
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategic design to create HER2-targeting proteins with target-binding peptides immobilized on a fibronectin type III domain scaffold†
Wanaporn Yimchuena,
Tetsuya Kadonosono *a,
Yumi Otaa,
Shinichi Satob,
Maika Kitazawaa,
Tadashi Shiozawaa,
Takahiro Kuchimaruc,
Masumi Takid,
Yuji Itoe,
Hiroyuki Nakamurab and
Shinae Kizaka-Kondoha
*a,
Yumi Otaa,
Shinichi Satob,
Maika Kitazawaa,
Tadashi Shiozawaa,
Takahiro Kuchimaruc,
Masumi Takid,
Yuji Itoe,
Hiroyuki Nakamurab and
Shinae Kizaka-Kondoha
aSchool of Life Science and Technology, Tokyo Institute of Technology, Yokohama 226-8501, Japan. E-mail: tetsuyak@bio.titech.ac.jp; Fax: +81-45-924-5848; Tel: +81-45-924-5848
bInstitute of Innovative Research, Tokyo Institute of Technology, Yokohama 226-8501, Kanagawa, Japan
cCenter for Molecular Medicine, Jichi Medical University, Shimotsuke 329-0498, Japan
dGraduate School of Informatics and Engineering, The University of Electro-Communications, Tokyo 182-8585, Japan
eGraduate School of Science and Engineering, Kagoshima University, Kagoshima 890-0065, Japan
First published on 17th April 2020
Abstract
Tumor-binding peptides such as human epidermal growth factor receptor 2 (HER2)-binding peptides are attractive therapeutic and diagnostic options for cancer. However, the HER2-binding peptides (HBPs) developed thus far are susceptible to proteolysis and lose their affinity to HER2 in vivo. In this report, a method to create a HER2-binding fluctuation-regulated affinity protein (HBP-FLAP) consisting of a fibronectin type III domain (FN3) scaffold with a structurally immobilized HBP is presented. HBPs were selected by phage-library screening and grafted onto FN3 to create FN3-HBPs, and the HBP-FLAP with the highest affinity (HBP sequence: YCAHNM) was identified after affinity maturation of the grafted HBP. HBP-FLAP containing the YCAHNM peptide showed increased proteolysis-resistance, binding to HER2 with a dissociation constant (KD) of 58 nM in ELISA and 287 nM in biolayer interferometry and specifically detects HER2-expressing cancer cells. In addition, HBP-FLAP clearly delineated HER2-expressing tumors with a half-life of 6 h after intravenous injection into tumor-bearing mice. FN3-based FLAP is an excellent platform for developing target-binding small proteins for clinical applications.
1. Introduction
The human epidermal growth factor receptor 2 (HER2) is a member of the human epidermal growth factor receptor (HER/EGFR/ErbB) family of receptor tyrosine kinases that plays vital role in human cancer.1 HER2 is a therapeutic target because of its role in the progression of many cancers, including breast cancer,2 gastric cancer3 and ovarian cancer.4 Accordingly, HER2-targeting monoclonal antibodies (mAbs) have already been approved by the Food and Drug Administration for treatment of HER2-positive cancers: trastuzumab for the treatment of breast cancer and gastric cancer;5 and pertuzumab for treatment of HER2-positive breast cancer.6 These mAbs show high stability in the body and effective treatment; however, their relatively large molecular weight (150 kDa) limits tissue penetration and production costs are high.7Tumor-binding peptides are a promising alternative to mAbs as they demonstrate greater tumor penetration because of their small molecular size.8 Various kinds of HER2-binding peptides (HBPs) have been identified, including a linear peptide KCCYSL,9 bicyclic peptides ACYLQDPNCDWWGPYCGGSG10 and a trastuzumab-derived cyclic peptide FCGDGFYACYMDV.11 Trastuzumab- and pertuzumab-derived peptides, QDVNTAVAW and EWVADVNPNSGGFIYNQYFK, respectively, have also been designed and used in tumor imaging.12,13 However, their clinical application is limited to use as diagnosis probes because short peptides are very sensitive to protease activity in circulation and tissues, resulting in reduced binding to targets. Therefore, proteins with proteolysis-resistant target-binding peptides may increase the applicability of tumor-binding peptides for clinical use.
Several strategies have been developed to increase proteolysis-resistance of peptides in vivo, including conjugation of peptides to macromolecules, including polyethylene glycol (PEG), the Fc domain, or serum albumin.14 Cyclization of peptides or grafting of peptide into cyclic peptide are also simple strategies to increase resistance to proteases.15,16 Recently, we found that the affinity and proteolysis-resistance of peptides was greatly improved after their structure was immobilized by grafting into a particular site of a protein scaffold.17 Based on this finding, we reported a computational design strategy for developing antibody mimetics named FLAP, which is fluctuation-regulated affinity protein (FLAP) having structurally immobilized mAb-derived peptide.18
In this study, we focused on the development of a HER2-targeting small protein with a structurally immobilized HBP, HBP-FLAP. The optimal HBP in an HBP-FLAP showed improved proteolysis resistance and exhibited specific HER2-binding in vitro and in vivo. These results provide new routes for developing therapeutically applicable target binding proteins.
2. Experimental section
2.1 Peptide screening
The T7 phage libraries displaying the random cyclic peptides X3CX8CX3 or X3CX9CX3, where X represents the randomized amino acids, were constructed using the T7 Select10-3b vector (Merck Millipore, Burlington, MA, USA), as described previously.19 HER2-Fc (R&D systems, Minneapolis, MN, USA) was biotinylated using a labeling kit (Dojindo, Kumamoto, Japan) and immobilized on streptavidin-conjugated magnetic beads (Tamagawa Seiki, Nagano, Japan). The beads were blocked with 0.5% BSA in PBS, incubated with the phage libraries of X3CX8CX3 (1 × 1010 pfu) or X3CX9CX3 (1 × 1010 pfu) for 1 h, washed 30 times with PBS with 1% Tween 20 (1% PBST) and then added to 7 mL of E. coli BLT5403 cells (Merck Millipore) in log phase growth. After incubation at 37 °C and bacteriolysis, the phages were recovered from the culture supernatant by centrifugation (9100 g for 10 min at 4 °C) and used for the next round of biopanning. Amino acid sequences of displayed cyclic peptides on selected T7 phage clones were analyzed by DNA sequencing.2.2 ELISA
For phage-ELISA, the wells of 96-well black plates (Thermo Fisher Scientific, Waltham, MA, USA) were coated with 1000 ng of HER2-Fc, streptavidin (Wako, Osaka, Japan), or IgG1-Fc (R&D systems) and blocked with 2% Perfect-Block (MoBiTec, Göttingen, Germany) in PBS for 1 h. Phages (1 × 1010 pfu) were added to each well and incubated for 1 h. After washing the plate, the bound phages were detected with the anti-T7 fiber tail antibody (Merck Millipore) as a primary antibody and the anti-mouse IgG HRP-linked antibody (Cell Signaling Technology, Danvers, MA, USA).For indirect ELISA, the wells were first coated with 50 ng of HER2-Fc and then blocked with 2% Perfect-Block in PBS for 2 h. After incubation with sample proteins for 1 h, the HRP-conjugated anti-His-tag antibody (Abcam, Cambridge, MA, USA) was incubated for 1 h with the samples and then treated with the QuantaRed Enhanced Chemifluorescent HRP Substrate kit (Thermo Fisher Scientific). The resulting fluorescence was measured using an Infinite F500 (Tecan, Mannedorf, Switzerland) with specific filters (Ex/Em = 535 nm/590 nm).
