
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric nano-vesicles via intermolecular action to load and orally deliver insulin with enhanced hypoglycemic effect†
Yumiao Hu‡
,
Juan Wang‡ and
Liyan Qiu‡ *
*
Ministry of Educational (MOE) Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: lyqiu@zju.edu.cn; Fax: +86 571 87952306; Tel: +86 571 87952306
First published on 24th February 2020
Abstract
To date few polymeric vesicles have been investigated to improve oral insulin (INS) absorption due to their limited loading capacity. Therefore, an amphiphilic polyphosphazene (PEOP) containing lipid-like octadecylphosphoethanolamine (OPA) groups and amino-modified poly(ethylene glycol) at the proper ratio was designed and synthesized in this study. It was found that PEOP can self-assemble into nano-vesicles, which displayed considerable loading capability for INS by taking advantage of the synergetic effect of the interaction between OPA and INS and the physical encapsulation by the aqueous lumen of the vesicles. Furthermore, PEOP vesicles can promote INS absorption across the subsequent lymphatic transport of PEOP vesicles after their uptake by the enterocytes in the gastrointestinal tract, and consequently achieve better hypoglycemic effects in vivo. These results suggested that PEOP vesicles have great potential as oral INS carriers for diabetes therapy.
Introduction
As a metabolic and chronic disease, diabetes mellitus keeps progressively increasing around the world. Severe hyperglycemia can cause many complications, such as cardiovascular disease, chronic kidney disease, retinopathy, or even cancer.1–4 At present, insulin (INS) therapy via subcutaneous injection still serves as the primary treatment of diabetes mellitus, which is associated with pain, trauma, local infection, stress and acupuncture phobia. The current demand for developing an alternative administration route of INS is focused on improving patient compliance as well as receiving effective blood glucose control at the normal level.5 Considering its convenience and safety, oral delivery of INS is a good choice.6,7 Moreover, the oral administration is considered to be the most physiological route because it is similar to the endogenous INS pathway and provides better glucose homeostasis.8 However, as a peptide hormone composed of 51 amino acids, oral delivery of INS faces a number of obstacles including degradation, poor penetration through the epithelial cell layer in the intestine, and consequently extremely low bioavailability.9,10Various methods have been adopted to improve the oral INS absorption in forms of conjugates with transferrin, hyaluronan, calcium phosphate, casein, polymer microspheres or nanoparticles, lipid nanosomes and solid lipid nanoparticles.11–15 Comparatively, biopolymer-based nano-vesicles have been rarely applied in oral delivery of peptide or protein. Polymeric nano-vesicle, also named as polymersome, is a typical type of nano-aggregations self-assembled by amphiphilic copolymers but different from micelles. As an unique characteristic, polymersome owns relatively large aqueous lumens surrounded by hydrophobic membrane, which provides proper space for water-soluble drug to load.16,17 However, in a number of reports, low levels of loading content and encapsulation efficiency were hardly avoided, especially for peptides or proteins. For instance, PEG-PLGA polymersomes functionalized with lactoferrin can facilitate the transport of the neuroprotective peptide S14G-humanin into the brain, but the loading content and encapsulation efficiency of S14G-humanin were only 0.78% and 21.78%, respectively.18 Also, polymersome-encapsulated hemoglobin was self-assembled from poly(ethylene oxide)-b-polycaprolactone or poly(ethylene oxide)-b-polylactide, the encapsulation efficiency of hemoglobin was less than 20%, when the hemoglobin/diblock copolymer weight ratios ranged in 1.2–1.5.19 The polymersomes formed from block copolymers of poly(ethylene glycol)-b-poly(propylene sulfide) could only encapsulate 37% of ovalbumin, 19% of bovine serum albumin, and 15% of bovine γ-globulin, respectively.20
Our group previously synthesized an amphiphilic β-cyclodextrin-centered triarm star polymer (mPEG2k-PLA3k)3-CD to self-assemble into nano-vesicles for DOX·HCl oral delivery.21 By taking advantage of inclusion action between β-CD groups and DOX·HCl, the high drug loading content of 8.78% and high encapsulation efficiency of 98.97% was consequently achieved. Benefited from this idea, in this study, we aimed to construct a novel polymeric vesicle based on amphiphilic graft polyphosphazenes to improve INS loading and its hypoglycemic effect after oral administration, which was expected to provide a good model for the oral delivery of therapeutic proteins (Scheme 1). As we know, lipids and proteins are two important components to form stable cell membrane structures in terms of the strong interaction between them. We designed a lipid-like molecule of octadecylphosphoethanolamine (OPA), which was linked as the hydrophobic groups to polyphosphazene backbones with amino-modified poly(ethylene glycol) (mPEG2k-NH2) as their hydrophilic segments. By choosing the proper graft ratio of mPEG2k-NH2 to OPA, the resultant amphiphilic polyphosphazene PEOP can self-assemble into nano-vesicles easily. More importantly, a strong intermolecular interaction occurred between the lipid-like OPA and INS, so that the PEOP vesicles displayed a satisfactory capability to load INS by simply dialysis method. Moreover, the PEOP vesicles can promote the oral absorption of INS in the gastrointestinal tract. We also performed comprehensive in vivo studies including pharmacokinetic studies, oral transport mechanism and hypoglycemic activity in rats.
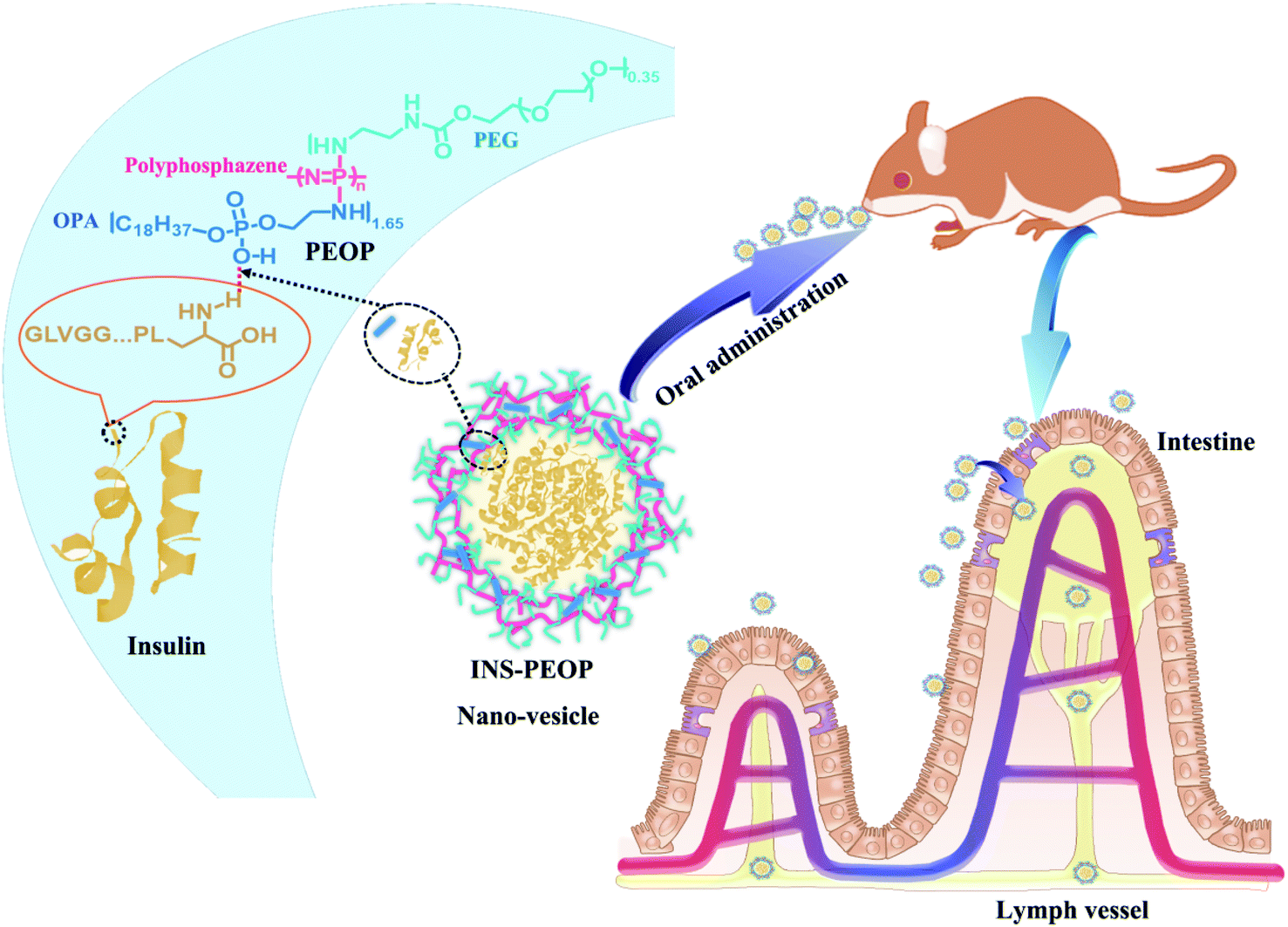 | ||
| Scheme 1 The formation of the INS-PEOP vesicles and applied for oral cure of diabetes mellitus. | ||
Experimental
Materials, cell lines and animals
Insulin with purity exceeding 98.0% was obtained from Wan-Bang Biochemical Medicine Co., Ltd (Xuzhou, China). Hexachlorocyclotriphosphazene (Acros Organics, Belgium) was purified by sublimation at 80–90 °C. Monomethoxy poly(ethylene glycol) (PEG, molecular weight 2000, Fluka, Germany) was azeotropic distilled with toluene before use. Aluminum chloride (99%) was purchased from Acros Organics (Belgium) and used without further purification, and all the other reagents were commercially available and used without further purification. Fetal bovine plasma (FBS), MEM medium, and trypsin–EDTA (0.5% trypsin, 5.3 mM EDTA tetrasodium) were purchased from JiNuo Biotechnology Company (Hangzhou, China).Madin–Darby canine kidney (MDCK) cell lines were obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China), cultured in MEM medium containing 10% FBS and 1% penicillin–streptomycin at 37 °C, 5% CO2, and humidified atmosphere.
