have attracted considerable attention due to their excellent performance in various practical catalytic reactions.21 Moreover, Ru is the most active metal among the three Group VIII metals.22 Several investigations have been performed to elucidate the adsorption and reaction of NO on Ru surfaces. Thomas presented two adsorption types for NO on the Ru (001) surface with different activation energies for dissociation.23 Reed reported chemical evidence that NO was primarily adsorbed associatively on Ru (101) at 300 K and suggested that the dissociation reaction was sensitive to the surface crystallography.24 To date, there is still a great deal of disagreement regarding the detailed NO structure at metal surfaces because of the distinction between molecular and dissociative adsorption. For example, an infrared absorption spectroscopic investigation of adsorbed NO/Ru (001) demonstrated the absence of spectral features associated with the triple-bridge bonded NO species.25 Low energy electron diffraction (LEED) and photoelectron diffraction studies demonstrated that assignment of the adsorption configuration of NO was ambiguous based on the frequencies of the NO stretching vibrational mode.26–28 However, to solve these problems, SERS as a “fingerprinting” information tool provides a direct and viable method for studying molecular adsorption at metal surfaces.
In this study, SERS investigations of the NO adsorption kinetics at gas–Ru interfaces were performed. A Ru layer was electrochemically deposited onto an Au nanoparticle monolayer film to form Au/Ru composite nanostructures. The giant SERS effect of the inner Au nanoparticles enhanced the surface Raman signal of NO adsorbed on the external Ru surface through the long distance enhancement effect. SERS with high surface sensitivity provides direct evidence at the molecular level. Thus, it allows good understanding and reinterpretation of the NO adsorption and dissociation behaviors. It is believed that the present SERS-based technique provides a new approach to screening real NO–metal catalytic systems at a fundamental level.
Experimental
Chemicals
Nitric oxide (99.99%) was obtained from Shanghai WuGang Gas Co. Ltd. and used without further purification. HAuCl4·4H2O, RuCl3, and sodium citrate were purchased from Shanghai Reagent Co. Ltd. Indium tin oxide (ITO) was purchased from CSG Holding Co. Ltd. Polyvinylpyrrolidone (PVP, Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was supplied by Sigma. All chemicals used were of analytical reagent grade. Milli-Q water (18.2 MΩ cm) was used for all experiments.
000) was supplied by Sigma. All chemicals used were of analytical reagent grade. Milli-Q water (18.2 MΩ cm) was used for all experiments.
Apparatus
The experiments were conducted by using a self-built gas-flow apparatus. The SERS substrate was fixed at the bottom of the glass chamber with a magnetic sheet stuck in the middle, while being held parallel to an optical glass viewport mounted on the detector channel. The objective lens of the Raman spectrometer can be extended up and down in the detection port. The gas flow was detected in situ by a digital barometer. A three-way valve was connected to the inlet of the chamber for controlling the flow rate. The sample chamber was evacuated to 10−3 torr by a mechanical pump before the SERS measurements.SERS experiments were performed by using a Jobin Yvon LabRam HR800 from HORIBA. The incident power of a He–Ne laser of 632.8 nm is ca. 5 mW on the SERS substrate. The objective lens was 50× with a working distance of 8 mm. The slit width and the confocal hole were 100 μm and 400 μm, respectively, to achieve a reasonable confocal performance. The electrochemical measurements were carried out on a CHI660B electrochemical workstation (Shanghai Chenhua) in a three-electrode system. All electrode potentials are quoted versus the saturated calomel electrode (SCE) and the measurements were made at room temperature.
The SERS substrate
A composite Au/Ru nanoparticle film supported on a 0.25 cm2 ITO surface was used as the SERS substrate. An Au nanoparticle monolayer film with excellent monodispersion was fabricated based on our previous studies.29 Briefly, Au nanoparticles with the diameter of about 35 nm were prepared using the seed growth method. Then, the Au monolayer film was assembled by the formation of binary colloidal monolayers at the air–water interface. Finally, it was transferred onto an ITO wafer by the dip-coating method, thus the thickness of the monolayer film is the same as the diameter of the Au nanoparticles. The ITO/Au substrate was then electrochemically polished at a potential of 1.4 V for 10 s and roughened in 0.1 M HCl for 30 s. The electrodeposition of the metal overlayers was performed by using 1 mM RuCl3 and 0.1 M HClO4 solutions. The Ru film was electrodeposited by the galvanostatic method at a constant current of 0.1 mA.Results and discussion
Optimization of the SERS activity of the Ru overlayers