For specificity evaluation, the wells were incubated overnight with HER2-Fc (50 ng/50 μL in PBS), EGFR-Fc (50 ng/50 μL in PBS; R&D systems), or 50 μL PBS, and applied to the indirect ELISA described above.
2.3 Molecular dynamics (MD) simulations
Initial coordinates of the fibronectin type III domain (FN3) protein were taken from the Protein Data Bank (PDB ID: 1TTG). The structures of linear peptides and FN3 mutants were generated using Discovery Studio 3.1 (Accelrys, San Diego, CA, USA). The systems were optimized via energy minimization and equilibrated with backbone restraints. Production runs were performed for at least 10 ns for trajectory analysis. All MD simulations were performed using the Amber 14 and 16 program packages20 on TSUBAME (Global Scientific Information Center, Tokyo Institute of Technology, Japan). The Amber ff14SB force fields and the GB/SA implicit solvent model were used. The time-step for MD simulations was set to 2 fs with the SHAKE algorithm. A nonbonded cutoff of 999.9 Å was used. The temperature was kept constant at 300 K using the Berendsen rescaling method. The average root-mean-square fluctuation (RMSF) of all hydrogen-free atoms during the final 5 ns of each production run were calculated to investigate the backbone fluctuations in each system using the cpptraj module.2.4 Plasmid construction and protein purification
The recombinant DNA experiment was carried out according to the necessary regulations based on the Tokyo Institute of Technology recombinant DNA experimental safety management regulations defined by the Tokyo Institute of Technology recombinant DNA experimental safety management committee. The cDNA encoding the fusion protein, consisting of a His-tag, GGGS linker and FN3 (H-FN3), was inserted into the multi-cloning site of the pGEX-6P-3 plasmid (GE Healthcare, Little Chalfont, UK). The cDNA encoding H-FN3 mutants, C-terminally CTag (GMAQIEVNCSNE)21-fused His-tagged FLAP (H-FLAP-C), or C-terminally CTag-fused H-FN3 (H-FN3-C) were constructed by site-directed mutagenesis using the H-FN3 cDNA as the template. The plasmid vectors expressing H-FN3, H-FN3 mutants, H-FN3-C and H-FLAP-C were transformed into E. coli BL21 (DE3) pLysS cells (Promega, Fitchburg, WI, USA). These proteins were expressed in the bacteria as GST fused proteins. GST-fused H-FN3, H-FN3 mutants, H-FN3-C and H-FLAP-C were purified from the supernatants of bacterial extracts using glutathione agarose (Sigma-Aldrich, St Louis, MO, USA). The monomeric purified proteins were passed through Superdex 200 Increase 10/300 GL columns (GE Healthcare), according to the manufacturer's instructions, to increase protein sample purity.2.5 Cell immunostaining
The human cervical cancer cell line HeLa, human neuroblastoma cell line SH-SY5Y, and human breast cancer cells expressing firefly luciferase SK-BR-3 were obtained from the RIKEN Bio-Resource Center (Tsukuba, Japan). The human gastric cancer cell line N87 was obtained from the JCRB Cell Bank (Osaka, Japan). Cells were maintained with 5% fetal bovine serum-Dulbecco's Modified Eagle's Medium (Nacalai Tesque, Kyoto, Japan) supplemented with penicillin (100 U mL−1) and streptomycin (100 μg mL−1) (Nacalai Tesque) in a 5% CO2 incubator at 37 °C and regularly checked for mycoplasma contamination by a mycoplasma test kit (Lonza, Basel, Switzerland). The cells (3.0 × 104 cells per well) were seeded on a slide chamber plate, cultured for 16 h and fixed by treatment with a 4% paraformaldehyde solution for 10 min at 25 °C. The cells were then blocked with 1% BSA-PBS for 1.5 h at room temperature. After the addition of 1 μM of purified proteins to the chamber, the cells were incubated for 16 h at 4 °C. The Alexa Fluor 488-conjugated mouse anti-His tag secondary antibody (Medical & Biological Laboratories, Aichi, Japan) was then used to fluorescently label the His-tagged proteins. The slide was mounted with mounting medium (DBS, Pleasanton, CA, USA) with 1/1000 of Hoechst 33342 (Enzo Life Sciences, Farmingdale, NY, USA). All photos were taken using a BZ-X700 microscope (Keyence, Osaka, Japan) with the filters of BZ-X filter GFP, OP-87763 (Ex: 470/40 nm, Em: 525/50 nm) and BZ-X filter DAPI, OP-87762 (Ex: 360/40 nm, Em: 460/50 nm).2.6 Flow cytometry
Single cell suspensions of HeLa, SH-SY5Y, SK-BR-3 and N87 cells (1.0 × 105 cells/100 μL) were incubated in PBS containing 1000 times-diluted Alexa Fluor 488 anti-human HER2 antibody (clone 24D2; BioLegend, San Diego, CA, USA) on ice in the dark for 30 min. Cells without immunostaining were used as a negative control. Flow cytometry was performed by a flow cytometer ec800 (Sony Biotechnology, San Jose, CA, USA).2.7 Proteinase K treatment
Fifty μM of FN3-p1.M.2 or the C-terminally elongated p1.M.2 peptide (YCAHNM-GSGSGK) was digested by 50 μU mL−1 proteinase K for 2 h in 10 mM Tris–HCl (pH 8.0) containing 10 mM EDTA. Sample aliquots were collected at various reaction times and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) or reversed-phase high-performance liquid chromatography (HPLC; InertSustainSwift C18 column; GL Sciences, Tokyo, Japan). The ratios of intact FN3-p1.M.2 and the C-terminally elongated p1.M.2 peptide were calculated based on the band intensity of the digested products on the SDS-PAGE gel and the HPLC peaks in the chromatogram, respectively.2.8 Preparation of HBP-FLAP and FN3 probes
HBP-FLAP-IR800 and FN3-IR800 monomer probes were prepared by covalent conjugation of a fluorescent dye IRDye 800CW-maleimide (LI-COR Biosciences, Lincoln, NE, USA) to a cysteine residue at the C-terminus of the CTag of H-FLAP-C. Twenty-five nmol of unlabeled H-FLAP-C or H-FN3-C was reacted with 50 nmol of IRDye 800CW-maleimide in PBS for 2 h at room temperature. PD-10 desalting columns (GE healthcare) were used to purify the reactants.2.9 Biolayer interferometry
The kinetics of HBP-FLAP binding to HER2 was studied using a ForteBio Octet Red (ForteBio, CA, USA) instrument. The assays were performed at 30 °C in 96-well black plates. HER2-Fc was biotinylated using a biotin labelling kit (Dojindo), and 100 nM biotinylated HER2-Fc in the kinetic buffer (0.1% BSA, 0.002% Tween-20 in PBS) was used to load the ligand onto the surface of streptavidin biosensors (ForteBio) for 300 s. After washing (30 s) and equilibrating (60 s) the biosensor, the association of the ligand on the biosensor to the analyte in solution (100–800 nM His-tagged HBP-FLAP in the kinetic buffer) was measured for 300 s. The dissociation of the interaction was subsequently measured for 300 s. Systematic baseline drift correction was done by subtracting the shift recorded for sensors loaded with ligand but incubated without analyte. Data analysis and curve fitting were done using Octet software version 11.0 (ForteBio). Experimental data were fitted with the binding equations available for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 interaction with local fitting and the mean ± standard error of the mean (SEM) values of kon and koff were calculated from the data of four different concentrations of analytes. The KD was calculated as the ratio of koff/kon.