Male Sprague Dawley rats (weighting 200 ± 20 g) were purchased from the Experimental Animal Center, Zhejiang Academy of Medical Sciences (Hangzhou, China) and kept under SPF conditions. The animals were given daily fresh diet with free access to water and acclimatized for 6 days prior to the experiments. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with the approval of the Scientific Investigation Board of Zhejiang University, Hangzhou, China.
Synthesis of poly[methoxy-poly(ethyleneglycol)/octadecylphosphoethanol-amine]phosphazene (poly[(mPEG)(OPA)phosphazene], PEOP) (Compound 9)
Firstly, octadecylphosphoethanolamine (OPA) was synthesized as shown in Scheme 2. Octadecanol (Compound 1, 10 g, 37 mmol) and 4 mL triethylamine were dissolved in anhydrous tetrahydrofuran (THF, 50 mL). Then a solution of phosphorous oxychloride (4.2 mL, 49.35 mmol) in 15 mL THF was added dropwise with vigorous stirring in ice bath. The reaction mixture was kept at room temperature with stirring to produce Compound 2. After 5 h, a solution of ethanolamine (2.7 mL, 44.4 mmol) and 12.4 mL triethylamine in anhydrous THF (30 mL) was added dropwise into the reaction mixture under vigorous stirring in ice bath. The reaction mixture was kept at room temperature with stirring for another 5 h. Then the reaction mixture was filtered by suction to remove precipitated triethylamine hydrochloride. The clear and slightly yellow filtrate containing Compound 3 was used directly for the next step. The filtrate was mixed with acetic acid (17 g, 283 mmol) and 25 mL deionized water and the mixture was heated at 70 °C under stirring for 6 h. A lot of white solid precipitated from the solution. Then the solid was collected by suction filtration and washed by acetone for two more times. The obtained compound was octadecylphosphoethanolamine (OPA, Compound 4), which was used directly for the next step.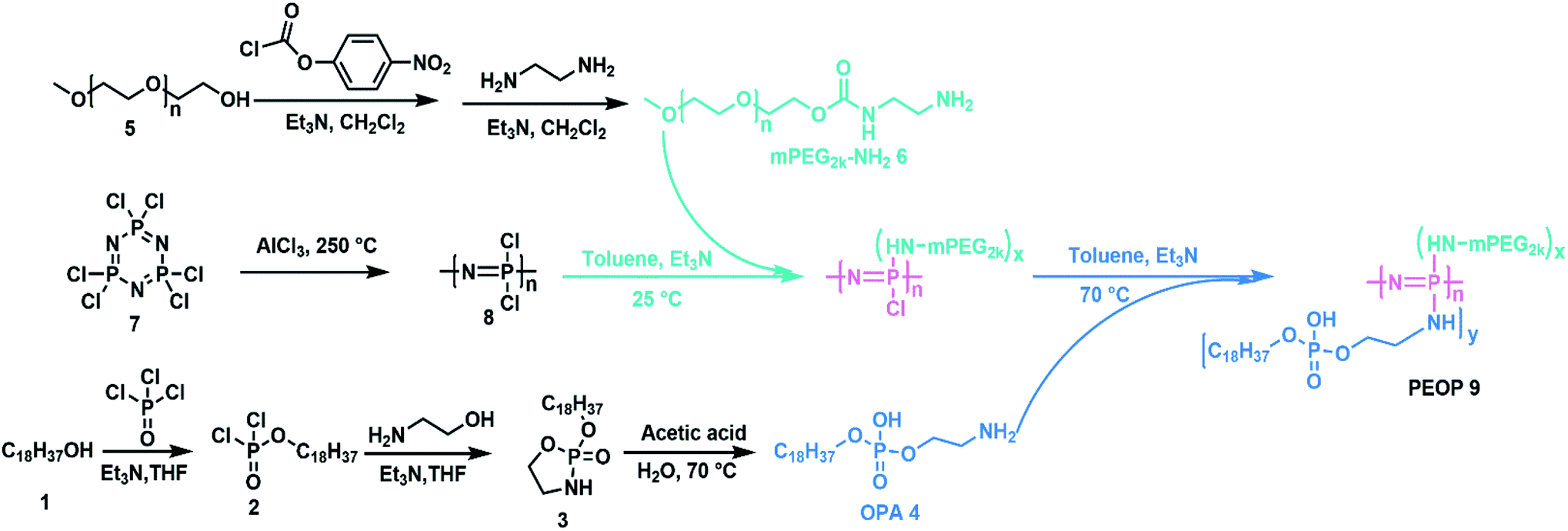 | ||
| Scheme 2 Synthetic route to the amphiphilic graft copolymers PEOP. | ||
Poly(dichlorophosphazene) was obtained by a ring-opening polymerization of hexachlorocyclotriphosphazene (Compound 7) at 250 °C for 4 h.22 mPEG2k-NH2 was prepared using a two-step protocol as described previously.23 Then the obtained poly(dichlorophosphazene) (0.3 g, 2.58 mmol –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–, Compound 8) was dissolved in 15 mL dry toluene, and a dehydrated mPEG2k-NH2 (0.65 g, 0.325 mmol, Compound 6) toluene solution containing equal molar trimethylamine was added dropwise. After 24 h, an excessive amount of OPA (4.06 g, 10.33 mmol, Compound 4) with an equal molar amount of triethylamine was slowly added to complete the substitution reaction for 72 h at 60 °C. The product was collected by precipitation in cold diethyl ether, followed by dialysis (MWCO 14.0 kDa, Spectrum Laboratories, Laguna Hills, CA) against deionized water. The freeze-dried PEOP was obtained finally.
P–, Compound 8) was dissolved in 15 mL dry toluene, and a dehydrated mPEG2k-NH2 (0.65 g, 0.325 mmol, Compound 6) toluene solution containing equal molar trimethylamine was added dropwise. After 24 h, an excessive amount of OPA (4.06 g, 10.33 mmol, Compound 4) with an equal molar amount of triethylamine was slowly added to complete the substitution reaction for 72 h at 60 °C. The product was collected by precipitation in cold diethyl ether, followed by dialysis (MWCO 14.0 kDa, Spectrum Laboratories, Laguna Hills, CA) against deionized water. The freeze-dried PEOP was obtained finally.
Characterization of PEOP
The formation of hydrophobic side group OPA and the resultant PEOP was confirmed by using a FTIR spectrometer (FT/IR-4100, JASCO, Japan).1H NMR spectrum were recorded on a NMR spectrometer (AC-80, Bruker Biospin., Germany) to characterize the chemical composition of OPA and PEOP. 20 mg mL−1 of PEOP in CDCl3 was used. 13C and 31P NMR spectrum were recorded on a NMR spectrometer (AC-80, Bruker Biospin., Germany) to further characterize PEOP. 50 mg mL−1 of PEOP in CDCl3 was used.
The molecular weight of PEOP was determined by gel permeation chromatography (GPC) performed on a Waters 515 HPLC Pump equipped with a Waters 2410 refractive index detector. The eluent was N,N-dimethylformamide (DMF) at a flow rate of 1.0 mL min−1. A series of low polydispersity polystyrene standards were employed for calibration. The software was Breeze 2.
Preparation and characterization of blank and INS-loaded PEOP vesicles
Blank vesicles were prepared via simple dialysis. PEOP copolymer was dissolved at 10 mg mL−1 concentration in DMF, and deionized water was added dropwise to PEOP copolymer solution at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio under magnetic stirring. This solution was put into dialysis bag (MWCO = 3500 Da) to dialyze against deionized water to remove DMF.
:1 volume ratio under magnetic stirring. This solution was put into dialysis bag (MWCO = 3500 Da) to dialyze against deionized water to remove DMF.
