
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAPI ionic liquids: probing the effect of counterion structure on physical form and lipid solubility†
Leigh Ford‡
 *a,
Erin Tay‡b,
Tri-Hung Nguyenc,
Hywel D. Williamsa,
Hassan Benameurc,
Peter J. Scammellsb and
Christopher J. H. Porter*de
*a,
Erin Tay‡b,
Tri-Hung Nguyenc,
Hywel D. Williamsa,
Hassan Benameurc,
Peter J. Scammellsb and
Christopher J. H. Porter*de
aOral Drug Delivery Innovation, Chemical Division, Lonza Pharma Biotech & Nutrition, Melbourne, Australia
bMedicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, Victoria 3052, Australia
cOral Drug Delivery Innovation, Chemical Division, Lonza Pharma Biotech & Nutrition, Strasbourg, France
dDrug Delivery, Disposition and Dynamics, Monash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, Victoria 3052, Australia
eARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Monash University, 381 Royal Parade, Parkville, Victoria 3052, Australia. E-mail: chris.porter@monash.edu; leigh.ford@lonza.com
First published on 31st March 2020
Abstract
Lipid based formulations (LBFs) are extensively utilised as an enabling technology in drug delivery. The use of ionic liquids (ILs) or lipophilic salts (LS) in drug delivery has also garnered considerable interest due to unique solubility properties. Conversion of active pharmaceutical ingredients (API) to ILs by pairing with an appropriately lipophilic counterion has been shown to decrease melting point of the salt complex and improve solubility in LBFs. However, the relationship between the structure of the counterion, the physicochemical properties of the resulting salts and solubility in LBFs has not been systematically explored. This study investigates the relationship between alkyl sulfate counterion structure and melting temperature (Tm or Tg) in addition to LBF solubility, utilizing cinnarizine and lumefantrine as model weakly basic APIs. Three series of structurally diverse alkyl sulfate counterions were chosen to probe this relationship. Pairing cinnarizine and lumefantrine with a majority of these alkyl sulfate counterions resulted in a reduction in melting temperature and enhanced solubility in model medium chain and long chain LBFs. The chain length of the alkyl sulfate plays a crucial role in performance, and consistently branched alkyl sulfate counterions perform better than straight chain alkyl sulfate counterions, as predicted. Most interestingly, trends in counterion performance were found to be consistent across two APIs with disparate chemical structures. The findings from this study will facilitate the design of counterions which enhance solubility of ionisable drugs and unlock the potential to develop compounds previously restrained by poor solubility.
Introduction
Lipid-based formulations (LBFs) are widely utilised to improve the oral exposure of an increasing number of poorly water-soluble drugs that are emerging from the drug discovery pipeline.1,2 LBFs increase oral exposure by circumventing drug dissolution, recruiting endogenous solubilisation processes, and by promoting the uptake of certain drugs into the lymphatic system, thereby avoiding hepatic first pass metabolism.3,4 In addition to their capacity to boost exposure, LBFs have a range of broader pharmaceutical benefits. These include: (i) improved uniformity of high potency/low dose drugs, (ii) fast onset of action, (iii) taste-masking, (iv) ability to deliver drugs with low melting points, (v) modified release profiles, (vi) increased drug permeability and (vii) capacity to meet the market needs of consumer preference,5–9 and often a combination of these. Notably, in these broader LBF applications, advantages may accrue for both poorly water soluble and water-soluble drugs.The use of ionic liquids (ILs) in drug development has received increasing interest in recent years due to their versatility and desirable physicochemical properties, such as improved thermal stability, low vapour pressure, and the ability to dissolve organic, inorganic, and polymeric materials.10 ILs may be defined as salts with a melting (Tm)/glass transition (Tg) point below 100 °C, with a subset of ILs referred to as room-temperature ionic liquids (RTILs), which are liquid at or below ambient temperature.11 The highly tunable nature of ILs (i.e. the ability to alter both the cation and the anion) has led to their use in a wide variety of applications ranging from catalysis12 to solar cells13 to drug delivery.14
The use of ILs in drug delivery has been described in the formation of novel microemulsions15 and use as permeation enhancers for transdermal delivery.16 More relevant to this work is the conversion of ionisable APIs themselves into ILs to improve delivery.17,18 Examples of this application also include instances where the counterion itself is bioactive leading to dual-function active pharmaceutical ingredient ionic liquids (API ILs).19–22 Ionic liquids are therefore an attractive option in drug delivery, in particular for their tendency to form amorphous or liquid salts circumventing issues of polymorphism11,20,22,23 and potentially enhancing solubility.24 When pairing an API with a counterion of low polarity in order to improve solubility in lipids the complex can be referred to as a lipophilic salt (LS) or hydrophobic ion pair (HIP).25–28 This alternate definition also recognises the fact that LSs with melting points above 100 °C (and therefore not strictly ionic liquids) may also have benefit.
API ILs when utilised in combination with LBFs have been shown previously to enhance drug exposure.14,29,30 The conversion to API ILs can enhance solubility in LBFs and ultimately result in greater oral exposure of the API, by exploiting the benefits of LBFs described earlier.25 This may be particularly important during drug development where toxicity screening of lead compounds requires delivery of large doses (up to 2000 mg kg−1) where dissolution of standard API forms may not be feasible.31 Despite the utility of API ILs, the relationship between counterion structure and IL physical properties has not been well established. The melting temperature of the API IL is largely determined by the strength of the crystal lattice where the stronger the forces holding the ions together, the higher this temperature. The relationship between salt structure and crystal lattice energy, and therefore melting temperature has been discussed elsewhere.24,32–34 Utilising thermodynamic considerations of the free energy of the solid and liquid phases leads to the following equation.
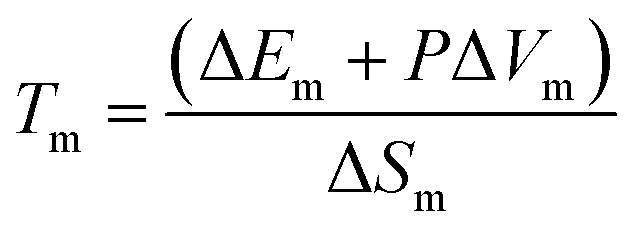
Studies by Seddon and co-workers have previously shown the relationship between the chain length of alkyl substituents on the properties of N-alkyl-methylimidazolium ILs ([Cnmim]+ X−).40 As predicted, the symmetrical imidazolium (N-methyl) IL had the highest melting point. As the length of the alkyl chain increased the melting point decreased until n = 10, beyond which the melting point started to increase rapidly, presumably as van der Waals forces became more significant. Similar trends were also observed in a series of imidazolium hexaflurophosphate ILs. Interestingly, in some cases, branched alkyl substituents with short chain lengths resulted in increased melting point, highlighting the difficulties in predicting properties from first principles.41
Reducing crystal lattice energy is, of course, only one aspect of increasing solubility in lipid systems. Interactions between the solute and solvent (in this case the IL and the lipid excipients) are also key and must overcome the energy required to break solute–solute interactions. Solubility thus follows the common adage ‘like dissolves like’.1 We hypothesise that by judicious choice of lipophilic counterion, it is possible to both reduce the crystal lattice energy and increase favourable interactions with lipid excipients, both factors combining to enhance solubility and therefore drug loading, generating a high performance API IL form. A systemic study of structure/solubility relationships has therefore been undertaken to address this aim.
In this study, the relationship between IL structure and physicochemical properties has been assessed using two model drugs, cinnarizine (1) and lumefantrine (2) (Fig. 1) that have been converted to similar salts by us and others.14,42,43 Compounds 1 and 2 are both ionisable and weakly basic (pKa's > 8), have melting points <150 °C and workable organic solvent solubility and are therefore excellent candidates for this study. API IL were formed by pairing the model drugs with three series of alkyl sulfate counterions. The first series examined the effect of counterion chain length within linear alkyl sulfates, the second series utilised branched counterions in the hope of increasing rotational freedom (increasing entropy), and the third series shifted the position of the head group along a C10 chain, reducing symmetry of the counterion with the aim of reducing the Madelung constant of the system. All compounds were evaluated for physical form, melting point and solubility of the API IL in a type IIIA LBF (as classified by Pouton)44 which is representative of a formulation type widely used in industry.
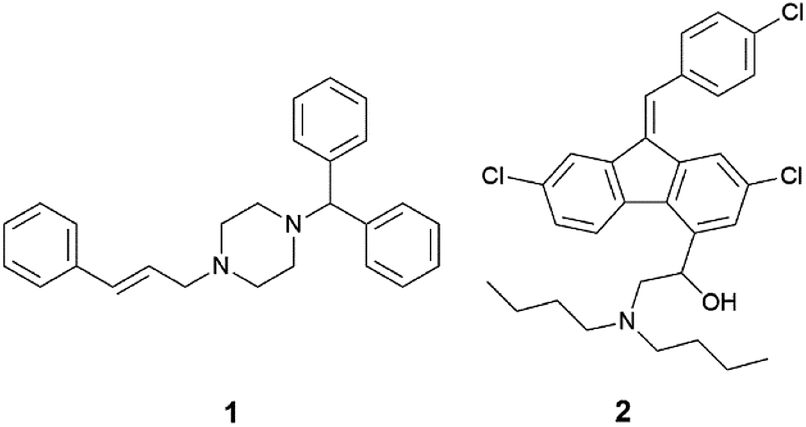 | ||
| Fig. 1 The two model APIs – cinnarizine (1) and lumefantrine (2). | ||
Alkyl sulfates were selected as counterions in this study in large part because of their low pKa. As the ILs formed in this study are protic (i.e. rely on proton transfer between ions), a large ΔpKa value between API and counterion increases the likelihood of salt formation and reduces the chance of dissociation in solution.45–47 Sulfates also have favourable charge distribution, reducing electrostatic interactions between the anion and cation, which promotes reduced melting temperature. Alkyl sulfates traditionally have low toxicity and appear in many commercial products, such as household cleaning products and cosmetics.48 Additionally, alkyl sulfates can be easily accessed through simple sulfation of a plethora of commercially available alcohols in one step.