:1 interaction with local fitting and the mean ± standard error of the mean (SEM) values of kon and koff were calculated from the data of four different concentrations of analytes. The KD was calculated as the ratio of koff/kon.
2.10 Mice and in vivo fluorescent imaging
All animal experiments were performed with the approval of the Animal Ethics Committees of Tokyo Institute of Technology (no. 2017004) and in accordance with the Ethical Guidelines for Animal Experimentation of Tokyo Institute of Technology. Male BALB/c nude mice were purchased from Oriental Yeast Co., Ltd. (Tokyo, Japan). All mice used were 6–9 weeks of age. For the subcutaneous xenograft model, N87 cells suspended in PBS (1.0 × 106 cells/20 μL) were mixed with an equal volume of Geltrex (Life Technologies) and injected into the forefoot of mice. Mice with subcutaneous tumors of 10–15 mm in diameter were used for experiments. To acquire fluorescence images, 300 pmol in 100 μL PBS of HBP-FLAP-IR800 or FN3-IR800 was intravenously injected into mice with subcutaneous tumors in the forefoot. The following conditions were used for image acquisition: excitation filter = 710 ± 15 nm; emission filter = 800 ± 10 nm; and exposure time = 1 s. The minimum and maximum photons per s per cm2 per sr of each image are indicated in each figure by a yellow-red bar scale.2.11 Statistical analysis
Data in this study are expressed as means ± standard error of the mean (SEM). Differences are analyzed by a two-sided Student's t-test; p values of <0.05 were considered as statistically significant.3. Results
3.1 Strategy for creating HBP-FLAP
Our previous research demonstrated that short peptides grafted in a particular region of protein scaffold were structurally immobilized and showed increased resistance to proteases.17 This finding prompted us to develop clinically applicable HER2-targeting small proteins with a structurally immobilized HBP. Fig. 1 shows the process for producing such protein HBP-FLAP. FN3, one of three types of internal repeats found in the plasma protein fibronectin, was selected as the scaffold because it is an endogenous protein domain with high stability and high solubility.22,23 In addition, previous calculations revealed that residues 51–56 of FN3 are a graft acceptor (GA) site in which the structure of grafted hexapeptides can be immobilized.18 FN3 has been used for the development of affinity proteins targeting various antigens by protein engineering,23,24 some of which have entered clinical trials,25 suggesting that FN3 is a promising scaffold for creating HBP-FLAP.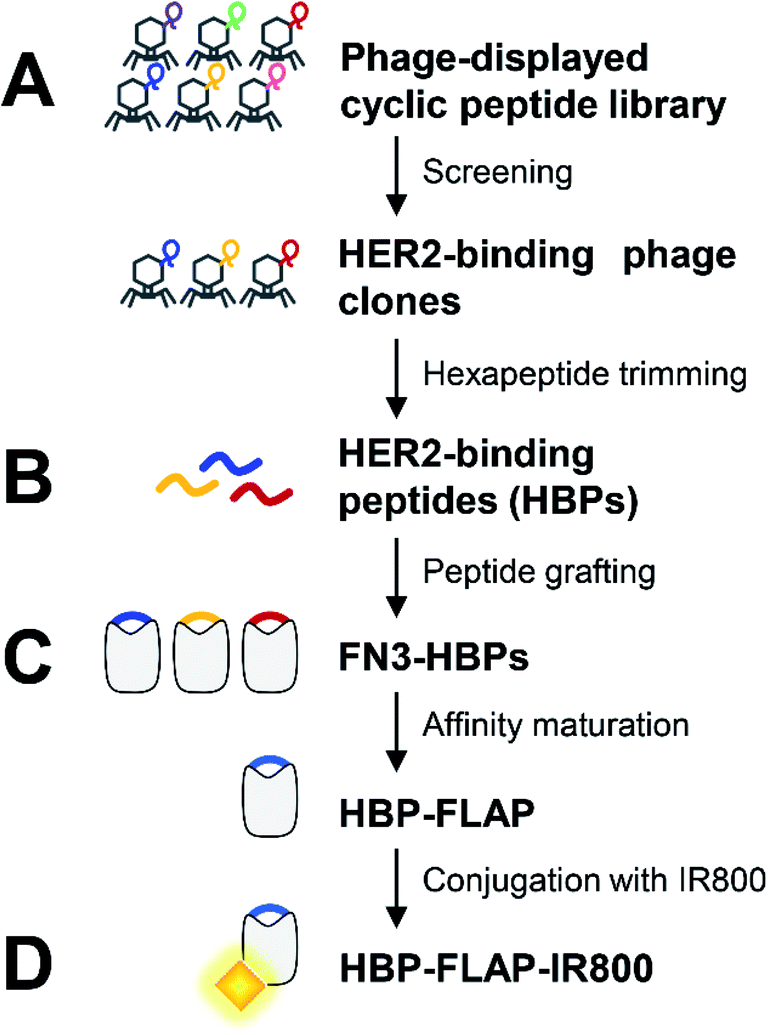 | ||
| Fig. 1 Overview of HBP-FLAP development by grafting HBP into a small protein scaffold for peptide stabilization. (A) The HBPs were isolated from a phage-displayed cyclic peptide library. (B) Hexapeptides trimmed from HBPs were grafted into the GA site of the FN3 scaffold to generate FN3-HBPs. (C) Binding affinity of FN3-HBP was improved by affinity maturation using alanine scanning and the FN3-HBP with the highest affinity was screened using a phage-displayed FN3-HBP mutant library to create the optimal HBP-FLAP. (D) The affinity-matured HBP-FLAP was conjugated with near infrared fluorescence dye IRDye 800CW (IR800) to give an in vivo imaging probe, HBP-FLAP-IR800. | ||
The HBPs were first isolated from a phage-displayed peptide library (Fig. 1A) and then some six consecutive amino acids (hexapeptides) in the isolated HBPs were selected and grafted into the GA site of FN3 to create FN3-HBPs (Fig. 1B). The binding affinity of the FN3-HBPs was improved by affinity maturation (Fig. 1C). The affinity-matured HBP-FLAP that showed the highest binding affinity among the tested FN3-HBPs was labeled with the near-infrared fluorescence dye IRDye 800CW and evaluated for target-binding ability in vitro and in vivo (Fig. 1D).
3.2 Design of a HER2-binding small proteins
Five T7 phage clones, P1–P5, displaying HBPs were enriched by eight rounds of biopanning against HER2-Fc from T7 phage libraries displaying random cyclic peptides X3CX8CX3 or X3CX9CX3, where X represents the randomized amino acids (Fig. 2A). Further evaluation of the phage clones by phage-ELISA revealed their target specificity: P1, P4 and P5 bind strongly and specifically, whereas P2 binds weakly to HER2-Fc. P3 binds to IgG-Fc non-specifically (Fig. 2B). Therefore, HBPs expressed in P1, P4 and P5 (hereafter named p1, p4 and p5) were used for the development of FN3-HBPs. Continuous hexapeptides p4.1–p4.4 and p5.1–p5.4 were prepared from p4 and p5, respectively (Table 1), and grafted into the GA site of FN3 (Fig. 2C). Note that p1 was originally displayed as a hexapeptide on T7 phage because the seventh codon is a stop codon. Computational evaluation by MD simulations confirmed a decrease in the average RMSF value. This indicates that all linear hexapeptides were immobilized in the GA site of FN3 (Fig. 2D). As a result, we obtained nine FN3-HBPs: FN3-p1, FN3-p4.1, FN3-p4.2, FN3-p4.3, FN3-p4.4, FN3-p5.1, FN3-p5.2, FN3-p5.3 and FN3-p5.4.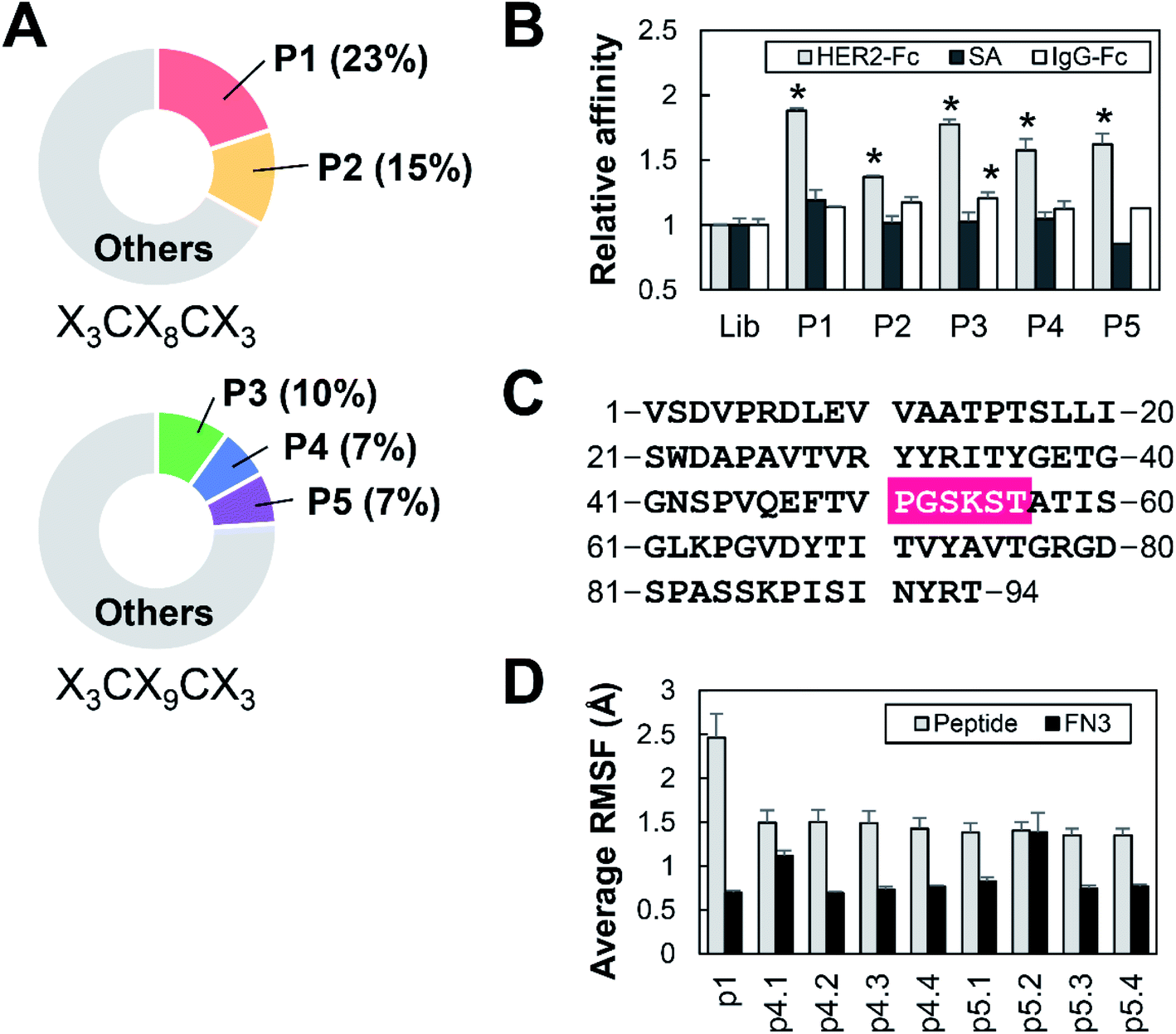 | ||
| Fig. 2 Preparation of HBP-FN3s. (A) The frequency of detection of HBPs during the phage library screening. Phage clones P1 and P2 were selected from an eight-amino acid cyclic peptide (X3CX8CX3) library. Phage clones P3–P5 were selected from a nine-amino acid peptide (X3CX9CX3) library. (B) The binding affinity of the indicated phage clones to HER2-Fc (grey), streptavidin (SA, black) and IgG-Fc (white) was measured by ELISA and the relative affinities to the control cyclic peptide library (Lib) are shown. n = 3, mean ± SEM, *p < 0.05 compared with Lib based on a Student's t-test. (C) Amino acid sequence of the FN3 domain. The HBP was grafted into FN3 by substitution at residues 51–56, which are highlighted in pink. (D) RMSF calculation of HBPs in linear form (grey) and grafted into the FN3 scaffold (black). | ||
| Peptides | Amino acid sequences |
|---|---|
| p1 | YCGCEM |
| p2 | PFICCRRPRGRRCEHG |
| p3 | SLFCWVRVM |
| p4 | AMVCTRARKPKSGCRRVG |
| p5 | VELCATAKHEVKSCLSFG |
| p4.1 | TRARKP |
| p4.2 | RARKPK |
| p4.3 | ARKPKS |
| p4.4 | RKPKSG |
| p5.1 | ATAKHE |
| p5.2 | TAKHEV |
| p5.3 | AKHEVK |
| p5.4 | KHEVKS |
| p1.1A | ACGCEM |
| p1.2A | YAGCEM |
| p1.3A | YCACEM |
| p1.4A | YCGAEM |
| p1.5A | YCGCAM |
| p1.6A | YCGCEA |
| p1.M.1 | YCICCM |
| p1.M.2 | YCAHNM |
| p1.M.3 | YCPVCM |
3.3 Affinity maturation of HER2-binding small proteins
Recombinant FN3-HBPs were purified by a bacterial protein expression system and used to determine the FN3-HBP with highest affinity to HER2. Two out of the nine FN3-HBPs, FN3-p4.1 and FN3-p5.2, were not expressed but the other seven FN3-HBPs were expressed successfully as soluble proteins and easily purified. Evaluation of the binding affinity to HER2 revealed that three out of the seven, FN3-p1, FN3-p4.3 and FN3-p4.4 bound to HER2 (Fig. 3A) with dissociation constants (KD) 280–500 nM (Table 2, ESI Fig. S1A†), indicating moderate binding affinity. Sequence optimization of p1 with the lowest KD value (KD = 280 nM) was performed to increase the affinity of FN3-p1. By single amino acid substitutions in p1 (YCGCEM) to alanine (A), six FN3-p1 mutants, FN3-p1.1A, FN3-p1.2A, FN3-p1.3A, FN3-p1.4A, FN3-p1.5A and FN3-p1.6A were generated (Table 1). Reduced binding affinity to HER2 was observed with FN3-p1.1A, FN3-p1.2A and FN3-p1.6A (Fig. 3B), indicating that the first (Y51), second (C52) and sixth (M56) residues were important for HER2 binding. Optimization of the third, fourth and fifth residues of p1 was performed using a T7 phage-displayed FN3-p1 mutant library with three randomized sequences 51-YCXXXM-56. After eight rounds of biopanning, three peptides were selected from the most frequently enriched FN3-p1 mutants: FN3-p1.M.1 (YCICCM), FN3-p1.M.2 (YCAHNM) and FN3-p1.M.3 (YCPVCM; Fig. 3C and Table 1). Amino acid substitutions made in the third, fourth and fifth residue positions showed minimal effect on structural fluctuations of Y51, C52 and M56 (Fig. 3D and E). Among the three affinity-matured FN3-p1 mutants, FN3-p1.M.2 showed the highest binding affinity toward HER2 (KD = 58 nM in ELISA and KD = 287 nM in biolayer interferometry; Table 2 and Fig. S1B†) and was selected for further characterization.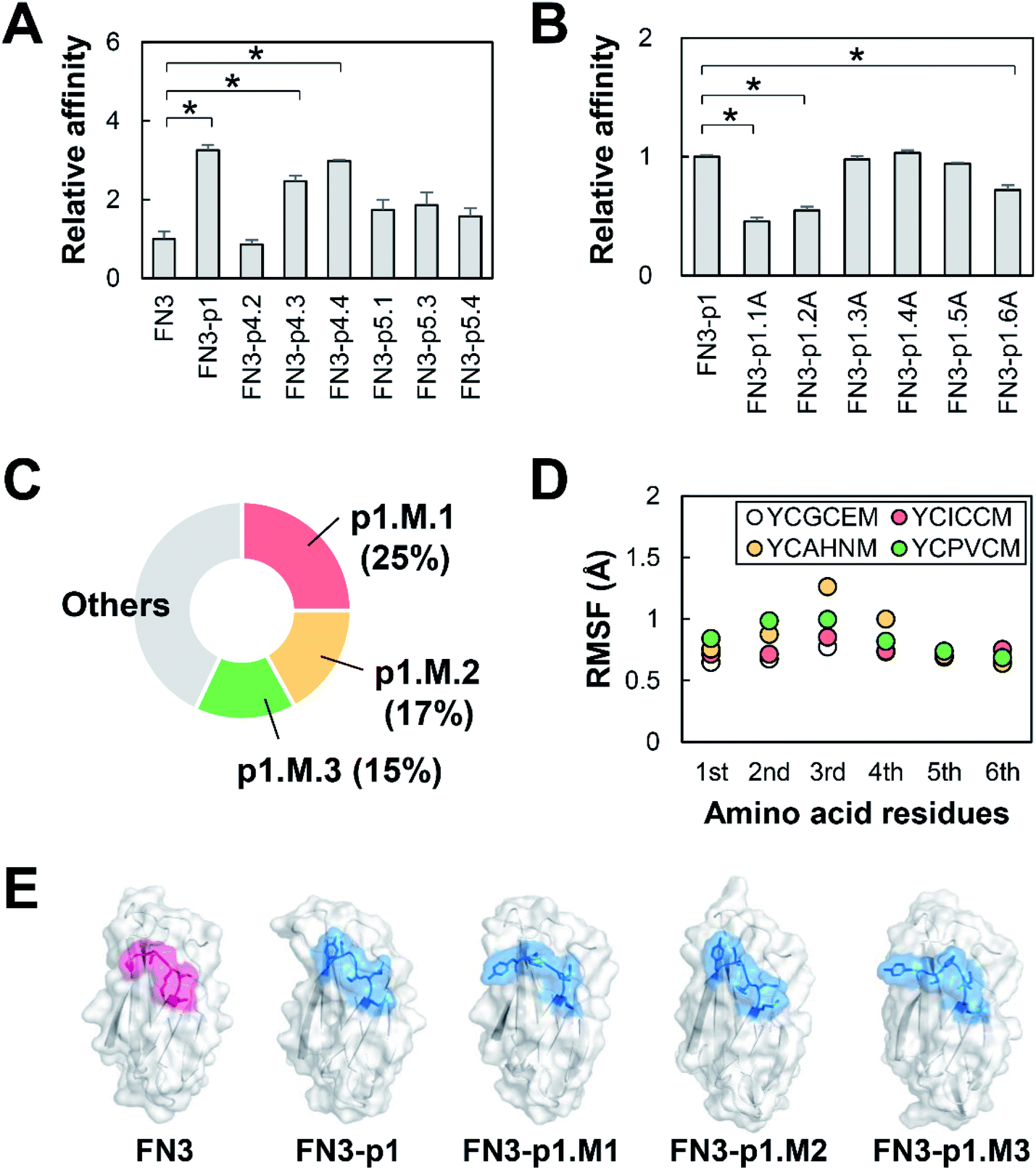 | ||
| Fig. 3 Affinity maturation of HER2-binding small proteins. (A) ELISA was used to measure the affinity of FN3-HBP mutants toward HER2-Fc. The mean ± SEM of three independent experiments is shown; *p < 0.05. (B) ELISA measurements showing the affinity of alanine substitution mutants of FN3-p1 toward HER2-Fc. The mean ± SEM of three independent experiments is shown; *p < 0.05. (C) The pie chart shows the frequency of detection of the HBP sequence (YCXXXM) in screening after affinity maturation. (D) Fluctuation of the residues of the indicated HBP sequences (YCXXXM) in FN3-p1. The RMSF values of each residue are shown. (E) Predicted structures of FN3, FN3-p1, and FN3-p1.M.1–M.3 showing the GA site (pink) and the grafted HBPs (blue). | ||
| FN3-HBPs | KD (nM) |
|---|---|
| FN3-p1 | 280 ± 23 |
| FN3-p4.3 | 450 ± 74 |
| FN3-p4.4 | 500 ± 150 |
| FN3-p1.M.1 | 68 ± 14 |
| FN3-p1.M.2 | 58 ± 5.0 |
| FN3-p1.M.3 | 110 ± 28 |
3.4 Characterization of HBP-FLAP in vitro
The proteolysis-resistance of p1.M.2 peptide (YCAHNM) was evaluated by treatment with proteinase K, which cleaves peptide bonds adjacent to the carboxyl group of the 1st (Y) and 3rd (A) residues (Fig. 4A). A C-terminally elongated synthetic p1.M.2 peptide (YCAHNM-GSGSGK) was used for easy detection of peptide-proteolysis by HPLC. As expected, the p1.M.2 peptide in FN3 was resistant to proteinase K treatment for 2 h, whereas the linear p1.M.2 was digested within 10 min (Fig. 4B and S2†).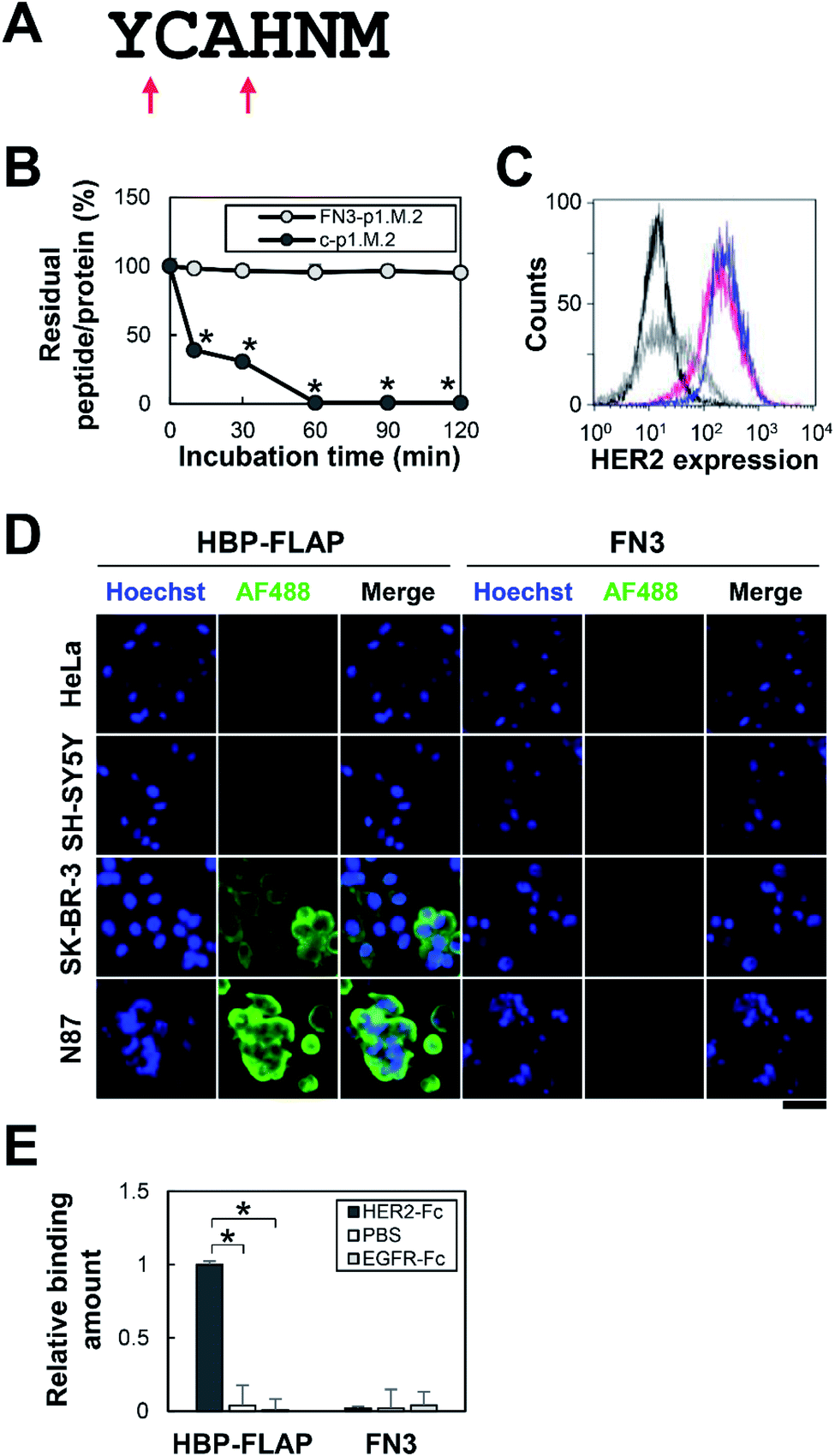 | ||
| Fig. 4 Proteolysis-resistance of the p1.M.2 peptide and binding specificity of HBP-FLAP to HER2-expressing cells. (A) Cleavage sites of the p1.M.2 peptide by proteinase K. Peptide bonds adjacent to the carboxyl group of the 1st (Y) and 3rd (A) residues are cleaved (red arrows). (B) Proteinase K resistance of p1.M.2 peptides. Proteinase K-treated FN3-p1.M.2 and the C-terminally elongated p1.M.2 peptide (c-p1.M.2) were analyzed at 10, 30, 60, 90 and 120 min. Percentage of intact FN3-p1.M.2 and the C-terminally elongated p1.M.2 peptide was calculated by SDS-PAGE and HPLC analysis, respectively. n = 3, mean ± SEM, *p < 0.05 compared with incubation time at 0 min based on a Student's t-test. (C) Flow cytometry analysis of HER2 expression on the cell surface of HeLa (black line), SH-SY5Y (gray line), SK-BR-3 (blue line), and N87 (magenta line) cells. Histograms are representative of duplicate samples. (D) Evaluation of HER2-binding affinity of HBP-FLAP to HeLa, SH-SY5Y, SK-BR-3 and N87 cells. HBP-FLAP and control FN3 were labeled with AF488 (green) and reacted with fixed cells. The nuclei were stained with Hoechst (blue). Scale bar = 50 μm. (E) Specific binding of HBP-FLAP to HER2. HBP-FLAP binding to HER2-Fc, non-coated well (PBS), or EGFR-Fc were evaluated by ELISA. The mean ± SEM of three independent experiments is shown; *p < 0.05. | ||
The binding specificity of FN3-p1.M.2, hereinafter referred to as HBP-FLAP, was then evaluated using cell lines expressing different levels of HER2. HeLa cells (no HER2 expression), SH-SY5Y cells (no HER2 expression), SK-BR-3 cells (HER2 expression), or N87 cells (HER2 expression) were incubated with the HBP-FLAP, followed by staining with a fluorescently labeled anti-His-tag antibody that binds to the His-tag at the N-terminus of FN3. The fluorescence intensity of the cells was observed to correlate with the expression level of HER2 (Fig. 4C and D). The specificity of HBP-FLAP for HER2-Fc was further confirmed by an ELISA using epidermal growth factor receptor (EGFR)-Fc-coated wells: HBP-FLAP hardly bound to EGFR-Fc-coated wells (Fig. 4E). These results showed that the HBP-FLAP has high specificity for HER2.
3.5 In vivo fluorescent imaging of a HER2-expressing tumor by HBP-FLAP
To assess the applicability of HBP-FLAP as a probe for targeting a HER2-expressing tumor in vivo, the HBP-FLAP and FN3 scaffold were labeled with a near-infrared fluorescent dye IR800CW (HBP-FLAP-IR800 and FN3-IR800; MW = 10 kDa) (Fig. 1D, 5A and S3†). In vivo imaging was performed at various post-injection (p.i.) times to monitor HBP-FLAP-IR800 and FN3-IR800 probes in N87 tumor-bearing nude mice. Although similar fluorescent intensities were observed in tumors at 0.5 h p.i. of the probes, higher fluorescent signals were detected at longer periods in tumors from mice injected with HBP-FLAP-IR800 (Fig. 5B and C). HBP-FLAP-IR800 specifically accumulated at HER2-expressing tumors with higher T/B ratios when compared with that of FN3-IR800 (Fig. 5D). As expected from the molecular weight of the probes, both probes were mainly excreted through the urinary system (Fig. 5B). These results suggested that binding to HER2 retained HBP-FLAP for longer periods in tumors and that the HBP-FLAP design is a promising strategy for developing clinically applicable small proteins using target-binding peptides.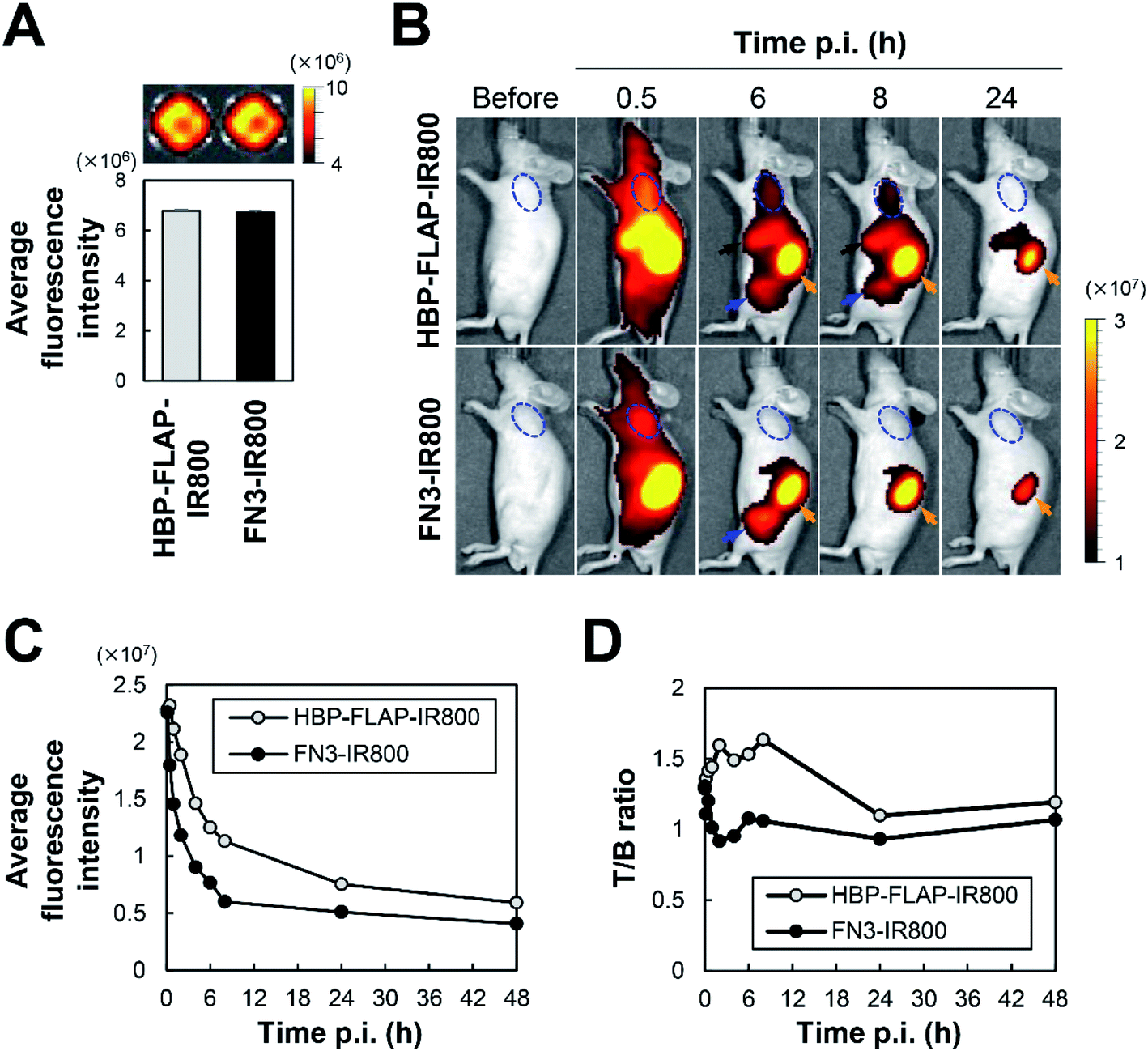 | ||
| Fig. 5 In vivo fluorescence imaging of a HER2-expressing tumor. (A) Average fluorescent intensity (photons per s per cm2 per sr) of HBP-FLAP-IR800 and FN3-IR800 probe before i.v. injection; n = 3, mean ± SEM. (B) Representative in vivo images of N87 tumor-bearing mice at the indicated times of p.i. of the probes shown in (A). A dashed blue circle indicates the tumor, a black arrow indicates the liver, an orange arrow indicates the kidney and the blue arrow indicates the bladder. (C) Average fluorescent intensity (photons per s per cm2 per sr) of HBP-FLAP-IR800 and FN3-IR800 probes in tumors. (D) The ratio of tumor (fluorescence intensity of tumors) versus background (fluorescence intensity of muscles) (T/B ratio) of HBP-FLAP-IR800 and FN3-IR800 probes at the indicated times of p.i. | ||
4. Discussion
In this study, we successfully developed an HBP-FLAP that specifically binds to HER2 (Table 2 and Fig. 4D) and showed applicability for targeting HER2-expressing tumors in vivo (Fig. 5). Although target-binding peptides/proteins selected by in vitro screening may bind to off-targets in vivo, HBP-FLAP likely binds to HER2 specifically because no detectable signal was observed in parts of the body other than HER2-expressing tumors and excretion organs (Fig. 5B). In the evaluation of FN3-HBPs (Fig. 2D and 3A), all grafted hexapeptides were immobilized into the FN3 scaffold and three FN3-HBPs bound to HER2, whereas the other FN3-HBPs may lose critical residues for binding. This result shows that the design method of FLAP is a promising strategy to develop target-binding proteins by grafting target-binding peptides to the GA site of the FN3 scaffold and can contribute to the development of clinically applicable target-binding proteins. In comparison to HBP, HBP-FLAP was more resistant to proteinase K (Fig. 4B). The HBP of HBP-FLAP must be exposed on the surface of the molecule because the GA site was selected because it is a solvent exposed loop in the FN3 structure.18 Therefore, the proteolysis resistance of the HBP in HBP-FLAP is not because of reduced protease access to the target sequence but is likely due to its immobilized structure, which restricts protease activity.The linear HER2-targeting peptide (KCCYSL)9 and cyclic peptide (FCGDGFYACYMDV)11 reported in other studies showed less serum stability, which might be caused by susceptibility to proteases. These peptides were PEGylated to increase resistance to proteases and the molecular mass, resulting in prolonged blood circulation.26,27 Another study demonstrated that conjugation of a bicyclic peptide to an albumin-binding peptide increased proteolysis-resistance.28 Our study reported a different strategy to improve the proteolysis-resistance of HBP by peptide grafting, which directly immobilized the conformation of the peptide.
The FLAP design might be a preferable strategy for developing clinically applicable proteolysis-resistant molecules from target-binding peptides when compared with cyclization or grafting into cyclic peptide approaches, because many proteins with a variety of molecular sizes and charges can be used as scaffolds for developing FLAPs. Therefore, we can optimize the in vivo retention time of FLAPs by using the appropriate scaffolds. In comparison to other strategies such as PEGylation, Fc-fusion and albumin-fusion, in which either the C- or N-terminus of the target-binding peptides is exposed, the FLAP design can mask both termini by covalent conjugation with the scaffold, suggesting that peptides in FLAP also show higher proteolysis-resistance to exo-proteases in the body when compared with that of peptides with other modifications.
We showed the application of HBP-FLAP as a diagnostic probe (in vitro immunostaining and in vivo imaging) in this study. To address the applicability of HBP-FLAP for tumor treatment, we need to further evaluate inhibition of HER2-signaling, cancer cell proliferation and tumor growth. Theragnostic probes by radioisotope- and near-infrared dye-conjugations are also possible applications of FLAPs.
5. Conclusion
We successfully developed a HER2-binding protein, HBP-FLAP, by applying the design method of FLAP. HBP-FLAP specifically binds to HER2 and has improved resistance to proteases. In addition, HBP-FLAP exhibited in vivo applicability as a HER2-expressing tumor-targeting probe. FLAP design can be applied to any other target-binding peptides. Thus this study provides a promising strategy for creating useful tools for basic, preclinical and clinical studies.Author contributions
S. K.-K. and T. K. designed and supervised the experiments. Y. O., T. K., M. K., T. S., M. T. and Y. I. provided T7 phage libraries and performed screenings. W. Y., Y. O., T. K., S. S., H. N. and T. Ku. validated the designed proteins. W. Y., T. K. and S. K.-K. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to Biomaterials Analysis Division, Technical Department, Tokyo Institute of Technology for their DNA sequence analyses. We also sincerely thank Dr Hiroshi Ueda, Institute of Innovative Research, Tokyo Institute of Technology for the support of biolayer interferometry. This research was partially supported by the JSPS KAKENHI, Grant Number JP19K06970 (to T. K.). We thank the Edanz Group (https://en-author-services.edanzgroup.com/) for editing a draft of this manuscript.Notes and references
- E. L. Nelson, Clin. Invest., 2014, 4, 705–728 CrossRef CAS.
- S. Loibl and L. Gianni, Lancet, 2017, 389, 2415–2429 CrossRef CAS.
- E. Van Cutsem, Y. J. Bang, F. Feng-yi, J. M. Xu, K. W. Lee, S. C. Jiao, J. L. Chong, R. I. López-Sanchez, T. Price, O. Gladkov, O. Stoss, J. Hill, V. Ng, M. Lehle, M. Thomas, A. Kiermaier and J. Rüschoff, Gastric Cancer, 2015, 18, 476–484 CrossRef CAS PubMed.
- A. H. Sims, A. J. M. Zweemer, Y. Nagumo, D. Faratian, M. Muir, M. Dodds, I. Um, C. Kay, M. Hasmann, D. J. Harrison and S. P. Langdon, Br. J. Cancer, 2012, 106, 1779–1789 CrossRef CAS PubMed.
- Y. J. Bang, E. Van Cutsem, A. Feyereislova, H. C. Chung, L. Shen, A. Sawaki, F. Lordick, A. Ohtsu, Y. Omuro, T. Satoh, G. Aprile, E. Kulikov, J. Hill, M. Lehle, J. Rüschoff and Y. K. Kang, Lancet, 2010, 376, 687–697 CrossRef CAS.
- J. Tabernero, P. M. Hoff, L. Shen, A. Ohtsu, M. A. Shah, K. Cheng, C. Song, H. Wu, J. Eng-Wong, K. Kim and Y. K. Kang, Lancet Oncol., 2018, 19, 1372–1384 CrossRef CAS PubMed.
- P. Chames, M. Van Regenmortel, E. Weiss and D. Baty, Br. J. Pharmacol., 2009, 157, 220–233 CrossRef CAS PubMed.
- S. Marqus, E. Pirogova and T. J. Piva, J. Biomed. Sci., 2017, 24, 1–15 CrossRef PubMed.
- N. G. Karasseva, V. V. Glinsky, N. X. Chen, R. Komatireddy and T. P. Quinn, J. Protein Chem., 2002, 21, 287–296 CrossRef CAS PubMed.
- P. Diderich and C. Heinis, Tetrahedron, 2014, 70, 7733–7739 CrossRef CAS.
- B. W. Park, H. T. Zhang, C. Wu, A. Berezov, X. Zhang, R. Dua, Q. Wang, G. Kao, D. M. O'Rourke, M. I. Greene and R. Murali, Nat. Biotechnol., 2000, 18, 194–198 CrossRef CAS PubMed.
- H. Honarvar, E. Calce, N. Doti, E. Langella, A. Orlova, J. Buijs, V. D'Amato, R. Bianco, M. Saviano, V. Tolmachev and S. De Luca, Sci. Rep., 2018, 8, 1–12 CrossRef CAS PubMed.
- X. Yang, Z. Wang, Z. Xiang, D. Li, Z. Hu, W. Cui, L. Geng and Q. Fang, PLoS Comput. Biol., 2017, 13, 1–22 Search PubMed.
- A. D. AlQahtani, D. O'Connor, A. Domling and S. K. Goda, Biomed. Pharmacother., 2019, 113, 108750 CrossRef CAS PubMed.
- A. C. Conibear, S. Chaousis, T. Durek, K. Johan Rosengren, D. J. Craik and C. I. Schroeder, Biopolymers, 2016, 106, 89–100 CrossRef CAS PubMed.
- C. K. Wang, C. W. Gruber, M. Cemazar, C. Siatskas, P. Tagore, N. Payne, G. Sun, S. Wang, C. C. Bernard and D. J. Craik, ACS Chem. Biol., 2014, 9, 156–163 CrossRef CAS PubMed.
- T. Kadonosono, E. Yabe, T. Furuta, A. Yamano, T. Tsubaki, T. Sekine, T. Kuchimaru, M. Sakurai and S. Kizaka-Kondoh, PLoS One, 2014, 9, 1–11 CrossRef PubMed.
- T. Kadonosono, W. Yimchuen, Y. Ota, K. See, T. Furuta, T. Shiozawa, M. Kitazawa, Y. Goto, A. Patil, T. Kuchimaru and S. Kizaka-Kondoh, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- T. Hatanaka, S. Ohzono, M. Park, K. Sakamoto, S. Tsukamoto, R. Sugita, H. Ishitobi, T. Mori, O. Ito, K. Sorajo, K. Sugimura, S. Ham and Y. Ito, J. Biol. Chem., 2012, 287, 43126–43136 CrossRef CAS PubMed.
- D. A. Case, D. S. Cerutti, T. E. I. Cheatham, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, D. Greene, N. Homeyer, S. Izadi, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2017, University of California, San Francisc, 2017 Search PubMed.
- J. Dong, T. Kojima, H. Ohashi and H. Ueda, J. Biosci. Bioeng., 2015, 120, 504–509 CrossRef CAS PubMed.
- D. Lipovšek, Protein Eng., Des. Sel., 2011, 24, 3–9 CrossRef PubMed.
- O. Hantschel, M. Biancalana and S. Koide, Curr. Opin. Struct. Biol., 2020, 60, 167–174 CrossRef CAS PubMed.
- L. Bloom and V. Calabro, Drug Discovery Today, 2009, 14, 949–955 CrossRef CAS PubMed.
- D. Schiff, S. Kesari, J. De Groot, T. Mikkelsen, J. Drappatz, T. Coyle, L. Fichtel, B. Silver, I. Walters and D. Reardon, Invest. New Drugs, 2015, 33, 247–253 CrossRef CAS PubMed.
- S. S. Guan, C. T. Wu, C. Y. Chiu, T. Y. Luo, J. Y. Wu, T. Z. Liao and S. H. Liu, J. Transl. Med., 2018, 16, 1–18 CrossRef PubMed.
- A. Bandekar, C. Zhu, A. Gomez, M. Z. Menzenski, M. Sempkowski and S. Sofou, Mol. Pharm., 2013, 10, 152–160 CrossRef CAS PubMed.
- L. Pollaro, S. Raghunathan, J. Morales-Sanfrutos, A. Angelini, S. Kontos and C. Heinis, Mol. Cancer Ther., 2015, 14, 151–161 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00427h |
| This journal is © The Royal Society of Chemistry 2020 |