INS-loaded PEOP (INS-PEOP) vesicles were prepared under the same operation. PEOP copolymer was dissolved at 10 mg mL−1 concentration in DMF, and INS was dissolved at 1 mg mL−1 concentration in hydrochloric acid (HCl, 0.01 M) and then the pH was adjusted to 7.0 using sodium hydroxide (NaOH, 1 M). INS solution was added dropwise to PEOP copolymer solution under magnetic stirring. The mixture was stirred at room temperature for 20 min. This solution was put into dialysis bag (MWCO = 300 kDa) to dialyze against deionized water to remove DMF and unencapsulated INS. Furthermore, FITC-labeled INS loaded PEOP (FITC-INS-PEOP) vesicles were prepared in the same way by dissolving FITC-INS in deionized water at the same concentration. FITC-INS was synthesized according to the procedure previously reported.24
The vesicles were characterized for particle size and zeta potential by dynamic light scattering (DLS) analysis with a Malvern Zetasizer Nano ZS90 (Malvern Instruments Ltd., U.K.). The morphology of blank or INS-PEOP vesicles were examined by transmission electron microscope (TEM, JEM-1230, JEOLLtd., Tokyo, Japan). To measure the drug loading content and encapsulation efficiency, vesicular suspensions were put into dialysis bag (MWCO 3500 Da) to dialyze against deionized water to remove DMF and then centrifuged at 12000 rpm for 30 min at 4 °C. Then the amount of free FITC-INS in the supernatant of fluorescence-labeled vesicles was measured by Microplate reader (SpectraMax M5, USA). The excitation and emission wavelengths were set at 488 and 520 nm, respectively. INS loading content and encapsulation efficiency were calculated according to the following formula:
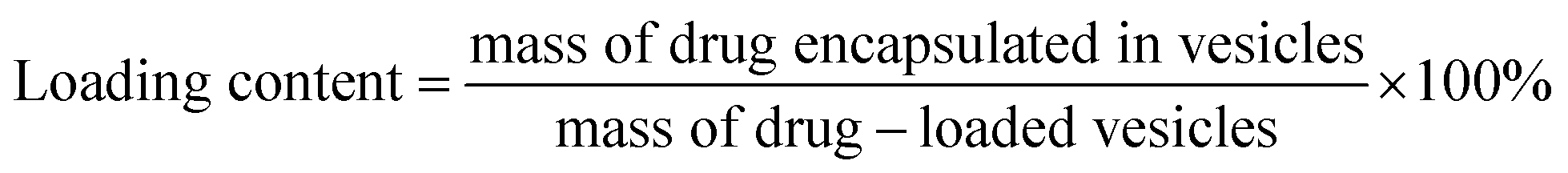
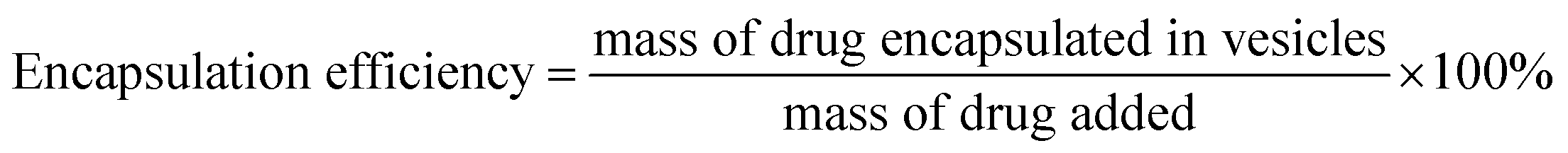
To explore the interaction mechanism between PEOP and INS, 2D-1H-NOESY NMR spectrum of 20 mg mL−1 PEOP and 10 mg mL−1 INS dissolved in pure DMSO-d6 were measured by a NMR spectrometer (AC-80, Bruker Biospin. Germany). Furthermore, FTIR spectrum of INS and OPA mixed at various weight ratios were measured by using a FTIR spectrometer (FT/IR-4100, JASCO, Japan).
In vitro INS release
In vitro release studies were conducted to estimate INS release from PEOP vesicles after oral administration. 1 mL of FITC-INS-PEOP vesicles (equal to 350 μg FITC-INS) were placed into dialysis diffusion bag (MWCO = 300 kDa), and then the dialysis diffusion bag was immersed in 5 mL simulated gastric fluid (SGF, pH 1.5) (USP34) and incubated for 2 h at 37 °C with constant shaking at 100 rpm. After 2 h, the medium was replaced by simulated intestinal fluid (SIF) (USP 34) at pH 6.8. At the predetermined time intervals, 50 μL FITC-INS-contained solution outside the dialysis diffusion bag was withdrawn and 50 μL fresh pre-heated buffer was added to replenish the removed sample to maintain a constant volume. The collected aliquots were analyzed by Microplate reader (SpectraMax M5, USA). All the tests were performed under sink conditions and conducted in triplicate.Cellular uptake investigation
For quantitative study, MDCK cells were seeded at a density of 1 × 105 cells per well into a 12-well culture plate and allowed to attach for 24 h until confluence. The medium was then removed and treated with fresh MEM containing free FITC-INS or FITC-INS-PEOP (10 μg mL−1) at 37 °C for 1 h and 4 h. After incubation, the cells were washed with PBS (pH 7.4), detached with trypsin–EDTA and resuspended in proper volume of PBS for flow cytometer analysis (Beckman Coulter, FC500MCL, US). Data from 10000 events were gated using forward and side scatter parameters to exclude debris and dead cells as well as control cells incubated with media alone as control for autofluorescence. All experiments were conducted in triplicate and data presented as means ± standard deviation.
Transport studies across MDCK cell monolayer
For the transport experiments, MDCK cells were seeded onto Transwell filters at a density of 2.5 × 105 cells per well and were allowed to grow and differentiate for 4 days. Cell monolayers were used when the net transepithelial electrical resistance (TEER) value exceeded 250 Ω cm2. The TEER value was measured by a Millicell-ERS volt-ohmmeter (Millipore Co., U.S.A.). The intrinsic resistance of the system (insert alone) was subtracted from the total resistance (cell monolayer plus insert) to yield the net value. Prior to the transport experiment, culture media were removed from the mature MDCK monolayers, and the monolayers were rinsed twice with 37 °C HBSS. After a 15 min incubation of both sides of the monolayers with HBSS (0.5 mL to apical side and 1.0 mL to basolateral side), 0.5 mL of HBSS solution containing free FITC-INS, FITC-INS-PEOP (10 μg mL−1) was applied to the apical side followed by addition of 1.0 mL of HBSS solution to the basolateral side for absorptive (AP to BL) transport, while secretory (BL to AP) transport was evaluated by adding 1.0 mL of drug solution in basolateral side and 0.5 mL of HBSS in apical side. Cells were incubated in a 37 °C shaking incubator. Aliquots of 200 μL were taken from the basolateral side (to study absorptive transport) and the apical side (to study secretory transport) at 20, 40, 60, 80, 100, and 120 min time intervals. The same volume of fresh preheated HBSS was added to keep the volume constant. The content of FITC-INS was detected by multifunctional microplate reader (SpectraMax M5, USA) at Ex/Em 488/520 nm. During the experiment, the integrity of the monolayers was assessed by means of TEER measurements. The apparent permeability coefficients (Papp) for free FITC-INS, FITC-INS-PEOP, and were calculated according to the following formula:where Papp is the permeability, dQ/dt is the apparent appearance rate of drug in the receiver side calculated using linear regression of amounts in receiver chamber versus time, C is the drug concentration in the donor chamber (10 μg mL−1), and S is the surface area of the monolayer (S = 1.13 cm2).
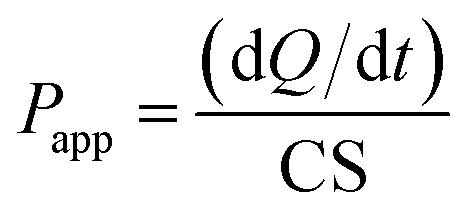
In vivo pharmacokinetics and intestinal lymphatic transport investigation
In order to monitor the plasma INS level in vivo, the diabetic rats (220 ± 20 g) were randomly divided into three groups (five rats per group, n = 5): Group 1, oral INS-PEOP vesicles in the INS dosage of 100 IU kg−1; Group 2, oral INS solution in the INS dosage of 100 IU kg−1; Group 3, subcutaneous (S.C.) injection of INS solution (3 IU kg−1). Blood samples were collected from the orbital veins prior to the administration and at different time intervals after the administration, and immediately centrifuged at 6500g for 5 min. The plasma INS concentration was determined using INS ELISA kits (Mercodia, Winston-Salem, USA). Each sample was measured in triplicate. The relative bioavailability (F%) was calculated based on the blood INS level according to the following equation:where AUC was the total area under the curve of plasma INS concentration vs. time curve calculated by Kinetica 4.4.1.
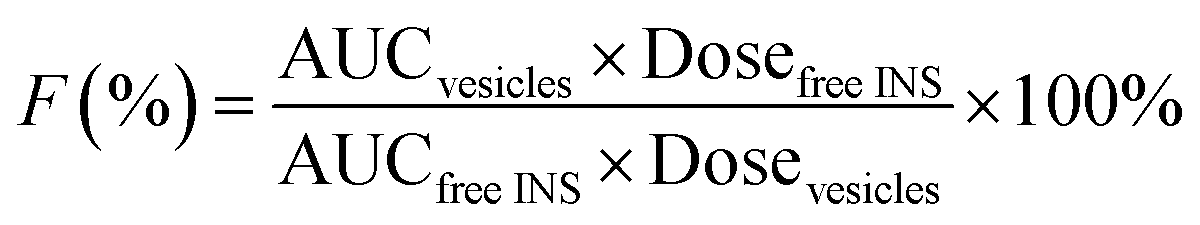
As a protein synthesis inhibitor, cycloheximide is known to inhibit the secretion of chylomicrons from the enterocyte, apparently through antimicrotubular effects, thus inhibiting lymphatic transport.25 For quantitative lymphatic transport investigation, ten diabetic rats were randomly divided into two groups (namely, Group 1 and Group 2). Each group received cycloheximide (3 mg kg−1) by intraperitoneal injection (i.p.) route. 1 h after injection, free INS and INS-PEOP vesicles were administered by oral gavage to rats for both groups, respectively. Blood samples were collected and plasma INS concentration was determined as mentioned earlier. Each sample was measured in triplicate.
For qualitative lymphatic transport investigation, nine normal male Sprague Dawley rats were randomly divided into three groups: normal saline (control group), oral FITC-INS-PEOP vesicles, and oral FITC-INS-PEOP vesicles after i.p. cycloheximide (3 mg kg−1) 1 h. 5 h after oral administration, rats were sacrificed and mesenteric lymph nodes were surgically removed. CLSM (LSM 510, Zeiss, Germany) was used to observe the fluorescence of FITC-INS-PEOP vesicles in the mesenteric lymph nodes homogenate among these groups. The excitation and emission wavelengths for CLSM observation were 488 nm and 520 nm, respectively.
In vivo hypoglycemic effect
The diabetic rats (220 ± 20 g) were built by injection of streptozotocin (65 mg kg−1) according to literature reported.13 Blood glucose level was determined using a glucose meter (ACCU-CHEK, Roche. Co. Ltd. Mannheim, Germany). The rats that exhibited fasting blood glucose level over 16.0 mM 1 week after treatment were considered to be diabetic rats. As shown previously, diabetic rats were randomly divided into three groups (five rats per group, n = 5). Blood glucose was thereafter measured prior to the administration and at different time intervals after the administration. Besides, we also measured blood glucose after blocking lymphatic transport. All animal experiments were conducted in accordance with the approval of Experimental Animal Ethical Committee of Zhejiang University.Results and discussion
Synthesis and characterization of PEOP
Amphiphilic graft PEOP with the appropriate graft ratio were synthesized by the sequential substitution reaction between mPEG2k-NH2/OPA and chloride atoms on the poly(dichlorophosphazene) backbone as shown in Scheme 2. The structure of OPA was confirmed by 1H spectroscopy (Fig. S1†) with the characteristic signals at 0.86 ppm (–CH3, a), 1.26 ppm (–CH2–, b), 3.07 ppm (–CH2–NH2, d), 3.84 ppm (–CH2–O–, c). The final polymeric product PEOP was characterized by 1H NMR, 13C NMR, 31P NMR, FT-IR, and GPC spectroscopy. According to Fig. 1A, the characteristic signals in 1H NMR spectrum contributed by the PEG segments were at 3.65 ppm (–CH2–) and 3.38 ppm (–OCH3), while those contributed by the OPA segments were at 1.26 ppm (–CH2–) and 0.86 ppm (–CH3). The grafting ratio of mPEG2k-NH2/OPA was calculated as 0.35/1.65 by the peak area ratio of –CH2– in PEG to –CH2– in OPA. The weight ratio of mPEG2k-NH2 for PEOP graft copolymer (fPEG (w)) was calculated using the following equation: fPEG (w) = A3.38 × MPEG/(A3.38 × MPEG + A0.86 × MOPA + (A0.86 + A3.38)MPPP/2), where A0.86 and A3.38 represent the peak areas of chemical shifts at 0.86 and 3.38 ppm in the 1H NMR spectrum of PEOP, respectively. Molar mass of PEG, OPA and repeated unit (NP) of polyphosphazene was represented as MPEG, MOPA and MPPP. The corresponding weight ratio of mPEG2k-NH2 (fPEG) in the copolymer PEOP was calculated as 49.9%. As shown in Fig. 1B, the characteristic signal contributed by the PEG segments was at 70 ppm, and those contributed by the OPA segments were at 32, 29.74, 22.67, and 14.19 ppm. Two types of phosphorus signals can be observed in the 31P NMR spectrum in Fig. 1C, including one contributed by the phosphorus in OPA segments at −11 ppm and the other contributed by the phosphorus in the polymer backbone at 0 ppm. In addition, the chlorine atoms linked to the backbone have been replaced completely, as there isn't sharp peak of P–Cl units at near −17 ppm. In the FT-IR spectrum shown in Fig. 1D, the characteristic peaks of 3437 (–O–H or –N–H stretching), 2917, 2842 (–C–H stretching), 1634 (–CO stretching), 1092 (–C–O– stretching), 1463 and 1241 (–PO–, –NP– stretching) can be observed. Based on the GPC chromatogram (Fig. S2†), the number-average molecular weight (Mn) of PEOP was 68782 with a polydispersity (PDI) 1.68. All these characterizations were in full agreement with the desired structure of PEOP.
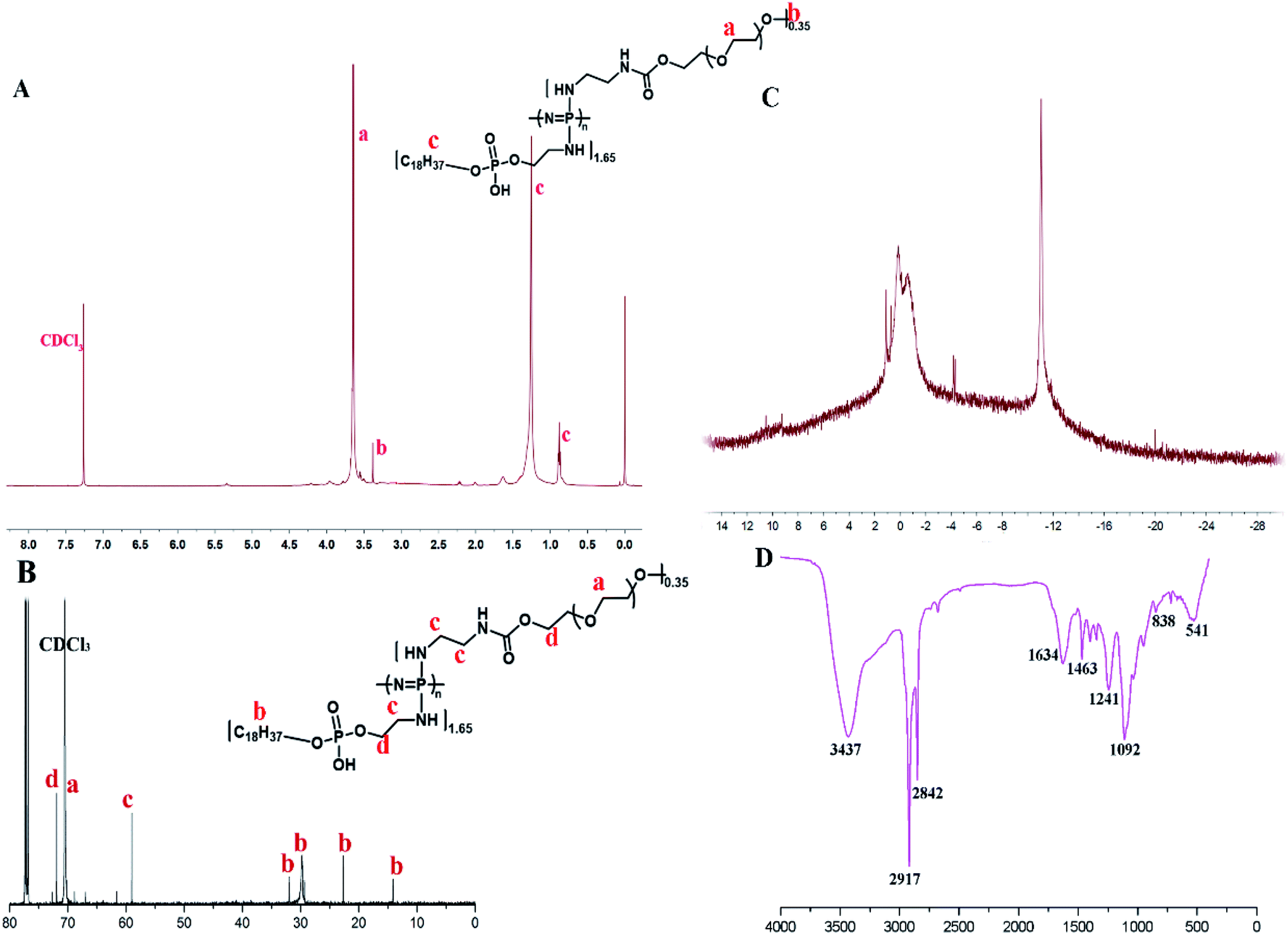 | ||
| Fig. 1 The characterization of PEOP by (A) 1H NMR spectrum (500 MHz, CDCl3), (B) 13C NMR spectrum (500 MHz, CDCl3), (C) 31P-NMR spectrum, and (D) FT-IR spectrum. | ||
Self-assembly behavior of PEOP
The self-aggregate of PEOP was obtained by dialysis against deionized water, which displayed a typical vesicle structure with a hollow core as shown in Fig. 2A. The size of blank PEOP vesicles measured by DLS was 223.5 with the narrow particle size distribution (PDI 0.133). The self-assembly behavior of polymers is a very interesting topic. For the amphiphilic diblock copolymers, the suitable weight percent of hydrophilic segments to form vesicles is in the range of 30–40%,26,27 over which copolymers are prone to form micelles. The self-assembly behavior of PEOP is quite similar to those amphiphilic diblock copolymers, except that the PEG proportion was significantly higher at the vesicle–micelle transition point. This phenomenon has been found in our previous work.22,28 The reason may be that the unique architecture of the graft amphiphilic copolymers makes the backbone more rigid and restricts the bending of the backbone, which facilitates the decrease of the curvature of the self-assembled membrane and results in the formation of vesicles. Herein, PEOP with relatively high fPEG of 49.9% was able to self-assemble to nano-vesicles indeed, again validating the conclusion based on our previous observations.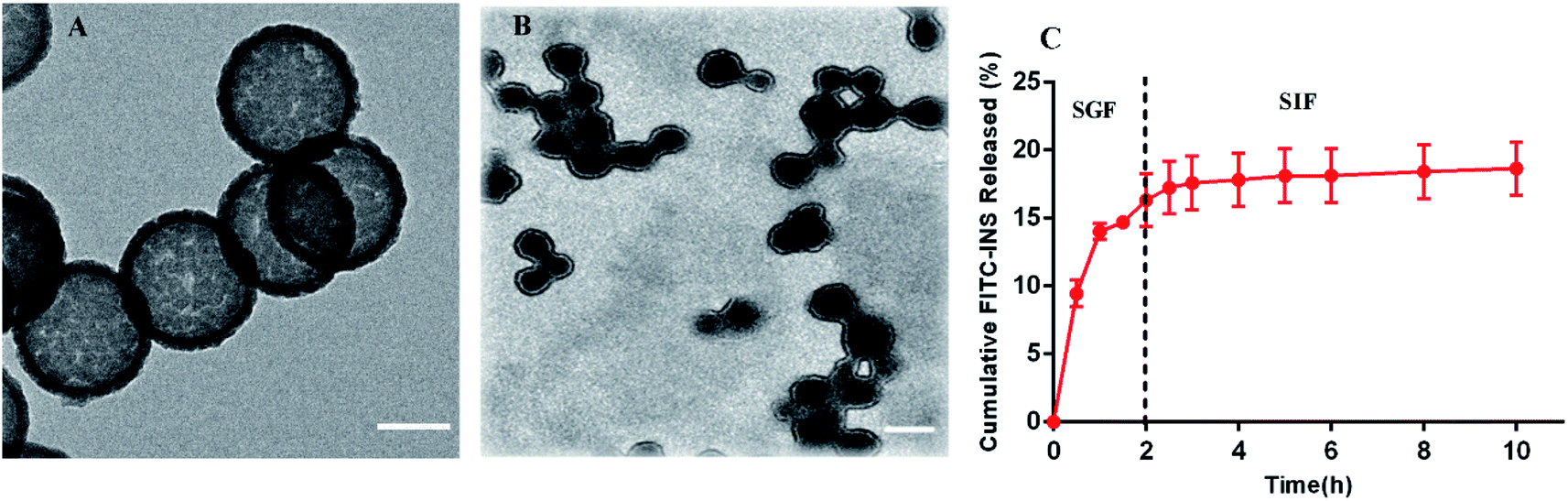 | ||
| Fig. 2 The TEM images of (A) blank PEOP vesicles (scale bar 100 nm) and (B) INS-PEOP vesicles (scale bar 250 nm). (C) FITC-INS released from FITC-INS-PEOP vesicles, data are means ± SD (n = 3). | ||
Drug loading
For the INS delivery application, we prepared INS-PEOP vesicles by dialysis and evaluated the drug loading capability of PEOP. The size of INS-PEOP vesicles measured by DLS were 245.0 nm with the PDI of 0.190. The TEM of INS-PEOP vesicles was shown in Fig. 2B, which appear like solid nanoparticles with a shell.29 In theory, the hydrophilic INS can be encapsulated into the aqueous cavum of vesicles. In fact, however, due to its bulk size, the INS loading is difficult to achieve. For example, the INS loading content of a glucose-responsive polymersome nanovesicle, assembled by a diblock copolymer consisting of poly(ethylene glycol) dissociates was only 2.5 wt%.30 Interestingly, PEOP vesicles displayed an excellent capability to encapsulate INS with a high INS encapsulation efficiency (84.7%) and loading content (8.47%). We supposed that this phenomenon may be attributed to certain strong molecule interactions between the hydrophobic OPA and INS.To verify our hypothesis, a 2D 1H NMR NOESY measurement of INS/OPA mixture was conducted. In general, if the spatial distance of two hydrogen atoms in the compounds is very close (less than 5 angstroms) and if one of the hydrogen atoms is irradiated by the hydrogen spectrum, the peak group area of another hydrogen will change, leading to the emergence of a new peak. This is termed the nuclear overhauser effect (NOE), and the NOESY spectrum can be applied for characterizing the interaction between the compounds effectively.31 As Fig. 3A shows, the highlighted dots in the blank boxes represent NOE cross peaks generated by H1 protons in the lipid-like hydrophobic OPA (Fig. S1 in “ESI†”, –CH2, H11.26 ppm) with H2 protons in INS (Fig. S3 in “ESI†”, H2 6.5–8.5 ppm). To further illustrate molecule interactions between the hydrophobic OPA and INS, we prepared a series of samples containing different ratios of the hydrophobic OPA and INS to examine their FT-IR spectrogram. As shown in Fig. 3B, the peak of the hydroxyl (3411 cm−1) and amino group (3304 cm−1) of the hydrophobic chain and INS can be easily observed by the FT-IR. After mixing the two compounds in the different proportions (1:1, 1:3, 1:5), the peak intensity of the hydroxyl and amino groups become weaker and weaker, and a new characteristic peak (3274 cm−1) was observed. Based on these results, the spontaneous interactions, mainly hydrogen bonding interaction, generated between the hydrophobic OPA and the INS was verified, which was supposed to be responsible for the high INS loading into the PEOP vesicles.32 On one hand, INS can be loaded into the aqueous cavity as it is water-soluble. On the other hand, INS can also be loaded into the hydrophobic membrane of the vesicles due to the intermolecular interactions between the hydrophobic OPA and INS. The synergetic effect can improve the INS-loading ability of the PEOP vesicles, that is the reason why PEOP vesicles can load INS by dialysis method with high loading content and encapsulation efficiency.
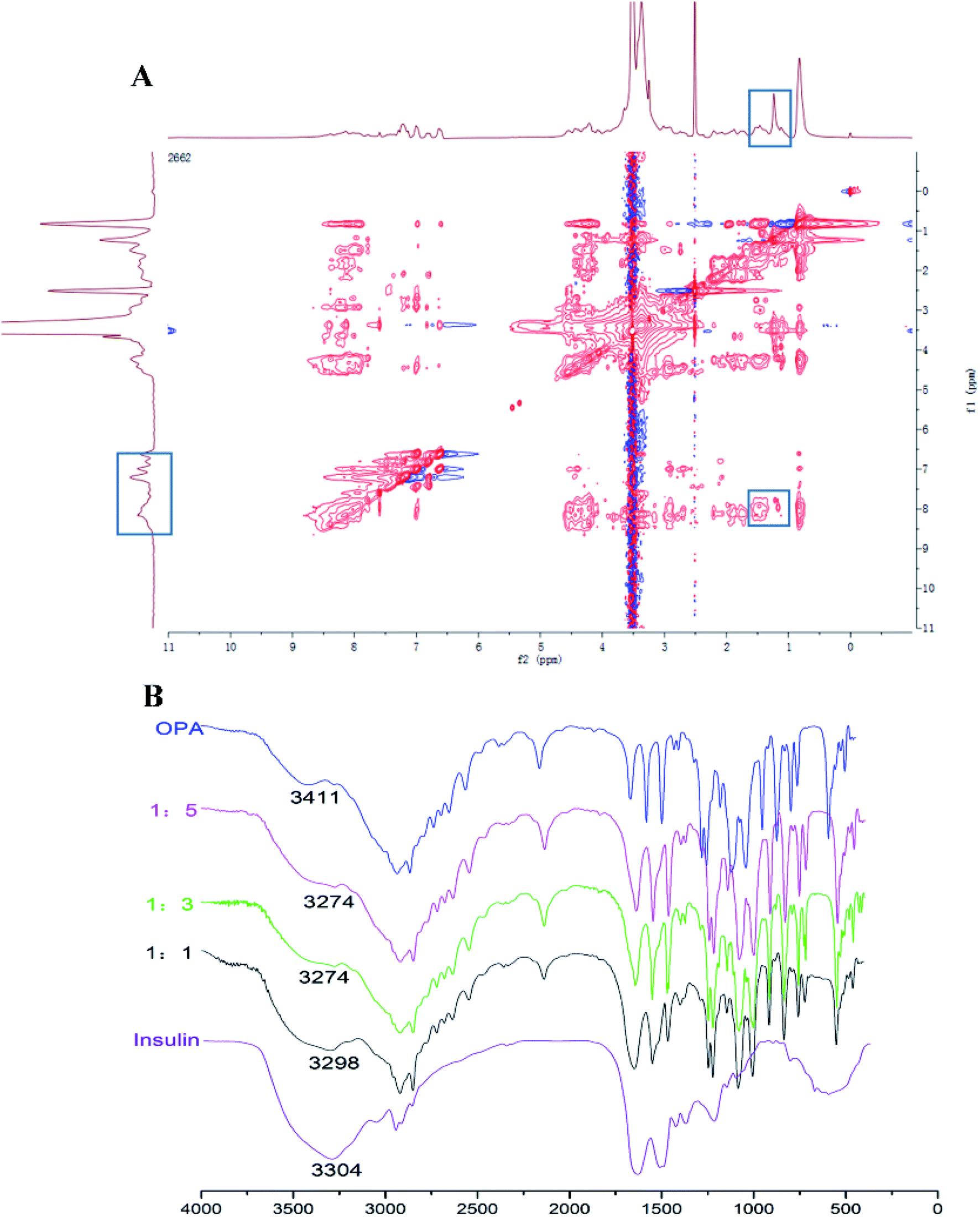 | ||
| Fig. 3 (A) 2D 1H–1H NOESY spectrum of PEOP and insulin mixture in DMSO-d6 (important NOE peaks are highlighted in solid blue rectangular). (B) FT-IR spectrum of insulin, the hydrophobic chain OPA, and their mixtures at the ratio of 1:1, 1:3, and 1:5. | ||
In vitro release investigation
In order to address the oral barrier of INS, INS-loaded nanoparticles have been extensively studied to provide drug protection and more physiological pathways.33–35 Therefore, in vitro release studies were conducted after oral administration of INS-PEOP vesicles under simulated gastrointestinal conditions. As shown in Fig. 2C, FITC-INS-PEOP vesicles were first investigated in SGF for 2 h, and then in SIF until the maximum release was achieved. A rapid increase in FITC-INS release was observed in SGF and the amount of FITC-INS released was about 16.32% in SGF (pH 1.5) for 2 h. After 2 h, the release medium was converted to SIF (pH 6.8), mimicking the transport of the intestinal environment. The amount of FITC-INS released was maintained at a relatively stable level for 8 h, reaching about 18.66% cumulative FITC-INS. The variation in FITC-INS release behavior of PEOP vesicles in SGF and SIF might be correlated with its pH-dependent solubility. The isoelectric point of INS is about 5.3. Therefore, INS released at the relatively rapid rate from vesicles through diffusion due to the relatively high solubility at the low pH of SGF although there was certain intermolecular force between INS and PEOP. However, this amphiphilic material might form a stable barrier to retain INS and limit its release from PEOP vesicles at neutral pH. The results indicate that when given orally, the amount of INS released from the vesicles in the stomach would be controlled at a relatively low level, while the remainder might provide a quite slow release in the physiological intestine. Thus, the in vitro release experiments confirmed the potential of PEOP vesicles to protect the drug from the hostile conditions of the gastrointestinal tract.Cell uptake and transport
The MDCK cell line, having clear apico-basal polarity and closed-up tight junctions, is a widely used model to mimic intestinal epithelial cells for epithelial biology investigation.36,37 To investigate INS uptake in MDCK cells after different incubation times, flow cytometry was applied.32 According to the results shown in Fig. 4A, when MDCK cells were treated with free FITC-INS, FITC-INS-PEOP vesicles for 1 h, the percentages of gated cells were 13.60% and 22.60%, respectively. 4 h later, the percentage of gated cells after FITC-INS-PEOP vesicles treatment was 40.65%, while only 16.86% of gated cells were observed after free FITC-INS treatment. These results indicate that MDCK cells had a higher uptake capacity to PEOP vesicles over time, and this might be attributed to the better ability of PEOP vesicles to enter cells via the endocytosis pathway.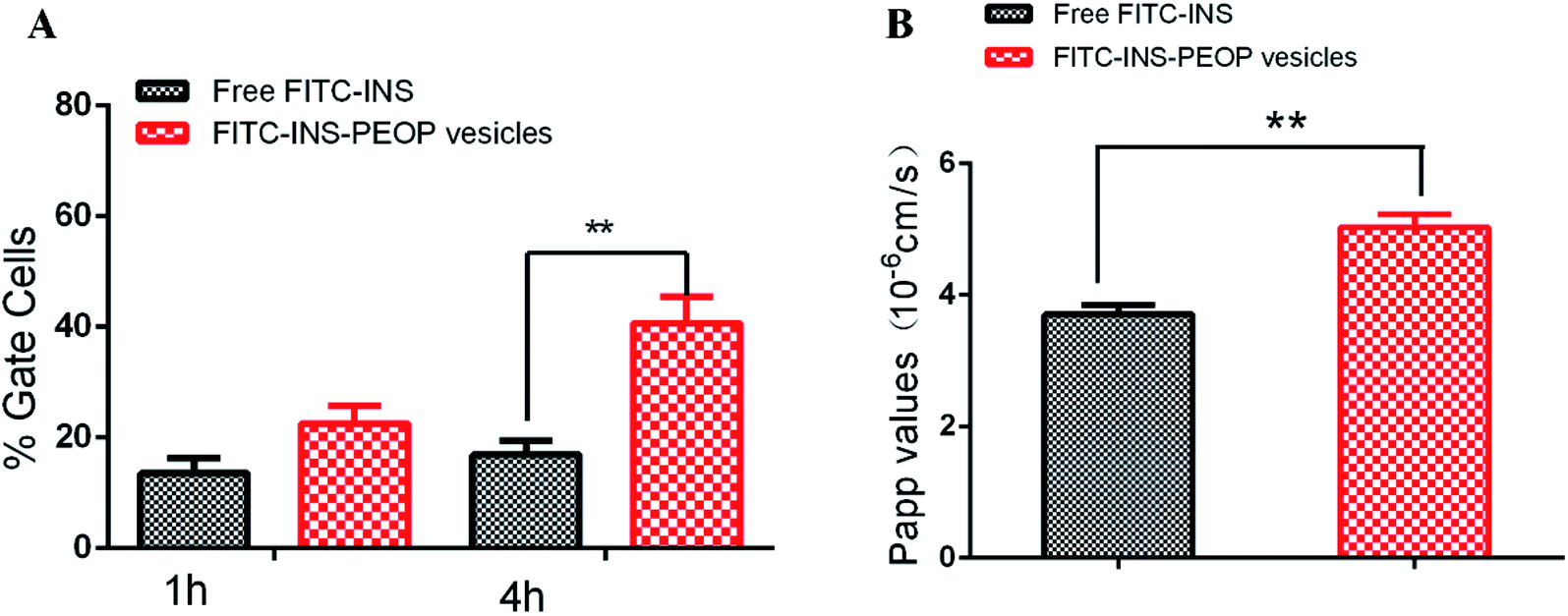 | ||
| Fig. 4 (A) Cellular uptake of free FITC-INS and FITC-INS-PEOP vesicles in MDCK (10 μg mL−1 FITC-INS) at different time points. (B) Apparent permeability coefficient (Papp) for free FITC-INS, FITC-INS-PEOP vesicles across MDCK monolayers. **, p < 0.01. Data are means ± SD (n = 3). | ||
The transport of the free FITC-INS and FITC-INS-PEOP vesicles was investigated by well-established MDCK monolayers with net transendothelial electrical resistance (TEER) over 250 Ω cm2. Generally, higher values of apparent permeability coefficients (Papp) indicate greater permeability across the intestinal barrier and optimal oral bioavailability. According to the results of the Fig. 4B, the Papp of free FITC-INS was 3.71 ± 0.24 × 10−6 cm s−1, and that of the FITC-INS-PEOP vesicles was 5.04 ± 0.35 × 10−6 cm s−1. Compared with the free FITC-INS sample, the Papp of FITC-INS-PEOP vesicles was approximately 1.4-fold higher. Moreover, it was observed that the TEER values of the MDCK monolayers were not changed after the incubation of the samples (Table S1†), which confirmed that the MDCK monolayers were intact. Therefore, it was indicated that PEOP vesicles can improve INS permeability across the intestinal epithelium, which would lead to the enhanced oral bioavailability.21,38 In addition, the intactness of MDCK monolayers provided the evidence for the good biocompatibility of PEOP nanovesicles.
In vivo pharmacokinetics and transport pathway
Plasma INS concentration after administration of different INS formulations was determined by ELISA. As shown in Fig. 5A, the subcutaneous (S.C.) injection of INS solution caused a sharp increase in the plasma INS concentration, which reached 82.41 mIU L−1 after 1 h and then decreased rapidly. In contrast, a gradual growth was observed after oral INS-PEOP vesicles (100 IU kg−1) and then a great peak of the plasma INS concentration (52.60 mIU L−1) was observed at 6 h. Thereafter, the plasma INS concentration slowly decreased, reaching baseline after 10 h. Besides, the oral administration of free INS was ineffectively, and the plasma INS concentration did not increase during the experiment. Accordingly, the area under the curve (AUC) of the plasma INS concentration curves was 320.31 mIU h L−1 for S.C. free INS solution, 255.51 mIU h L−1 for oral INS-PEOP vesicles, and only 5.40 mIU h L−1 for oral free INS solution. Most importantly, the relative bioavailability of oral INS-PEOP vesicles was 47.32 times higher than that of oral free INS solution. It indicated that PEOP vesicles can effectively improve the bioavailability of insulin after oral administration.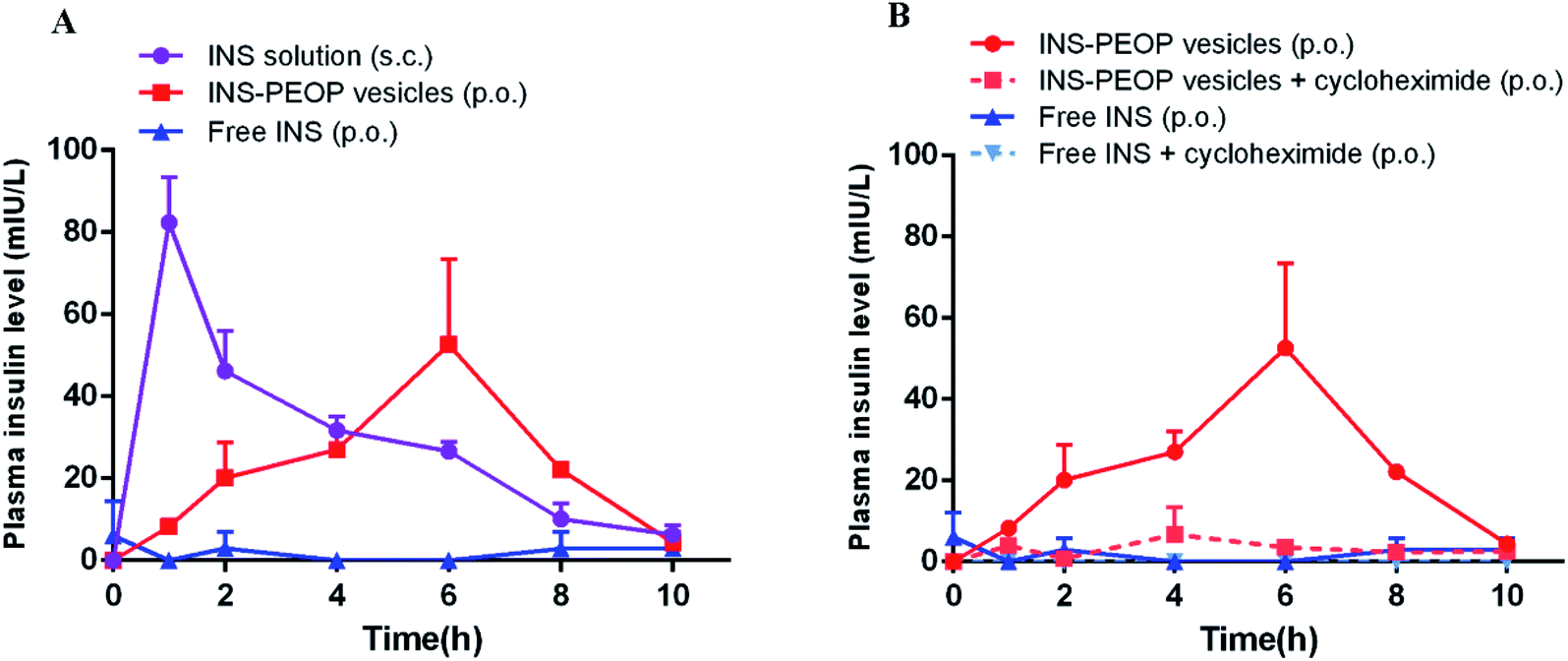 | ||
| Fig. 5 (A) Variation of plasma insulin concentration of diabetic rats after orally administering INS-PEOP vesicles or insulin solution at dose of 100 IU kg−1, or subcutaneous injection with insulin solution at 3 IU kg−1. (B) The plasma concentration of insulin after oral administration of INS-PEOP vesicles or insulin solution at dose of 100 IU kg−1 to nontreated and cycloheximide-treated rats. Data are means ± SD (n = 5). | ||
Based on the above results, we further investigated the subsequent transport of PEOP vesicles after their uptake into the enterocytes. As a chemical inhibitor, cycloheximide was used to inhibit the lymphatic transport pathway without causing non-specific damage to other active and passive absorption pathways.39–41 As shown in Fig. 5B, when INS-PEOP vesicles were orally administered to rats treated with cycloheximide, the plasma INS concentration was significantly reduced compared with that of PEOP vesicles without cycloheximide. Similarly, the plasma INS concentration of rats treated with cycloheximide could not be determined after orally administering free INS solution at dose of 100 IU kg−1. These results indicated that cycloheximide blocked a large amount of INS absorption of PEOP vesicles and demonstrate that the oral absorption of INS-PEOP vesicles is mainly mediated by the lymphatic transport system.