An understanding of the structure/solubility relationship of counterions utilised in the synthesis of API ILs is expected to facilitate the design of counterions tailored for specific APIs to promote solubility and to unlock utility in LBFs whilst maintaining low toxicity.
Results and discussion
Synthesis and characterisation
Synthesis of cinnarizine·HCl14. Cinnarizine free base (6.5 g, 17.6 mmol) was dissolved in diethyl ether (300 mL) with vigorous stirring and HCl (8.85 mL, 2.0 M in diethyl ether) was added slowly at room temperature. The resulting mixture was allowed to stir at ambient temperature for 4 h. The cloudy solution was filtered under vacuum and the white precipitate was washed with ice cold portions of diethyl ether (total volume – 100 mL). The resulting white solid was dried under high vacuum (7.05 g, 99%).
Synthesis of lumefantrine·HCl. Lumefantrine free base (5.21 g, 9.85 mmol) was suspended in diethyl ether (300 mL) with vigorous stirring and HCl (4.95 mL, 2.0 M in diethyl ether) was added slowly at room temperature. The resulting mixture was protected from the light and allowed to stir at ambient temperature for 4 h. The cloudy solution was filtered under vacuum and the yellow precipitate was washed with ice cold portions of Et2O (total volume – 100 mL). The resulting yellow solid was dried under house vacuum protected from the light (5.45 g – 98%).
API·HCl (1 mmol) and sodium alkyl sulfate (1 mmol) or sodium docusate (1 mmol) were weighed out, and solids were dissolved in a biphasic solution of CH2Cl2 and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 – 100 mL). The resulting mixture was stirred vigorously at ambient temperature overnight (protected from light in the case of lumefantrine salts). This biphasic mixture was transferred to a separating funnel and the organic phase was collected. The aqueous phase was further extracted (2 × 50 mL – CH2Cl2). The combined organics were backwashed with cold distilled water (20 mL) until a negative AgNO3 (0.02 M aq.) precipitate test result was obtained. The organic solution was dried with Na2SO4, filtered and concentrated in vacuo. The resulting material was placed under high vacuum until uniform mass was obtained.
:1 – 100 mL). The resulting mixture was stirred vigorously at ambient temperature overnight (protected from light in the case of lumefantrine salts). This biphasic mixture was transferred to a separating funnel and the organic phase was collected. The aqueous phase was further extracted (2 × 50 mL – CH2Cl2). The combined organics were backwashed with cold distilled water (20 mL) until a negative AgNO3 (0.02 M aq.) precipitate test result was obtained. The organic solution was dried with Na2SO4, filtered and concentrated in vacuo. The resulting material was placed under high vacuum until uniform mass was obtained.
| LBF | Component | (% w/w) |
|---|---|---|
| Medium chain SEDDS | Glyceryl tricaprylate/caprate | 30% |
| Glycerol monocaprylocaprate | 30% | |
| Polyoxyl 35 castor oil | 30% | |
| Ethanol | 10% | |
| Long chain SEDDS | Soybean oil | 30% |
| Glyceryl monolinoleate | 30% | |
| Polyoxyl 35 castor oil | 30% | |
| Ethanol | 10% |
Physical characterisation of cinnarizine salt forms
Three series of counterions were prepared by systematically increasing either alkyl chain length in linear alkyl sulfates (series one) and branched alkyl sulfates (series two). The position of the head group was also varied (series three) in order to probe the effect this had on packing, and by extension API IL properties. Cinnarizine was chosen as the primary model compound due to its favourable pKa and moderate Tm.The lipophilic salts were prepared utilising a biphasic metathesis reaction, from the API·HCl salt and the sodium salt of the counterion, to generate pure (>95%) product without further purification. Series one probed the performance of ILs containing linear alkyl sulfates with varying chain lengths (increasing by two carbons from C6 to C18). We predicted that salts with sulfates bearing alkyl chain lengths similar to those found in the lipidic excipients would show favourable solubility (and potentially physical stability) in the lipid formulations (i.e. C8–C10 for MC-SEDDS and C16–C18 for LC-SEDDS). Series two, which consisted of branched alkyl sulfate counterions, could be compared with the corresponding compounds from series one (with identical numbers of carbons) to determine the overall effect of branching, but additionally allowed evaluation of the optimal chain length within the branched series. It was predicted that branching of the alkyl chains would frustrate packing in the crystal lattice in line with previous observations,14,50 but also maximise entropy within the system.2 The third series was prepared in order to determine how varying the position of the sulfate group along a C10 chain (affecting the Madelung constant) would alter crystal packing and by extension solubility. The physical properties of the salt forms were assessed by hot stage microscopy in order to evaluate the effect of counterion on physical form (crystalline or amorphous) and glass transition (amorphous) or melting (crystalline) point, the data are presented in Tables 2–7.
|
|||
|---|---|---|---|
| # | Name | Physical state | Tm/Tg (°C) |
| a Cinnarizine 1-octadecyl sulfate showed signs of crystallization after long term storage. | |||
| 1 | Free base | Crystalline | 122–123 |
| 1a | Hydrochloride | Crystalline | 207 |
| 1b | 1-Hexyl sulfate (C6) | Amorphous | 35–36 |
| 1c | 1-Octyl sulfate (C8) | Viscous oil | <25 |
| 1d | 1-Decyl sulfate (C10) | Viscous oil | <25 |
| 1e | 1-Dodecyl sulfate (C12) | Viscous oil | <25 |
| 1f | 1-Tetradecyl sulfate (C14) | Viscous oil | <25 |
| 1g | 1-Hexadecyl sulfate (C16) | Viscous oil | <25 |
| 1h | 1-Octadecyl sulfate (C18) | Viscous oil | <25a |
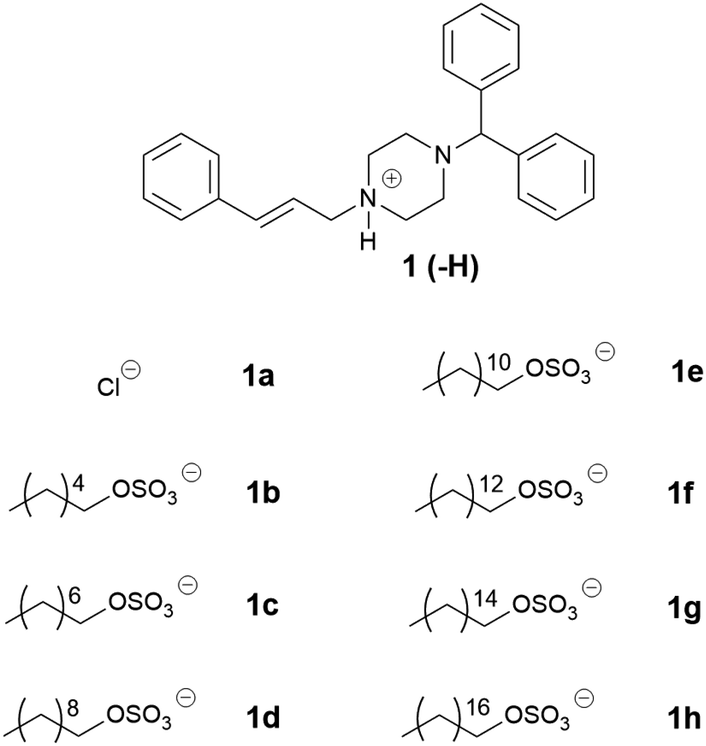
The physical properties of the first series of cinnarizine ILs are summarised in Table 2. As expected cinnarizine hydrochloride (1a) had the highest melting temperature (201–210 °C), due to strong electrostatic interactions between the protonated API and the chloride ion, which is small and has a non-diffuse charge, followed by cinnarizine free base (1) (122–123 °C). For the cinnarizine IL series, only the 1-hexyl sulfate (1b) was a solid at room temperature (melting point: 35–36 °C). The short chain hexyl sulfate was likely unable to disrupt packing in the crystal lattice to the extent of the other counterions, hence the higher melting point and solid physical state. All other cinnarizine (C8–C18) sulfates (1c–h) were viscous liquids at room temperature, precluding more detailed analysis of the effect of structure on melting point. Cinnarizine 1-octyldecyl sulfate (1h) showed signs of crystallization after long term storage (>6 months at −20 °C), suggesting the possibility of a more stable (and less soluble) crystalline form. It remains possible that the other isolated ILs may also have more thermodynamically stable crystal forms, however, we were unable to isolate such forms of the other salts (over 6 months storage at −20 °C).
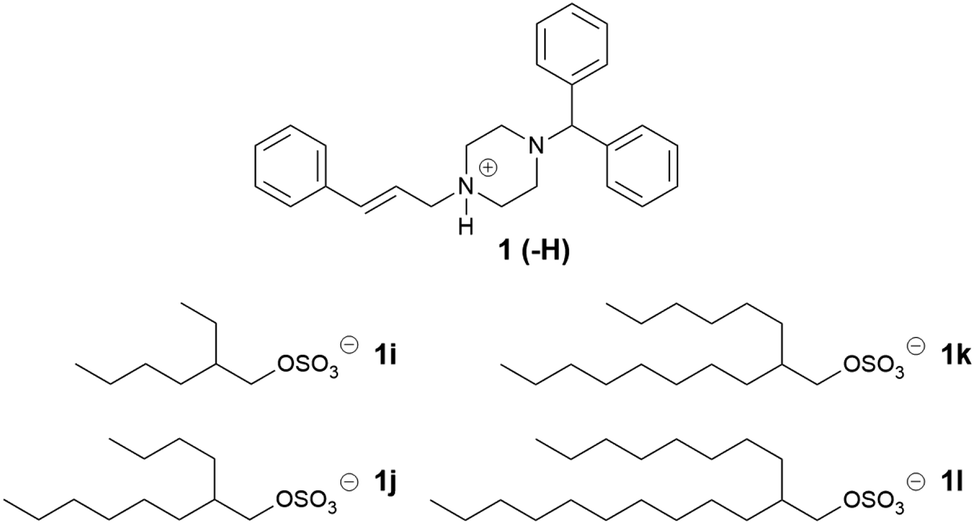
Four counterions were designed for the second series (Table 3), increasing the chain length at each branch by two carbons. The alkyl sulfates were obtained in one step from the corresponding alcohol. Of the ILs formed, only cinnarizine 2-ethyl-1-hexyl sulfate (1i) was a solid at room temperature, and had a melting point of 40–42 °C, no birefringence was observed under polarized microscopy, suggesting an amorphous nature. Interestingly, the branched cinnarizine 2-ethyl-1-hexyl sulfate was a semi-solid, whereas the linear 1-octyl sulfate was a liquid. This was unexpected, particularly considering the increased solubility of the branched compound in LC-SEDDS (see below). The reason for the increased melting point of cinnarizine 2-ethyl-1-hexyl sulfate is not currently understood. The 2-butyl-1-octyl sulfate (1j), 2-hexyl-1-decyl sulfate (1k), and 2-octyl-1-dodecyl sulfate (1l) ILs were viscous liquids at room temperature. The longer alkyl chain sulfates in 1j, 1k, 1l provide more extensive branching and likely contributed to the liquid state of these salts as the counterions were better able to disrupt packing of the crystal lattice. The evidence that a crystal form of 1l exists (formed on long term storage), may be explained by increased van der Waals interactions due to the longer alkyl chain.