After the substrate was placed in the reactor, the chamber was evacuated to less than 10−3 torr and then filled with NO gas with a flow rate of about 15 mL min−1. Generally, it was not possible to detect the species adsorbed on the pure Ru surface because of its poor SERS activity. Alternatively, using the borrowing strategy, the Raman signal of molecules adsorbed on the Ru surface was significantly enhanced by the SERS effect from the Au nanoparticles inside. However, the thickness of the Ru layers on the Au nanoparticles became a critical factor for the SERS effect. It is essential to consider dual effects in investigations of the adsorption at Ru surfaces. A thicker Ru layer on the Au nanoparticles allowed the material to exhibit similar metallic properties to bulk Ru. Unfortunately, a thicker layer of Ru brings about significant damping of the SERS effect originating from the Au nanoparticles underneath. A thinner layer resulted in a slight difference in the electronic state of the Ru overlayer compared with the bulk material. However, the high catalytic activity originates from the first several atomic layers rather than the total bulk metal, and nanostructures are always employed in heterogenous catalysis. Therefore, it is essential to balance the SERS effect and the metallic properties of Ru in the fabrication of Au/Ru films. As seen in Fig. 1, electrochemical depositions of Ru with different durations were performed to examine the optimal Ru thickness for SERS detection, and SERS spectra were collected after the adsorption of NO gas for 2 min and 8 min. Two peaks in the relatively low frequency region were observed at ca. 250 cm−1 and 315 cm−1 with a broader FWHM (full width at half maximum) (Fig. 1A and B). Mamede and co-workers presented the Raman assignments of various N-containing adsorbed species over Pd and Pt surfaces during the CO–NO reaction. The band at 240 cm−1 was attributed to the Pt–NO stretching mode.30 Weaver reported real-time voltammetric SERS to reveal the electrochemically formed oxides on Pt and the band at 250 cm−1 was tentatively attributed to the M–NO vibration.31 Neyman investigated the high-resolution electron energy loss spectroscopy (EELS) of NO/Ru (001) adsorption complexes by using the orbital local density functional model, defining 250 cm−1 as ω(Ru–NO) of the Ru–NO cluster.32 Therefore, it was reasonable to assign this bands to the Ru–NO bond. The intensity of the peak at ca. 250 cm−1 was dependent on the deposition duration of the Ru overlayer and the adsorption time of NO, i.e. it increased at longer deposition times, and reached a maximum when the duration was about 13 s (as shown in Fig. 1C). Therefore, an Au/Ru film fabricated with this optimal process was used as the appropriate SERS substrate in the following studies. By a rough estimation based on the electrochemical reaction, the preferred thickness of Ru is about two layers for the deposition duration of 13 s. The occurrence of the band at 315 cm−1 indicated the complexity of the surface adsorption of NO, and the detailed investigation and assignments are presented in the next section. Indeed, when the thickness of the Ru layer was increased in the electrodeposition process, the following two negative effects should be pointed out: (i) it resulted in more lattice mismatch,33 which contributed to stronger forces between NO molecules and increasing probability of NO desorption from the surface. This could be understood in terms of the blue-shift in the Ru–NO peak frequency from 253 cm−1 to 257 cm−1. (ii) It caused a damping of the SERS effect due to the weakening long distance enhancement.34,35 | ||
| Fig. 1 Optimization of Ru layer thickness for different deposition times. SERS spectra of NO with different adsorption times of (A) 2 min and (B) 8 min were selected. (C) Statistical diagram of the Ru–NO band for different deposition times. | ||
NO adsorption on Au/Ru film
In order to obtain deeper insight into the behaviour and kinetics of NO adsorption, time dependent spectra were acquired following NO dosage. The time shown for each spectrum in Fig. 2 refers to the end of each accumulation period. | ||
| Fig. 2 Time dependent SERS spectra of an Au/Ru substrate exposed to NO gas. (A) Low frequency region; (B) high frequency region. (a) 2 min; (b) 6 min; (c) 10 min; (d) 14 min; (e) 18 min; (f) 22 min; (g) 26 min; (h) 30 min; (i) 34 min; (j) 38 min. | ||
The corresponding spectra including the intramolecular N–O and the metal–NO vibrational modes are presented separately as shown in Fig. 2. In the low frequency region (Fig. 2A), the spectra were dominated by an intense band at 250 cm−1 assigned to Ru–NO, together with broader and weak bands at 315 cm−1 and 470 cm−1. The spectral feature of the Ru–NO band at 250 cm−1 changed unambiguously on increasing the duration of NO introduction, i.e. a gradual downward trend in intensity and a red-shift in frequency were observed. This mainly originated from a reinforcement of the intramolecular interactions of NO. The spectral feature of the N–O stretching vibrational mode was affected by the dynamic dipole–dipole coupling effect between the adsorbed NO molecules, which induced a blue-shift of the N–O band frequency, i.e. a strengthening of the intramolecular interactions of NO. Weaver and co-workers observed the dissociation of NO on a metal surface to produce atomic nitrogen.36,37 The band located at about 315 cm−1 was associated with the adsorption of atomic nitrogen, and it was assigned to the Ru–N stretching vibrational mode. Its intensity increased quickly in the initial stage of NO introduction followed by a slight change over the whole duration (Fig. 3B). Based on the above fact, it was reasonable to assume that the NO dissociation reaction occurred and produced atomic nitrogen. The observation of the band at about 470 cm−1 was also associated with the Ru–NO bond, and it was demonstrated that it originated from the other product obtained during NO dissociation.
 | ||
| Fig. 3 Peak intensity and frequency–time profiles of NO adsorbed on Au/Ru surfaces in the low frequency region. (A) 250 cm−1 band; (B) 315 cm−1 band. | ||
Although the structure of the diatomic NO molecule is simple, understanding the surface adsorption and the reaction of NO at Ru surfaces still remains a significant challenge. A rather simple view of NO adsorption was obtained from NO dissociation by Rhodin and co-workers.38 They demonstrated that the NO adsorption chemistry on metal surfaces was very complex, and a large number of NO adsorption configurations were proposed. They assigned a broad range of N–O stretching frequencies, ranging from 1300 cm−1 to 1900 cm−1, resulting in difficulties in assigning a specific adsorption configuration. Brown has reported the observed vibrational frequencies and assignments of surface adsorbates.39 In particular, the adsorption of NO was classified into four configurations of 3-fold bridge (with three Ru atoms), 2-fold bridge (with two Ru atoms), top and bent, giving rise to different vibrational frequencies. Fig. 2B presents the real-time SERS spectra in the high frequency region simultaneously. According to calculations and experiments,32,39–42 the bands observed in this region were assigned as follows: the band at 1345 cm−1 was attributed to the 3-fold bridge site; the band at 1600 cm−1 was assigned to the 2-fold bridge site; and the band at 1830 cm−1 was associated with the top site (linearly-bonded) configurations. As shown in Fig. 3A and 4A, the frequency of the Ru–NO band was red-shifted from 253 cm−1 to 245 cm−1, while the νN–O bands were blue shifted in the initial stage, indicating enhancement of the intermolecular forces with increasing NO adsorption. One could find that the intensities of the bands associated with the 3-fold and 2-fold configurations were gradually decreased (Fig. 4B). The intensity of the band for top adsorption exhibited a similar decreasing tendency in the later stage. The decrease in the intensities of the NO associated bands indicated that the active adsorption sites were partially occupied by various other structures such as dissociation products during the NO exposure time. In addition, a slightly broad feature centred at ca. 470 cm−1 was observed after NO exposure. It was attributed to the formation of Ru–O due to oxidation of the Ru surface. It was consistent with the formation of RuO2,43 resulting in the decrease in intensity as mentioned above. It also demonstrated that the dissociation of NO on Ru surfaces produced atomic nitrogen to form Ru–N and oxygen species to form Ru–O.
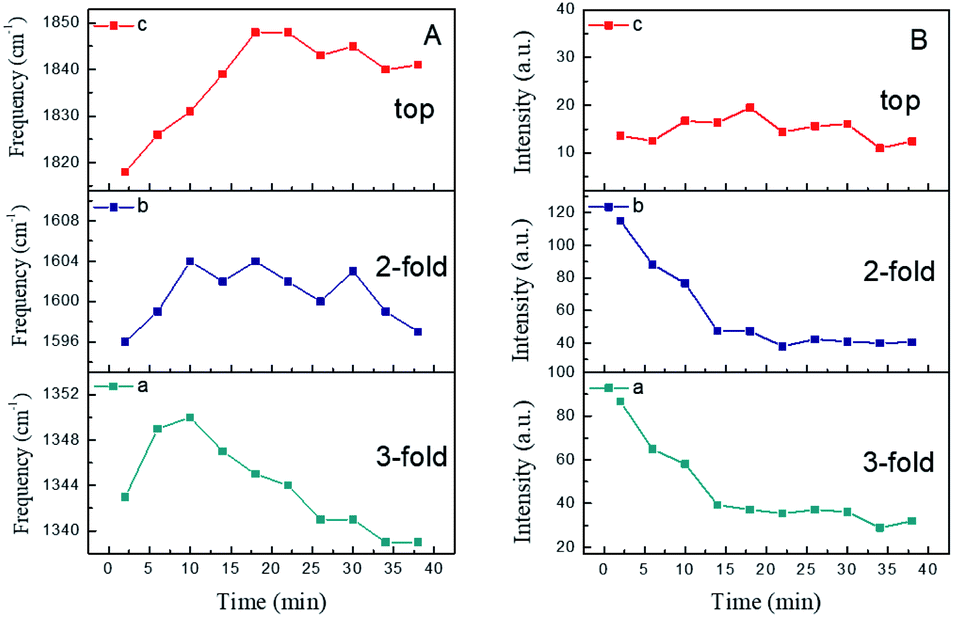 | ||
| Fig. 4 Time dependent peak frequency and intensity profiles of NO. (A) Frequencies and (B) intensities. (a) 3-fold; (b) 2-fold; (c) top. | ||
Generally, molecular adsorption on a surface involves the preferred sites, the axial geometry and interatomic spacing, which play critical roles in molecular reactivity. With further examination, the changes in frequency and intensity (Fig. 4) suggested a dynamic change in the NO structures. As mentioned above, three peaks were observed at 1345 cm−1, 1600 cm−1 and 1830 cm−1. With increasing NO exposure time, the peak at 1344 cm−1 was blue-shifted to 1350 cm−1 in the first 10 min due to increasing adsorption of NO molecules in the 3-fold bridge configuration. After that, its frequency was evidently red-shifted from 1350 cm−1 to 1340 cm−1 during the next 25 min.
This suggested that the number of adsorbed molecules in the 3-fold configuration decreased along with its dipole moment. For a clear description, it is worth comparing the frequencies and intensities of different N–O bands. In the case of the 2-fold bridge and top sites in the relatively high frequency region, it was found that the peaks increased slowly and took a long time to achieve a stable state. Additionally, the band at about 1818 cm−1 assigned to top site adsorption was blue-shifted by 29 cm−1 to 1847 cm−1 and its intensity increased in the first 20 min. However, for the same duration, the intensities of the bands associated with the 3-fold and 2-fold bridge configurations decreased significantly (as shown in Fig. 4Ba and b).
Obviously, the intensities of the bands relevant to the 3-fold and 2-fold bridge configurations decreased remarkably during the initial 15 min. Nevertheless, the band attributed to top site adsorption exhibited relatively small changes in intensity, and the absolute intensity of this mode was weaker than that of the other two bridge modes. Though the dynamic dipole coupling caused a screening effect on the electric field, the decrease in the intensities of the 2-fold and 3-fold bands is attributed to the replacement of active adsorption sites and the transformation of adsorption modes. As the number of bridge bonded molecules gradually decreases, the corresponding screening effect of dipole–dipole coupling on the electric field is reduced. This results in minor fluctuations in both the 2-fold and 3-fold bands in the later stage. Moreover, it should be noted that the intensity of the 2-fold band was found to be damped even more than that of the 3-fold band before stabilization. This indicated that 2-fold bonded molecules have a closer distance between them than 3-fold bonded molecules, resulting in a stronger molecular force as well as the screening effect.
However, compared to other weakly adsorbed dipolar molecules like carbon monoxide (CO), NO has only one unpaired π-electron. Therefore, the dissociation energy (6.529 eV) of the free molecule is much lower than that of CO (11.152 eV). This brings about richer chemisorption states and surface reactions of NO molecules on metal surfaces.39 Hence, there is a great probability of finding both molecular and dissociated NO species on Ru surfaces. NO donates its 2π* electron to the surface or accepts electron density from the surface into the half-filled 2π* orbital. On the one hand, it could be assumed that the dissociated NO species occupied the adsorption sites which blocked the adsorption of NO molecules. As a consequence, the N and O species remained on the Ru surface together with a small amount of top site adsorption. On the other hand, the bridge bonding could be replaced by top site adsorption with varying coverage.39,44 Since the lower frequency bands (1345 cm−1 and 1600 cm−1) became saturated before the equilibrium adsorption of the top state (1830 cm−1), these N–O bands were assumed to be due to different adsorbed species. Therefore, it was concluded that conversion of the adsorption configuration from bridge to top occurred in the initial stage, followed by mixed adsorption involving bridge and top configurations of NO and dissociated species.
Moreover, good understanding of the origination of this conversion is highly desired. A similar phenomenon has also been observed in NO adsorbed on Pt surfaces.45,46 It suggested that top site adsorption occurred at high coverage when the lateral repulsion between neighbouring bridged NO molecules became larger than that between top site adsorbed NO molecules. Meanwhile, various theoretical simulations have been performed to elucidate what caused the dramatic alteration in the vibrational spectral features. King reported that the 3-fold bridge sites required stronger adsorption energy than any of the others for NO on Pt (111),47 i.e. the sequence of the stability of adsorption configurations is: 3-fold > 2-fold > top. DFT calculations on NO adsorption on Pd and Rh surfaces demonstrated that the 3-fold sites became the most stable at lower coverage.48–50 At higher coverage, the 3-fold and top configurations coexisted/coadsorbed on both Pd and Rh surfaces. Consequently, adsorption energies and coverages are the dominant reasons for the above-mentioned experimental facts.
The adsorption of NO at different pressures
Actually, the effect of total pressure on the adsorption behaviour is also considered as an important issue. To clearly describe this effect, controlled pressure dependent SERS measurements were performed at pressure differences varying from −80 kPa to −20 kPa for an adsorption time of 10 min. Some changes in the SERS spectra acquired at lower pressures were found compared with those obtained at atmospheric pressure. As seen in Fig. 5, a broad yet prominent feature at ca. 315 cm−1 appeared at the pressure difference of −80 kPa, due to the adsorbed N atoms from the dissociation process, while the N–O bands were blue-shifted obviously. However, an extremely weak feature appeared at 255 cm−1, demonstrating the occurrence of NO adsorption. Most notably, there was a significant increase in the intensity of the νRu–NO feature and a red-shift tendency for νRu–N with growing total NO pressure. | ||
| Fig. 5 SERS spectra of NO adsorption at various pressure differences relative to atmospheric pressure: (A) low frequency region; (B) high frequency region. (a) −80 kPa; (b) −60 kPa; (c) −40 kPa; (d) −20 kPa; (e) 0 kPa. | ||
The NO pressure dependent frequency and intensity profiles are presented in Fig. 6. The frequencies of the bands assigned to both Ru–NO and Ru–N (Fig. 6A) exhibited significant red-shift due to the increase in dipole–dipole coupling through NO molecular repulsion. Specifically, the relative intensities of I315 cm−1/I250 cm−1 gradually diminished with further increasing NO pressure (Fig. 6B). This indicates that there is competitive adsorption and/or co-adsorption between the NO dissociative chemisorption products and chemisorbed NO. The more intense the NO dose, the stronger the adsorption capacity of chemisorbed NO. The spectral features in the high frequency region (Fig. 5B) also revealed that the intensity of the N–O vibrational mode increased with the increase in pressure, along with a blue-shift in frequency. The blue-shift is primarily a result of the vibrational dipolar coupling of increasing numbers of parallel and adjacent NO molecules. However, at higher pressures, NO molecular adsorption was anticipated to be facilitated and dissociation was deactivated, and so the adsorption process became dominant. This issue has indeed been the focus of several reports in the literature and has been found to be coverage or pressure sensitive.51–53 For example, Oh et al. claimed that the Rh (111) surface was dominated by adsorbed nitrogen under UHV conditions,54 while Hecker assumed a NO-dominated surface on silica-supported Rh at near-ambient pressures.55 Furthermore, it has been found that at higher pressure or coverage, the larger repulsions between adatoms blocked dissociative adsorption. From an energetic point of view, molecular NO is thermodynamically more stable than dissociated NO at higher coverage.56 This suggested that NO dissociation required empty sites, and a two-step dissociation mechanism was proposed accordingly, that is, the NO adsorbed on the metal surface as it was introduced, and then reoriented to the “side-on” configuration, which caused the O atom to interact with the surface. Finally, the dissociation of “side-on” NO molecules produced N and O adatoms. As illustrated in Fig. 7, the dissociation intermediate is a “side-on” NO species. This process appears to be relevant for NO dissociation on Ni, Rh, Ru, and Ir.57,58 The above spectral features were in good agreement with the proposed dissociation mechanism.
 | ||
| Fig. 6 Peak frequencies (A) and relative intensities (B) of the NO features as a function of pressure difference in the low frequency region. | ||
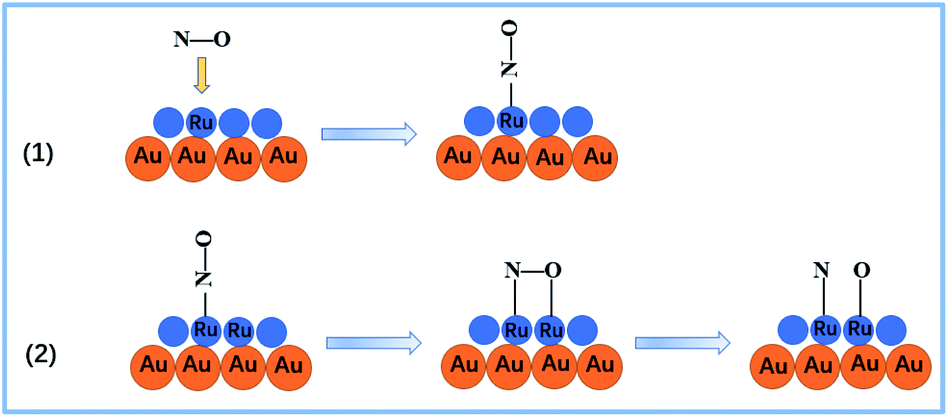 | ||
| Fig. 7 Schematic diagram of the NO adsorption and dissociation processes.57 | ||
To verify this assumption, further investigations on the changes in frequencies and intensities have been performed in the high frequency region. In general, increasing NO pressure led to a gradual increase in intensity and frequency for the νN–O of the adsorbed species. Fig. 8B presents the intensity ratios of the three distinct N–O features relative to the minimum at −80 kPa NO. It was shown that the intensity of the 3-fold configuration exhibited a more rapid increase compared with the other two N–O features. Fig. 8A presents the frequency changes of the different adsorption sites for molecular NO. The frequency of the 3-fold band was blue-shifted dramatically with increasing pressure compared with the other two adsorption configurations, and then changed slightly at higher pressures. It should be noted that the 2-fold band showed an intense blue-shift at relatively higher pressure, while top adsorption maintained steady growth over an extended pressure range. This illustrates clearly that the bridge-bonded sites, especially the 3-fold bridging sites, became the priority adsorption sites at lower pressures. NO was more inclined to adsorb in the 3-fold configuration, while the 2-fold and top adsorption sites were less occupied. However, both bridge-bonded and linear-bonded sites were occupied over the whole pressure range. Furthermore, one can assume that the dissociation of linear NO is inhibited at higher pressure due to the lack of sites for the reaction products. This is beneficial for obtaining deeper insight into the monotonic increase in linear adsorption of NO at high pressure.
 | ||
| Fig. 8 Pressure dependent frequency (A) and peak intensity (B) profiles of NO in the high frequency region. (a) 3-fold; (b) 2-fold; (c) top. | ||
Above all, the SERS spectral features enabled us to propose that reconstructions occur during NO adsorption and the dissociation product has a negative effect on the NO adsorption process. It was concluded that the effect of pressure played an important role in molecular adsorption and dissociation. It is worth noting that molecular adsorption and dissociation were in direct competition at different pressures.
The dissociation of NO, especially the top adsorption configuration, was inhibited at higher pressures by the lack of active sites for the reaction products. Obviously, the present studies demonstrated that it was possible to obtain detailed information about the adsorption of NO on Ru surfaces. This was beneficial for developing a powerful technique for an overall understanding of gas adsorption and surface reaction processes.
NO dissociation process at Au/Ru film
As mentioned above, NO is a more reactive species in comparison with CO. NO adsorption on metal surfaces is undoubtedly a complicated process involving molecular adsorption and dissociation. Brown and King found that molecular non-dissociative adsorption only normally occurred on the surfaces of metals in the last few groups of the transition metal series together with the noble metals Cu, Ag and Au.39 Some surfaces, like W, exhibit activities for full dissociation. Nevertheless, metallic Ru is located at the borderline between molecular adsorption and dissociation.The survey results obtained by using LEED and TPD indicated that NO adsorption, for instance, on Ru (001) at room temperature, was dissociative initially, and further adsorption occurred in the molecular state at higher coverages.59,60 Additionally, previous investigations on NO adsorption at other metal surfaces assisted in developing general insights for understanding how NO interacts with the surfaces of Au/Ru films. For example, NO adsorption on Rh served as a reference due to its high similarity to Ru in chemical properties. Goodman assumed that at low coverage NO prefers the 3-fold bridge sites and then the molecules in the 3-fold hollow sites dissociate and fill the hollow sites.61
The present observations demonstrated that the initial adsorption of NO onto a Ru surface occurred dissociatively and was followed by partial oxidation of the surface. The real-time spectra presented a distinct νRu–N band during the first several minutes, suggesting rapid dissociation of NO and precedence over adsorption of molecular NO. Williams has investigated the dissociation process of NO on Rh and claimed that it took about 40 seconds to observe the νRh–N band at room temperature.62
It was concluded that a 3-fold bridge molecular NO species was the precursor before dissociation, as the 3-fold bridge adsorption sites were occupied prior to the others. At lower coverage of N and O adatoms, most NO adsorbed firstly into the (vacant) bridged sites, which were energetically preferred. After that, the dissociation products occupied the same adsorption sites as the bridged state of molecular NO. It was suggested that the dissociation products and molecular NO filled the 3-fold sites, and this made the incoming NO molecules adsorb at the top sites. Besides, the oxygen adatoms produced during NO dissociation adsorbed in the hollow sites to cause the oxidation of the Ru surface. This indicated that N adatoms diffused into the near-surface metal region rather than the oxide of Ru. This was confirmed by the observation of attenuation of the intensity of the Ru–N stretching mode after a period of time.
Finally, the dissociation of the linearly adsorbed NO is inhibited at higher pressure or coverage by the lack of sites for the reaction products. Two proposed procedures for NO adsorption and dissociation are demonstrated as follows (also see the schematic diagram in Fig. 7):
| NO + Ru ↔ Ru–NOads | (1) |
| Ru–NOads + Ru → Ru–Nads + Ru–Oads | (2) |
This indicated that two adjacent active sites were involved in the surface dissociation. The top linear adsorption of NO occurred at high pressure or coverage, which resulted in the absence of adjacent sites essential for dissociation. Unfortunately, a rigorous quantitative analysis is blocked by the lack of accurate information regarding surface coverage, although some preliminary assumptions about the relationship between surfaces coverage and SERS intensity have been made.62 With molecular adsorption to saturation, the desorption processes were definitely necessary to vacate the active sites before the occurrence of dissociation.
Conclusions
The NO adsorption and dissociation behaviour on the Ru surface has been investigated using well-designed SERS substrates and the borrowing strategy. An Au monolayer nanoparticle film covered with continuous Ru layers was employed as the substrate. Internal SERS with high sensitivity and a metallic surface with practical application were integrated into a composite of Au/Ru. Firstly, the competitive relationship between molecular adsorption and dissociation was demonstrated under higher pressure of NO gas. Secondly, 3-fold sites are preferred for dissociation over the other structures and conversion from bridge-bonded sites to top linear-bonded sites occurs. Finally, a two-step dissociation mechanism and the steps of NO adsorption were proposed accordingly. However, the dissociation process, especially for linear NO, was site dependent and blocked at higher pressure or coverage.Although the present study constituted a preliminary survey, the results suggested that the SERS technique with appropriate attractive metal overlayers provided a significant and possibly even valuable means to explore adsorptive behaviour and kinetics at gas–solid interfaces. In some respects, the ability of SERS to access sensitively the metal surface vibrational region represents a major additional virtue of this technique. However, the SERS technique appears to be uniquely capable of monitoring the low frequency band for adsorbed atomic nitrogen and thereby elucidating its role in the reaction pathway. Additionally, an enticing virtue of this technique is its ability to acquire time dependent spectra straightforwardly and it has the benefit of allowing the observation of interfaces with relatively weak surface enhancing properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is financially supported by the National Natural Science Foundation of China (21773166 and 21673152), the Natural Science Fundamental Research Project of Jiangsu Colleges and Universities (18KJD150005), Priority Academic Program Development of Jiangsu (PAPD) and Project of Scientific and Technologic Infrastructure of Suzhou (SZS201708).References
- J. P. Eiserich, M. Hristova, C. E. Cross, A. D. Jones, B. A. Freeman and B. Halliwell, Nature, 1998, 391, 393–397 CrossRef CAS PubMed.
- A. Law, S. Gauthier and R. Quirion, Brain Res. Rev., 2001, 35, 73–96 CrossRef CAS.
- W. Xu, L. Z. Liu and M. Loizidou, Cell Res., 2002, 12, 311–320 CrossRef PubMed.
- C. T. Lee, H. Y. Lee and Y. S. Chiu, IEEE Sens. J., 2016, 16, 1 Search PubMed.
- D. J. Stuehr, J. Santolini, Z. Q. Wang, C. C. Wei and S. Adak, J. Biol. Chem., 2004, 279, 36167–36170 CrossRef CAS PubMed.
- Y. Li, Q. Liu, X. S. Liang, Q. Xiao, Y. J. Fang and Y. H. Wu, Sens. Actuators, B, 2016, 230, 405–410 CrossRef CAS.
- D. A. Jose, N. Sharma, R. Sakla, R. Kaushik and S. Gadiyaram, Methods, 2019, 168, 62–75 CrossRef PubMed.
- H. L. Wang, M. Li, B. Wang, M. Wang, I. Kurash, X. Z. Zhang and W. Y. Feng, Anal. Bioanal. Chem., 2016, 408, 5479–5488 CrossRef CAS PubMed.
- Z. H. Taha, Talanta, 2003, 61, 3–10 CrossRef CAS PubMed.
- R. Nims, J. Darbyshire and D. Wink, Methods, 1995, 7, 48–54 CrossRef CAS.
- J. H. Yoon, J. W. Shin, J. P. Lim, M. Mohsen, G. B. Bapurao, T. Lee and J. W. Choi, Colloids Surf., B, 2017, 159, 729–736 CrossRef CAS PubMed.
- M. D. Brown and M. H. Schoenfisch, Chem. Rev., 2019, 119, 11551–11575 CrossRef CAS PubMed.
- R. A. Hoebe, C. H. V. Oven, T. W. J. Gadella, P. B. Dhonukshe, C. J. F. Van Noorden and E. M. M. Manders, Nat. Biotechnol., 2007, 25, 249–253 CrossRef CAS PubMed.
- X. Wang, J. Su, H. Chen, G. D. Li, Z. Shi, H. Zou and X. Zou, ACS Appl. Mater. Interfaces, 2017, 9, 16335–16342 CrossRef CAS.
- Z. Q. Tian, B. Ren, J. F. Li and Z. L. Yang, Chem. Commun., 2007, 3514–3534 RSC.
- R. Jiang, M. Zhang, S. L. Qian, F. Yan, L. Q. Pei, S. Jin, L. B. Zhao, D. Y. Wu and Z. Q. Tian, J. Phys. Chem. C, 2016, 120, 16427–16436 CrossRef CAS.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- C. J. Zhang, E. M. You, Q. Jin, Y. X. Yuan, M. M. Xu, S. Y. Ding, J. L. Yao and Z. Q. Tian, Chem. Commun., 2017, 53, 6788–6791 RSC.
- J. Cui, K. Hu, J. J. Sun, L. L. Qu and D. W. Li, Biosens. Bioelectron., 2016, 85, 324–330 CrossRef CAS PubMed.
- L. L. Li, Y. M. Si, B. S. He and J. S. Li, Talanta, 2019, 205, 120116–120120 CrossRef CAS PubMed.
- S. Hosokawa, H. Kanai, K. Utani, Y. Taniguchi, Y. Saito and S. Imamura, Appl. Catal., B, 2003, 45, 181–187 CrossRef CAS.
- T. Lopez, P. Bosch, M. Asomoza and R. Gomez, J. Catal., 1992, 133, 247–259 CrossRef CAS.
- G. E. Thomas and W. H. Weinberg, Phys. Rev. Lett., 1978, 41, 1181–1184 CrossRef CAS.
- P. D. Reed, C. M. Comrie and R. M. Lambert, Surf. Sci., 1978, 72, 423–432 CrossRef CAS.
- B. E. Hayden, K. Kretzschmar and A. M. Bradshaw, Surf. Sci., 1983, 125, 366–376 CrossRef CAS.
- M. C. Asensio, D. P. Woodruff, A. W. Robinson, K. M. Schindler, P. Gardner and D. Ricken, Chem. Phys. Lett., 1992, 192, 259–264 CrossRef CAS.
- S. Aminpirooz, A. Schmalz, L. Becker and J. Haase, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 6337–6340 CrossRef CAS PubMed.
- L. D. Mapledoram, A. Wander and D. A. King, Chem. Phys. Lett., 1993, 208, 409–413 CrossRef CAS.
- Q. H. Guo, M. M. Xu, Y. X. Yuan, R. A. Gu and J. L. Yao, Langmuir, 2016, 32, 4530–4537 CrossRef CAS PubMed.
- A. S. Mamede, G. Leclercq, E. Payen, P. Granger and J. Grimblot, J. Mol. Struct., 2003, 651–653, 353–364 CrossRef CAS.
- Y. Zhang, X. P. Gao and M. J. Weaver, J. Phys. Chem., 1993, 97, 8656–8663 CrossRef CAS.
- K. M. Neyman, N. Rosch, K. L. Kostov, P. Jakob and D. Menzel, J. Chem. Phys., 1994, 100, 2310–2321 CrossRef CAS.
- J. F. Li, Z. L. Yang, B. Ren, G. K. Liu, P. P. Fang, Y. X. Jiang, D. Y. Wu and Z. Q. Tian, Langmuir, 2006, 22, 10372–10379 CrossRef CAS PubMed.
- J. W. Hu, S. Chen, R. P. Johnson, X. D. Lin, Z. L. Yang and A. E. Russell, J. Phys. Chem. C, 2013, 117, 24843–24850 CrossRef CAS.
- X. M. Fan, W. J. Zou, R. A. Gu and J. L. Yao, Chem. J. Chin. Univ., 2008, 29, 130–134 CAS.
- T. Wilke, X. P. Gao and M. J. Weaver, Langmuir, 1991, 7, 714–721 CrossRef CAS.
- A. A. Tolia, C. T. Williams, M. J. Weaver and C. G. Takoudis, Langmuir, 1995, 11, 3438–3445 CrossRef CAS.
- G. Broden, T. N. Rhodin, C. Brucker, R. Benbow and Z. Hurych, Surf. Sci., 1976, 59, 593–611 CrossRef CAS.
- W. A. Brown and D. A. King, J. Phys. Chem. B, 2000, 104, 2578–2595 CrossRef CAS.
- P. Jakob, M. Stichler and D. Menzel, Surf. Sci., 1997, 370, L185–L192 CrossRef CAS.
- K. Kostov, P. Jakob and D. Menzel, Surf. Sci., 1995, 331–333, 11–17 CrossRef CAS.
- F. Eh, S. Ladas, S. Kennou, A. Siokou and R. Imbihl, Surf. Sci., 1996, 35, 253–258 Search PubMed.
- H. Y. H. Chang, C. G. Takoudis and M. J. Weaver, J. Catal., 1997, 172, 336–345 CrossRef.
- N. sukahara, K. Mukai, Y. Yamashita, J. Yoshinobu and H. Aizawa, Surf. Sci., 2006, 600, 3477–3483 CrossRef.
- J. Raymond, R. J. Gorte and J. L. Gland, Surf. Sci., 1981, 102, 348–358 CrossRef.
- W. A. Brown, R. K. Sharma and D. A. King, J. Phys. Chem. B, 1998, 102, 5303–5308 CrossRef CAS.
- Q. Ge and D. A. King, Chem. Phys. Lett., 1998, 285, 15–20 CrossRef CAS.
- M. Perez-Jigato, K. Somasundram, V. Termath, N. C. Handy and D. A. King, Surf. Sci., 1997, 380, 83–90 CrossRef CAS.
- M. Bertolo and K. Jacobi, Surf. Sci., 1990, 226, 207–220 CrossRef CAS.
- C. T. Kao, G. S. Blackman, M. A. Van Hove, G. A. Somorjai and C. M. Chan, Surf. Sci., 1989, 224, 77–96 CrossRef CAS.
- D. S. King and R. R. Cavanagh, J. Chem. Phys., 1982, 76, 5634–5636 CrossRef CAS.
- S. E. Bialkowski, J. Chem. Phys., 1983, 78, 600–601 CrossRef CAS.
- H. J. Kreuzer, S. H. Payne, P. Jakob and D. Menzel, Surf. Sci., 1999, 424, 36–54 CrossRef CAS.
- S. H. Oh, G. B. Fisher, J. E. Carpenter and D. W. Goodman, J. Catal., 1986, 100, 360–376 CrossRef CAS.
- W. C. Hecker and A. T. Bell, J. Catal., 1983, 84, 200–215 CrossRef CAS.
- W. Erley, Surf. Sci., 1988, 205, L771–L776 CrossRef CAS.
- A. Sandell, A. Nilsson and N. Martensson, Surf. Sci., 1991, 251–252, 971–978 CrossRef CAS.
- J. S. Villarrubia and W. Ho, J. Chem. Phys., 1987, 87, 750–764 CrossRef CAS.
- P. Feulner, S. Kulkarni, E. Umbach and D. Menzel, Surf. Sci., 1980, 99, 489–506 CrossRef CAS.
- W. Stenzel, H. Conrad, B. E. Hayden and A. M. Bradshaw, J. Electron Spectrosc. Relat. Phenom., 1983, 29, 261 CrossRef.
- W. T. Wallace, Y. Cai, M. S. Chen and D. W. Goodman, J. Phys. Chem. B, 2006, 110, 6245–6249 CrossRef CAS.
- C. T. Williams, A. A. Tolia, M. J. Weaver and C. G. Takoudis, Chem. Eng. Sci., 1996, 51, 1673–1682 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |