DOI:
10.1039/D0RA00377H
(Review Article)
RSC Adv., 2020,
10, 9563-9578
Synthesis and application in asymmetric catalysis of P-stereogenic pincer–metal complexes
Received
14th January 2020
, Accepted 21st February 2020
First published on 5th March 2020
Abstract
P-stereogenic pincer–metal complexes are one of the most interesting pincer type organometallic compounds. Many kinds of this type of complexes were synthesized and used as catalysts in asymmetric catalysis. On the basis of our work in this field, this paper reports the recent progress in P-stereogenic pincer chemistry, including the synthesis of P-stereogenic pincer ligands, the synthesis of P-stereogenic pincer–metal complexes, and the achievements in P-stereogenic pincer–metal complex catalyzed asymmetric synthesis.
 Yijun Xiang | Yijun Xiang was born in Hunan Province, China. She is currently conducting her undergraduate studies under the supervision of Prof. Xing Zheng and Dr Zehua Yang at the University of South China. |
 Qianyi Ge | Qianyi Ge was born in Hebei Province, China. She received her Bachelor's degree in the major of pharmaceutical engineering from Weifang University, China in 2015. And she is currently pursuing her Master's degree in the major of Pharmacy at the University of South China under the supervision of Dr Zehua Yang. Her current research interests include organometallic chemistry as well as chiral ligand design and synthesis. |
 Shulei Wu | Shulei Wu was born in Hunan Province, China. She is currently studying at the University of South China as an undergraduate student in the major of Pharmacy, and will receive her Bachelor's degree in 2020. She will continue her Master's studies under the supervision of Dr Zehua Yang. Asymmetric catalysis is her major research field. |
 Xing Zheng | Xing Zheng received his PhD in Chemistry from Donghua University, China. He also worked as a postdoctoral fellow at Arizona State University and Wayne State University. He moved back to the University of South China where he was promoted to Professor in 2010. His scientific interests are the area of organofluorine chemistry, medicinal chemistry and natural product chemistry. |
 Zehua Yang | Zehua Yang received his PhD in Pharmacy from Shanghai Jiao Tong University, China in 2015. And then worked at the University of South China as a lecturer. His scientific interests are in the area of asymmetric synthesis, organometallic chemistry, and medicinal chemistry. |
1. Introduction
As early as 1971, Nelson et al. reported a PNP type tridentate ligand 2,6-di(diphenylphosphinoethyl)pyridine and coordinated it with iron(II), cobalt(II), and nickel(II).1 van Koten et al. then synthesized a NNN type tridentate ligand and proposed the concept of “pincerlike” to describe it in 1986.2 Since then, with the development of the application of such complexes in metal catalysis,3 pincer chemistry has gradually become a research hotspot in organometallic chemistry. Among these, the chiral pincer4 is particularly remarkable.
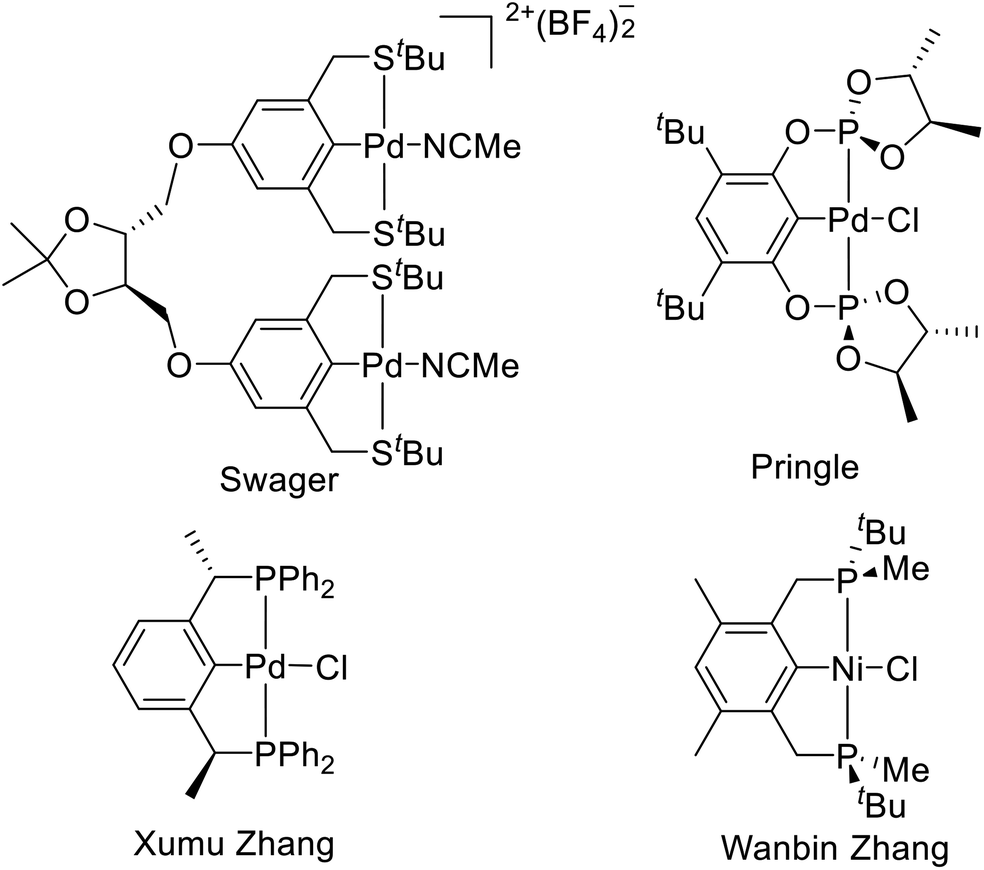
Chiral pincer ligands can be divided into four types according to the different positions of chiral centers in the ligand (Fig. 1). (1) Substituents as chiral centers on the pincer skeleton.5 Among them, the binuclear pincer–palladium complexes reported by Swager et al. are representative.5i (2) Substituents as auxiliary chiral centers on the coordination atom,6 such as the chiral palladium bis(phosphite) pincer complexes reported by Pringle et al.7 (3) Substituents as chiral centers at the benzylic position.8 The PCP type pincer–Pd complexes developed by X. Zhang et al. are typical representatives of this kind of chiral pincer.9 (4) P-stereogenic chiral pincer, like the PCP type pincer–Ni complexes reported by Wanbin Zhang et al.10 This paper will focus on the P-stereogenic chiral pincer complexes.
 |
| | Fig. 1 Represent complexes of four type chiral pincers. | |
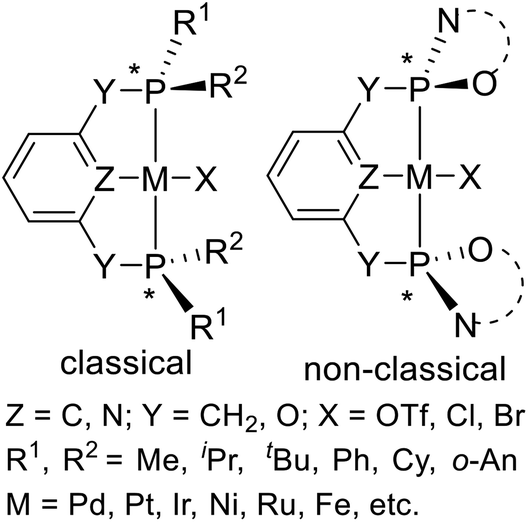
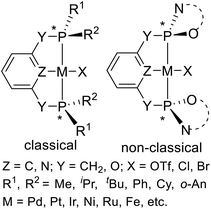
The classical P-stereogenic pincer complex is a compound with four different substituents on the phosphorus atoms (Fig. 2, left). The substituents on phosphorus include methyl (Me), isopropyl (iPr), tertiary butyl (tBu), cyclohexyl (Cy), phenyl (Ph), and ortho-anisyl (o-An). In addition, there is a class of non-classical11 P-stereogenic pincer complexes: the two heteroatoms (N or O) which bond to the pincer's phosphorus atoms are joined by chiral carbon chains to form heterocyclics (Fig. 2, right). In this paper, we will summarize the recent progress of synthesis and application in asymmetric catalysis of both classical and non-classical P-stereogenic chiral pincer complexes.
 |
| | Fig. 2 The P-stereogenic pincer–metal complexes. | |
2. Synthesis of P-stereogenic pincer ligands
2.1 Synthesis of the classical P-stereogenic pincer ligands
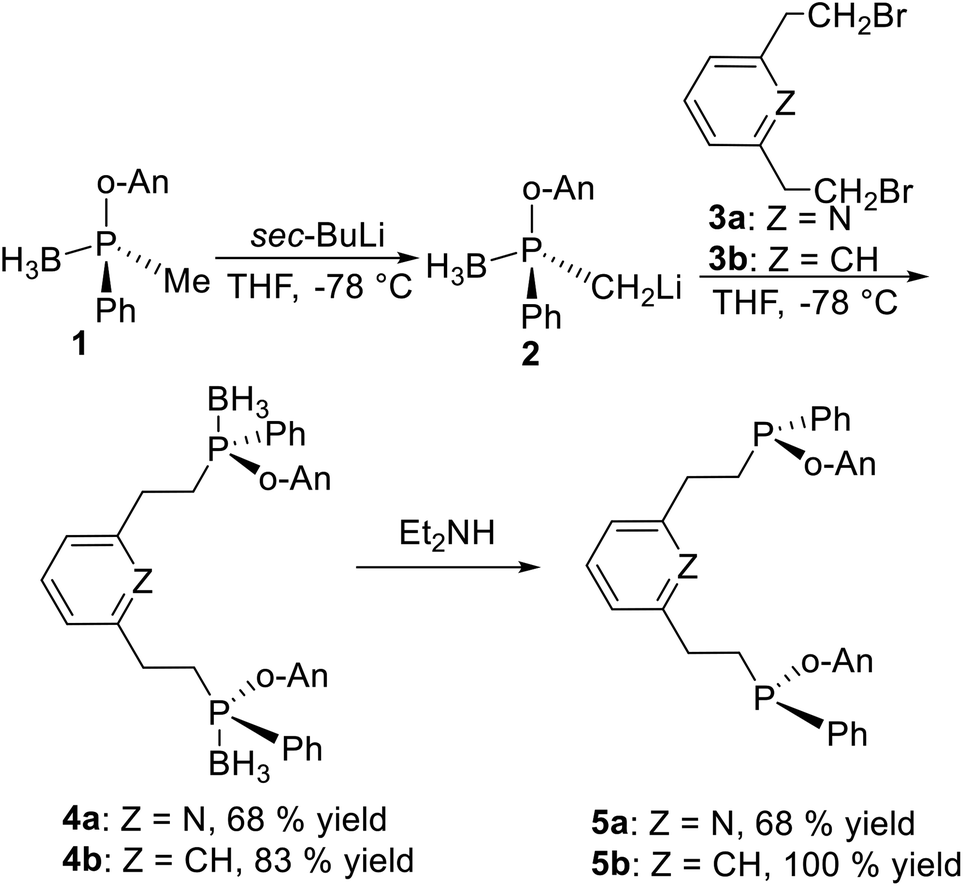
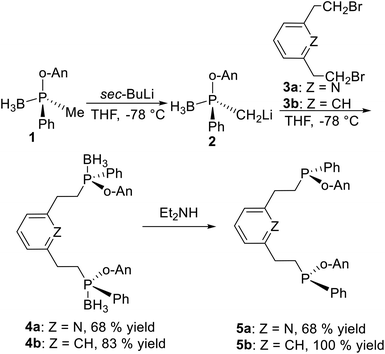
2.1.1 Condensation of P-stereogenic synthons and halohydrocarbons. The basic procedure to produce pincer type P-stereogenic ligands is based on the synthesis of P-stereogenic synthons, that have been well summarized by Mezzetti et al.12 It includes three pathways: ephedrine derived 1,2,3-oxazaphospholidine boranes,13 enantioselective deprotonation with sparteine,14 and menthyl phosphinates.15 After obtaining the P-stereogenic synthons, the most common method to synthesize the classical P-stereogenic pincer ligands is by condensation of P-stereogenic synthons and halohydrocarbons.Pioneering work was reported by X. Zhang et al.16 Deprotonation of chiral synthon 1 with sec-BuLi in situ generates anion 2 (Scheme 1), which reacts with 2,6-bis(bromomethyl)pyridine 3a or the 2,6-bis(bromomethyl)benzene 3b to form 4a and 4b in high yields, respectively. After removing the borane groups from 4a and 4b, ligand 5a and 5b are obtained in pure form. This is a classical strategy to synthesize the classical P-stereogenic pincer ligands.
 |
| | Scheme 1 Synthesis of P-stereogenic pincer ligands. | |
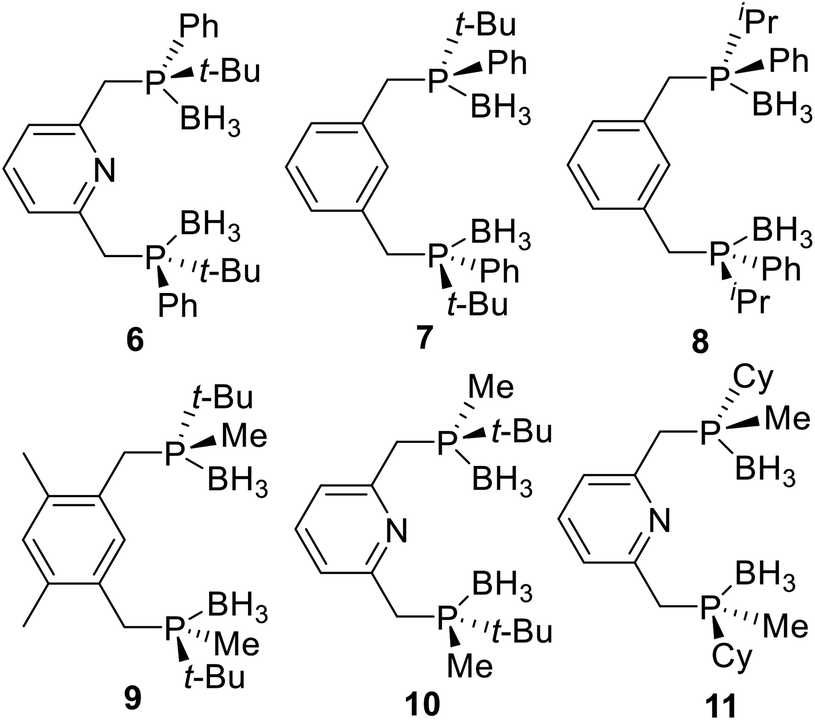
Following, many P-stereogenic pincer ligands were synthesized by this method (Fig. 3), such as the PNPtBu,Ph (6) type pincer ligand reported by Livinghouse17 and Castillón,18 the PCPtBu,Ph (7) and the PCPiPr,Ph (8) type pincer ligands reported by van Koten19 and Morales-Morales,20 the PCPtBu,Me (9) and the PNPtBu,Me (10) type pincer ligands reported by Wanbin Zhang,10,21 and the PNPCy,Me (11) type pincer ligand reported by Mezzetti.4n,22 Since alkyl substituted phosphines are sensitive to both air and moisture, these ligands are protected by boranes, and the boranes need to be removed first when coordinate them with metals.
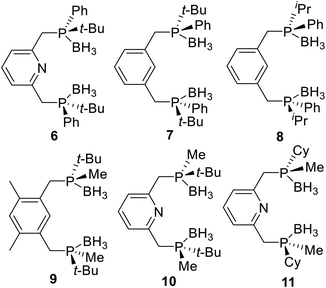 |
| | Fig. 3 Classical P-stereogenic pincer ligands synthesized by condensation of P-stereogenic synthons and halohydrocarbons. | |
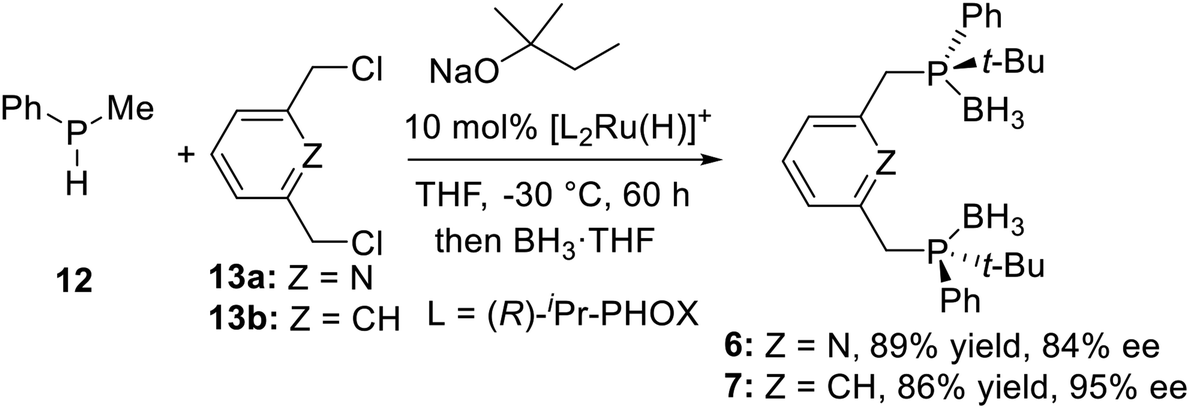
2.1.2 Asymmetric catalytic synthesis. In 2006, Toste et al.23 developed a new procedure to synthesize P-stereogenic pincer ligands: a nucleophilic ruthenium phosphido complex mediates asymmetric catalytic synthesis (Scheme 2). They used the (R)-iPr-PHOX–Ru complex as catalyst, catalyzed the alkylation of methylphenylphosphine 12 by 2,6-bis(chloromethyl)pyridine 13a and 1,3-bis(chloromethyl)benzene 13b to obtain the PNPtBu,Ph (6) and PCPtBu,Ph (7) pincer ligands with 84% (6) and 95% ee (7) (Scheme 2), respectively.
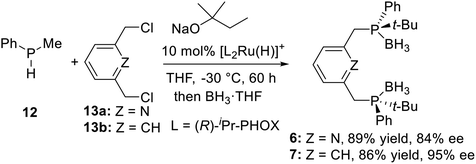 |
| | Scheme 2 Synthesis of P-stereogenic pincer ligands by asymmetric catalytic synthesis. | |
This enantioselective alkylation reaction provided an efficient access for useful and synthetically challenging P-stereogenic pincer ligands in a single step from secondary phosphines and alkyl halides. Analogously, Duan et al.8j developed a PCP type pincer–Pd complex catalyzed asymmetric alkylation of methylphenylphosphine with alkyl halides, which could be efficiently used to synthesize the P-stereogenic pincer ligands.
2.2 Synthesis of the non-classic P-stereogenic pincer ligands
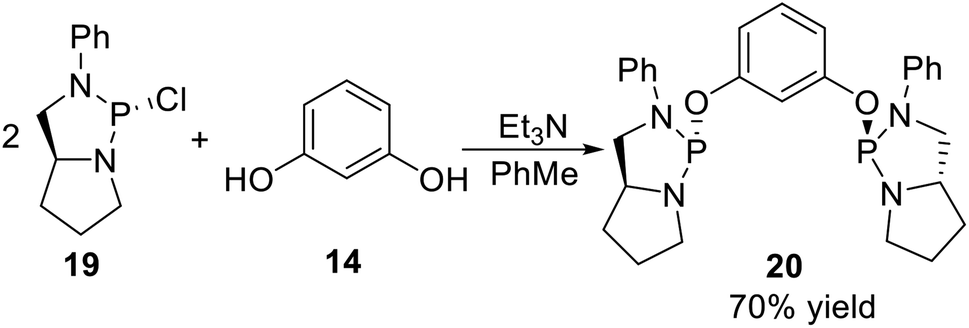
All the non-classical P-stereogenic pincer ligands reported so far are POCOP type pincer ligands. The synthesis of this kind of ligand was through phosphorylation of resorcinol derivatives by P-stereogenic synthons, such as the pincer type phosphodiamidite ligand reported by Gavrilov11 in 2009. They used the (2R,5S)-2-chloro-3-phenyl-1,3-diaza-2-phosphabicyclo[3.3.0]octane (19) as the P-stereogenic synthon, adopting a one-step phosphorylation of resorcinol 14 in the presence of Et3N and PhMe (Scheme 4), and gave the target ligand 20 in 70% yield.
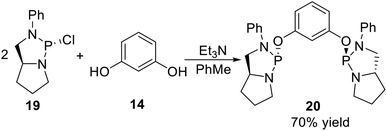 |
| | Scheme 4 Synthesis of the non-classic P-stereogenic pincer ligands reported by Gavrilov. | |
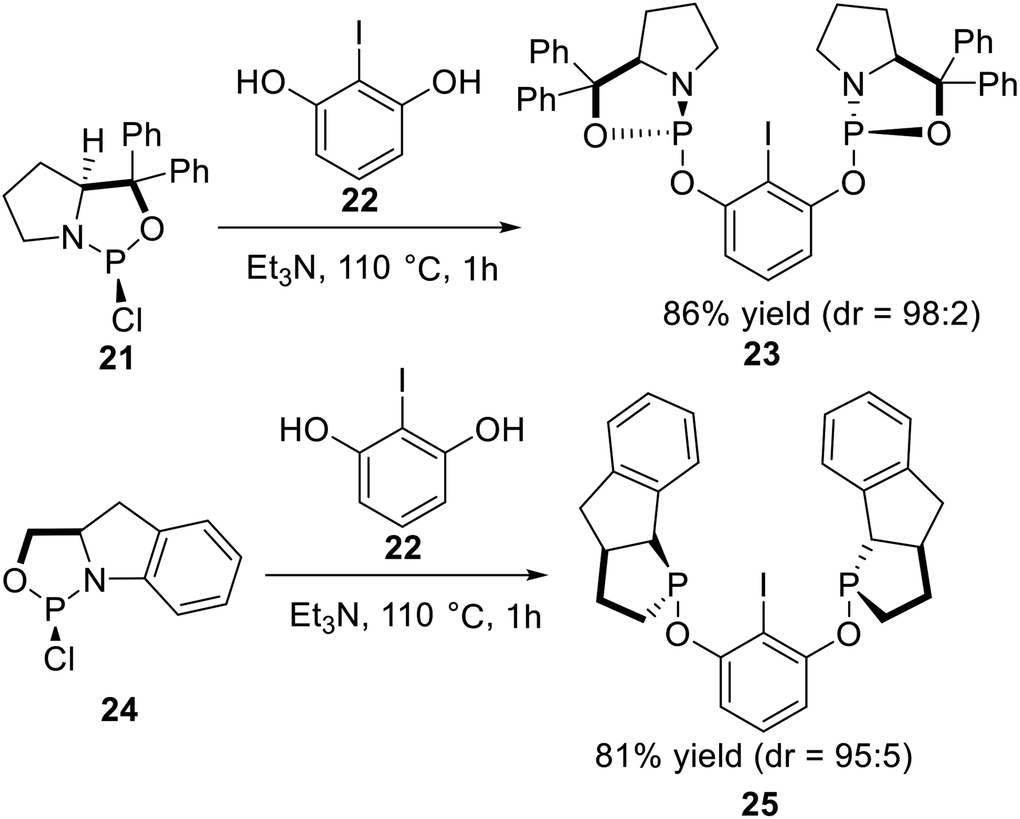
In 2010, Gebbink et al.25 reported a series of novel P-stereogenic bis-phosphoramidite pincer ligands derived from chiral amino alcohols. The optically active amino alcohol was phosphonated by PCl3 in the presence of triethylamine to afford the corresponding phosphorochloridate adduct 21, which was subsequently coupled with 2-iodoresorcinol 22 in toluene at 110 °C to yield P-stereogenic pincer arene ligand 23 in good yield (Scheme 5, top). The diastereomeric ratio (dr) of a sample of crude 23 in solution after filtration was roughly estimated as 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 by comparing the integral values in the 31P NMR of the two phosphoramidite diastereoisomers. Following a similar synthesis route as reported for 23, the corresponding phosphorochloridate 24 was subsequently reacted with 2-iodoresorcinol 22 at 110 °C to yield pincer arene ligand 25 in good yield and acceptable purity after a simple filtration (Scheme 5, bottom). The diastereomeric ratio of crude 25 in solution after filtration was roughly estimated at 95:5 by comparing the integral values of the 31P NMR signals.
:2 by comparing the integral values in the 31P NMR of the two phosphoramidite diastereoisomers. Following a similar synthesis route as reported for 23, the corresponding phosphorochloridate 24 was subsequently reacted with 2-iodoresorcinol 22 at 110 °C to yield pincer arene ligand 25 in good yield and acceptable purity after a simple filtration (Scheme 5, bottom). The diastereomeric ratio of crude 25 in solution after filtration was roughly estimated at 95:5 by comparing the integral values of the 31P NMR signals.
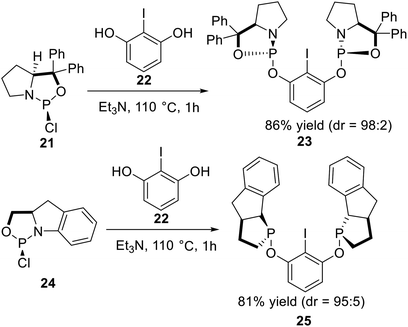 |
| | Scheme 5 Synthesis of the non-classic P-stereogenic pincer ligands reported by Gebbink. | |
3. Synthesis of P-stereogenic pincer complexes
Pincer ligands are a kind of tridentate ligands with strong rigidity, which can easily form stable complexes with transition metals. P-stereogenic pincer ligands coordinate with transition metals in four main ways: C–H activation, oxidative addition, transmetalation, and direct coordination.
3.1 Synthesis of P-stereogenic pincer complexes via C–H activation
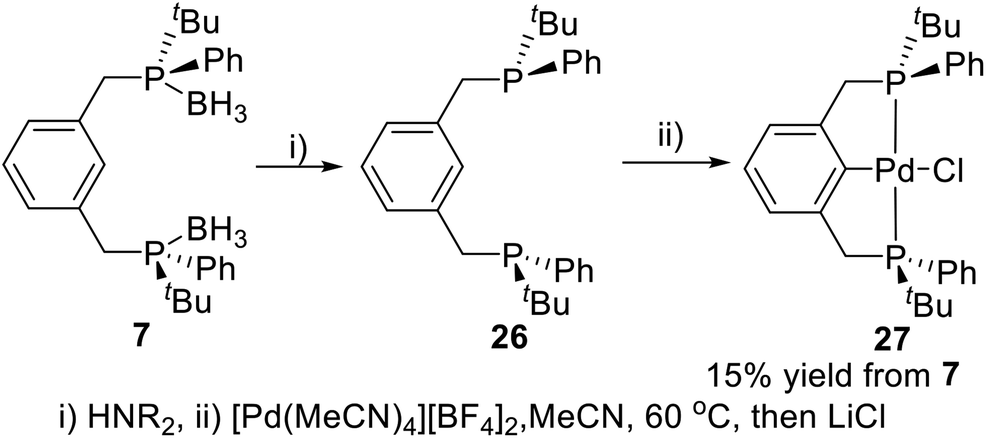
Metalation of pincer ligands by appropriate metal precursors via C–H activation is an efficient approach to synthesize P-stereogenic pincer–metal complexes. van Koten and coworkers reported the first P-stereogenic pincer–metal complex in 2001 by C–H activation.19a The borane protected P-stereogenic pincer ligand 7 was deprotected in alkaline conditions to give ligand 26 (Scheme 6). Stirring the freshly deprotected ligand 26 with [Pd(MeCN)4][BF4]2 in MeCN generated the P-stereogenic pincer–Pd complex 27 through C–H activation in 15% yield from 7. The complex 27 has been characterized by multinuclear (1H, 13C, 31P) NMR and polarimetry. Crystallization of 27 from CH2Cl2/hexane produced single crystals suitable for X-ray crystallography. The X-ray crystallography data showed that this P-stereogenic pincer–Pd complex had approximately a C2 symmetry structure.
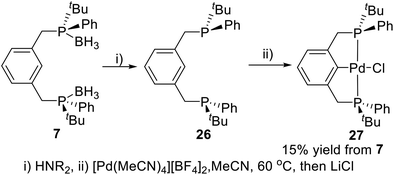 |
| | Scheme 6 Synthesis of P-stereogenic pincer–Pd complex via C–H activation reported by van Koten. | |
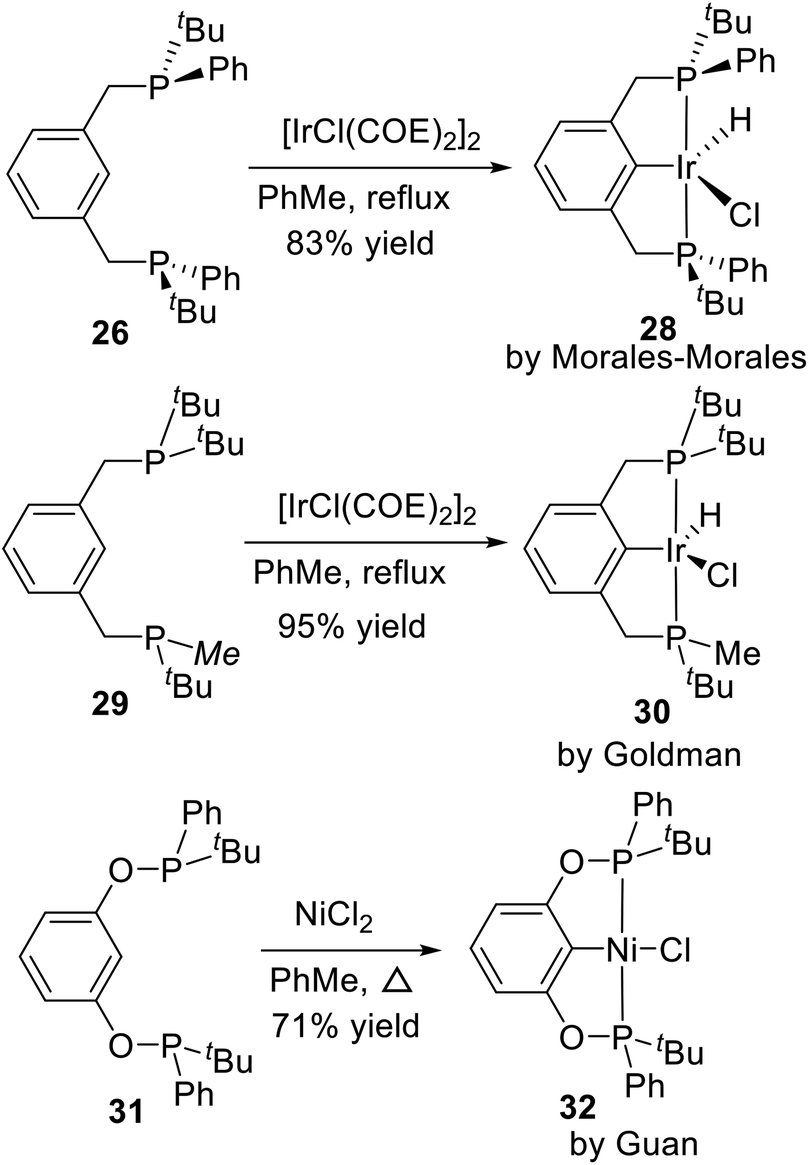
This route is the most common method to synthesize P-stereogenic pincer–metal complexes, and many complexes were synthesized by this method. Later in 2002, Morales-Morales et al.20 reported the Ph and tBu substituted P-stereogenic PCP type pincer ligand 26 and its palladium complex 27. He also used the ligand 26 to coordinate with [IrCl(COE)2]2 and obtained a PCP type P-stereogenic pincer–Ir complex 28 via C–H activation (Scheme 7). Goldman et al.26 synthesized an unsymmetrical PCP type P-stereogenic pincer–Ir complex 30 based on this approach in 2009. In this complex, one phosphorus is substituted by two tBu, the other phosphorus is substituted by one tBu and one Me. He only made the racemic complex. Similarly, the racemic POCOP type P-stereogenic pincer–Ni complex 32 reported by Guan et al.24 was synthesized by the same approach.
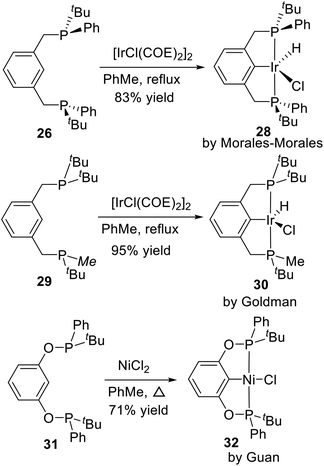 |
| | Scheme 7 Synthesis of P-stereogenic pincer–Ir and pincer–Ni complexes by C–H activation. | |
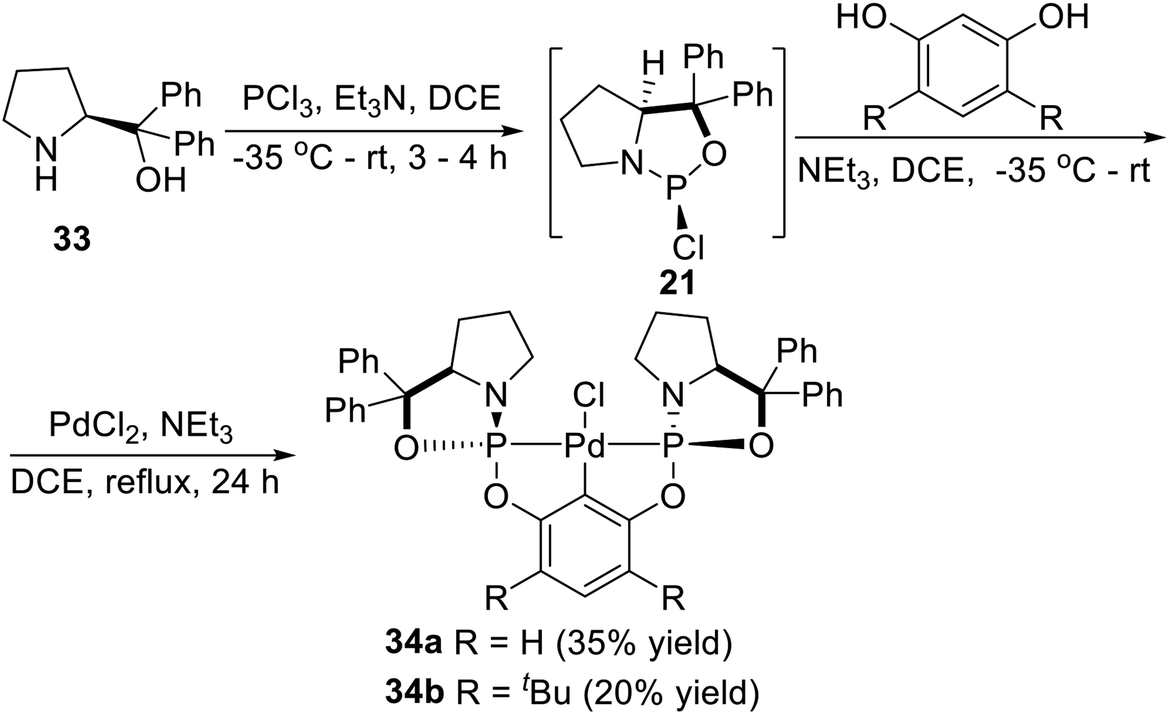
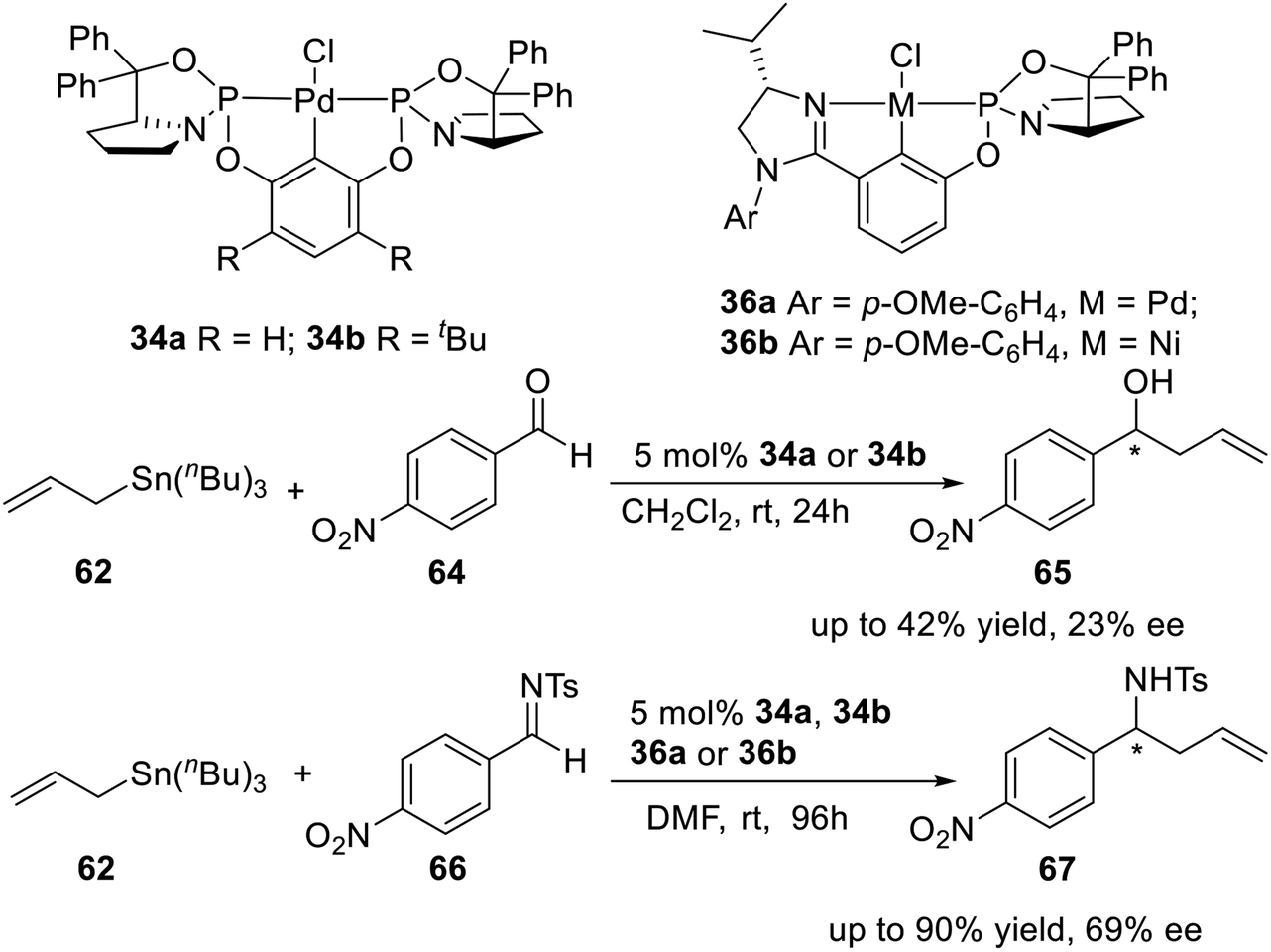
In 2010, Song et al.27 synthesized a series of P-stereogenic pincer complexes from easily available starting materials in a four-component (including diphenylprolinol, PCl3, resorcinol, and PdCl2), one-pot manner, as shown in Scheme 8. Firstly, the optically active amino alcohol (S)-diphenyl(pyrrolidin-2-yl)methanol (33) was phosphonated with PCl3 in the presence of triethylamine in DCE (1,2-dichloroethane) to afford the expected phosphorochloridate adduct (21). Then the adduct reacted in situ with resorcinol, followed by treatment with PdCl2. The pure air- and moisture-stable complex 34a was successfully obtained in 35% isolated yield (based on resorcinol) as a white solid after chromatography on silica gel. Using more hindered 4,6-di-tert-butylbenzene-1,3-diol instead of resorcinol as a backbone, the complex 34b was obtained in 20% yield with a similar process.
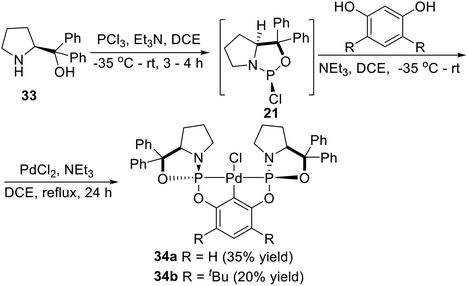 |
| | Scheme 8 Synthesis of P-stereogenic pincer–Pd complexes via C–H activation reported by Song. | |
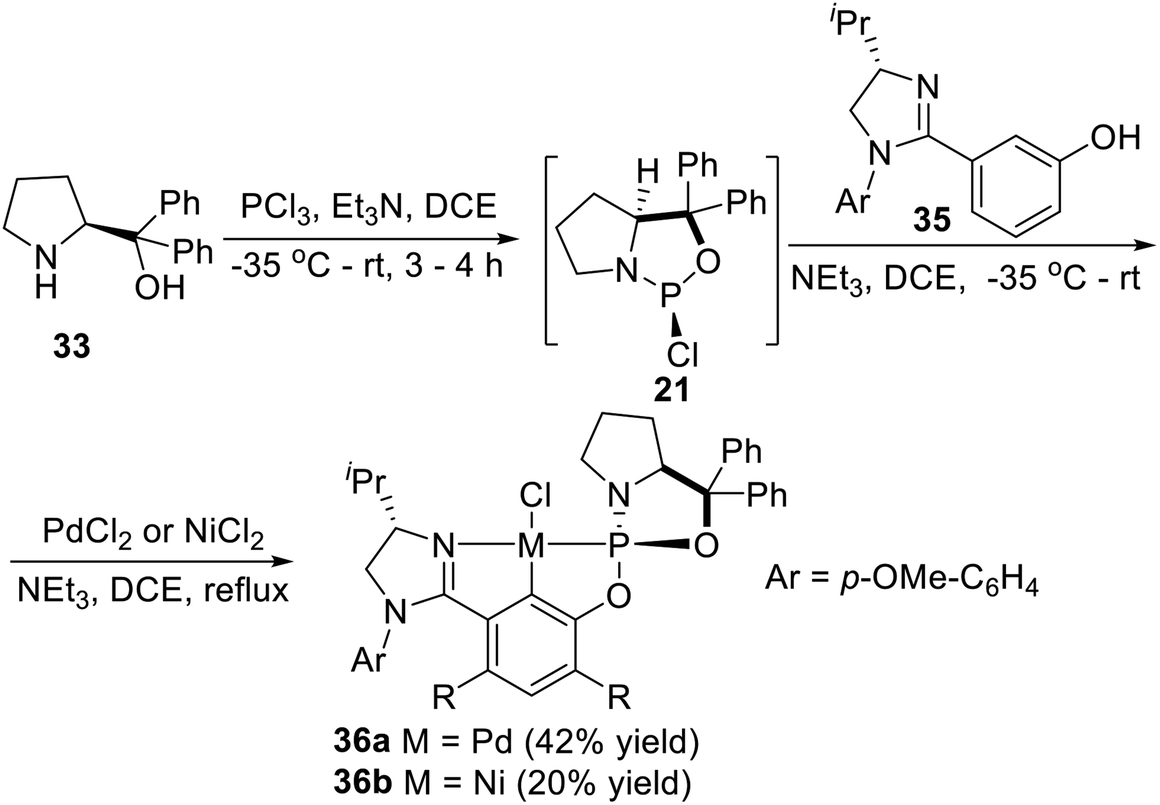
Displacement of one phospholidine cycle at the 2- or 6-position of the central aryl ring in the symmetrical complexes 34a and 34b by a chiral imidazoline unit, Song et al.6i,27 also synthesized an unsymmetrical P-stereogenic pincer–Pd complex and a pincer–Ni complex (Scheme 9). Following a synthetic route similar to that for complexes 34a and 34b, the adduct obtained from treatment of (S)-diphenyl(pyrrolidin-2-yl)methanol with PCl3 was reacted in situ with the imidazolinyl-containing m-phenol derivative 35. The subsequent palladation also proceeded in situ by the addition of PdCl2. The unsymmetrical Pd(II) complex 36a was successfully isolated as a yellow solid after chromatography on silica gel in 42% yield. Nickelation of the related preligand with NiCl2 instead of PdCl2, the corresponding PCN–pincer Ni(II) complex 36b could also be obtained via C–H activation as a pale yellow solid after chromatography on silica gel, albeit in a lower yield (20%).
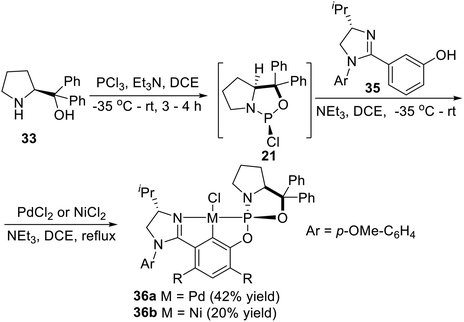 |
| | Scheme 9 Synthesis of the unsymmetrical P-stereogenic pincer–metal complexes via oxidative addition reported by Song. | |
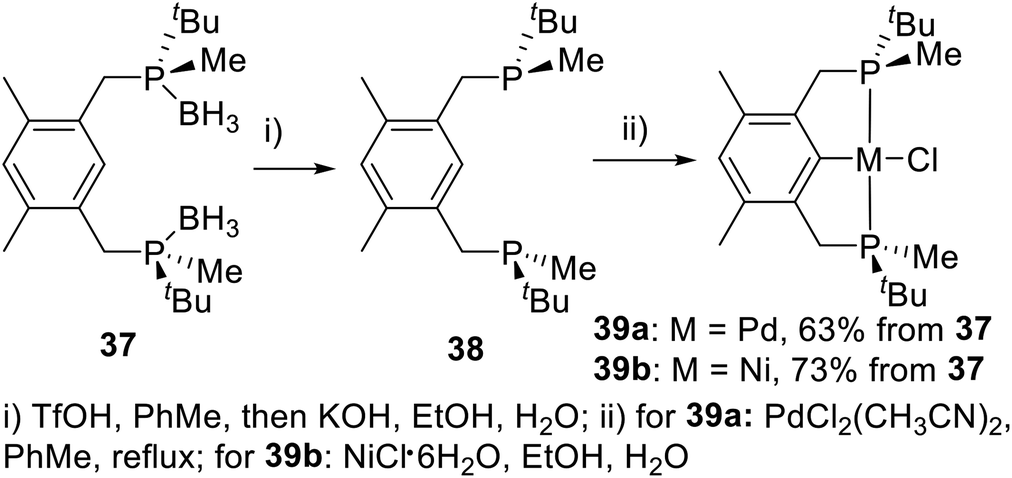
In 2013, Wanbin Zhang et al.21a developed a novel PCP type P-stereogenic pincer ligand 37 with P(tBu)Me as the chiral center (Scheme 10). In this ligand, the phosphorus was substituted by one Me and one tBu, with a very large steric difference. The P-stereogenic PCP-type pincer–metal complexes were prepared in two steps using an optimal “one-pot” procedure. First, the boranes in compound 37 were removed using trifluoromethanesulfonic acid (TfOH) in degassed toluene, followed by reaction with aqueous KOH in degassed ethanol to produce bisphosphine 38. Ligand 38 was then directly reacted with bis(acetonitrile)dichloropalladium(II) or nickel(II) chloride hexahydrate10 via C–H bond activation in toluene or a mixed solvent system of ethanol and water to give the P-stereogenic PCP type pincer–Pd complex 39a or pincer–Ni complex 39b. The total yields from 37 to 39a and 39b were 63% and 73%, respectively. The resulting solid is stable to air and moisture and does not require any special storage procedures.
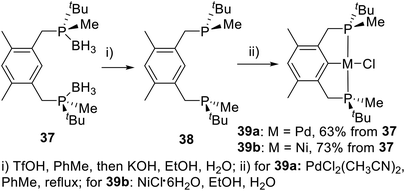 |
| | Scheme 10 P-stereogenic pincer complexes reported by Wanbin Zhang. | |
3.2 Synthesis of P-stereogenic pincer complexes via oxidative addition
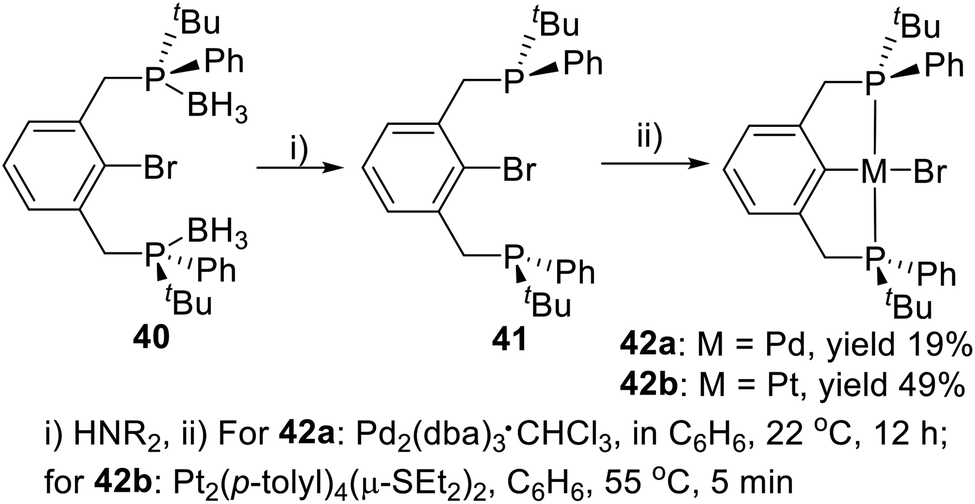
Except for the tertiary butyl and phenyl substituted PCP type P-stereogenic pincer ligand 7, van Koten and coworkers19a also synthesized a similar ligand (40) with a bromine substituted on the iso-carbon (Scheme 11). The phosphine–boranes in 40 can be deprotected with an excess of various types of amines to give ligand 41. The freshly deprotected 41 dissolved in benzene and reacted with Pd2(dba)3·CHCl3 over the course of 12 h to generate the pincer–Pd complex 42a via oxidative addition in 19% yield. The product was filtered through silica in ether. Correspondingly, react the deprotected ligand 41 with Pt2(p-tolyl)4(μ-SEt2)2 in benzene generated the pincer–Pt complex 42b in 49% yield.
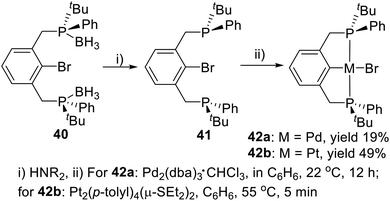 |
| | Scheme 11 Synthesis of P-stereogenic pincer complexes via oxidative addition reported by van Koten. | |
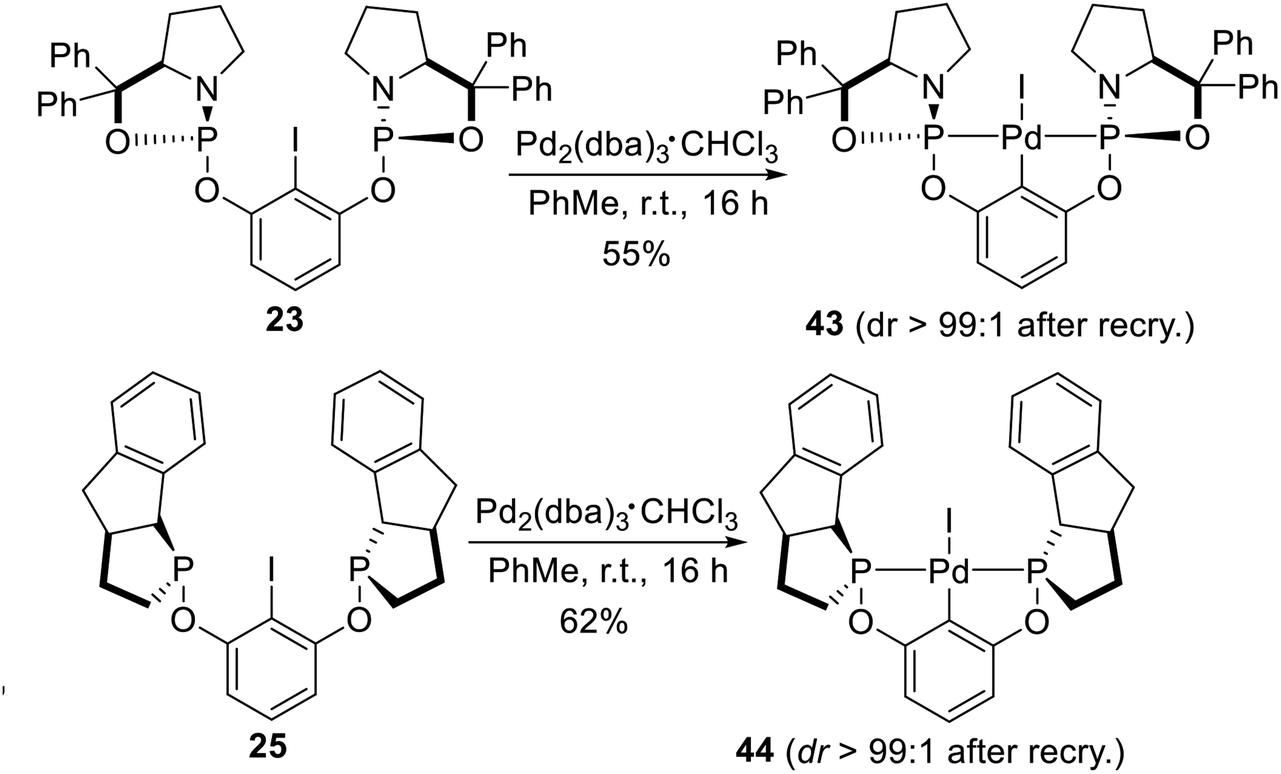
The chiral amino alcohol derived bis-phosphoramidite non-classical pincer–Pd complexes reported by Gebbink et al.25 were synthesized by oxidative addition too. The ligand 23 and 25 have an iodine substituted on the iso-carbon (Scheme 12). Palladation of pincer ligand 23, with complete retention of the stereospecificity, was achieved via an oxidative addition reaction of 23 with a zerovalent Pd species (i.e., Pd2(dba)3·CHCl3) under mild conditions (rt, 16 h). The pincer–palladium complex 43 was obtained in reasonable yield (55%) and with excellent diastereoselectivity (dr > 99:1) after fractional crystallization from concentrated CH2Cl2 solution by the addition of hexanes. Following a similar synthesis route as that reported for 43, palladation of the resulting pincer aryl iodide ligand 25 was smoothly achieved through oxidative addition with Pd2(dba)3·CHCl3 under mild conditions (rt, 16 h). Complex 44 was obtained in promising yield (62%) with excellent diastereoselectivity (dr > 99:1) after fractional recrystallization from CH2Cl2/hexanes.
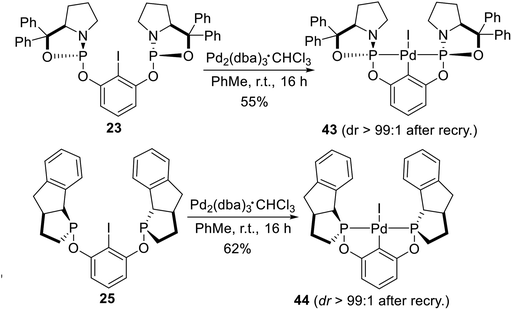 |
| | Scheme 12 Synthesis of P-stereogenic pincer complex via oxidative addition reported by Gebbink. | |
3.3 Synthesis of P-stereogenic pincer complexes via transcyclometalation
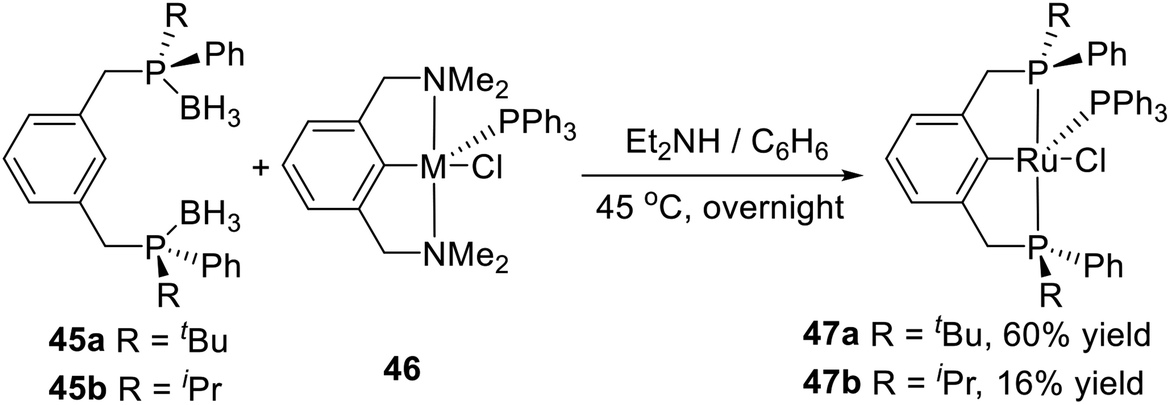
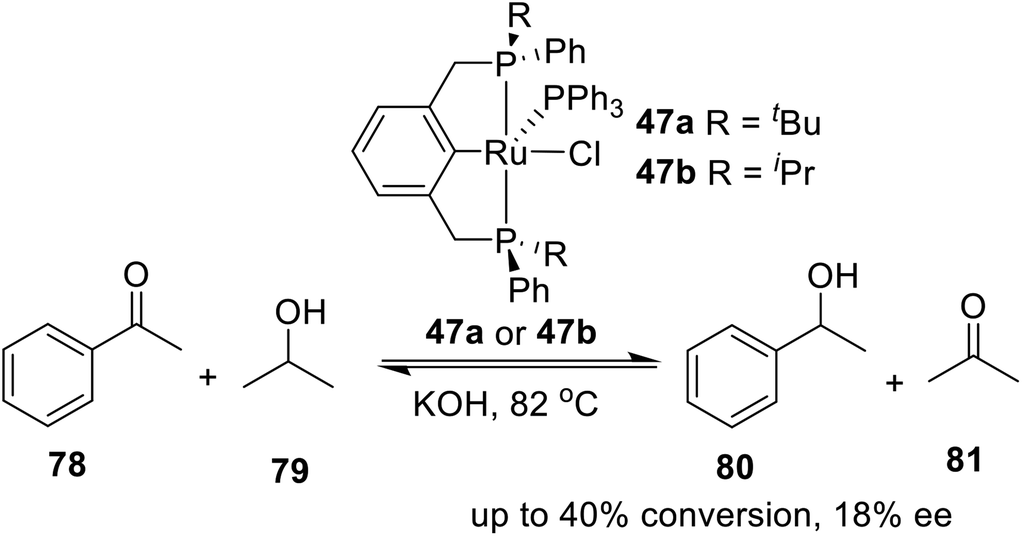
In 2005, van Koten et al. synthesized two kinds of PCP type P-stereogenic pincer–Ru complexes in a direct one-pot synthesis,19b the deprotection of the prepared phosphine–boranes 45a or 45b and the subsequent transcyclometalation (Scheme 13). Deprotection of the phosphine–boranes was easily accomplished by overnight heating of a benzene solution of 45a or 45b in the presence of an excess of Et2NH at 45 °C. Complex 46 was then added and stirred for 24 h. Complex 47a was isolated as a relatively air-stable, ink-blue solid in 60% yield. Complex 47b was obtained pure in low yield (16%) after several purification steps as an air-sensitive, deep green, crystalline solid. This report provided a new approach to synthesize pincer–metal complexes.
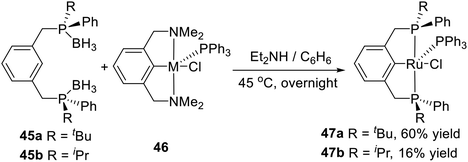 |
| | Scheme 13 Synthesis of P-stereogenic pincer complexes via transcyclometalation. | |
3.4 Synthesis of P-stereogenic pincer complexes via direct coordination
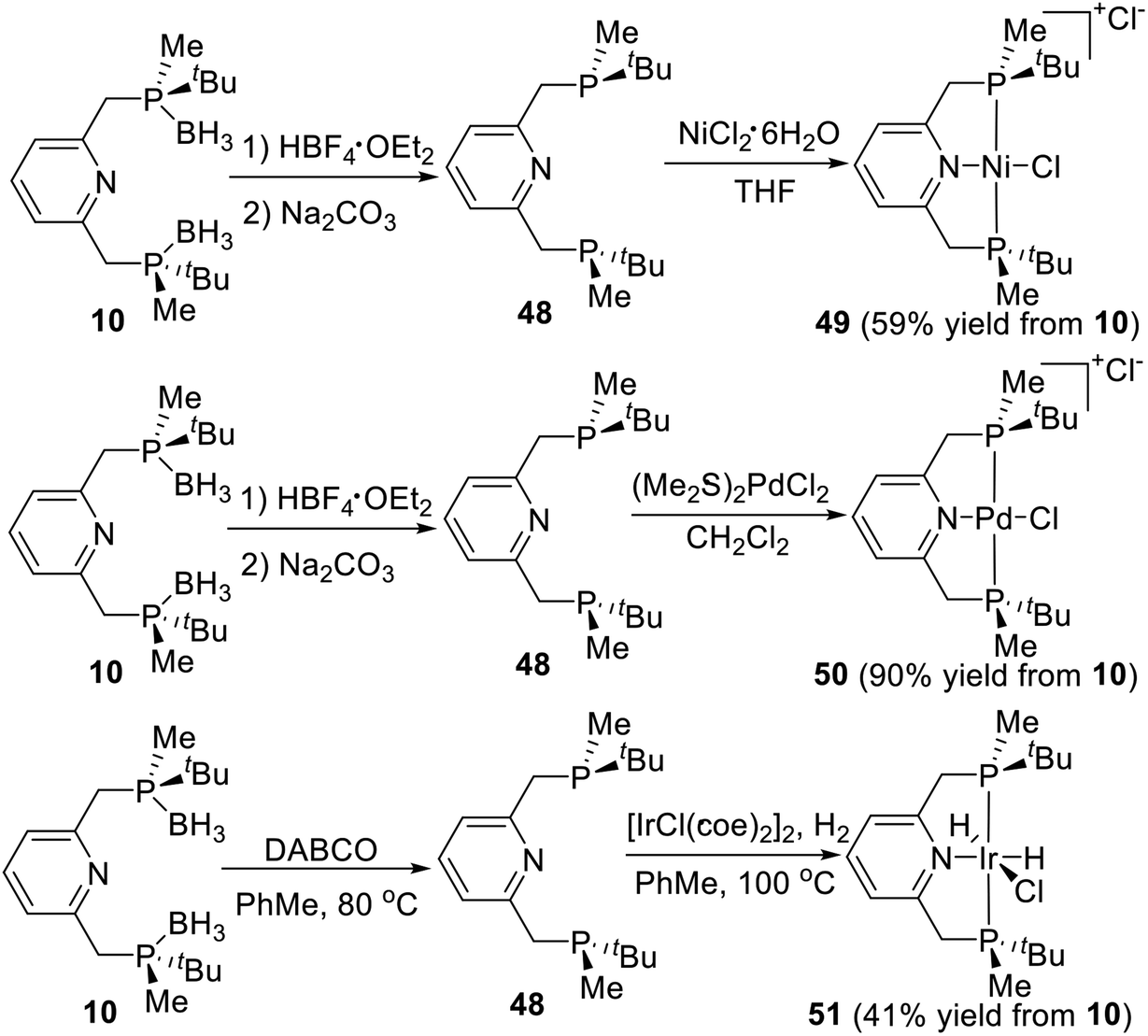
PNP type pincer ligand is a deeply researched tridentate ligand, which is easy to coordinate with transition metals directly. Wanbin Zhang et al.21d adopted the Livinghouse's deprotection method28 to prepare a series of P-stereogenic PNP type pincer–metal complexes over two high-yielding steps (Scheme 14). Thus, the borane groups were removed via the reaction of (R,R)-2,6-bis-[(boranato(tert-butyl)methylphosphino)methyl]pyridine (10) with tetrafluoroboric acid diethyl ether in degassed dry dichloromethane, followed by treatment with degassed 10% Na2CO3 solution to produce the resulting bisphosphine pyridine 48. Ligand 48 directly reacted with nickel(II) chloride hexahydrate in THF at ambient temperature to afford the cationic complex 49 as a red solid,10 which was obtained in 59% yield from 10. The ligand 50 then reacted directly with (Me2S)2PdCl2 in degassed dry dichloromethane to form the P-stereogenic pincer–Pd complex 50 as an orange solid in 90% total yield.21d The ligand 50 reacted with [IrCl(coe)2]2 under hydrogen pressure to give the pincer–iridium complex 51 as a white solid in 41% yield.21c All of the resulting solid products are stable to air and moisture and required no special storage precautions.
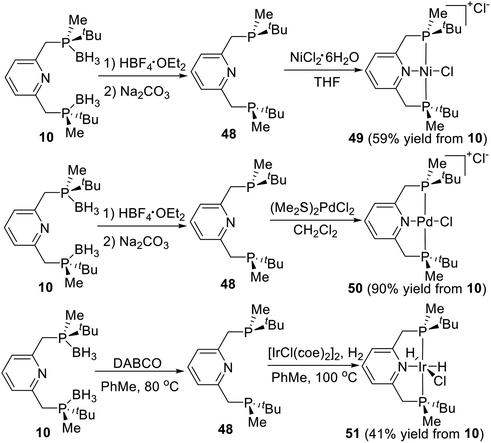 |
| | Scheme 14 Synthesis of P-stereogenic pincer complex via direct coordination reported by Wanbin Zhang. | |
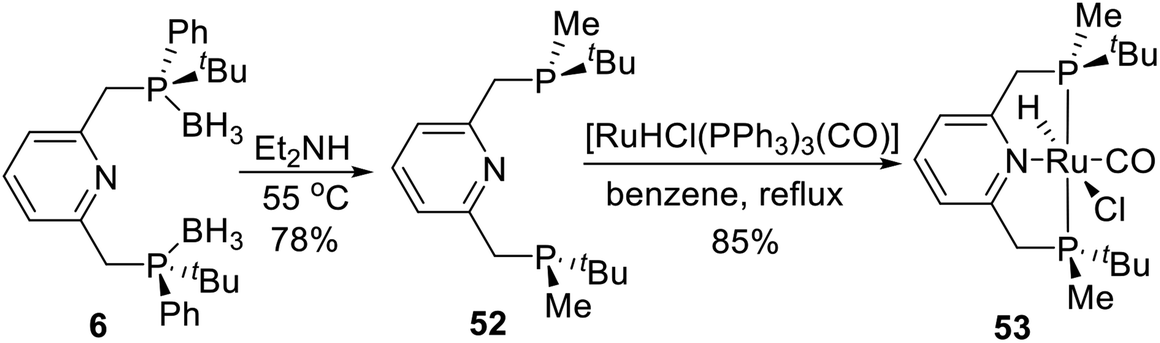
In 2015, Castillón et al.29 reported a novel P-stereogenic PNPtBu,Ph ruthenium complex via direct coordination the pincer ligand and ruthenium precursor. The straightforward synthesis of 53 was accomplished in two steps, starting from phosphine–borane complex 6 (Scheme 15). After trying many deprotection methods, ligand 52 was finally obtained in good yield (78%) by reaction with an excess of diethylamine and purification by preparative TLC inside a glove-box. The use of alumina instead of silica plates and careful anhydrous handling were crucial in order to obtain reproducible experiments. The resulting ligand was treated with [RuHCl(PPh3)3(CO)] in refluxing benzene to afford Ru complex 53 in 85% yield as a pale-yellow solid.
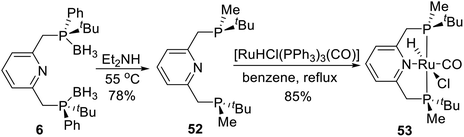 |
| | Scheme 15 Synthesis of P-stereogenic pincer complex via direct coordination reported by Castillón. | |
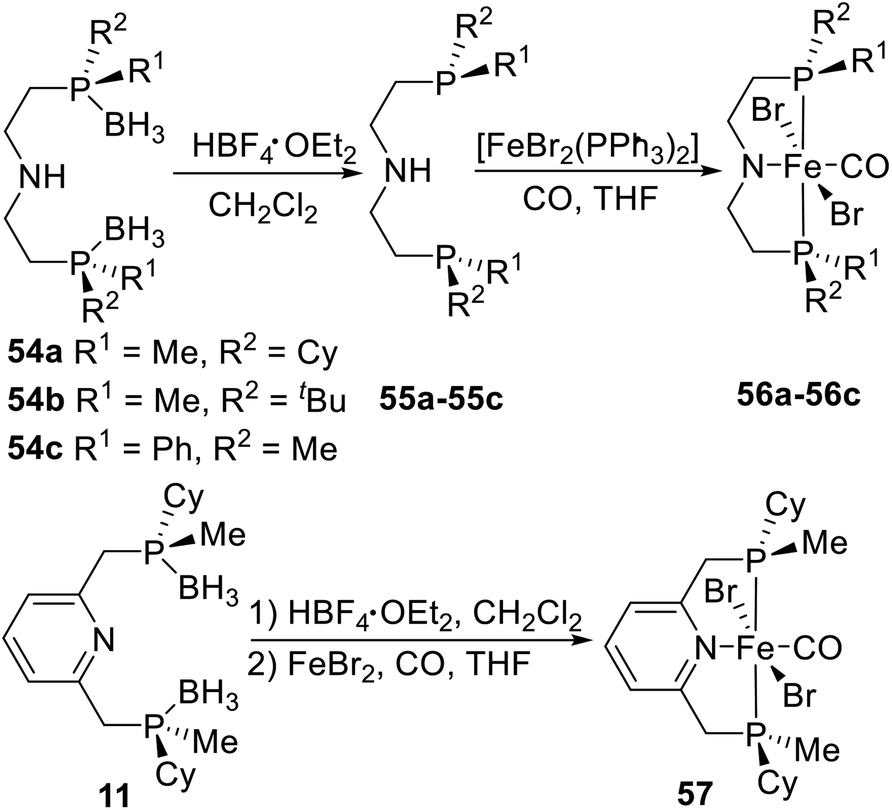
Recently, Mezzetti et al.4n,22 synthesized a series of PN(H)P and PNP type pincer–Fe complexes by a similar coordination strategy. The borane-protected pincer ligands 54a–54c were indefinitely stable upon storage in air at room temperature and were deboronated with HBF4·OEt2 in dichloromethane before complexation (Scheme 16). After workup, the resulting pincer ligands 55a–55c were reacted with [FeBr2(PPh3)2] in THF to give the target P-stereogenic pincer–Fe complexes.22 In addition the PN(H)P pincer ligand, they also synthesized the pyridine based PNP pincer ligand 11. Phosphine–borane 11 was deprotected with HBF4·OEt2 in dichloromethane. Treatment of the free ligand with FeBr2 under a CO atmosphere (1.1 atm) gave the deep blue dibromocarbonyl complex 57, which was precipitated with pentane, filtered in air, and purified by washing with water, ethanol, diethyl ether, and pentane.4n Complex 57 is perfectly stable toward air and moisture both in solution and in the solid state. No decomposition was observed after storing solid samples in air at room temperature for several months.
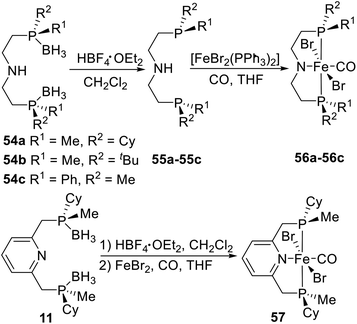 |
| | Scheme 16 Synthesis of P-stereogenic pincer complex via direct coordination reported by Mezzetti. | |
4. P-stereogenic pincer–metal complexes catalyzed asymmetric reaction
The most important application of P-stereogenic pincer–metal complexes is as chiral metal catalysts to catalyze various asymmetric synthetic reactions. At present, there have been many reports on this field.
4.1 Asymmetric allylic alkylation
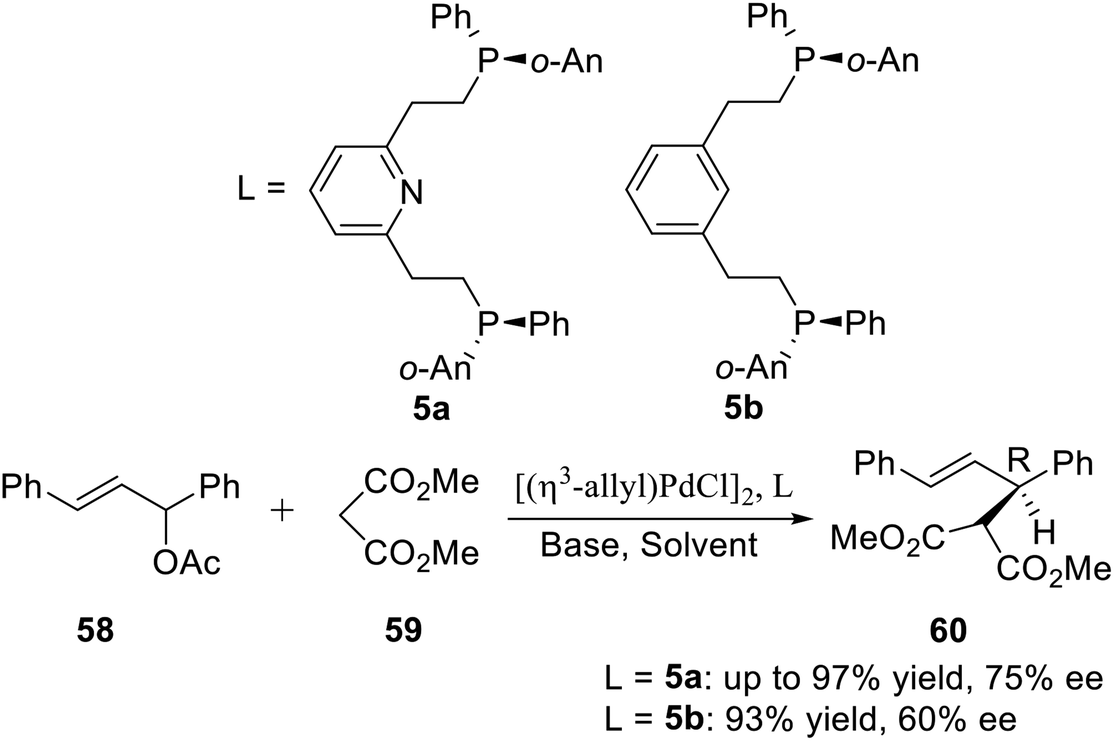
Allylic alkylation catalyzed by Pd complexes is an extremely versatile carbon–carbon bond forming reaction. In order to achieve substrate generality, the search for efficient ligand systems continues to receive considerable attention. X. Zhang et al.16a used the P-stereogenic pincer–Pd complex generated by ligand 5a or 5b with [(η3-allyl)PdCl]2 in situ to catalyze the asymmetric allylic alkylation between 1,3-diphenyl-2-propenyl acetate and dimethyl malonate (Scheme 17). After examining various bases and solvents, their results indicated that the combination of BSA and KOAc in benzene gave the best enantioselectivity (75% ee). This was the first report about the P-stereogenic pincer-complex catalyzed asymmetric reaction. The results showed that the P-stereogenic pincer-catalyst is a very promising chiral catalyst.
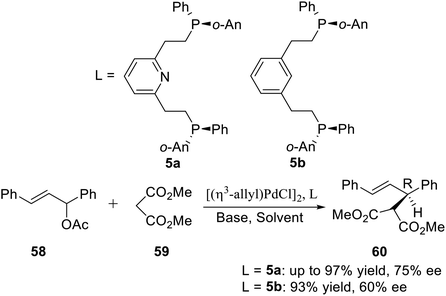 |
| | Scheme 17 PNPo-An,Ph pincer–Pd complexes catalyzed asymmetric allylic alkylation. | |
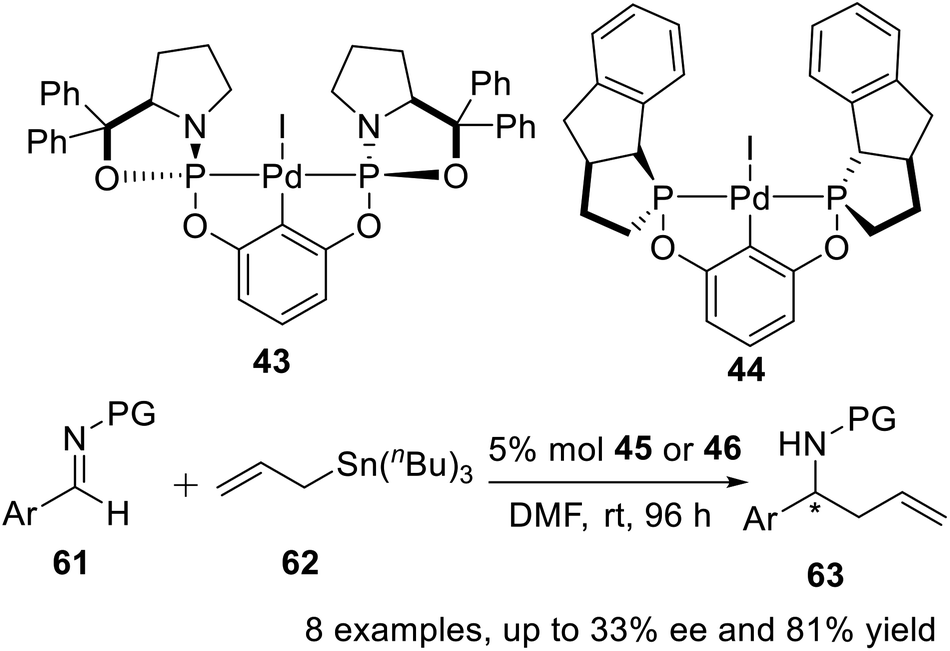
Gebbink et al.25 reported two novel P-stereogenic pincer–metal complexes in 2010, and embarked on testing the stereocontrolling potential of the two novel complexes 43 and 44. These tests were carried out for the reaction of allyltributyltin with sulfonimines, i.e., with protected aryl aldimines, at room temperature in dry DMF without additives (Scheme 18). The bis-phosphoramidite pincer–palladium complexes 43 and 44 were active catalysts for asymmetric homoallylation of sulfonimines, where low (up to ee 33%) or no enantioselectivity was observed for reactions catalyzed by 43 and 44, respectively. Preliminary catalytic results revealed that enantiomeric excess values varied by using differently functionalized sulfonimines, suggesting that both electronic properties and steric congestion of sulfonimines affected the transition state of the electrophilic attack of the 1η-allyl Pd intermediate in the allylation.
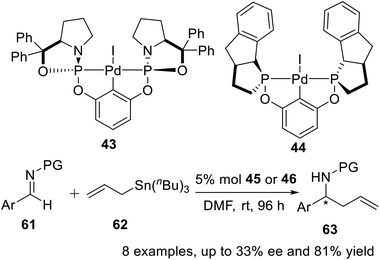 |
| | Scheme 18 POCOP pincer–Pd complexes catalyzed asymmetric homoallylation of sulfonimines. | |
The same year, Song and coworkers6i,27 synthesized four P-stereogenic pincer–Pd and pincer–Ni complexes 34a, 34b, 36a, and 36b (Scheme 19), in which the P-stereogenic pincer ligands were coordinated to palladium (34a, 34b and 36a) or nickel (36b). They used these complexes to catalyze the asymmetric allylation of 4-nitrobenzaldehyde or 4-nitrobenzenesulfonimine. They obtained up to 42% yield and 23% ee to 4-nitrobenzaldehyde and up to 90% yield and 69% ee to 4-nitrobenzenesulfonimine, respectively.
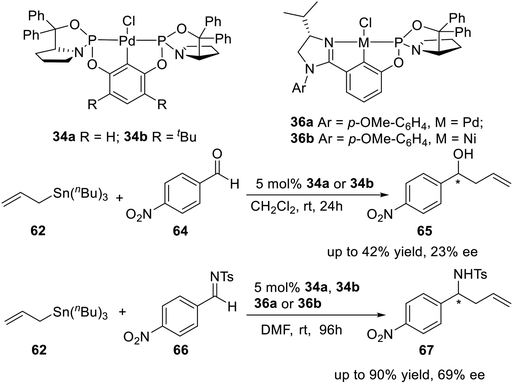 |
| | Scheme 19 P-stereogenic pincer–Pd and pincer–Ni complexes catalyzed asymmetric allylation of aldehydes and sulfonimines. | |
4.2 Asymmetric hydrosilylation
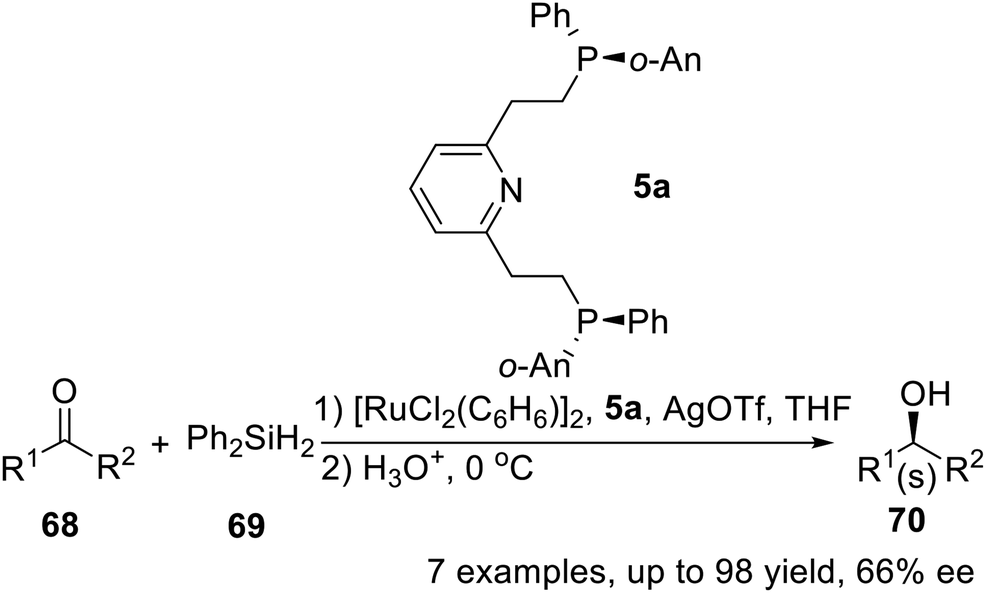
In 1997, X. Zhang and his team conducted a follow-up study on P-stereogenic pincer ligand 5a, which was employed as chiral ligand combined with Ru to catalyze the asymmetric hydrosilylation of several aryl alkyl ketones.16b They had performed asymmetric hydrosilylations of ketones under optimum conditions (Scheme 20). Typically, 1 mol% of ruthenium catalyst was used with 2.2 mol% of the chiral tridentate ligand. Enantioselectivities ranging from 47 to 66% were observed and the reactions occurred with excellent conversions (isolated yields from 85 to 98%). These values are the best results reported to date with ruthenium catalysts.
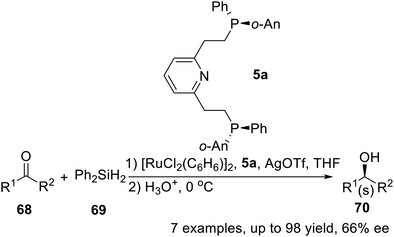 |
| | Scheme 20 PNPo-An,Ph pincer–Ru complexes catalyzed asymmetric hydrosilylation. | |
4.3 Asymmetric aldol condensation
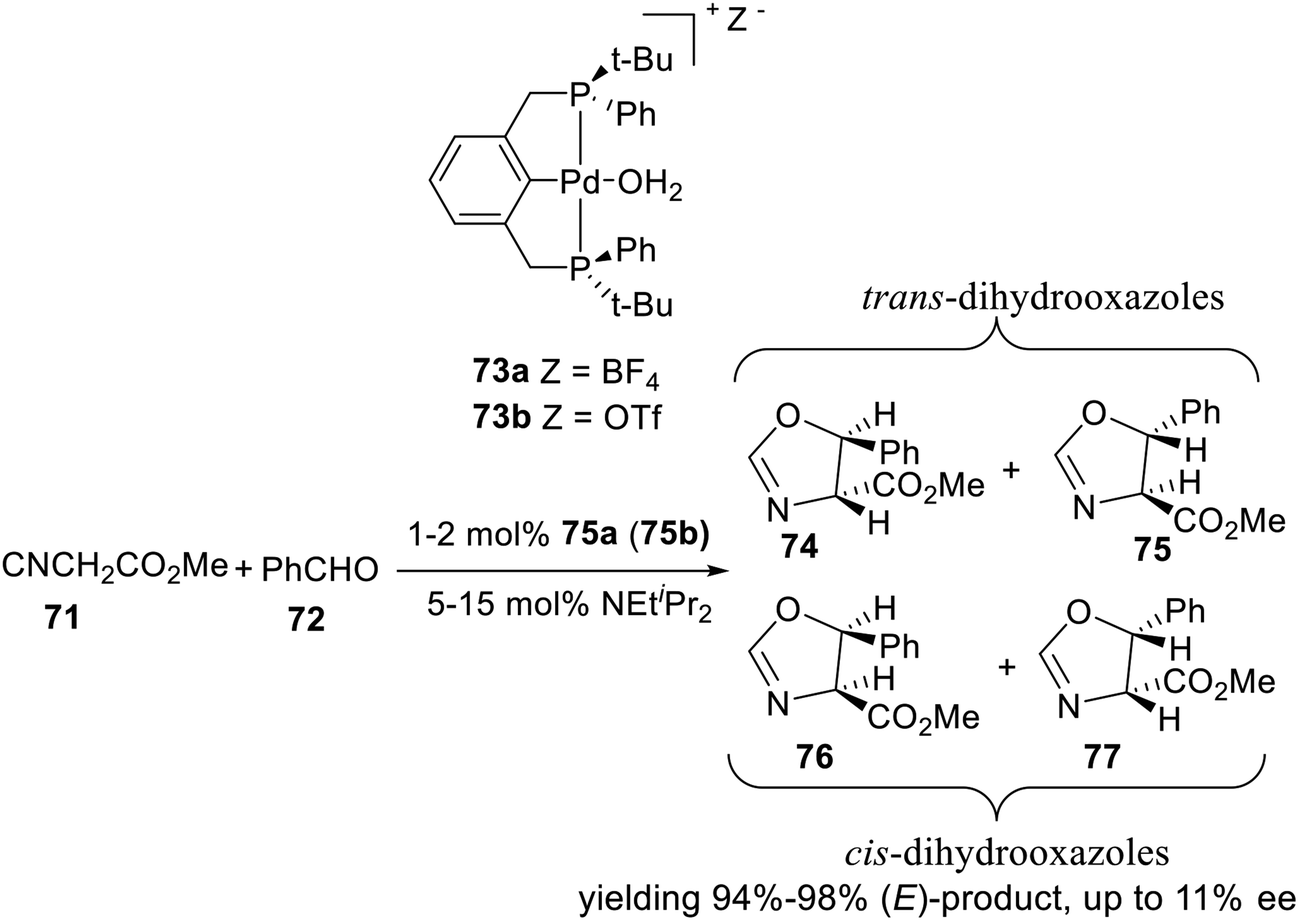
The asymmetric aldol condensation of methyl 2-isocyanoacetate and benzaldehyde is an effective reaction to synthesize chiral dihydrooxazoles. This reaction is interesting both because it is a potential route to β-hydroxy-amino acids, and because it involves the formation of a C–C bond with simultaneous creation of two chiral centers, resulting in four possible stereoisomers. It is thus a useful test reaction for exploring the chiral induction provided by new, chiral Lewis acid catalysts. In 2001, van Koten and coworkers synthesized two P-stereogenic pincer–Pd complexes 73a and 73b (Scheme 21), and did an initial test reaction between methyl 2-isocyanoacetate and benzaldehyde. Indeed, for the Pd complexes 73, the diastereoisomeric ratio was found to be higher, yielding 94–98% (E)-product. However, the enantiomeric excess was never found to be higher than 11%. Variations of the concentration of catalyst, aldehyde, and base did not appreciably affect the enantioselectivity, nor did changing the reaction solvent from CH2Cl2 to THF or toluene.
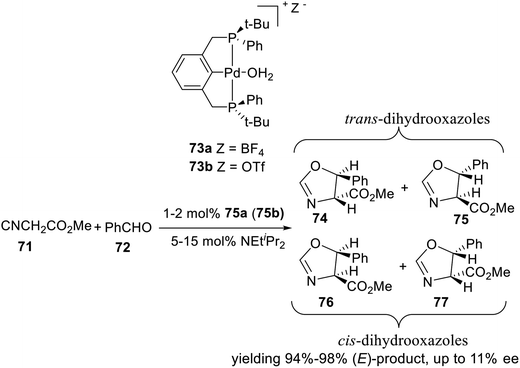 |
| | Scheme 21 Aldol condensation of methyl 2-isocyanoacetate and benzaldehyde. | |
4.4 Asymmetric hydrogen transfer reaction
In 2005, van Koten and coworkers19b designed and synthesized a kind of P-stereogenic PNPtBu,Ph ruthenium pincer complex and applied it in asymmetric reduction of ketones with propan-2-ol. As a result, a moderate yield (up to 40%) and low enantioselectivity (up to 18%) were obtained (Scheme 22). Apparently, the chiral pocket of the catalyst precursors allows for transfer of dihydrogen to both faces of the substrate equally well. Moreover, epimerization at the stereogenic P-centers under the conditions applied during the catalytic experiments cannot be excluded. The preliminary catalysis results showed that although the novel chiral ruthenium complexes were moderately active, the chiral induction was lost upon prolonged reaction.
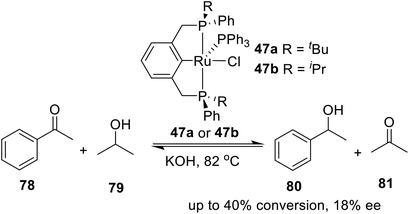 |
| | Scheme 22 Hydrogen transfer reaction of acetophenone by ruthenium. | |
4.5 Asymmetric Michael addition
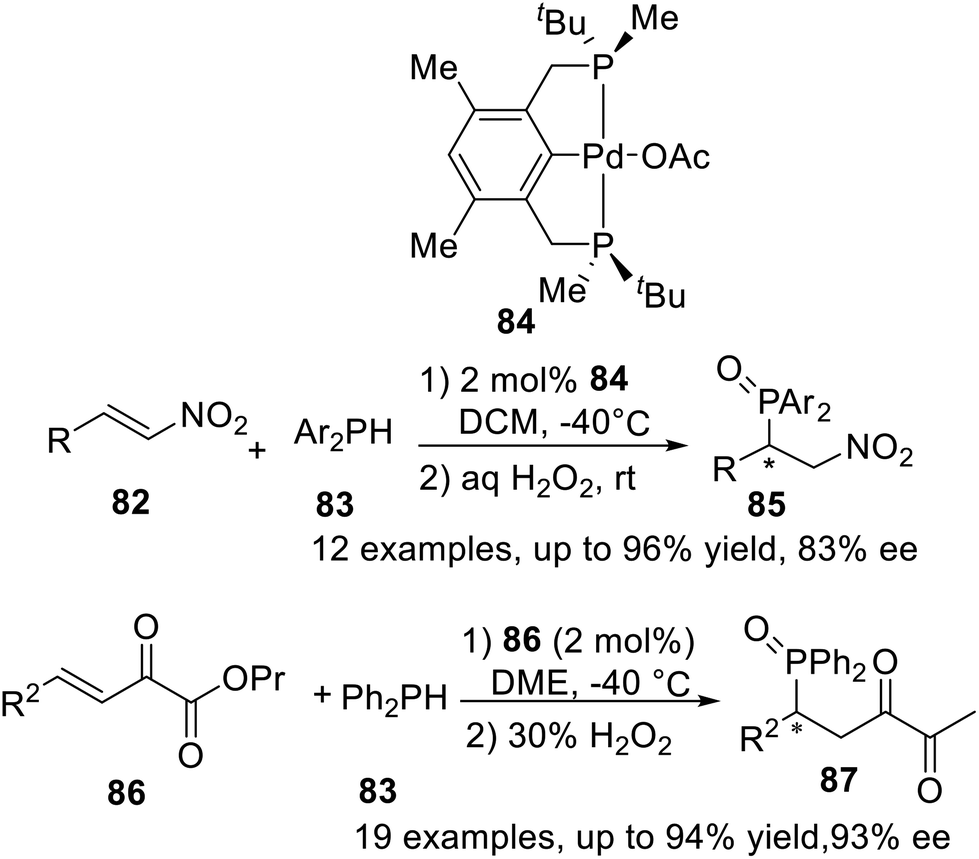
The catalytic asymmetric construction of P–C bonds is considered to be one of the most powerful methods for the preparation of chiral organophosphorus compounds. In 2013, Wanbin Zhang et al.21a used the P-stereogenic PCP type pincer–Pd complex 84 to catalyze the asymmetric Michael addition of diarylphosphines to nitroalkenes, obtained up to 96% yield and 83% ee within 12 examples (Scheme 23). And then in 2015,21b the asymmetric Michael addition of diphenylphosphine to β,γ-unsaturated α-keto esters was successfully processed by the same catalyst, obtained in up to 94% yield and 93% ee.
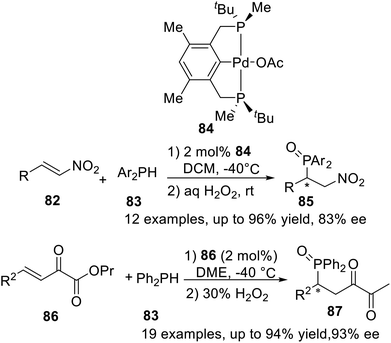 |
| | Scheme 23 Pincer–Pd catalyzed asymmetric Michael addition of diarylphosphine to nitroalkenes and β,γ-unsaturated α-ketoesters. | |
In 2015, Wanbin Zhang et al. developed a series of new P-stereogenic pincer–nickel complexes in 55–84% yields by using a flexible synthetic approach.10 These complexes were fully characterized by 1H NMR, 13C NMR, 31P NMR, 19F NMR, and/or single-crystal X-ray diffraction. These complexes were shown to be active catalysts for the aza-Michael addition of α,β-unsaturated nitriles (Scheme 24), providing the products in good to excellent yields (up to 99%) and with moderate enantiomeric excesses (up to 46% ee). Notably, the PCP complex exhibited higher catalytic activity in the aza-Michael addition than the PNP complexes.
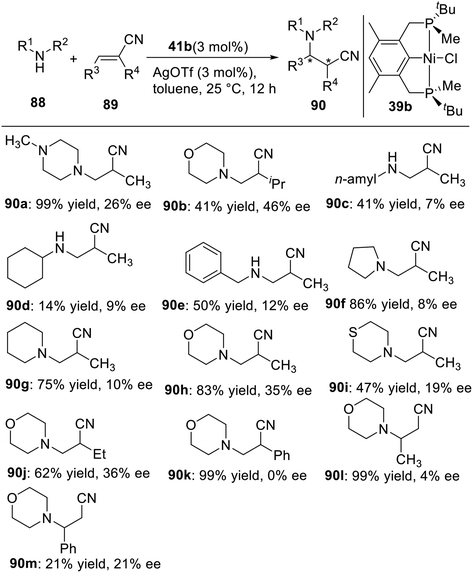 |
| | Scheme 24 P-stereogenic pincer–Ni catalyzed asymmetric aza-Michael reaction. | |
4.6 Asymmetric hydrogenation
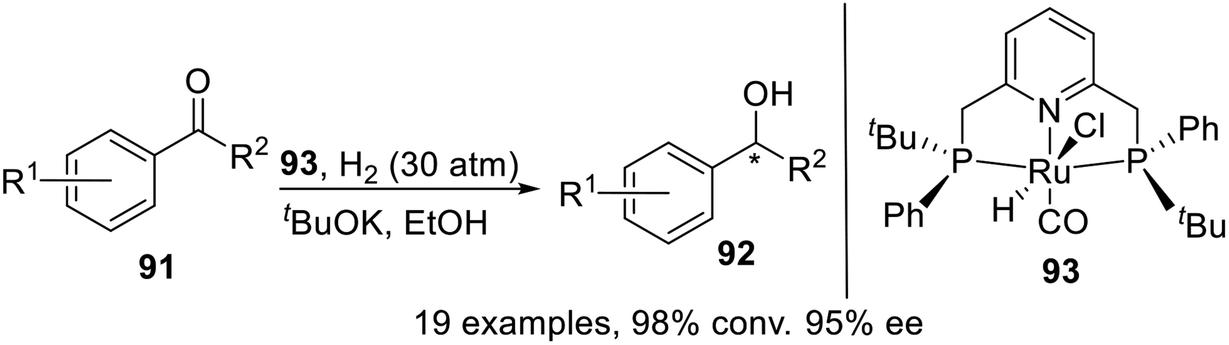
In 2015, Castillón et al.18 synthesized and characterized the first P-stereogenic PNPtBu,Ph–Ru complex which had been proven to be an efficient catalyst for the asymmetric reduction of a variety of aromatic and heterocyclic ketones (Scheme 25). Although the enantioselectivities obtained were not superior to those reported for other commercially available catalysts, this catalyst, active under very mild operating conditions, could be of significant interest in the chemoselective reduction of ketones in the presence of typical functional groups.
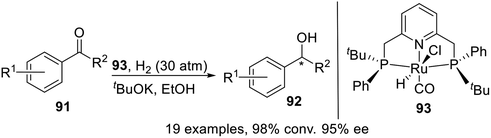 |
| | Scheme 25 PNPtBu,Ph Pincer–Ru complexes catalyzed asymmetric hydrogenation. | |
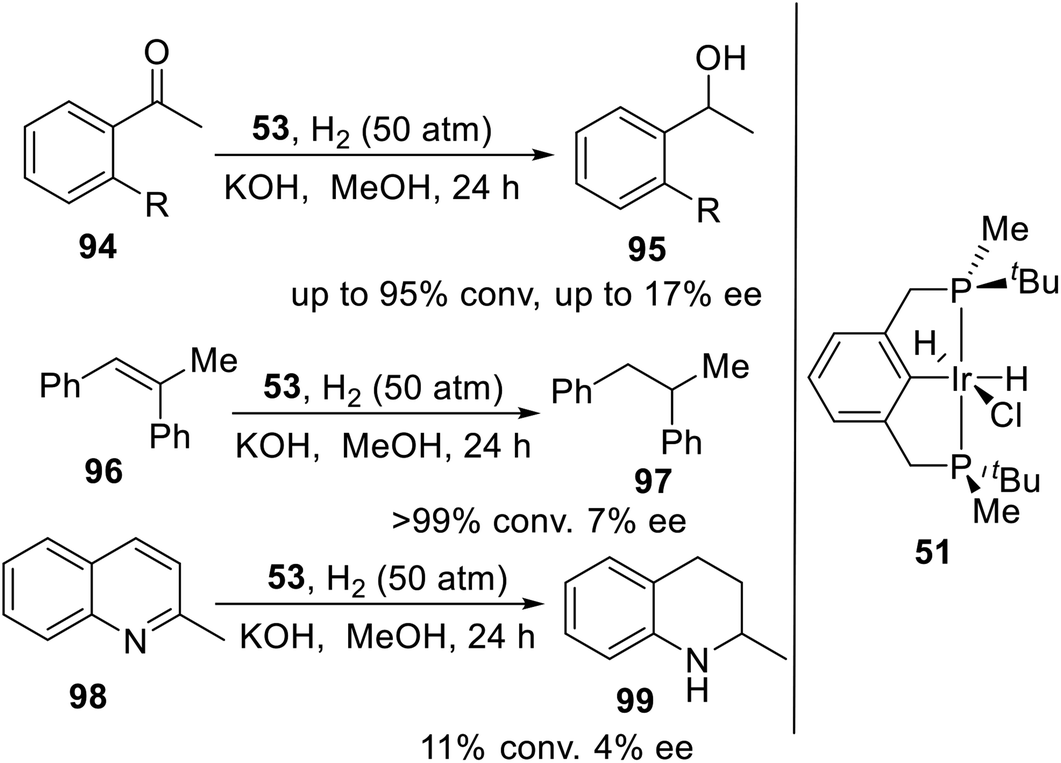
Wanbin Zhang and coworkers21c had designed and synthesized a novel P-stereogenic pincer–iridium complex 53 in reasonable yields using a short synthetic route. This complex was used as catalyst in the hydrogenation of ketones, olefins and quinoline derivatives to provide the desired products with moderate to excellent conversions (up to 99%) and up to 17% enantiomeric excess (Scheme 26).
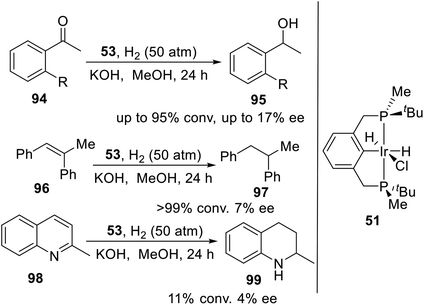 |
| | Scheme 26 PNPtBu,Me pincer–Ir complexes catalyzed asymmetric hydrogenation. | |
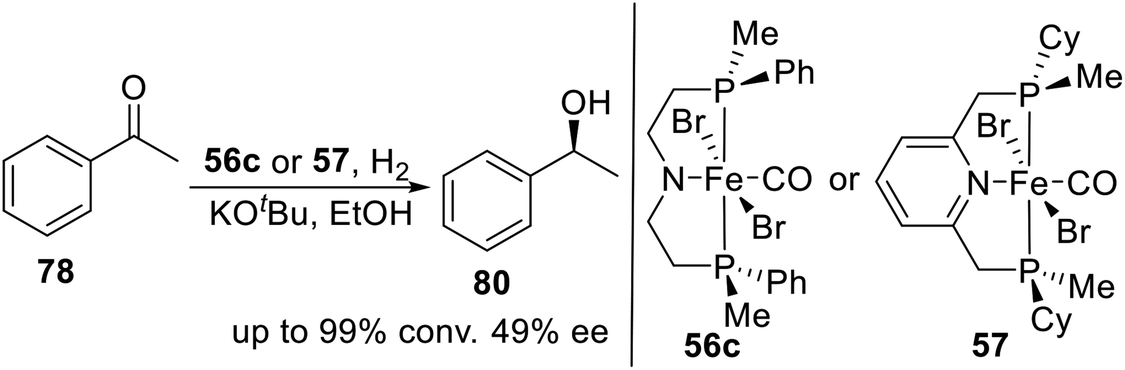
In 2018, Mezzetti et al. developed a series of P-stereogenic PN(H)P pincer ligands and their iron(II) derivatives by DFT-driven ligand design.22 These complexes were efficient catalysts in asymmetric hydrogenation of acetophenone (Scheme 27). In the same year, they reported a tridentate, P-stereogenic, C2-symmetric PNP pincer ligand and its iron(II) complex 57.4n In the presence of base, bromocarbonylhydride 57 catalyzes the hydrogenation of acetophenone to (S)-1-phenylethanol with 49% ee. The density functional theory (DFT) calculations show that the outer-sphere monohydride mechanism reproduces the experimentally observed sense of induction (S) and enantioselectivity, whereas the dihydride and inner-sphere pathways predict the formation of the R enantiomer.
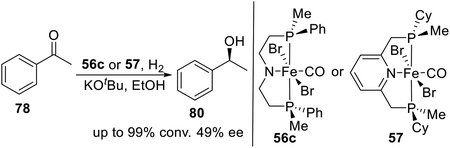 |
| | Scheme 27 P-stereogenic pincer–Fe catalyzed asymmetric hydrogenation of acetophenone. | |
4.7 Intramolecular hydroamination
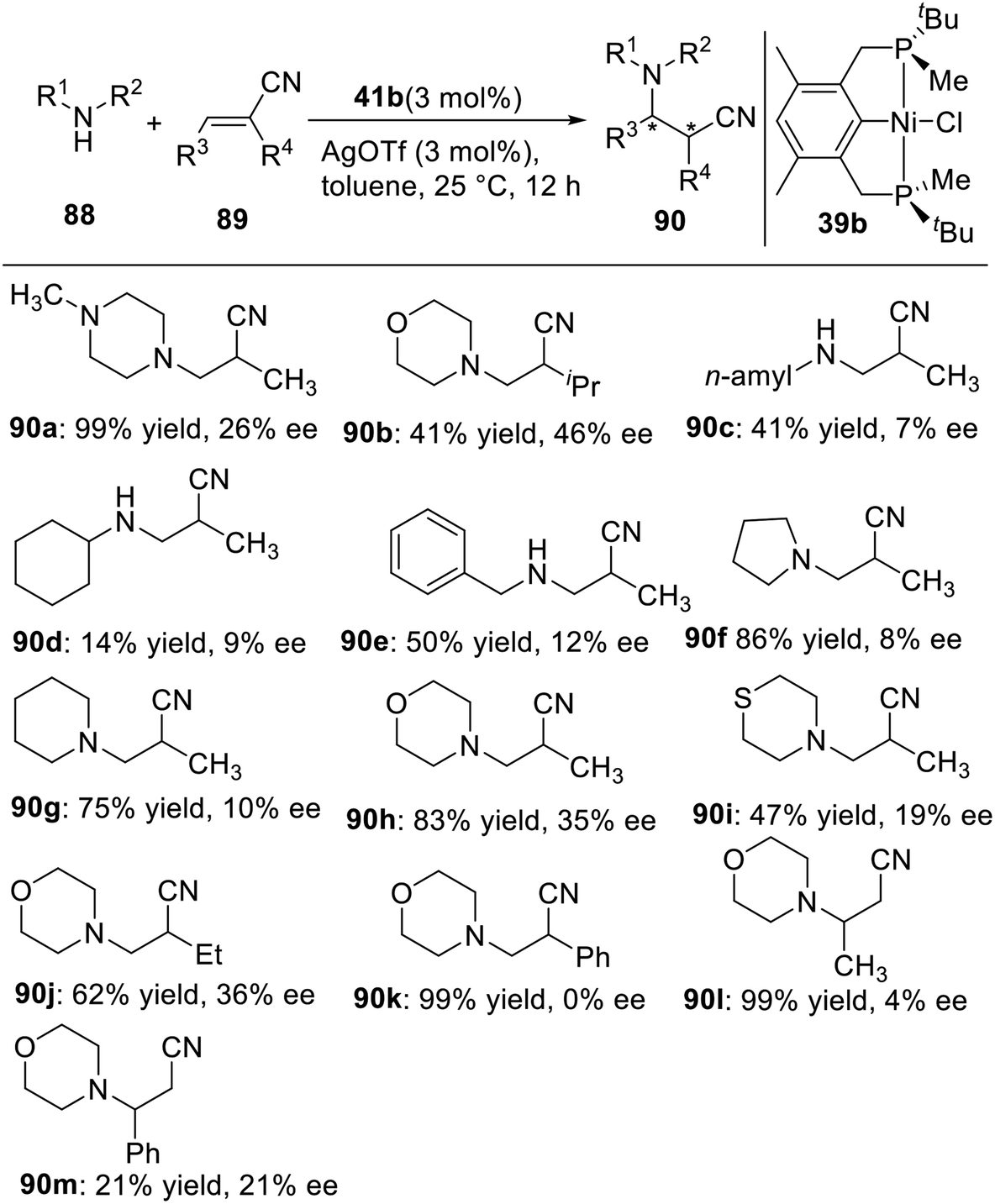
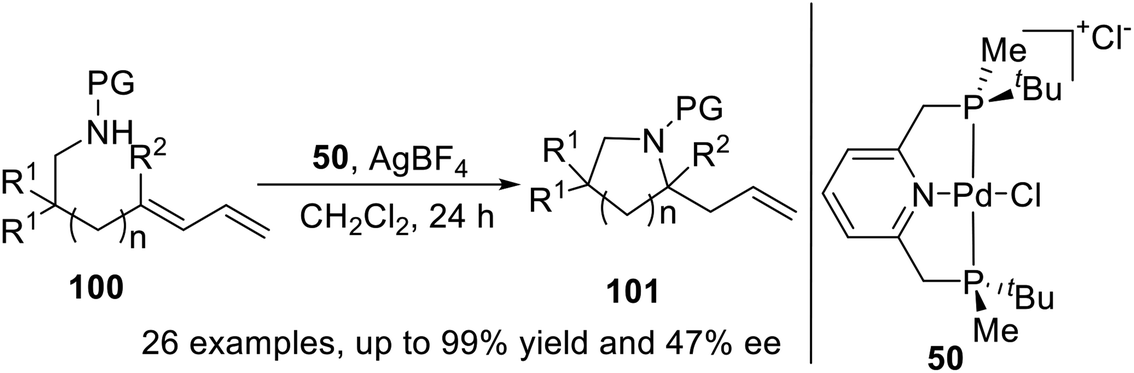
In 2015, Wanbin Zhang and coworkers21d prepared a novel P-stereogenic PNP pincer–Pd complex from optically pure 2,6-bis[(boranato(tert-butyl)-methylphosphino)methyl]pyridine and used it in the asymmetric intramolecular hydroamination of amino-1,3-dienes (Scheme 28). The desired products were obtained in high yields (up to quantitative yield) and with excellent regioselectivities (>99:1) and up to moderate enantioselectivities (up to 47% ee). The absolute configuration of an enantioenriched product was determined by X-ray crystallography studies.
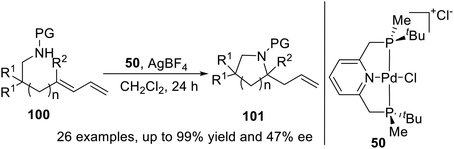 |
| | Scheme 28 P-stereogenic pincer–Pd catalyzed asymmetric intramolecular hydroamination. | |
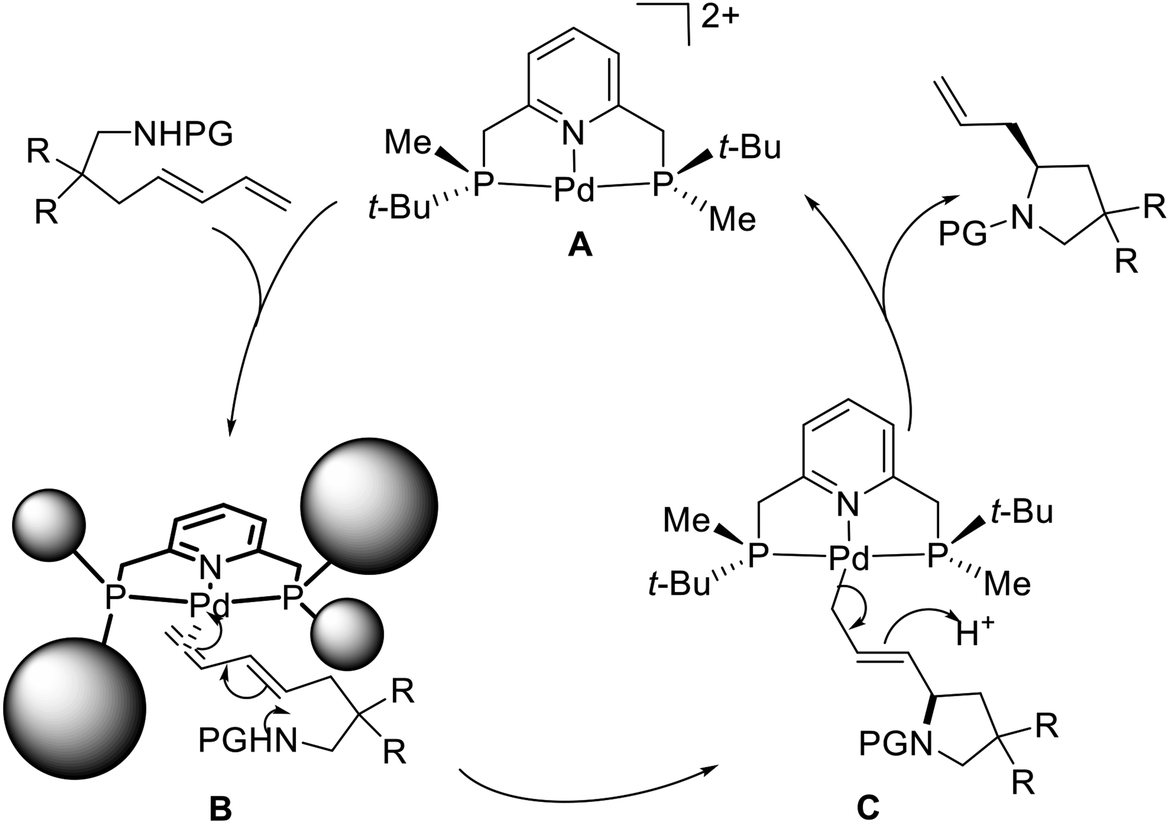
A proposed mechanism has been suggested to explain the excellent regioselectivity of the intramolecular hydroamination (Scheme 29).21d Initially, the chiral pincer-type catalyst 50 reacts with AgBF4 to give an activated catalytic molecule A. This cationic palladium species interacts with the terminal double bond of the amino-1,3-diene in close proximity the methyl group located on the phosphorus atom due to the strong steric hindrance of the tert-butyl group, affording the square planar π-complex B. The intermediate B undergoes an intramolecular C–N bond formation to produce the η1-allyl-palladium complex C, which is consistent with Michael's discovery of the isolated η1-allyl-palladium intermediate.30 Subsequent protonation and cleavage of the Pd–C bond produce the allylic-type S-configuration product and regenerate the catalyst A. In the absence of a bulky substituent, such as a tert-butyl group, the internal double bond of the amino-1,3-diene can also coordinate to the Pd atom of A, eventually leading to the undesired propenyl-type product.31 Because of the remote distance (four bonds) between the palladium atom and the reaction site in B, the moderate 43% ee obtained for the allylic-type pyrrolidine derivative presents a very promising result.
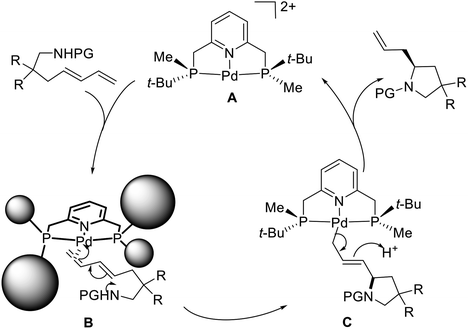 |
| | Scheme 29 Proposed reaction pathway of P-stereogenic pincer–Pd catalyzed asymmetric intramolecular hydroamination. | |
4.8 Asymmetric allylic sulfonylation and deracemization
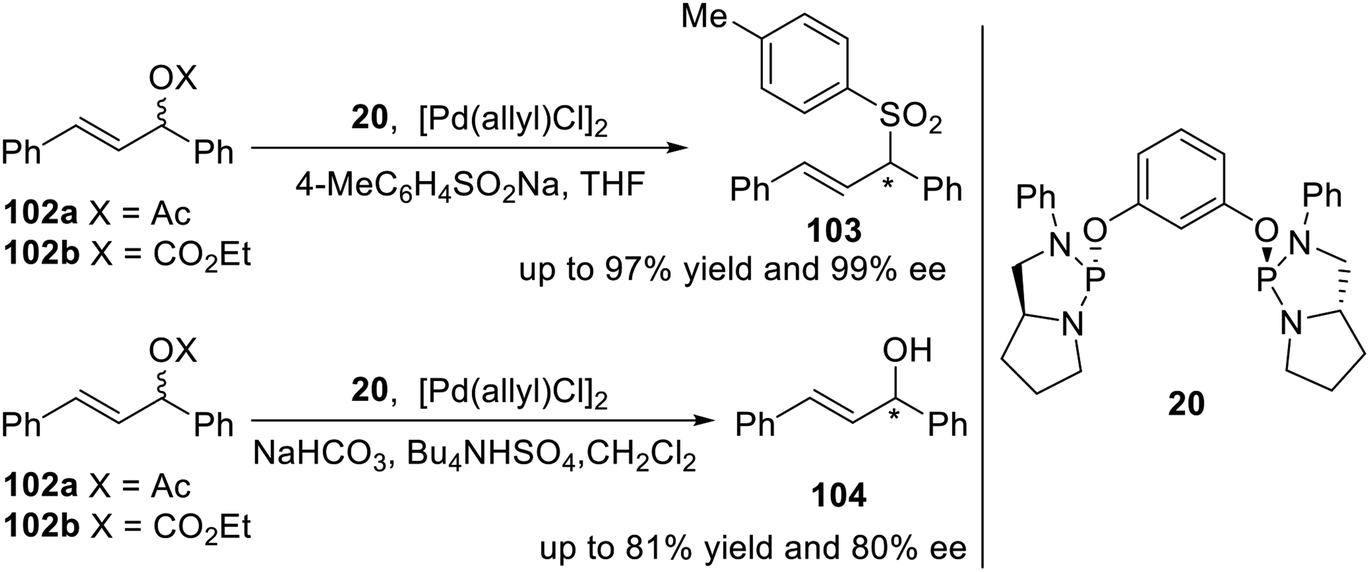
Gavrilov et al. reported a pincer type phosphodiamidite ligand 20 containing chiral phosphorus atoms11 in 2009. To estimate the stereodifferentiating ability of ligand 20 a test reaction of Pd-catalyzed enantioselective allylic sulfonylation of 102a and 102b was used (Scheme 30, top). As a result, nearly quantitative chemical yield and stereo-chemical outcome (up to 97% yield and 99% ee) were obtained. Ligand 20 was also involved in an important Pd-catalyzed deracemization reaction of compound 102b (Scheme 30, bottom) opening access to valuable optically active allylic alcohols, including chalcol 104. The catalytic system [Pd(allyl)Cl]2/2L (L = 20) provided good conversion of 102b (81%) and enantiomeric excess of (R)-104 (80%).
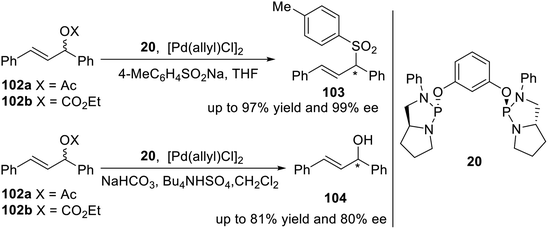 |
| | Scheme 30 P-stereogenic pincer–Pd catalyzed asymmetric allylic sulfonylation and deracemization. | |
5. Conclusion and outlook
Since the pioneering work reported by X. Zhang et al. about P-stereogenic pincer ligands in 1996, the chemistry of pincer compounds has achieved great progress. Many strategies have been developed to synthesize P-stereogenic pincer ligands, including classical and non-classical P-stereogenic pincer ligands. The P-stereogenic pincer complexes could be synthesized by C–H activation, oxidative addition, transcyclometalation, and direct coordination in good yields. These kinds of complexes were efficient catalysts in asymmetric catalysis. Many asymmetric reactions could be catalyzed by them, such as allylic alkylation, hydrosilylation, aldol condensation, hydrogen transfer reaction, Michael addition, hydrogenation, hydroamination, allylic sulfonylation and deracemization. In most of the reactions, excellent yields but moderate enantioselectivities were obtained. Further studies should focus on the design and synthesis of P-stereogenic pincer complexes with high asymmetric induction effects.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Natural Science Foundation of Hunan Province (Grant No. 2018JJ3426); the young and middle-aged innovative personnel training project of the Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study (Grant No. 0223-0002-0002000-52); the Undergraduate Research Learning and Innovative Experiment Project (Grant No. 2018XJXZ199, 2018XJXZ349); the guiding project of Hengyang Science and Technology Department (Grant No. S2018F9031015299); the open fund for innovation platform of Hunan Provincial Department of Education (Grant No. 19K079); and China Scholarship Council (Grant No. CSC201908430070).
Notes and references
- W. S. J. Kelly, G. H. Ford and S. M. Nelson, J. Chem. Soc. A, 1971, 388–396 RSC.
- J. Terheijden, G. van Koten, W. P. Mul, D. J. Stufkens, F. Muller and C. H. Stam, Organometallics, 1986, 5, 519–525 CrossRef CAS.
-
(a) J. I. Van Der Vlugt, Angew. Chem., Int. Ed., 2010, 49, 252–255 CrossRef CAS PubMed;
(b) J. Choi, A. H. R. Macarthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed;
(c) N. Selander and K. J. Szabo, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed;
(d) T. Robert and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 5216–5218 CrossRef CAS PubMed;
(e) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS;
(f) S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS;
(g) M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2016, 55, 7892–7896 CrossRef CAS;
(h) A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed;
(i) Z. Wang, G. A. Solan, W. Zhang and W.-H. Sun, Coord. Chem. Rev., 2018, 363, 92–108 CrossRef CAS;
(j) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS.
-
(a) Z. Zuo, S. Xu, L. Zhang, L. Gan, H. Fang, G. Liu and Z. Huang, Organometallics, 2019, 38, 3906–3911 CrossRef CAS;
(b) L. Zhang, Y. Tang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2019, 58, 4973–4977 CrossRef CAS PubMed;
(c) W. S. Tay, X.-Y. Yang, Y. Li, S. A. Pullarkat and P.-H. Leung, Dalton Trans., 2019, 48, 4602–4610 RSC;
(d) S. Nakamura, A. Tokunaga, H. Saito and M. Kondo, Chem. Commun., 2019, 55, 5391–5394 RSC;
(e) H. Liu, H. Yuan and X. Shi, Dalton Trans., 2019, 48, 609–617 RSC;
(f) R. Huber, A. Passera and A. Mezzetti, Chem. Commun., 2019, 55, 9251–9266 RSC;
(g) M. Garbe, Z. Wei, B. Tannert, A. Spannenberg, H. Jiao, S. Bachmann, M. Scalone, K. Junge and M. Beller, Adv. Synth. Catal., 2019, 361, 1913–1920 CrossRef CAS;
(h) N. Deak, O. Thillaye Du Boullay, I.-T. Moraru, S. Mallet-Ladeira, D. Madec and G. Nemes, Dalton Trans., 2019, 48, 2399–2406 RSC;
(i) T. Arai, K. Araseki and J. Kakino, Org. Lett., 2019, 21, 8572–8576 CrossRef CAS PubMed;
(j) J. Yan, Y.-B. Wang, Z.-H. Zhu, Y. Li, X. Zhu, X.-Q. Hao and M.-P. Song, Organometallics, 2018, 37, 2325–2334 CrossRef CAS;
(k) Q. Wan, X.-S. Xiao, W.-P. To, W. Lu, Y. Chen, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 17189–17193 CrossRef CAS PubMed;
(l) F. W. Seidel, S. Friess, F. W. Heinemann, A. Chelouan, A. Scheurer, A. Grasruck, A. Herrera and R. Dorta, Organometallics, 2018, 37, 1160–1171 CrossRef CAS;
(m) C. H. Schiwek, V. Vasilenko, H. Wadepohl and L. H. Gade, Chem. Commun., 2018, 54, 9139–9142 RSC;
(n) R. Huber, A. Passera and A. Mezzetti, Organometallics, 2018, 37, 396–405 CrossRef CAS;
(o) X. Chen, Z. Cheng, J. Guo and Z. Lu, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed;
(p) X. Chen, Z. Cheng, J. Guo and Z. Lu, Nat. Commun., 2018, 9, 3939 CrossRef;
(q) J. Wenz, H. Wadepohl and L. H. Gade, Chem. Commun., 2017, 53, 4308–4311 RSC;
(r) V. Vasilenko, C. K. Blasius, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2017, 56, 8393–8397 CrossRef CAS PubMed;
(s) J. S. Marcum, C. C. Roberts, R. S. Manan, T. N. Cervarich and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 15580–15583 CrossRef CAS PubMed;
(t) M. Kondo, M. Omori, T. Hatanaka, Y. Funahashi and S. Nakamura, Angew. Chem., Int. Ed., 2017, 56, 8677–8680 CrossRef CAS PubMed;
(u) M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS PubMed;
(v) J.-K. Liu, J.-F. Gong and M.-P. Song, Org. Biomol. Chem., 2019, 17, 6069–6098 RSC.
-
(a) N. Zhao, G. Hou, X. Deng, G. Zi and M. D. Walter, Dalton Trans., 2014, 43, 8261–8272 RSC;
(b) S. Bonnet, J. Li, M. A. Siegler, L. S. Von Chrzanowski, A. L. Spek, G. van Koten and R. J. M. Klein Gebbink, Chem.–Eur. J., 2009, 15, 3340–3343 CrossRef CAS PubMed;
(c) E. T. J. Strong, S. A. Cardile, A. L. Brazeau, M. C. Jennings, R. Mcdonald and N. D. Jones, Inorg. Chem., 2008, 47, 10575–10586 CrossRef CAS PubMed;
(d) V. F. Kuznetsov and D. G. Gusev, Organometallics, 2007, 26, 5661–5666 CrossRef CAS;
(e) V. F. Kuznetsov, A. J. Lough and D. G. Gusev, Inorg. Chim. Acta, 2006, 359, 2806–2811 CrossRef CAS;
(f) M. Q. Slagt, S.-E. Stiriba, H. Kautz, R. J. M. K. Gebbink, H. Frey and G. van Koten, Organometallics, 2004, 23, 1525–1532 CrossRef CAS;
(g) V. F. Kuznetsov, A. J. Lough and D. G. Gusev, Chem. Commun., 2002, 2432–2433 RSC;
(h) A. a. D. Tulloch, A. A. Danopoulos, G. J. Tizzard, S. J. Coles, M. B. Hursthouse, R. S. Hay-Motherwell and W. B. Motherwell, Chem. Commun., 2001, 1270–1271 RSC;
(i) R. Gimenez and T. M. Swager, J. Mol. Catal. A: Chem., 2001, 166, 265–273 CrossRef CAS.
-
(a) O. Cohen, O. Grossman, L. Vaccaro and D. Gelman, J. Organomet. Chem., 2014, 750, 13–16 CrossRef CAS;
(b) D. Monge, A. Bermejo, J. Vazquez, R. Fernandez and J. M. Lassaletta, Arkivoc, 2013, 33–45 Search PubMed;
(c) N. Grueger, L.-I. Rodriguez, H. Wadepohl and L. H. Gade, Inorg. Chem., 2013, 52, 2050–2059 CrossRef CAS PubMed;
(d) M. E. El-Zaria, H. Arii and H. Nakamura, Inorg. Chem., 2011, 50, 4149–4161 CrossRef CAS PubMed;
(e) C. Del Pozo, A. Corma, M. Iglesias and F. Sanchez, Green Chem., 2011, 13, 2471–2481 RSC;
(f) R. B. Bedford, Y.-N. Chang, M. F. Haddow and C. L. Mcmullin, Dalton Trans., 2011, 40, 9034–9041 RSC;
(g) A. I. Aranda Perez, T. Biet, S. Graule, T. Agou, C. Lescop, N. R. Branda, J. Crassous and R. Reau, Chem.–Eur. J., 2011, 17, 1337–1351 CrossRef CAS PubMed;
(h) B.-S. Zhang, W. Wang, D.-D. Shao, X.-Q. Hao, J.-F. Gong and M.-P. Song, Organometallics, 2010, 29, 2579–2587 CrossRef CAS;
(i) J.-L. Niu, Q.-T. Chen, X.-Q. Hao, Q.-X. Zhao, J.-F. Gong and M.-P. Song, Organometallics, 2010, 29, 2148–2156 CrossRef CAS.
- R. A. Baber, R. B. Bedford, M. Betham, M. E. Blake, S. J. Coles, M. F. Haddow, M. B. Hursthouse, A. G. Orpen, L. T. Pilarski, P. G. Pringle and R. L. Wingad, Chem. Commun., 2006, 3880–3882 RSC.
-
(a) J. Lu, J. Ye and W.-L. Duan, Chem. Commun., 2014, 50, 698–700 RSC;
(b) C. Li, Q.-L. Bian, S. Xu and W.-L. Duan, Org. Chem. Front., 2014, 1, 541–545 RSC;
(c) J. Huang, M.-X. Zhao and W.-L. Duan, Tetrahedron Lett., 2014, 55, 629–631 CrossRef CAS;
(d) X.-Q. Hao, J.-J. Huang, T. Wang, J. Lv, J.-F. Gong and M.-P. Song, J. Org. Chem., 2014, 79, 9512–9530 CrossRef CAS PubMed;
(e) T. Wang, J. Niu, S. Liu, J. Huang, J. Gong and M. Song, Adv. Synth. Catal., 2013, 355, 927–937 CrossRef CAS;
(f) T. Wang, X.-Q. Hao, J.-J. Huang, J.-L. Niu, J.-F. Gong and M.-P. Song, J. Org. Chem., 2013, 78, 8712–8721 CrossRef CAS;
(g) S. Nakamura, K. Hyodo, M. Nakamura, D. Nakane and H. Masuda, Chem.–Eur. J., 2013, 19, 7304–7309 CrossRef CAS;
(h) Z. Lu, S. Abbina, J. R. Sabin, V. N. Nemykin and G. Du, Inorg. Chem., 2013, 52, 1454–1465 CrossRef CAS PubMed;
(i) J. Lu, J. Ye and W.-L. Duan, Org. Lett., 2013, 15, 5016–5019 CrossRef CAS PubMed;
(j) C. Li, W. Li, S. Xu and W. Duan, Chin. J. Org. Chem., 2013, 33, 799–802 CrossRef CAS.
- J. M. Longmire and X. Zhang, Tetrahedron Lett., 1997, 38, 1725–1728 CrossRef CAS.
- Z. Yang, D. Liu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Organometallics, 2015, 34, 1228–1237 CrossRef CAS.
- K. N. Gavrilov, E. A. Rastorguev, A. A. Shiryaev, T. B. Grishina, A. S. Safronov, S. E. Lyubimov and V. A. Davankov, Russ. Chem. Bull., 2009, 58, 1325–1327 CrossRef CAS.
- R. Huber, A. Passera and A. Mezzetti, Chem. Commun., 2019, 55, 9251–9266 RSC.
- S. Juge, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 233–248 CrossRef CAS.
- K. Nagata, S. Matsukawa and T. Imamoto, J. Org. Chem., 2000, 65, 4185–4188 CrossRef CAS PubMed.
- Q. Xu, C.-Q. Zhao and L.-B. Han, J. Am. Chem. Soc., 2008, 130, 12648–12655 CrossRef CAS PubMed.
-
(a) G. Zhu, M. Terry and X. Zhang, Tetrahedron Lett., 1996, 37, 4475–4478 CrossRef CAS;
(b) G. Zhu, M. Terry and X. Zhang, J. Organomet. Chem., 1997, 547, 97–101 CrossRef CAS.
- B. Wolfe and T. Livinghouse, J. Am. Chem. Soc., 1998, 120, 5116–5117 CrossRef CAS.
- I. Arenas, O. Boutureira, M. I. Matheu, Y. Diaz and S. Castillón, Eur. J. Org. Chem., 2015, 2015, 3666–3669 CrossRef CAS.
-
(a) B. S. Williams, P. Dani, M. Lutz, A. L. Spek and G. van Koten, Helv. Chim. Acta, 2001, 84, 3519–3530 CrossRef CAS;
(b) S. Medici, M. Gagliardo, S. B. Williams, P. A. Chase, S. Gladiali, M. Lutz, A. L. Spek, K. G. P. M. Van and K. G. van, Helv. Chim. Acta, 2005, 88, 694–705 CrossRef CAS.
- D. Morales-Morales, R. E. Cramer and C. M. Jensen, J. Organomet. Chem., 2002, 654, 44–50 CrossRef CAS.
-
(a) B. Ding, Z. Zhang, Y. Xu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Org. Lett., 2013, 15, 5476–5479 CrossRef CAS PubMed;
(b) Y. Xu, Z. Yang, B. Ding, D. Liu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Tetrahedron, 2015, 71, 6832–6839 CrossRef CAS;
(c) Z. Yang, X. Wei, D. Liu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, J. Organomet. Chem., 2015, 791, 41–45 CrossRef CAS;
(d) Z. Yang, C. Xia, D. Liu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Org. Biomol. Chem., 2015, 13, 2694–2702 RSC;
(e) Y. Liu, B. Ding, D. Liu, Z. Zhang, Y. Liu and W. Zhang, Res. Chem. Intermed., 2017, 43, 4959–4966 CrossRef CAS.
- R. Huber, A. Passera, E. Gubler and A. Mezzetti, Adv. Synth. Catal., 2018, 360, 2900–2913 CrossRef CAS.
- V. S. Chan, I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 2786–2787 CrossRef CAS PubMed.
- A. Adhikary, J. A. Krause and H. Guan, Organometallics, 2015, 34, 3603–3610 CrossRef CAS.
- J. Li, M. Lutz, A. L. Spek, G. P. M. Van Klink, G. van Koten and R. J. M. Klein Gebbink, Organometallics, 2010, 29, 1379–1387 CrossRef CAS.
- S. Kundu, Y. Choliy, G. Zhuo, R. Ahuja, T. J. Emge, R. Warmuth, M. Brookhart, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2009, 28, 5432–5444 CrossRef CAS.
- J.-L. Niu, X.-Q. Hao, J.-F. Gong and M.-P. Song, Dalton Trans., 2011, 40, 5135–5150 RSC.
- L. Mckinstry and T. Livinghouse, Tetrahedron Lett., 1994, 35, 9319–9322 CrossRef CAS.
- I. Arenas, O. Boutureira, M. I. Matheu, Y. Diaz and S. Castillón, Eur. J. Org. Chem., 2015, 3666–3669 CrossRef CAS.
- J. M. Pierson, E. L. Ingalls, R. D. Vo and F. E. Michael, Angew. Chem., Int. Ed., 2013, 52, 13311–13313 CrossRef CAS PubMed.
- H. Yamamoto, I. Sasaki, S. Shiomi, N. Yamasaki and H. Imagawa, Org. Lett., 2012, 14, 2266–2269 CrossRef CAS PubMed.
Footnote |
| † These two authors contributed equally to this article. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*