|
|||
|---|---|---|---|
| # | Name | Physical state | Tm/Tg (°C) |
| a Cinnarizine 2-octyl-1-dodecyl sulfate showed signs of crystallization after long term storage. | |||
| 1 | Free base | Crystalline | 122–123 |
| 1a | Hydrochloride | Crystalline | 207 |
| 1i | 2-Ethyl-1-hexyl sulfate | Amorphous | 40–42 |
| 1j | 2-Butyl-1-octyl sulfate | Viscous oil | <25 |
| 1k | 2-Hexyl-1-decyl sulfate | Viscous oil | <25 |
| 1l | 2-Octyl-1-dodecyl sulfate | Viscous oil | <25a |
Series three of the cinnarizine ILs (varying sulfate position) had very similar melting points (Table 4). Only the 1-decyl sulfate (1d) was a viscous liquid at room temperature, while the remaining salts were semi-solid, but became less viscous upon heating to 27–29 °C. This was surprising as these counterions were expected to disrupt the crystal lattice more than the straight chain 1-decyl sulfate due to increased asymmetry. All salts in this series were amorphous as determined a lack of birefringence under cross-polarised light microscopy.
|
|||
|---|---|---|---|
| # | Name | Physical state | Tm/Tg (°C) |
| 1 | Free base | Crystalline | 122–123 |
| 1a | Hydrochloride | Crystalline | 207 |
| 1d | 1-Decyl sulfate | Viscous oil | <25 |
| 1m | 2-Decyl sulfate | Amorphous | 27–28 |
| 1n | 3-Decyl sulfate | Amorphous | 27–28 |
| 1o | 4-Decyl sulfate | Amorphous | 27–29 |
| 1p | 5-Decyl sulfate | Amorphous | 27–29 |
Equilibrium solubility of cinnarizine salts in model lipid-based formulations
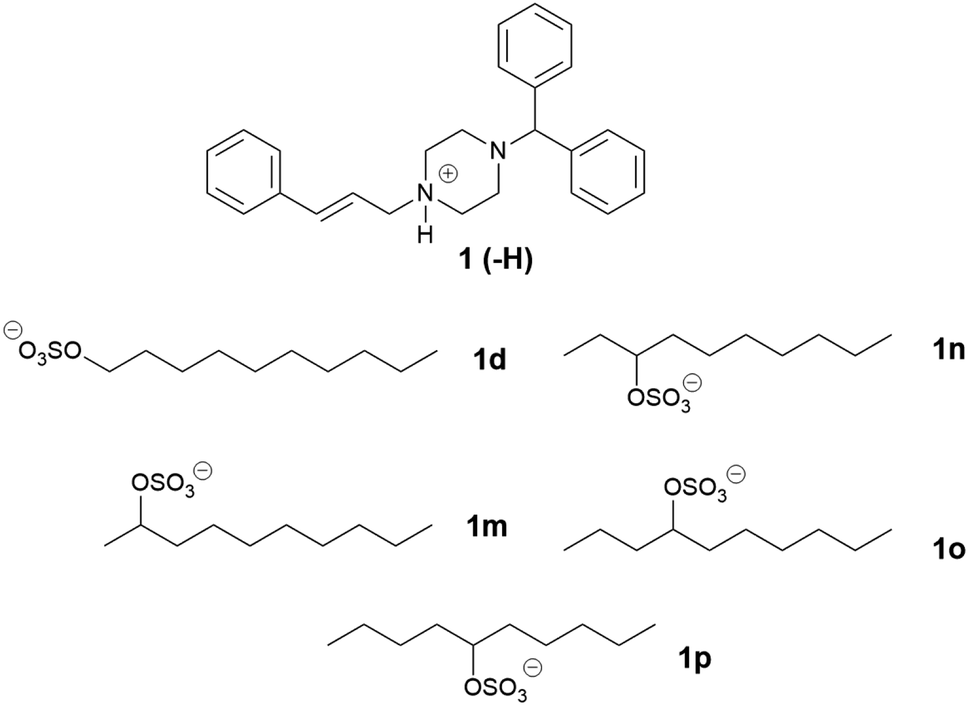
The equilibrium solubility of the cinnarizine salt forms was measured in model medium-chain (MC-SEDDS) and long-chain (LC-SEDDS) self-emulsifying drug delivery systems (compositions shown in Table 1). The equilibrium solubility values for the linear alkyl sulfate series of cinnarizine compounds are shown in Fig. 2, note that for highly miscible salts solubility was defined as the measured concentration at 1:1 mass API IL: LBF.
 | ||
| Fig. 2 Equilibrium solubility in MC-SEDDS and LC-SEDDS for cinnarizine (1), cinnarizine HCl (1a) and cinnarizine series one sulfates (1-hexyl sulfate 1b, 1-octyl sulfate 1c, 1-decyl sulfate 1d, 1-dodecyl sulfate 1e, 1-tetradecyl sulfate 1f, 1-hexadecyl sulfate 1g and 1-octadecyl sulfate 1h). Data are expressed as solubility in cinnarizine free base equivalents, and are n = 3, mean ± SD, except for cinnarizine 1-hexyl sulfate (1b) and cinnarizine 1-octyl sulfate (1c) in MC-SEDDS, and cinnarizine 1-decyl sulfate in both MC- and LC-SEDDS (1d), which were n = 1. *IL was miscible in formulation. # addition of IL to formulation formed 3 immiscible layers, so comparative data could not be obtained. | ||
As predicted, cinnarizine hydrochloride (1a) had the lowest solubility in both MC- and LC-SEDDS, followed by the free base (1). The values were consistent with previously published values14 and reflect their higher melting points relative to the IL forms. Cinnarizine 1-hexyl sulfate (1b), 1-octyl sulfate (1c), and 1-decyl sulfate (1d) were miscible in MC-SEDDS. These IL forms have alkyl chains with similar lengths to those found in glyceryl tricaprylate/caprate and glycerol monocaprylocaprate (C8–C10) and are expected to have favourable interactions with the excipients. As the chain length increased past C10 the solubility values in MC-SEDDS decreased, likely as intermolecular van der Waals interactions between counterions became more significant. Given the miscibility of the shorter chain cinnarizine ILs in MC-SEDDS, it was not possible to differentiate between them. The reduced solubility of the ILs in LC-SEDDS, however, provided more discrimination. Of the straight chain alkyl sulfates, only the 1-decyl sulfate (1d) was miscible in LC-SEDDS. Interestingly C10 was the optimal chain length for linear sulfates paired with cinnarizine in both LBFs despite the different chemical structure of the constituent excipients, suggesting the API IL structure impacts solubility more than the formulation composition. The C10 alkyl chain may represent a compromise between favourable non-polar interactions before van der Waals forces become prevalent. The 1-octyl sulfate (1c) and 1-dodecyl sulfate (1e) had similar solubility values with both exhibiting higher solubility than either the free base (1) or the hydrochloride salt (1a) (but lower than 1d). In general, solubility values in model lipid formulations decreased dramatically as chain length of the counterion increased past C12, representing the length beyond which van der Waals interactions of the API IL with other molecules of API IL become favoured over interaction with the excipients. Consistent with previous work,14 there was generally greater solubility of the API IL in MC-SEDDS when compared to LC-SEDDS,51 though 1d and 1e showed similar performance in both formulations reflecting the higher inherent solubility of these salts and favourable interactions with the excipient sidechains.
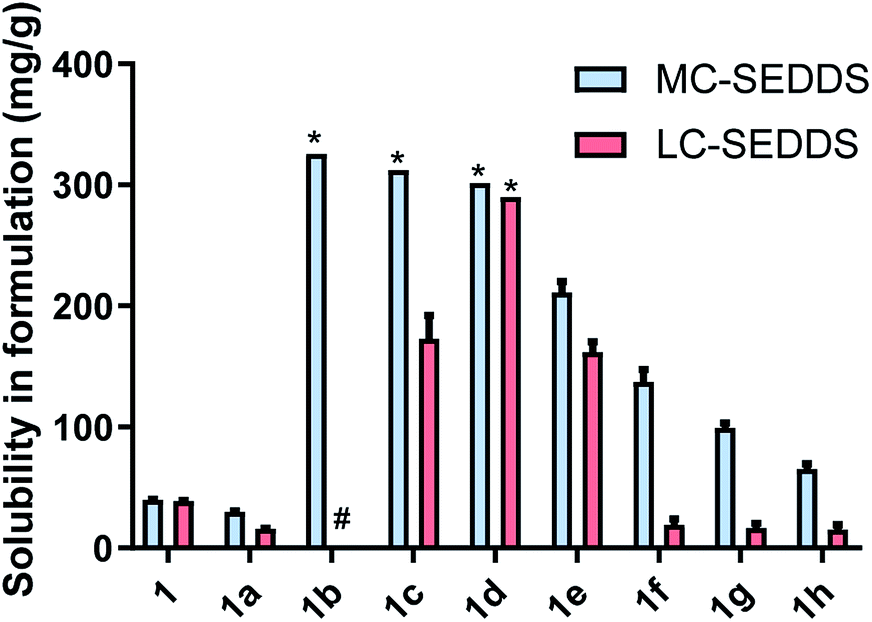
For the branched alkyl sulfates (series two), the 2-ethy-1-hexyl sulfate (1i), 2-butyl-1-octyl sulfate (1j), and 2-hexyl-1-decyl (1k) sulfate were miscible in MC-SEDDS, indicating that branching in the alkyl chain increased solubility, likely through disruption of packing in the crystal lattice. The branched alkyl sulfates all showed high solubility values in LC-SEDDS (relative to series one salts) while only cinnarizine 2-hexyl-1-decyl sulfate (1k) was found to be miscible. Cinnarizine 2-octyl-1-dodecyl sulfate (1l) had the lowest solubility in the series. Interestingly, this compound showed signs of crystallization on long term storage (>6 months) suggesting the presence of a more stable crystalline form, consistent with other long chain alkyl sulfate salts (Fig. 3).