In order to further verify the lymphatic accumulation of PEOP vesicles, fluorescence examination on the change of mesenteric lymph nodes was performed following orally administering FITC-INS-PEOP vesicles. As shown in Fig. 6A, there was no green fluorescence of FITC in the blank mesenteric lymph nodes. 5 h after oral FITC-INS-PEOP vesicles, the fluorescence in the mesenteric lymph nodes of the rats i.p. with cycloheximide (Fig. 6C) was significant reduced compared with that of the rats without chylomicron flow blockade (Fig. 6B). This phenomenon reveals that considerable accumulation of FITC-INS-PEOP vesicles in the mesenteric lymph nodes and then INS was absorbed into the systemic circulation via the lymphatic system.
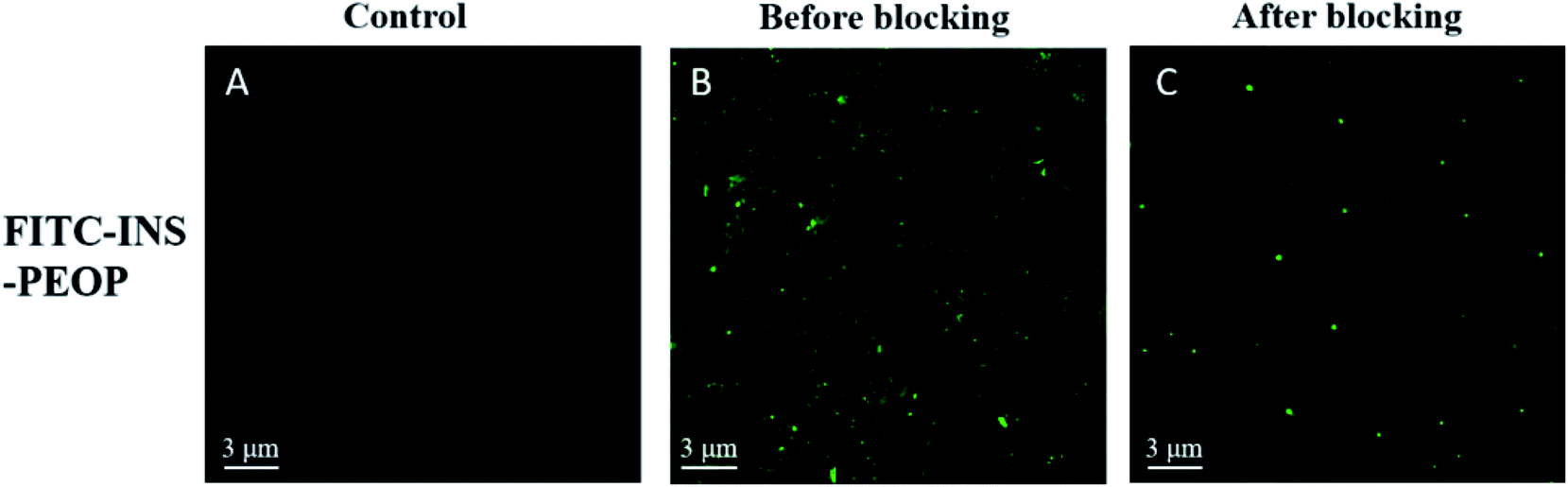 | ||
| Fig. 6 CLSM images of various groups: (A) blank mesenteric lymph nodes, (B) FITC-INS-PEOP vesicles in mesenteric lymph nodes without cycloheximide blocking, and (C) FITC-INS-PEOP vesicles in mesenteric lymph nodes after cycloheximide blocking. | ||
Generally, upon oral administration, lipid-based carrier-encapsulated drug can enter the lymphatic transport system through the following two mechanisms: (1) through M cells and gut-associated lymphoid tissue that compose of lymphoid follicles forming Peyer's patches, (2) stimulating chylomicron production and transport by triglyceride-rich lipoproteins.42 In the latter, the triglyceride-rich lipoproteins preferentially enter the lymphatic capillaries, in which cells are arranged in overlapped way with the gap.41 Based on the results from lymphatic transport studies, therefore, it could be speculated that the lymphatic transport pathway played an important role in the absorption of INS via PEOP vesicles into the systemic circulation.
In vivo hypoglycemic effect
We then investigated the hypoglycemic effect of INS-PEOP vesicles by oral administration to diabetic rats. As shown in Fig. 7, the blood glucose level before drug administration (0 h) was 100% per group, and the changes in blood glucose level (of initial%) of diabetic rats over time were shown after oral administration of various formulations or S.C. injection of free INS solution. The S.C. injection of free INS solution caused a significant decrease in blood glucose level that peaked (up to a 72% decrease) after 1 h, but then the blood glucose level rapidly returned to high level subsequently. In contrast, the oral administration of the free INS (100 IU kg−1) failed to reduce the blood glucose level of the rats due to its instability and poor absorption in the gastrointestinal tract.13 Notably, the oral administration of INS-PEOP vesicles produced a gradual decrease in blood glucose level and the maximum reduction in glucose level (up to 40%) was observed at 6 h. Although the reduction rate of blood glucose level was lower than that by S.C. injection of free INS solution, the blood glucose level decreased gradually after oral intake of INS-PEOP vesicles, and the hypoglycemic effect was maintained for approximately 8 h. This might be due the sustained release of INS from INS-PEOP vesicles (Fig. 2C). We also investigated the effect of cycloheximide on hypoglycemic action of oral insulin delivery. Accordingly, although the hypoglycemic profiles fluctuated up and down (Fig. 7), the in vivo hypoglycemic effect was basically disappeared after chylomicron flow blockade induced by cycloheximide.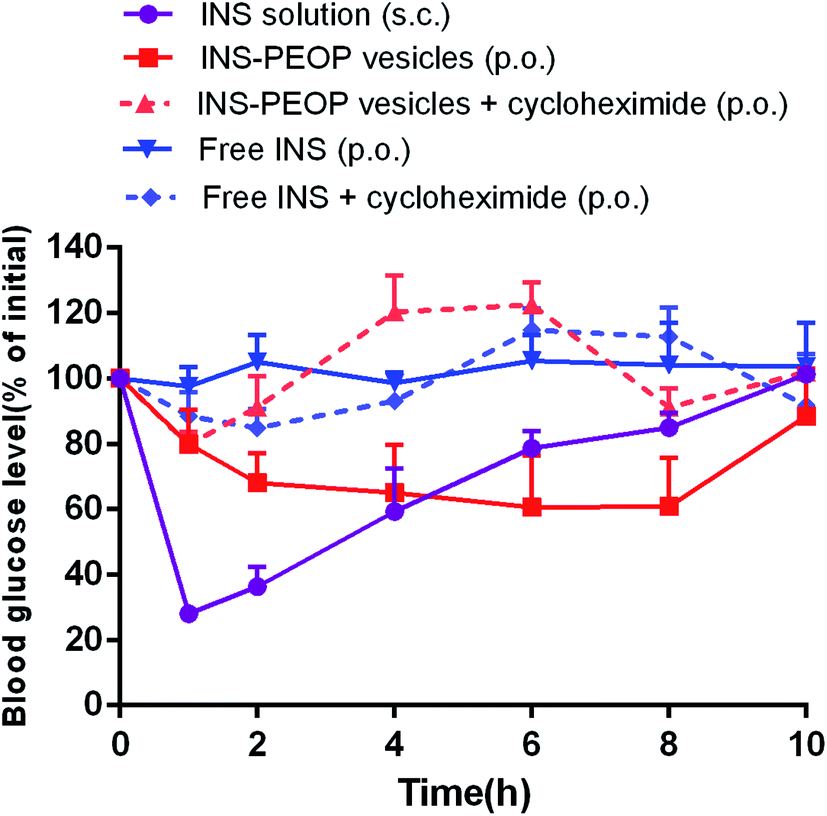 | ||
| Fig. 7 Variation of blood glucose levels of diabetic rats (nontreated and cycloheximide-treated rats) after orally administering INS-PEOP vesicles or insulin solution at dose of 100 IU kg−1, subcutaneous injection with insulin solution at 3 IU kg−1. Data are means ± SD (n = 5). | ||
According to the above results, PEOP nano-vesicle as an oral INS carrier could provide the following advantages for achieving a favorable hypoglycemic effect in vivo. Firstly, in virtue of the synergetic effect of the interaction between lipid-like hydrophobic OPA of PEOP and INS and the physical encapsulation by the aqueous vesicle lumens, PEOP nano-vesicle effectively improved the loading content and encapsulation efficiency of INS. Secondly, as an oral INS carrier, PEOP vesicle can maintain INS inside the nano-vesicles (Fig. 2C) and protect INS in the gastrointestinal tract to a certain extent. Thirdly, in vitro cell uptake study (Fig. 4) and in vivo pharmacokinetics investigation (Fig. 5) confirmed that PEOP nano-vesicles significantly promoted INS absorption mainly through lymphatic transport system when compared with INS solution. All these suggest that PEOP nano-vesicle as a novel polymeric vesicle has great potential for oral delivery of therapeutic peptide or protein agents.
Conclusion
In this work, we synthesized a novel amphiphilic graft polyphosphazenes that can self-assemble into vesicles by the control of the hydrophilic fraction. The vesicles can be used as carriers of INS due to their strong intermolecular interaction according to the loading content measurement results. However, the PEOP vesicles as an oral INS carrier displayed stronger capability to load INS, effectively protect INS in the gastrointestinal tract and promote the absorption of INS. According to the animal experiments, we found that the PEOP vesicles could improve the oral bioavailability of INS and finally resulted in the better diabetes treatment effect through the lymphatic transport pathway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81872825).Notes and references
- K. L. Cavanaugh, Clin. Diabetes, 2007, 25, 90–97 CrossRef.
- E. Giovannucci, D. M. Harlan, M. C. Archer, R. M. Bergenstal, S. M. Gapstur, L. A. Habel, M. Pollak, J. G. Regensteiner and D. Yee, Ca-Cancer J. Clin., 2010, 60, 207–221 CrossRef PubMed.
- P. Hillon, B. Guiu, J. Vincent and J.-M. Petit, Gastroenterol. Clin. Biol., 2010, 34, 529–533 CrossRef CAS.
- E. J. Gallagher and D. Leroith, Physiol. Rev., 2015, 95, 727–748 CrossRef CAS.
- K. El-Sayed, M. Mariko, O. Yoshinori and T. Kozo, Adv. Drug Delivery Rev., 2007, 59, 1521–1546 CrossRef.
- T. W. Wong, J. Drug Targeting, 2010, 18, 79–92 CrossRef CAS PubMed.
- C. Mei-Chin, S. Kiran, C. Ko-Jie and S. Hsing-Wen, Biomaterials, 2011, 32, 9826–9838 CrossRef.
- F. P. Kennedy, Drugs, 1991, 42, 213–227 CrossRef CAS PubMed.
- G. P. Carino and E. Mathiowitz, Adv. Drug Delivery Rev., 1999, 35, 249–257 CrossRef CAS PubMed.
- M. A. Lopes, B. A. Abrahim, S. A. Raquel, V. Francisco, C. R. Rodrigues and A. J. Ribeiro, Curr. Pharm. Biotechnol., 2014, 15, 629–638 CAS.
- E. Déat-Lainé, V. Hoffart, G. Garrait, J. F. Jarrige, J. M. Cardot, M. Subirade and E. Beyssac, Pharm. Res., 2013, 30, 721–734 CrossRef PubMed.
- E. Lee, J. Lee and S. Jon, Bioconjugate Chem., 2010, 21, 1720–1723 CrossRef CAS PubMed.
- S. Wei, Z. Xi, L. Min, L. Lian, Z. Jiaju, S. Wei, Z. Zhirong and H. Yuan, ACS Nano, 2015, 9, 2345–2356 CrossRef PubMed.
- J. Sheng, L. Han, J. Qin, G. Ru, R. Li, L. Wu, D. Cui, P. Yang, Y. He and J. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 15430–15441 CrossRef CAS PubMed.
- S. Neha, S. Mohammad-Ali, A. Francisca, Z. Hongbo, E. M. Mäkilä, K. Jussi, S. Bruno, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2014, 35, 7172–7179 CrossRef PubMed.
- D. A. Christian, S. Cai, D. M. Bowen, Y. Kim, J. D. Pajerowski and D. E. Discher, Eur. J. Pharm. Biopharm., 2009, 71, 463–474 CrossRef CAS PubMed.
- G. R. Dakwar, I. A. Hammad, M. Popov, C. Linder, S. Grinberg, E. Heldman and D. Stepensky, J. Controlled Release, 2012, 160, 315–321 CrossRef CAS PubMed.
- Y. Yuan, J. Xinguo, G. Shuyu, F. Liang, Z. Yanqiang and P. Zhiqing, Nanoscale, 2014, 6, 3250–3258 RSC.
- H. A. Shahid Rameez and A. F. Palmer, Bioconjugate Chem., 2008, 19, 1025–1032 CrossRef PubMed.
- C. P. O’Neil, T. Suzuki, D. Demurtas, A. Finka and J. A. Hubbell, Langmuir, 2009, 25, 9025–9029 CrossRef.
- M. Y. Hu, Y. R. Shen, L. Zhang and L. Y. Qiu, Biomacromolecules, 2016, 17, 1026–1039 CrossRef CAS PubMed.
- Z. Cheng, L. Qiu and K. Zhu, Polymer, 2009, 50, 1173–1177 CrossRef.
- J. Xu, Q. Zhao, Y. Jin and L. Qiu, Nanomedicine, 2014, 10, 349–358 CrossRef CAS PubMed.
- X. W. Zhang, J. P. Qi, Y. Lu, W. He, X. Y. Li and W. Wu, Nanomedicine, 2014, 10, 167–176 CrossRef CAS.
- A. Dahan and A. Hoffman, Eur. J. Pharm. Sci., 2005, 24, 381–388 CrossRef CAS.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- B. Li, G. Chen, F. Meng, T. Li, J. Yue, X. Jing and Y. Huang, Polym. Chem., 2012, 3, 2421–2429 RSC.
- C. Zheng, X. P. Yao and L. Y. Qiu, Macromol. Biosci., 2011, 11, 338–343 CrossRef CAS PubMed.
- M. Gao, X. Zhu, L. Wu and L. Qiu, Biomacromolecules, 2016, 17, 2199–2209 CrossRef CAS PubMed.
- W. Tai, R. Mo, J. Di, V. Subramanian, X. Gu, J. B. Buse and Z. Gu, Biomacromolecules, 2014, 15, 3495–3502 CrossRef CAS PubMed.
- I. K. Voets, A. de Keizer, P. de Waard, P. M. Frederik, P. H. H. Bomans, H. Schmalz, A. Walther, S. M. King, F. A. M. Leermakers and M. A. C. Stuart, Angew. Chem., Int. Ed., 2006, 45, 6673–6676 CrossRef CAS PubMed.
- J. Wang, M. X. Xu, X. J. Cheng, M. Kong, Y. Liu, C. Feng and X. G. Chen, Carbohydr. Polym., 2016, 136, 867–874 CrossRef CAS PubMed.
- Z. H. Zhang, Y. L. Zhang, J. P. Zhou and H. X. Lv, Int. J. Nanomed., 2012, 7, 3333–3339 CAS.
- L. Han, Y. Zhao, L. Yin, R. Li, Y. Liang, H. Huang, S. Pan, C. Wu and M. Feng, AAPS PharmSciTech, 2012, 13, 836–845 CrossRef CAS PubMed.
- G. Sharma, A. R. Sharma, J. S. Nam, G. P. C. Doss, S. S. Lee and C. Chakraborty, J. Nanobiotechnol., 2015, 13, 74 CrossRef PubMed.
- S. Tavelin, J. Taipalensuu, F. Hallböök, K.-S. Vellonen, V. Moore and P. Artursson, Pharm. Res., 2003, 20, 373–381 CrossRef CAS PubMed.
- E. Duizer, A. J. Gilde, C. H. Versantvoort and J. P. Groten, Toxicol. Appl. Pharmacol., 1999, 155, 117–126 CrossRef CAS PubMed.
- M. Lopes, N. Shrestha, A. Correia, M. A. Shahbazi, B. Sarmento, J. Hirvonen, F. Veiga, R. Seica, A. Ribeiro and H. A. Santos, J. Controlled Release, 2016, 232, 29–41 CrossRef CAS PubMed.
- A. Dahan and A. Hoffman, Eur. J. Pharm. Sci., 2005, 24, 381–388 CrossRef CAS PubMed.
- M. L. Lind, J. Jette, H. René and M. Anette, Eur. J. Pharm. Sci., 2008, 35, 211–218 CrossRef CAS PubMed.
- F. Gao, Z. Zhang, H. Bu, Y. Huang, Z. Gao, J. Shen, C. Zhao and Y. Li, J. Controlled Release, 2011, 149, 168–174 CrossRef CAS PubMed.
- A. A. Khan, J. Mudassir, N. Mohtar and Y. Darwis, Int. J. Nanomed., 2013, 8, 2733–2744 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00382d |
| ‡ Yumiao Hu and Juan Wang share first authorship. |
| This journal is © The Royal Society of Chemistry 2020 |