 | ||
| Fig. 3 Equilibrium solubility in MC-SEDDS and LC-SEDDS for cinnarizine (1), cinnarizine HCl (1a) and cinnarizine series two sulfates (2-ethyl-1-hexyl sulfate 1i, 2-butyl-1-octyl sulfate 1j, 2-hexyl-1-decyl sulfate 1k and 2-octyl-1-dodecyl sulfate 1l). Data are n = 3, mean ± SD. *IL was miscible in formulation. | ||
All cinnarizine ILs utilising counterions from the series three were miscible in both MC-SEDDS and LC-SEDDS (data in ESI†). This is in spite of the fact that the melting temperatures were higher than many ILs in other series. In an effort to better determine how the placement of the sulfate group in the alkyl chain affected crystal packing and therefore solubility, the solubility of all these salts was also evaluated in glyceryl tricaprylate/caprate, the hypothesis being that solubility in the single excipient would be lower and allow better discrimination of the API IL solubility. The free base (1) and hydrochloride salt (1a) solubility were also included as a reference (Fig. 4). Surprisingly 1 showed high solubility in glyceryl tricaprylate/caprate, while the hydrochloride salt (1a) was practically insoluble.
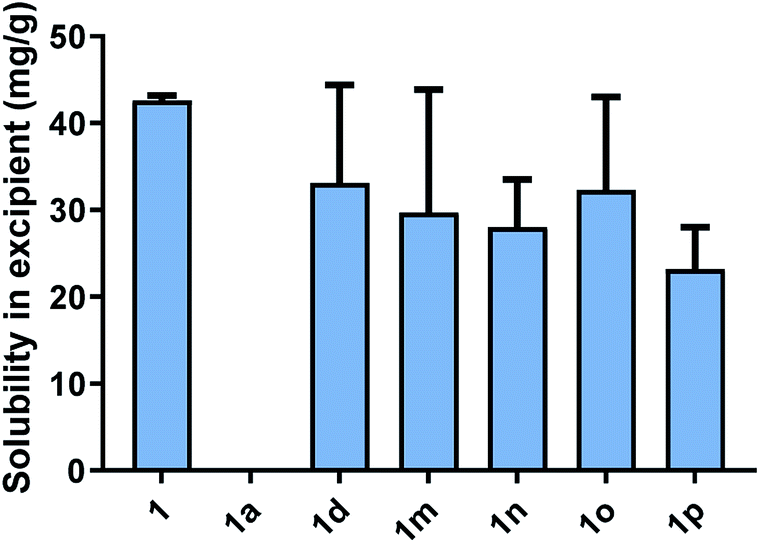 | ||
| Fig. 4 Equilibrium solubility of cinnarizine (1), cinnarizine (1a) and cinnarizine series three sulfates (1-decyl sulfate 1d, 2-decyl sulfate 1m, 3-decyl sulfate 1n, 4-decyl sulfate 1o and 5-decyl sulfate 1p) in glyceryl tricaprylate/caprate. Data are n = 3, ±SD. | ||
For the ILs, there was a great degree of variability in individual data sets and a clear solubility advantage was not observed with cinnarizine series three salts under these conditions. It was further hypothesised that pairing these counterions with an API with inherently lower solubility might allow differentiation between the counterions (see later).
Fig. 5 outlines the solubility of all the examined cinnarizine compounds, allowing for comparison of counterions grouped by # of carbons across different series. As all cinnarizine C10 sulfates were miscible in MC-SEDDS and LC-SEDDS, only one data set is shown. The cinnarizine compounds are displayed with increasing number of carbons from left to right, and with the straight chain length ILs (designated with an L) shown first, followed by the branched chain counterions (designated with a B). The coloured groupings signify counterions with the same number of carbons. There was a decreasing trend in solubility as the number of carbons in the counterion chain increased past C10. Additionally, as predicted, branching of the counterion resulted in higher solubility in LBF when compared to the straight chain counterparts. This effect was most dramatic for the C16 salts in LC-SEDDS, where cinnarizine 2-hexyl-1-decyl sulfate (1k) had a solubility of 261 mg g−1 and 249 mg g−1 in MC-SEDDS and LC-SEDDS, respectively, whereas the linear variant cinnarizine 1-hexadecyl sulfate (1g) had a greatly reduced solubility of 99 and 17 mg g−1 in MC-SEDDS and LC-SEDDS, respectively. Interestingly, no significant improvement was observed in MC-SEDDS or LC-SEDDS when branching occurred in the C12 IL (1e and 1j). This may reflect the fact that the solubility of the linear cinnarizine 1-dodecyl sulfate was considerably higher than cinnarizine 1-hexadecyl sulfate.
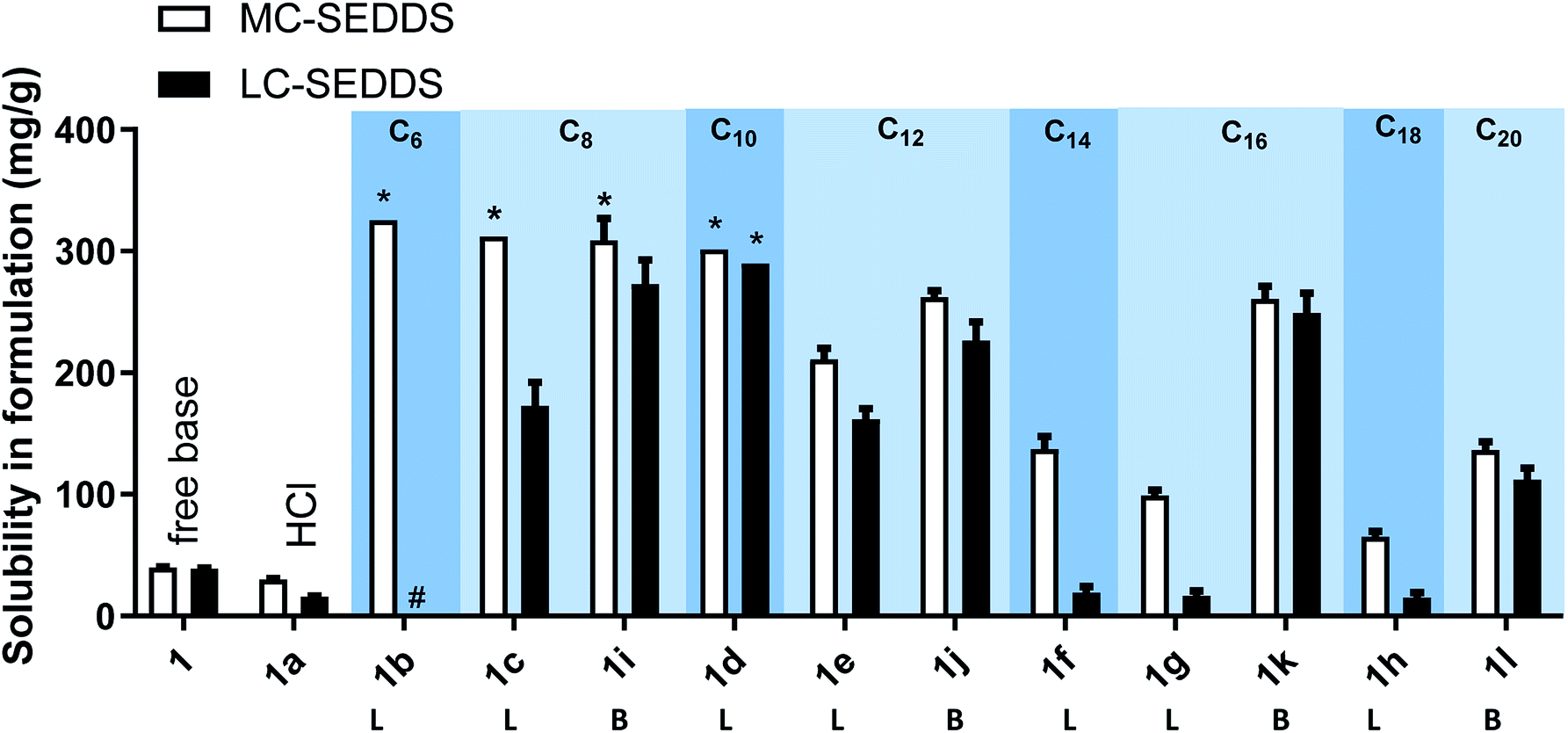 | ||
| Fig. 5 Equilibrium solubility in MC-SEDDS and LC-SEDDS of all cinnarizine compounds. As all of the third series of cinnarizine compounds were miscible, only one IL is shown. L – refers to linear alkyl sulfates. B – refers to branched alkyl sulfates. All data are n = 3, ±SD, except for cinnarizine 1-hexyl sulfate and cinnarizine 1-octyl sulfate in MC-SEDDS, and all cinnarizine decyl sulfate in both MC-SEDDS and LC-SEDDS, which were n = 1. *IL was miscible in formulation. # addition of IL to formulation formed three immiscible layers, so data was not obtained. Cinnarizine docusate is miscible in MC-SEDDS and LC-SEDDS formulations (unpublished data). | ||
Given the results outlined above, subsequent studies sought to determine whether the structural trends observed with cinnarizine were conserved with another API. This was expected to highlight the degree to which the structure of the API had an effect on overall LS properties and performance compared with counterion structure. If the API structure was found to have minimal impact, this would allow for more general selection of counterions when generating API IL forms.
Lumefantrine was chosen as a second model API and paired with counterions selected from series one to three, which showed favourable performance with cinnarizine. The physical characterization data for these compounds are compiled in Tables 5–7
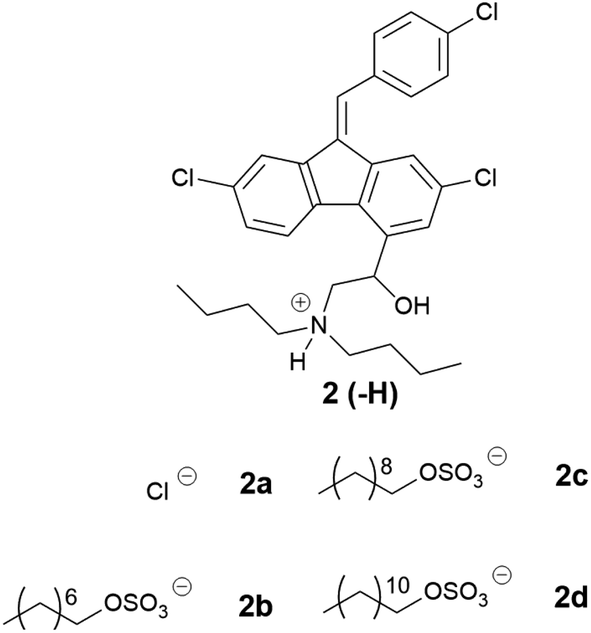
Three of the counterions from the linear alkyl series with cinnarizine were selected for pairing with lumefantrine as they improved solubility in the formulations to the greatest extent (Table 5). Unlike cinnarizine ILs, most lumefantrine salts were solid at room temperature, suggesting greater difficulty in disrupting the lumefantrine crystal lattice, in spite of the fact that the melting points of lumefantrine HCl (2a) and free base (2) were similar to that of cinnarizine. Lumefantrine hydrochloride (2a) had the highest melting range (180–200 °C), followed by the free base (2) (133–140 °C). Lumefantrine 1-octyl sulfate (2b) had the highest melting range among the ILs. Presumably, the C8 alkyl chain disrupted crystal packing less than the longer chain analogues. The 1-decyl sulfate (2c) and 1-dodecyl sulfate (2d) salts had similar melting ranges, 111–125 °C and 115–128 °C, respectively, suggesting a small window of improvement with linear alkyl sulfate salts of lumefantrine. All compounds in Table 5 showed birefringence under cross-polarised light microscopy, indicating that all compounds possessed some degree of crystallinity, consistent with a lack of crystal lattice disruption.
|
|||
|---|---|---|---|
| # | Name | Physical state | Tm/Tg (°C) |
| 2 | Free base | Crystalline | 133–140 |
| 2a | HCl | Crystalline | 180–200 |
| 2b | 1-Octyl sulfate | Crystalline | 147–155 |
| 2c | 1-Decyl sulfate | Crystalline | 111–125 |
| 2d | 1-Dodecyl sulfate | Crystalline | 115–128 |
Examining the branched series of lumefantrine salts (Table 6), the 2-hexyl-1-decyl sulfate (2g) had the highest melting range (133–135 °C), which may be explained by greater van der Waals interactions of the longer alkyl chains. The truncated 2-butyl-1-octyl sulfate (2f) had a reduced melting point range (117–136 °C), followed by 2-ethyl-1-hexyl sulfate (2e) (111–119 °C), here the branched C8 alkyl sulfate had a lower melting/glass transition point than the linear 1-octyl sulfate, in contrast to the trend observed for the cinnarizine ILs.
|
|||
|---|---|---|---|
| # | Name | Physical state | Tm/Tg (°C) |
| 2 | Free base | Crystalline | 133–140 |
| 2a | HCl | Crystalline | 180–200 |
| 2e | 2-Ethyl-1-hexyl sulfate | Crystalline | 111–119 |
| 2f | 2-Butyl-1-octyl sulfate | Crystalline | 117–136 |
| 2g | 2-Hexyl-1-decyl sulfate | Crystalline | 133–135 |
| 2h | Docusate | Amorphous | 52–60 |
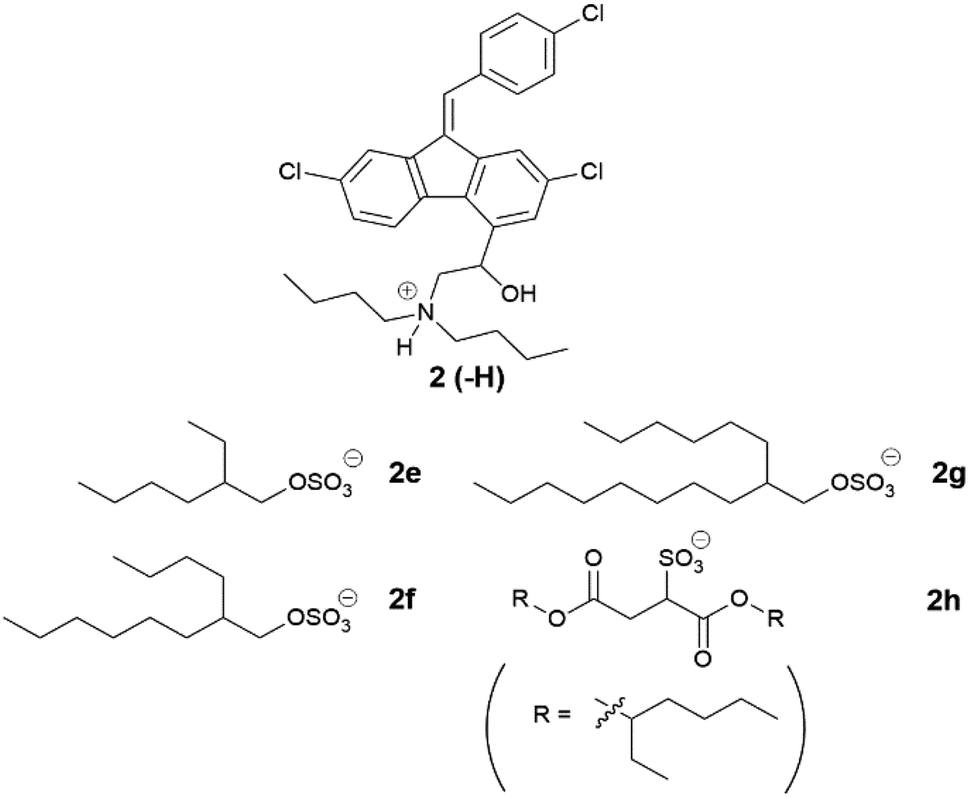
Interestingly 2f had a similar melting range to the dodecyl sulfate IL (2d), a trend also observed in the cinnarizine salts. Lumefantrine docusate (2h) is shown for comparison as it contains sidechains with similar branching to that in this series (c.f. 2e). 2h had the lowest melting point in this series, highlighting the ability of the docusate to disrupt crystal packing.17,18,47,52,53 Only the docusate salt lacked birefringence under cross-polarised light microscopy, while the other three lipophilic salts displayed varying degrees of crystallinity.
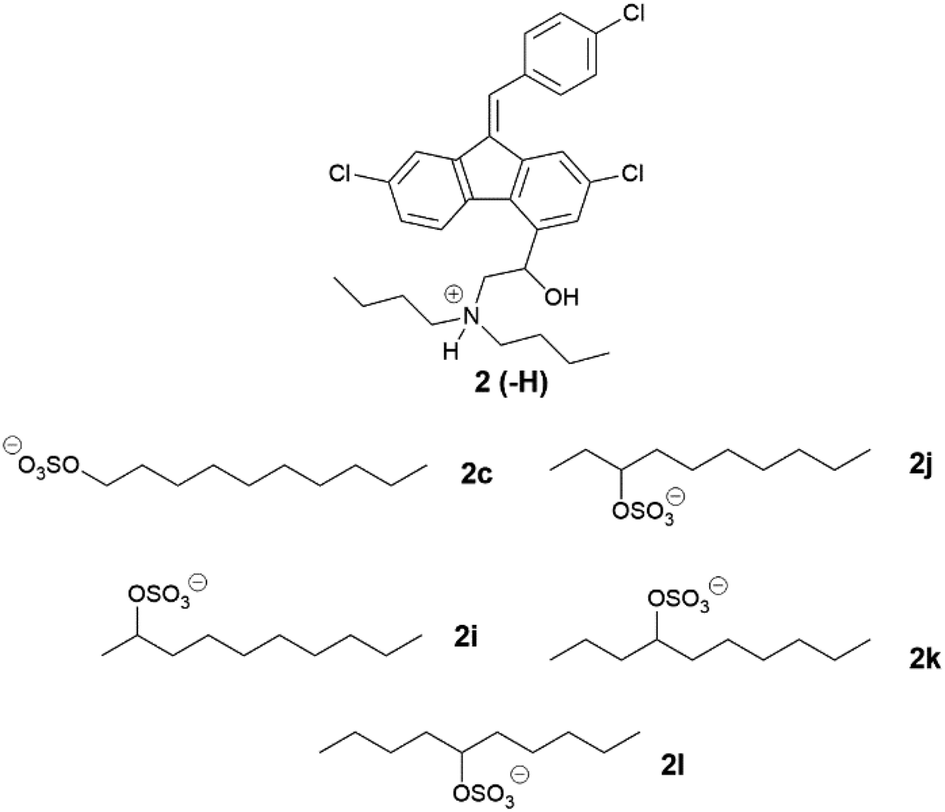
Unlike their cinnarizine counterparts, all lumefantrine ILs from series three were solid at room temperature (Table 7). Lumefantrine 1-decyl sulfate (2c) (111–125 °C) had a similar melting range to the 2-decyl sulfate salt (2i) (107–121 °C), and both displayed birefringence, indicating they were crystalline. The 5-decyl sulfate IL (2l) however, had a considerably lower melting range (51–53 °C), and this was also the case for both the 3-decyl sulfate salt (2j) (42–46 °C) and the 4-decyl sulfate salt (2k) (40–47 °C). Lumefantrine compounds 2j, 2k and 2l lacked birefringence under cross-polarised light microscopy, indicating that they were amorphous. For lumefantrine salts in series three the differences in melting point were more significant. In this case, moving the position of the sulfate group down the ten-carbon chain resulted in a decrease in melting/glass transition point, most notably when moving from the 2- position to the 3-position. There was a slight increase in glass transition point when moving from the 4-position to the 5-position. Shifting the sulfate group along the ten-carbon chain is expected to increase asymmetry in the counterion and appears to have significantly disrupted packing in the crystal lattice, thus decreasing melting/glass transition point. The 5-decyl sulfate counterion is likely more symmetrical than the 3- or 4-decyl sulfate, which may explain the slight increase in melting point. This data suggests that the position of the head group is highly important in crystal packing in the case of lumefantrine and that having the sulfate group situated towards the centre of the alkyl chain, in effect forming a more branched counterion, is more likely to disrupt packing.
|
|||
|---|---|---|---|
| # | Name | Physical state | Tm/Tg (°C) |
| 2 | Free base | Crystalline | 133–140 |
| 2a | HCl | Crystalline | 180–200 |
| 2c | 1-Decyl sulfate | Crystalline | 111–125 |
| 2i | 2-Decyl sulfate | Crystalline | 107–121 |
| 2j | 3-Decyl sulfate | Amorphous | 42–46 |
| 2k | 4-Decyl sulfate | Amorphous | 40–47 |
| 2l | 5-Decyl sulfate | Amorphous | 51–53 |
Equilibrium solubility of lumefantrine salts in model lipid-based formulations
From the linear alkyl sulfate series, the 1-octyl sulfate, 1-decyl sulfate, and 1-dodecyl sulfate counterions were selected and the solubility of the corresponding salts of lumefantrine were assessed. In contrast to the cinnarizine data, where the cinnarizine ILs generally had higher solubility in LBF than either the free base or hydrochloride salt, the lumefantrine ILs from series one showed little solubility advantage (Fig. 6). Indeed, lumefantrine free base (2) showed the greatest solubility in the formulations, with the exception of lumefantrine 1-decyl sulfate (2c) in MC-SEDDS. It is possible that due to the highly planar and aromatic structure of lumefantrine, disruption of crystal packing may be more difficult than is the case with cinnarizine. This is consistent with the relatively small reduction in melting point of series one lumefantrine ILs when compared to the free base. In contrast, the salts of cinnarizine from series one resulted in marked decreases in melting point, consistent with larger increases in LBF solubility.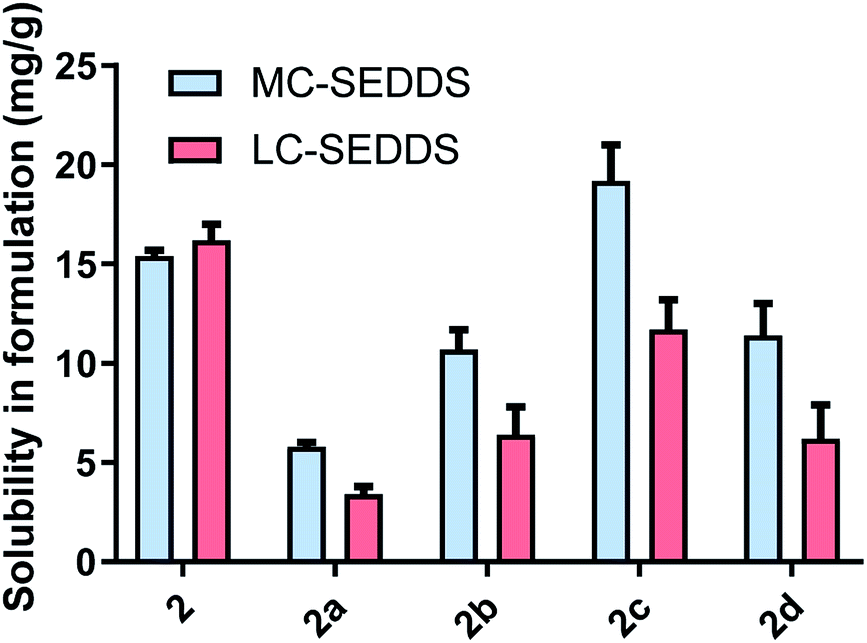 | ||
| Fig. 6 Equilibrium solubility in MC-SEDDS and LC-SEDDS for lumefantrine (2), lumefantrine HCl (2a), and lumefantrine series one sulfates (1-octyl sulfate 2b, 1-decyl sulfate 2c and 1-dodecyl sulfate 2d). Data are n = 3, ±SD. | ||
Within the lumefantrine IL series, however, the trends in solubility for the 1-octyl sulfate (2b), 1-decyl sulfate (2c), and 1-dodecyl sulfate salts (2d) in both MC-SEDDS and LC-SEDDS were similar to cinnarizine, such that lumefantrine 1-decyl sulfate had the highest solubility in MC-SEDDS compared to the other lumefantrine compounds. This is consistent with the hypothesis that the C10 counterion has the most favourable interactions with the lipid excipients. It also suggests that counterion structure/solubility trends may be consistent, independent of drug structure.
For the branched series (Fig. 7), lumefantrine 2-ethyl-1-hexyl sulfate (2e) and lumefantrine 2-hexyl-1-decyl sulfate (2g) had higher solubilities in MC-SEDDS and LC-SEDDS compared to the free base and hydrochloride salt. Similar to the cinnarizine counterparts (1i and 1k) both had higher solubility in both MC-SEDDS and LC-SEDDS compared to the 2-butyl-1-octyl sulfate IL in LC-SEDDS, again suggesting the trends may be independent of drug structure. The high solubility of 2g is surprising given its relatively high melting point and number of carbons (and considering the low solubility of the linear C12 sulfate 2d and therefore the expectation that a linear C16 sulfate would be lower still). Interestingly, this counterion also performed well for cinnarizine. Lumefantrine 2-ethyl-1-hexyl sulfate (2e) outperformed the linear C8 alkyl sulfate (2b) in both MC-SEDDS and LC-SEDDS presumably due to its ability to disrupt packing and increased entropy on melting.
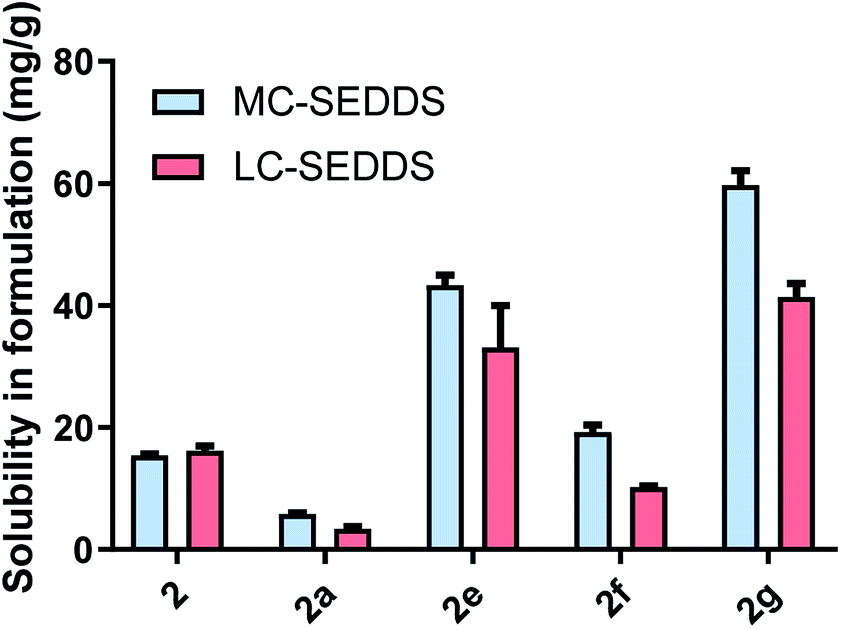 | ||
| Fig. 7 Equilibrium solubility in MC-SEDDS and LC-SEDDS for lumefantrine (2), lumefantrine HCl (2a) and lumefantrine series two sulfates (2-ethyl-1-hexyl sulfate 2e, 2-butyl-1-octyl sulfate 2f and 2-hexyl-1-decyl sulfate 2g). Data are n = 3, ±SD. | ||
The solubility of lumefantrine docusate (2h) was also evaluated to explore the effects of more extensive branching on solubility (see later in Fig. 9). We have shown previously that branching acts to disrupt packing and increase solubility, the docusate counterion has two branched alkyl ester substituents and was therefore expected to disrupt crystal packing to an even greater degree than simple branched alkyl sulfates. Consistent with this hypothesis, and the depressed melting point of lumefantrine docusate, the docusate IL was miscible in both MC-SEDDS and LC-SEDDS. This is in spite of the presence of the ester groups which are potential H-bond donors and are often undesirable (due to strong interactions in the solid state) in traditional ILs. In the context here in lipid formulations, the ester groups present in the docusate counterion do not appear to be problematic and in fact may form favourable interactions with the excipients and enhance solubility.27 When compared to the straight chain lipophilic salts with the same number of carbons, the solubility of the branched ILs was therefore higher in both systems (see Fig. 9), again consistent with the trends observed with the equivalent cinnarizine salts. The effect of branching of the counterion on IL solubility was further exemplified by lumefantrine docusate, which was miscible in both MC-SEDDS and LC-SEDDS, consistent with its low glass transition point.
As all cinnarizine ILs from the series three were miscible in both formulations, these counterions were paired with lumefantrine in an attempt to increase solubility and better differentiate performance (Fig. 8). Apart from lumefantrine 1-decyl sulfate (2c), all series three lumefantrine ILs displayed higher solubilities in LBF compared to both the free base and hydrochloride salt. In general, solubility increased as the position of the sulfate group moved further down the carbon chain, with the highest solubility for lumefantrine 4-decyl sulfate (2k) (66 mg g−1 in MC-SEDDS and 53 mg g−1 in LC-SEDDS), before decreasing when the sulfate group was in the 5-position (2l). This is consistent with the effect caused by greater branching in the counterion structure. Notably, whilst lumefantrine 4-decyl sulfate showed the highest solubility, the sodium salt of 4- and 5-decyl sulfate showed signs of decomposition on long term storage at 25 °C, this occurred via displacement of the sulfate group by water, liberating the parent alcohol and sulfuric acid, as determined by comparison with 1H NMR spectrum of the decomposed counterion with the pure parent alcohol, specifically the appearance of a multiplet at 3.55–3.62 ppm (CDCl3) and a noted decrease in pH (0–1) of the material. This decomposition was also observed in the salt form on long term storage (25 °C). The instability of the counterions may therefore limit practical use. Interestingly, however, this was not observed in long term storage of the cinnarizine salts. Fig. 9 shows all lumefantrine ILs grouped by # of carbons, highlighting the benefit of branched alkyl sulfates.
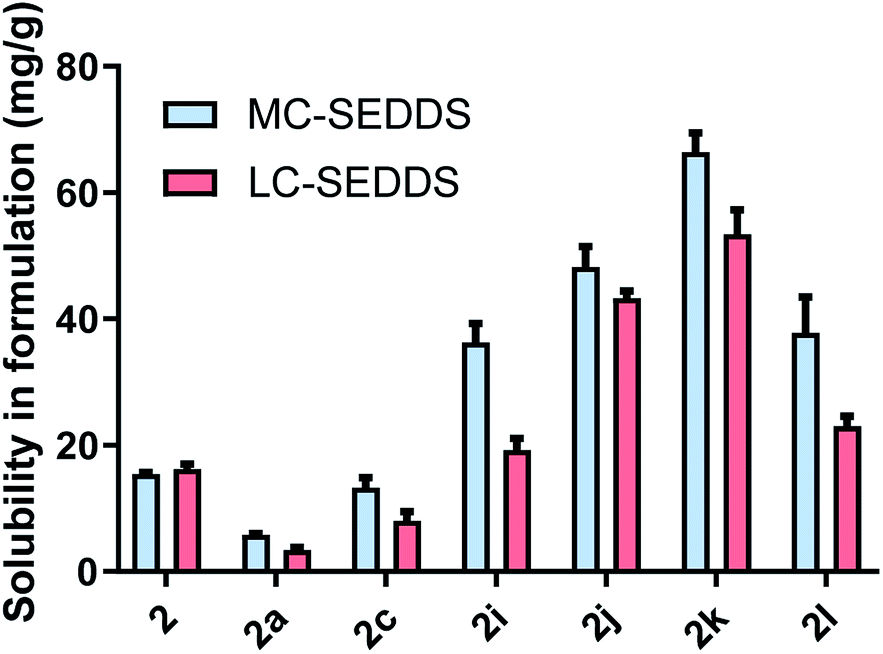 | ||
| Fig. 8 Equilibrium solubility in MC-SEDDS and LC-SEDDS for lumefantrine (2), lumefantrine HCl (2a) and lumefantrine series three sulfates (1-decyl sulfate 2c, 2-decyl sulfate 2i, 3-decyl sulfate 2j, 4-decyl sulfate 2k and 5-decyl sulfate 2l). Data are n = 3. | ||
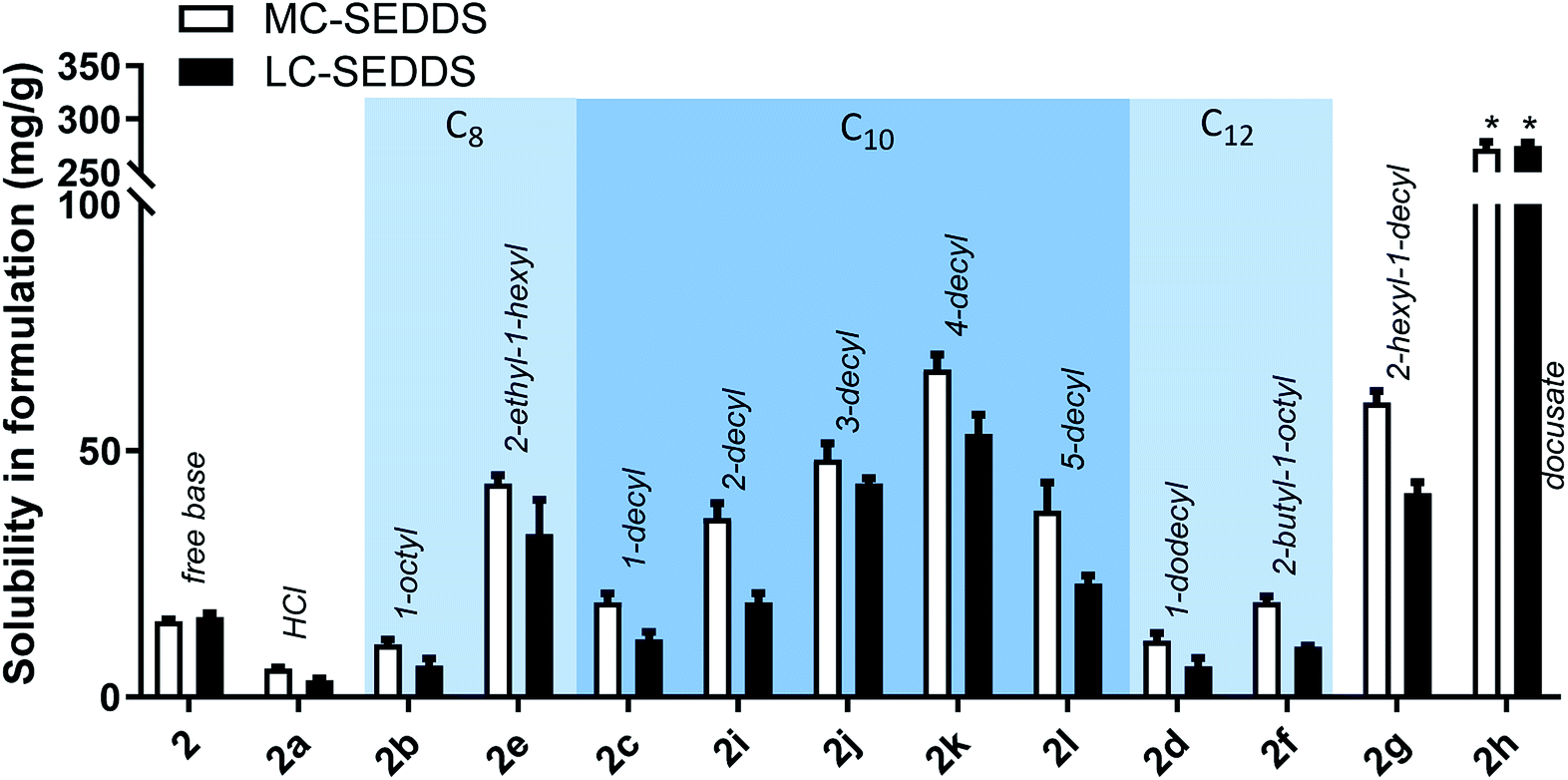 | ||
| Fig. 9 Equilibrium solubility of all lumefantrine compounds in MC-SEDDS and LC-SEDDS separated by # of carbons in the counterion. Data are n = 3, ±SD. *IL was miscible in formulation. | ||
Conclusions
Isolation of two weakly basic drugs as ILs with alkyl sulfate counterions in many cases reduced the melting point relative to the free base and enhanced drug solubility in LBF. The counterion structure played a key role in the physicochemical properties of the salt, as predicted by thermodynamic considerations. For cinnarizine and lumefantrine ILs, the reduction in melting point was more pronounced with C8–C12 alkyl chains. C10 was the optimal chain length for counterions for both drugs and increasing linear chain length past C12 generally resulted in reduced solubility.While the majority of the cinnarizine lipophilic salts were liquid at room temperature, this was not the case for the lumefantrine ILs, in spite of the fact that the melting points of the APIs are similar. For lumefantrine, the ILs with branched counterions consistently had lower melting points and higher solubilities than their straight-chain counterparts, reflecting the increase in entropy in the crystal lattice resulting from higher degrees of freedom of the counterion.
Solubility trends for cinnarizine and lumefantrine salt forms generally reflected trends in melting point, where the salts with lower melting/glass transition points generally had higher solubility in LBF [one exception being cinnarizine 2-ethyl-hexyl sulfate (1i) vs. cinnarizine 1-octyl sulfate (1c)] (a plot of Tm/Tg vs. solubility for lumefantrine salts can be found in ESI†). For both APIs, increasing branching of the counterion and increasing asymmetry typically reduced melting point and increased solubility in LBFs.
In summary, this study provides new insights into the complex relationship between IL counterion structure, API IL physical properties and solubility in model LBFs. The data confirm the need for rational counterion selection to realise the full potential of the IL form. Interestingly, many structure/solubility trends were consistent across two APIs, further highlighting that certain counterions (typically C10 counterions and those with branched structures) may be effective in significantly improving IL solubility in LBFs across a broad range of API structures. LBF solubility was enhanced up to 8-fold for cinnarizine ILs compared to the free base, and up to 18-fold for lumefantrine docusate compared to the free base form. This highlights the power of ILs to improve lipid solubility and unlocks the potential to deliver greater doses of API despite poor inherent solubility.
Conflicts of interest
This article describes intellectual property in the use of ionic liquids/lipophilic salts in drug delivery that has been assigned to Lonza. Authors L. F., H. D. W. and H. B. are from Lonza.Acknowledgements
Erin Tay is funded by an Australian Postgraduate Award. We kindly thank Dr Jason Dang for acquiring all HRMS data. We thank Colin Pouton and Vincent Jannin for helpful discussions related to this work.Notes and references
- H. D. Williams, N. L. Trevaskis, S. A. Charman, R. M. Shanker, W. N. Charman, C. W. Pouton and C. J. H. Porter, Pharmacol. Rev., 2013, 65, 315–499 CrossRef PubMed.
- O. M. Feeney, M. F. Crum, C. L. McEvoy, N. L. Trevaskis, H. D. Williams, C. W. Pouton, W. N. Charman, C. A. S. Bergstrom and C. J. H. Porter, Adv. Drug Delivery Rev., 2016, 101, 167–194 CrossRef CAS PubMed.
- C. J. H. Porter, N. L. Trevaskis and W. N. Charman, Nat. Rev. Drug Discovery, 2007, 6, 231–248 CrossRef CAS PubMed.
- H. Mu, R. Holm and A. Müllertz, Int. J. Pharm., 2013, 453, 215–224 CrossRef CAS PubMed.
- D. J. Hauss, Adv. Drug Delivery Rev., 2007, 59, 667–676 CrossRef CAS PubMed.
- E. T. Cole, D. Cadé and H. Benameur, Adv. Drug Delivery Rev., 2008, 60, 747–756 CrossRef CAS PubMed.
- V. Jannin, J. Musakhanian and D. Marchaud, Adv. Drug Delivery Rev., 2008, 60, 734–746 CrossRef CAS PubMed.
- E. V. Hersh, L. M. Levin, S. A. Cooper, G. Doyle, J. Waksman, D. Wedell, D. Hong and S. A. Secreto, Clin. Ther., 2000, 22, 1306–1318 CrossRef CAS PubMed.
- R. G. Strickley and R. Oliyai, in Solvent Systems and Their Selection in Pharmaceutics and Biopharmaceutics, ed. P. Augustijns and M. Brewster, Springer, New York, NY, 2007, vol. VI Search PubMed.
- D. Dobler, T. Schmidts, I. Klingenhofer and F. Runkel, Int. J. Pharm., 2013, 441, 620–627 CrossRef CAS PubMed.
- A. Balk, U. Holzgrabe and L. Meinel, Eur. J. Pharm. Biopharm., 2015, 94, 291–304 CrossRef CAS PubMed.
- C. P. Mehnert, Chem.–Eur. J., 2005, 11, 50–56 CrossRef PubMed.
- M. Gorlov and L. Kloo, Dalton Trans., 2008, 2655–2666 RSC.
- Y. Sahbaz, H. D. Williams, T. H. Nguyen, J. Saunders, L. Ford, S. A. Charman, P. J. Scammells and C. J. Porter, Mol. Pharm., 2015, 12, 1980–1991 CrossRef CAS PubMed.
- S. Kandasamy, M. Moniruzzaman, M. Sivapragasam, M. R. Shamsuddin and M. I. A. Mutalib, Sep. Purif. Technol., 2018, 196, 149–156 CrossRef CAS.
- D. Monti, E. Egiziano, S. Burgalassi, P. Chetoni, C. Chiappe, A. Sanzone and S. Tampucci, Int. J. Pharm., 2017, 516, 45–51 CrossRef CAS PubMed.
- W. L. Hough, M. Smiglak, H. Rodriguez, R. P. Swatloski, S. K. Spear, D. T. Daly, J. Pernak, J. E. Grisel, R. D. Carliss, M. D. Soutullo, J. H. Davis and R. D. Rogers, New J. Chem., 2007, 31, 1429–1436 RSC.
- W. L. Hough and R. D. Rogers, Bull. Chem. Soc. Jpn., 2007, 80, 2262–2269 CrossRef CAS.
- W. L. Hough-Troutman, M. Smiglak, S. Griffin, W. Matthew Reichert, I. Mirska, J. Jodynis-Liebert, T. Adamska, J. Nawrot, M. Stasiewicz, R. D. Rogers and J. Pernak, New J. Chem., 2009, 33, 26–33 RSC.
- J. L. Shamshina, P. S. Barber and R. D. Rogers, Expert Opin. Drug Delivery, 2013, 10, 1367–1381 CrossRef CAS PubMed.
- K. Bica, C. Rijksen, M. Nieuwenhuyzen and R. D. Rogers, Phys. Chem. Chem. Phys., 2010, 12, 2011–2017 RSC.
- J. Stoimenovski and D. R. MacFarlane, Chem. Commun., 2011, 47, 11429–11431 RSC.
- J. Stoimenovski, D. R. MacFarlane, K. Bica and R. D. Rogers, Pharm. Res., 2010, 27, 521–526 CrossRef CAS PubMed.
- P. M. Dean, J. Turanjanin, M. Yoshizawa-fujita, D. R. Macfarlane and J. L. Scott, Cryst. Growth Des., 2009, 9, 1137–1145 CrossRef CAS.
- H. D. Williams, L. Ford, A. Igonin, Z. Shan, P. Botti, M. M. Morgen, G. Hu, C. W. Pouton, P. J. Scammells, C. J. H. Porter and H. Benameur, Adv. Drug Delivery Rev., 2019, 142, 75–90 CrossRef CAS PubMed.
- H. D. Lu, P. Rummaneethorn, K. D. Ristroph and R. K. Prud'homme, Mol. Pharm., 2018, 15, 216–225 CrossRef CAS PubMed.
- J. Griesser, G. Hetenyi, M. Moser, F. Demarne, V. Jannin and A. Bernkop-Schnurch, Int. J. Pharm., 2017, 520, 267–274 CrossRef CAS PubMed.
- S. H. Choi and T. G. Park, Int. J. Pharm., 2000, 203, 193–202 CrossRef CAS PubMed.
- H. D. Williams, L. Ford, S. Han, K. J. Tangso, S. F. Lim, D. M. Shakleford, D. T. Vodak, H. Benameur, C. W. Pouton, P. J. Scammells and C. J. H. Porter, Mol. Pharm., 2018, 15, 5678–5696 CrossRef CAS PubMed.
- Y. Sahbaz, T. H. Nguyen, L. Ford, C. L. McEvoy, H. D. Williams, P. J. Scammells and C. J. H. Porter, Mol. Pharm., 2017, 14, 3669–3683 CrossRef CAS PubMed.
- S. Parasuraman, J. Pharmacol. Exp. Ther., 2011, 2, 74–79 CAS.
- D. R. MacFarlane, M. Kar and J. M. Pringle, Fundamentals of Ionic Liquids: From Chemistry to Applications, Wiley, 2017 Search PubMed.
- A. R. Katritzky, R. Jain, A. Lomaka, R. Petrukhin, U. Maran and M. Karelson, Cryst. Growth Des., 2001, 1, 261–265 CrossRef CAS.
- A. S. Prakash, J. Excipients Food Chem., 2011, 2, 28–40 Search PubMed.
- E. I. Izgorodina, U. Bernard, P. Dean, J. Pringle and D. Macfarlane, Cryst. Growth Des., 2009, 9, 4834–4839 CrossRef CAS.
- H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2004, 108, 16593–16600 CrossRef CAS.
- H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2005, 109, 6103–6110 CrossRef CAS PubMed.
- J. A. P. Coutinho, P. J. Carvalho and N. M. C. Oliveira, RSC Adv., 2012, 2, 7322–7346 RSC.
- E. I. Izgorodina, Theoretical Approaches to Ionic Liquids: From Past History to Future Directions, 2012 Search PubMed.
- I. López-Martin, E. Burello, P. N. Davey, K. R. Seddon and G. Rothenberg, ChemPhysChem, 2007, 8, 690–695 CrossRef PubMed.
- W. M. Reichert, J. D. Holbrey, R. P. Swatloski, K. E. Gutowski, A. E. Visser, M. Nieuwenhuyzen, K. R. Seddon and R. D. Rogers, Cryst. Growth Des., 2007, 7, 1106–1114 CrossRef CAS.
- K. Patel, V. Sarma and P. Vavia, Daru, J. Pharm. Sci., 2013, 21, 27–37 CrossRef CAS PubMed.
- N. M. Pinkerton, A. Grandeury, A. Fisch, J. Brozio, B. U. Riebesehl and R. K. Prud'homme, Mol. Pharm., 2013, 10, 319–328 CrossRef CAS PubMed.
- C. W. Pouton, Eur. J. Pharm. Sci., 2006, 29, 278–287 CrossRef CAS PubMed.
- M. Yoshizawa, W. Xu and C. A. Angell, J. Am. Chem. Soc., 2003, 125, 15411–15419 CrossRef CAS PubMed.
- J. Stoimenovski, P. M. Dean, E. I. Izgorodina and D. R. Macfarlane, Faraday Discuss., 2011, 154, 335–352 RSC.
- D. N. Moreira, N. Fresno, R. Pérez-Fernández, C. P. Frizzo, P. Goya, C. Marco, M. A. P. Martins and J. Elguero, Tetrahedron, 2015, 71, 676–685 CrossRef CAS.
- A. Wibbertmann, I. Mangelsdorf, K. Gamon and R. Sedlak, Ecotoxicol. Environ. Saf., 2011, 74, 1089–1106 CrossRef CAS PubMed.
- R. Mogul, E. Johansen and T. R. Holman, Biochemistry, 2000, 39, 4801–4807 CrossRef CAS PubMed.
- H. D. Williams, Y. Sahbaz, L. Ford, T. H. Nguyen, P. J. Scammells and C. J. H. Porter, Chem. Commun., 2014, 50, 1688–1690 RSC.
- H. D. Williams, P. Sassene, K. Kleberg, J.-C. Bakala-N'Goma, M. Calderone, V. Jannin, A. Igonin, A. Partheil, D. Marchaud, E. Jule, J. Vertommen, M. Maio, R. Blundell, H. Benameur, F. Carrière, A. Müllertz, C. J. H. Porter and C. W. Pouton, J. Pharm. Sci., 2012, 101, 3360–3380 CrossRef CAS PubMed.
- M. Morgen, A. Saxena, X.-Q. Chen, W. Miller, R. Nkansah, A. Goodwin, J. Cape, R. Haskell, C. Su, O. Gudmundsson, M. Hageman, A. Kumar, G. S. Chowan, A. Rao and V. K. Holenarsipur, Eur. J. Pharm. Biopharm., 2017, 117, 212–223 CrossRef CAS PubMed.
- F. Alves, F. S. Oliveira, B. Schröder, C. Matos and I. M. Marrucho, J. Pharm. Sci., 2013, 102, 1504–1512 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, characterization details, and additional figures. See DOI: 10.1039/d0ra00386g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |