
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NaV6O15 microflowers as a stable cathode material for high-performance aqueous zinc-ion batteries
Runxia Liab,
Chao Guanb,
Xiaofei Biana,
Xin Yub and
Fang Hu *b
*b
aSchool of Materials Science and Engineering, Dongguan University of Technology, Dongguan 523808, China
bSchool of Materials Science and Engineering, Shenyang University of Technology, Shenyang, 110870, China. E-mail: hufang25@126.com
First published on 13th February 2020
Abstract
Reversible aqueous zinc-ion batteries (ZIBs) have great potential for large-scale energy storage owing to their low cost and safety. However, the lack of long-lifetime positive materials severely restricts the development of ZIBs. Herein, we report NaV6O15 microflowers as a cathode material for ZIBs with excellent electrochemical performance, including a high specific capacity of ∼300 mA h g−1 at 100 mA g−1 and 141 mA h g−1 maintained after 2000 cycles at 5 A g−1 with a capacity retention of ∼107%. The high diffusion coefficient and stable tunneled structure of NaV6O15 facilitate Zn2+ intercalation/extraction and long-term cycle stability.
1. Introduction
To alleviate the increasingly severe energy crisis and climate-related challenges, the use of safe, green, reliable, and economical energy resources such as wind, solar, and water power is becoming a focus worldwide.1–3 Rechargeable battery technology provides a promising option for storing energy from these renewable energy systems. However, commercial Li-ion batteries cannot solve the problem of large-scale energy storage because of the limited lithium resources, high cost, safety issues, and environmental impact of toxic electrolyte.4–8 Recently, rechargeable aqueous Zn-ion batteries (ZIBs) have been introduced as prospects for large-scale electrochemical energy storage due to their intrinsic safety (no flammable organic electrolytes), easy assembly, low cost, and high ionic conductivity (two orders of magnitude higher than those of organic electrolytes).9–14To date, layered vanadium-based compounds have displayed better electrochemical performance than manganese-based compounds such as MnO2 and ZnMn2O4.15,16 Among these compounds, sodium or potassium layered vanadate (e.g., Na2V6O16·3H2O, Na5V12O32, Na1.1V3O7.9, KV3O8, and K2V6O16·1.57H2O) are candidate cathode materials in ZIBs.17–20 The large lattice spacing in the layered structure and the “pillar” effect of Na+/K+ between the vanadium oxygen interlayers might be responsible for the high capacity and good cycle performance, respectively. However, due to the weak interaction between the triconnected oxygen atoms on the layered surface and the Na+ or K+ ions along with the strong electrostatic interaction between the inserted Zn2+ and the unstable layers, the interlayer spacing in layered vanadate decreases, and the structure is destroyed.20,21 Vanadium-based compounds with tunneled structures (e.g., Na0.76V6O15, Na0.33V2O5, K2V8O21, and K0.25V2O5) have also been reported as cathode materials for ZIBs.20–22 The stable tunneled structure provides an effective diffusion path for Zn2+ insertion and avoids structural damage, resulting in excellent cycle performance. Mai et al. prepared Na0.33V2O5 nanowires with a high capacity of 367 mA h g−1 at 100 mA g−1 and long-term cycle stability with a capacity retention of over 93% for 1000 cycles.22 Kim et al. synthesized NaV6O15 nanorods that delivered a high discharge capacity of 427 mA h g−1 at 50 mA g−1 and a capacity retention of 65% over 300 cycles at 1 A g−1.23
In our past work, high-performance NaV6O15 microflowers were successfully synthesized as a cathode material for Li/Na-ion batteries.24 Herein, NaV6O15 microflowers are also demonstrated to be a competitive cathode material for ZIBs based on the following parameters: a high initial specific capacity of ∼300 mA h g−1 at 100 mA g−1; 141 mA h g−1 maintained after 2000 cycles at 5 A g−1; and a capacity retention of ∼107%. Meanwhile, the storage mechanism of the cathode was also investigated.
2. Experimental
NaV6O15 microflowers were fabricated by a simple hydrothermal method. NaOH (0.24 g), Na2CO3 (0.053 g), and NH4VO3 (0.234 g) were dissolved together in 40 mL of deionized water under magnetic stirring at 80 °C for 20 min. When the solution was cooled to room temperature, 1 mL of 3% H2O2 was added into the solution. Subsequently, the pH of the solution was adjusted to 1.9 via the dropwise addition of dilute hydrochloric acid (2 M). The orange solution was then transferred to a 50 mL Teflon-lined sealed autoclave and maintained at 200 °C for 24 h. After centrifugation, the yellow precursors were washed three times with distilled water and alcohol followed by drying for 10 h. NaV6O15 powders were finally obtained by heating the precursors at 400 °C in air for 4 h.The crystal structure of the as-prepared product was evaluated by X-ray diffraction (XRD; XRD-7000, Shimadzu) with a Cu Kα X-ray source. The morphology of the sample was investigated by scanning electron microscopy (SEM) with a Hitachi-4800 scanning electron microscope. X-ray photoelectron spectroscopy (XPS) was conducted using an Escalab250 spectrometer equipped with an Al Kα source. Galvanostatic charge–discharge experiments were performed using a battery testing system (Neware CT-3008) in the voltage window of 0.2–1.6 V. Cyclic voltammetry (CV) and galvano-static intermittent titration technique (GITT) measurements were carried out using a Bio-Logic VSP-300 multichannel.
Our cathode electrode for testing was prepared by mixing 70 wt% active material, 20 wt% acetylene black, and 10 wt% polytetrafluoro-ethylene (PTFE) binder to form a homogeneous mud pie, which was then rolled into a film on a carbon paper. The loading density of active material in the film was approximately 1 mg cm−2. Finally, the film was dried at 60 °C under vacuum for 10 h. To assemble the 2032-type coin cell, we choose 3 M Zn(CH3F3SO3)2 aqueous solution as the electrolyte to obtain good electrochemical performance. Metallic zinc foil was used as the anode, and glass microfiber served as the separator.
3. Results and discussion
The XRD pattern of the NaV6O15 microflowers is shown in Fig. 1a. All the diffraction peaks can be well indexed to the monoclinic layered NaV6O15 phase (PDF #24-1155), and no other impurities are detected. An SEM image of microflower-like NaV6O15 with a diameter of ∼5 μm is shown in Fig. 1b. The microflower is composed of nanorods with lengths of ∼2 μm and widths of ∼300 nm. Fig. 1c and d show the crystalline structure of NaV6O15. The layered NaV6O15 is composed of VO6 octahedra and VO5 square pyramids along the b-axis, and the layers are bonded with single-connected oxygen atoms; this structure is more stable than those of other layered vanadium oxides.25,26 The interlayer sodium ions along the a-axis act as pillars to further increase the stability of the layered vanadium oxide during Zn insertion/extraction.27,28 The tunneled structure along the b-axis (Fig. 1d) provides additional space, which also facilitates Zn insertion/extraction. | ||
| Fig. 1 (a) XRD pattern and (b) SEM image of NaV6O15 microflowers. (c and d) Crystalline structure of NaV6O15. | ||
Fig. 2a shows the cyclic voltammogram of the NaV6O15 electrode at a scan rate of 0.1 mV s−1 in the first three cycles. Two reduction/oxidation peaks appeared at approximately 0.82 V/1.01 V and 0.51 V/0.75 V, which can be attributed to the V5+/V4+ and V4+/V3+ redox couples in NaV6O15, respectively.29 The redox couple at approximately 1.10 V/1.25 V might indicate a phase transformation of the NaV6O15 electrode. The CV curves remained similar after the first cycle, demonstrating the good structural stability and high electrochemical reversibility of the electrode. The galvanostatic charge–discharge profiles of the NaV6O15 electrodes with Zn at a current density of 100 mA g−1 are shown in Fig. 2b. The NaV6O15 electrode delivered an initial specific discharge/charge capacity of 297/293 mA h g−1 with a high coulombic efficiency (98.6%). After the first three cycles, a higher discharge/charge capacity of 348/347 mA h g−1 was obtained with ∼100% coulombic efficiency. The similar discharge/charge profiles compared to the first cycle also demonstrate the good electrochemical stability of the NaV6O15 electrode.
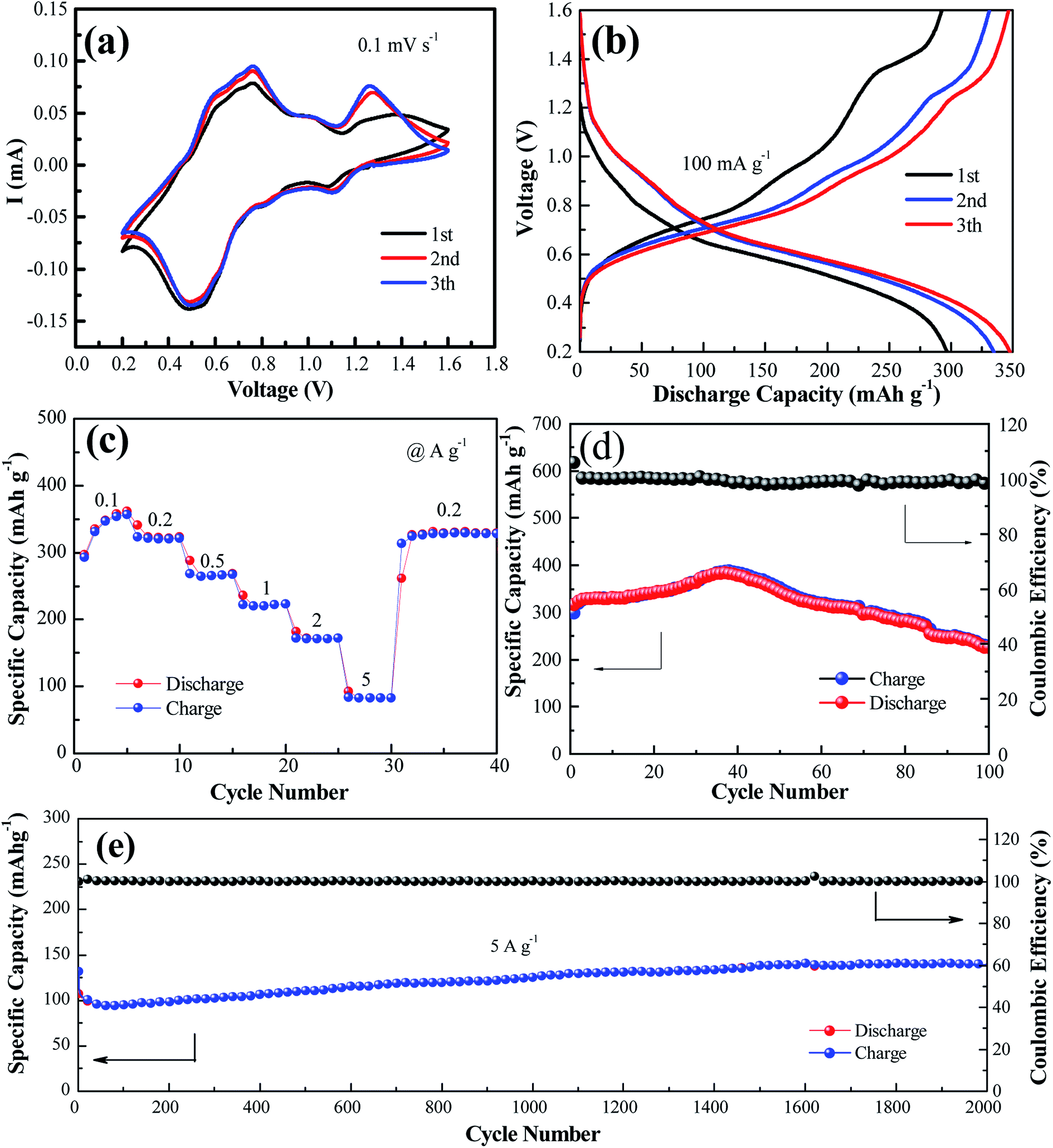 | ||
| Fig. 2 (a) CV profiles measured at a scan rate of 0.1 mV s−1. (b) Charge/discharge profiles at 0.1 A g−1 for the initial three cycles. (c) Rate performance and (d) cycling performance and coulombic efficiency at the current of 0.1 A g−1. (e) Long-term cycling performance at a high rate of 5.0 A g−1. | ||
The rate capabilities of the NaV6O15 electrodes were also investigated (Fig. 2c). The average discharge capacities were 346, 321, 265, 222, and 83.2 mA h g−1 at current densities of 0.1, 0.2, 0.5, 1.0, 2.0, and 5.0 A g−1, respectively. When the current density returned to 0.2 A g−1, a capacity of 329 mA h g−1 was recovered, indicating the good reversibility of the NaV6O15 electrode. The cycling stability of the electrode at 100 mA g−1 and 5 A g−1 was evaluated (Fig. 2d and e, respectively). The initial discharge capacity at 100 mA g−1 increased from 298 to 389 mA h g−1 (maximum capacity) after approximately 40 cycles. The reason for this is discussed later in detail. After 100 cycles, the capacity was reduced to 225 mA h g−1 with a capacity retention of 57.8% and 98% coulombic efficiency; this might explain the partial dissolution of the active material and poor electronic conductivity.30 At the higher current density of 5 A g−1, an initial discharge capacity of 132 mA h g−1 was obtained. After 2000 cycles, the NaV6O15 electrode still maintained a discharge capacity of 141 mA h g−1 with a high coulombic efficiency of ∼100% and a capacity retention of ∼107%.
To explore the mechanism of the increased capacity and stable cycle performance of the NaV6O15 electrode, ex situ XRD and XPS were carried out, respectively. To more clearly observe the phase shift or transformation of NaV6O15 during the discharge/charge process, the electrodes were washed with alcohol under ultrasonic treatment to remove the byproduct before ex situ XRD analysis, as reported previously.17,31 Fig. 3a shows the ex situ XRD spectra of the NaV6O15 electrode collected at different discharge/charge states in the first cycle at a current density of 100 mA g−1. When the electrode was discharged to 0.5 V, new peaks at 2θ = 6.5°, 12.9°, and 19.5° appeared; these peaks can be assigned to the set of (00l) reflections from a layered ZnxV2O5·nH2O phase.32,33 After discharging to 0.2 V, three obvious reflection peaks attributed to the (002), (104), and (106) planes of the initial NaV6O15 electrode were slightly shifted from 2θ = 12.1°, 29.0°, and 41.4° to higher positions of 12.3°, 29.2°, and 41.6°, respectively. These shifts might be due to the strong electrostatic interaction between intercalated Zn2+ and the tunnel-structured [V6O15]−.34,35 During the subsequent charging process, the ZnxV2O5·nH2O phase disappeared, and the above lattice planes of the NaV6O15 electrode shifted back to the low positions of 12.2°, 29.1°, and 41.5°, respectively, demonstrating the stability of the NaV6O15 structure and the high reversibility of the Zn2+ intercalation/extraction process in the NaV6O15 electrode. To provide further insight into the structural stability of the NaV6O15 electrode during long-term cycling, the structure and morphology of the electrode were respectively evaluated by XRD and SEM after 1000 cycles at a current density of 5 A g−1 (Fig. 3b and c, respectively). Compared to the initial NaV6O15 electrode, the main reflection peaks attributed to the (002), (104), and (106) planes remained, and the morphology of the NaV6O15 nanorods was well maintained, demonstrating the stable structure of the NaV6O15 electrode during long-term cycling.
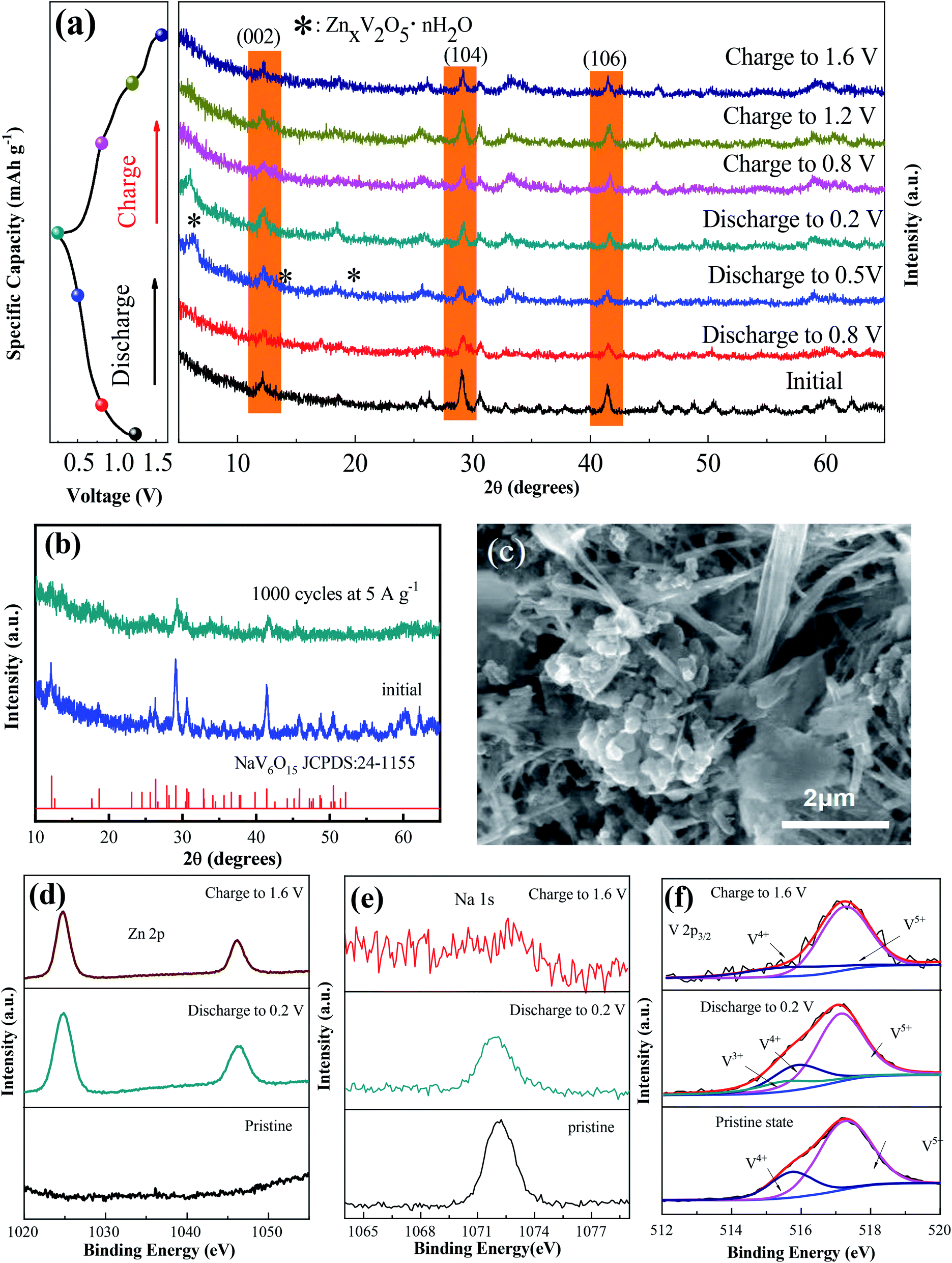 | ||
| Fig. 3 (a) Ex situ XRD patterns of NaV6O15 during the first discharge/charge process at a current density of 0.1 A g−1. (b and c) XRD pattern and SEM image of NaV6O15 after 2000 cycles at 5 A g−1. (d–f) Ex situ high-resolution XPS spectra of Zn, Na, and V in the initial, fully discharged, and charged states, respectively. | ||
Ex situ XPS was conducted to explore the evolution of the valence states of Zn, Na, and V during Zn insertion/deintercalation in the first cycle. As shown in Fig. 3d, no Zn signal was detected from the pristine electrode. After the first discharge, two strong peaks located at 1023.3 eV (Zn 2p3/2) and 1046.5 eV (Zn 2p1/2) appeared, indicating the insertion of Zn2+. When charged to 1.6 V, the intensity of the Zn2+ peak decreased but did not disappear, indicating the incomplete extraction of Zn2+. Meanwhile, the intensity of the Na 1s signal was gradually reduced after discharging to 0.2 V and charging to 1.6 V (Fig. 3e). This means that some Na+ was displaced by the insertion of Zn2+ ions during the discharge process and extracted during the charge process, which might explain the reduction/oxidation peak that appeared at approximately 1.10 V/1.25 V. Therefore, the capacity increase observed during the following cycles might be attributed to the larger proportion of extracted Na+ compared to displaced Zn2+.
In the V 2p3/2 region (Fig. 3f), the initial state of V 2p3/2 can be divided into two peaks located at 517.3 and 515.7 eV, corresponding to V5+ and V4+, respectively.36,37 With the intercalation of Zn2+ and the reduction of NaV6O15, a new peak located at 515.3 eV appeared, which can be attributed to V3+. Meanwhile, the V5+ component decreased, which could be ascribed to the reduction of V4+ to V3+ and V5+ to V4+.38 After charging to 1.6 V, the signals of V3+ and V4+ almost disappeared, also indicating the extraction of Na+ from the structure of NaV6O15. Combined with the ex situ XRD results, it can be concluded that the tunnel structure of [V6O15]− is stable despite the displacement of Zn2+ by Na+ and the extraction of Na+ during the discharge/charge process.
To further understand the electrochemical reaction kinetics of the NaV6O15 microflower-like electrode, the CV curves were measured at various scanning rates (0.1–1.0 mV s−1) to investigate the pseudocapacitive-controlled and diffusion-controlled processes (Fig. 4a). As the scanning rate increased, the peaks gradually broadened, while the curve shapes remained similar. Based on the sweep voltammetry test data, the electrochemical kinetic process can be described by the following equation:39
| i = avb | (1) |
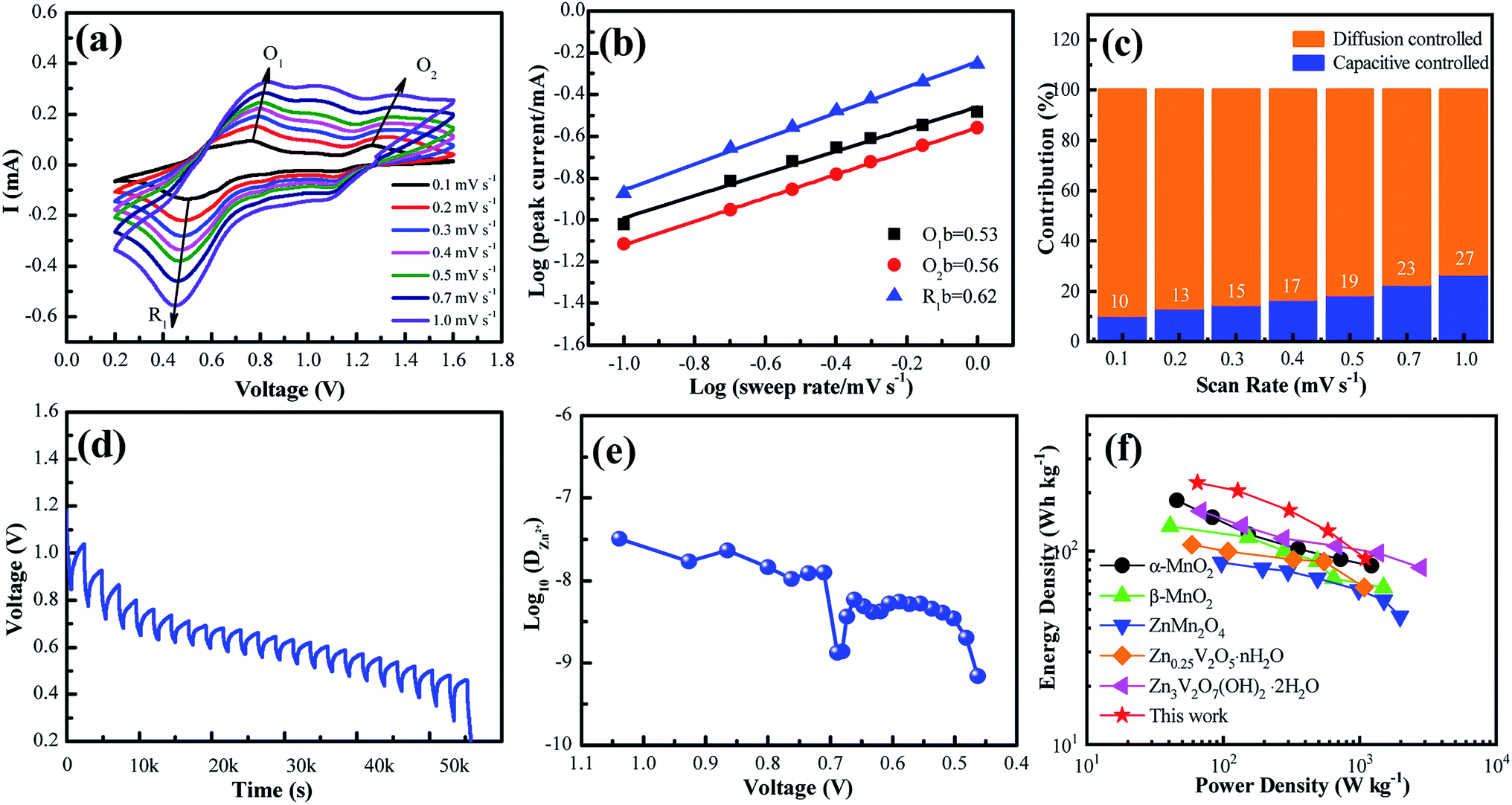 | ||
| Fig. 4 (a) CV curves at different scan rates ranging from 0.1–1.0 mV s−1. (b) Plots of log(i) vs. log(n) for specific peak currents at different scan rates. (c) Proportions of capacitive-controlled and diffusion-controlled capacities for NaV6O15 at different voltage scan rates. (d) Voltage vs. time profile for GITT measurement. (e) Calculated Zn2+ diffusion coefficient as a function of voltage. (f) Ragone plots of NVO in comparison with other reported electrodes for ZIBs. | ||
GITT was used to calculate the diffusion coefficient of zinc ions (DZn). As shown in Fig. 4d, the cell was discharged at a constant current of 0.1 A g−1 for 10 min followed by a 60 min open-circuit step to bring the voltage back to equilibrium. This process was repeated until the voltage reached 0.2 V. The DZn value of the NaV6O15 electrode (Fig. 4e) was evaluated by the following equation:42
 | (2) |
The Ragone plots of the NaV6O15 electrode are shown in Fig. 4f. The specific energy and power values were considered based on the active mass of the cathode electrode. Compared to α-MnO2,43 β-MnO2,44 ZnMn2O4,16 Zn0.25V2O5·nH2O,45 and Zn3V2O7(OH)2·nH2O,46 the NaV6O15 electrode exhibited a competitive energy density and power density (225 W h kg−1 at 65 W kg−1). When the power density was as high as 1100 W kg−1, the energy density remained at a high value of 91 W h kg−1, indicating that the NaV6O15 microflowers have a high-power capability for ZIBs.
4. Conclusions
We hydrothermally synthesized NaV6O15 microflowers with high specific capacity (∼300 mA h g−1 at 100 mA g−1) and excellent cycling stability (141 mA h g−1 maintained after 2000 cycles at 5 A g−1) in aqueous ZIBs. Ex situ XRD and XPS analyses demonstrated that the NaV6O15 electrode has a stable and reversible structure during the zinc intercalation/extraction process, even after long-term cycling. The findings show that NaV6O15 microflowers are a promising cathode material for aqueous ZIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (51674168) and the Nature Science Fund of Liaoning Province (20180550200).References
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed.
- N. T. Suen, S. F. Huang, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Q. Zhao, W. W. Huang, Z. Q. Luo, L. J. Liu, Y. Lu, Y. X. Li, L. Li, J. Y. Hu, M. Ma and J. Chen, Sci. Adv., 2018, 4, 1761 CrossRef PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636 CrossRef CAS PubMed.
- B. Scrosti, J. Hassoun and Y. K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 RSC.
- G. J. May, A. Davidson and B. Monahov, J. Energy Storage, 2018, 15, 145–157 CrossRef.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Y. G. Wang, J. Yi and Y. Y. Xia, Adv. Energy Mater., 2012, 2, 830–840 CrossRef CAS.
- P. He, M. Y. Yan, G. B. Zhang, R. M. Sun, L. N. Chen, Q. Y. An and L. Q. Mai, Adv. Energy Mater., 2017, 7, 601920 Search PubMed.
- Y. Yang, Y. Tang, G. Fang, L. Shan, J. Guo, W. Zhang, J. Zhou, C. Wang, L. Wang and S. Liang, Energy Environ. Sci., 2018, 11, 3157–3162 RSC.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- S. Zhao, B. Han, D. T. Zhang, Q. Huang, L. Xiao, L. B. Chen, D. G. Ivey, Y. D. Deng and W. F. Wei, J. Mater. Chem. A, 2018, 6, 5733–5739 RSC.
- D. Selvakumaran, A. Q. Pan, S. Q. Liang and G. Z. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 400930 Search PubMed.
- D. W. Xu, B. H. Li, C. G. Wei, Y. B. He, H. D. Du, X. D. Chu, X. Y. Qin, Q. H. Yang and F. Y. Kang, Electrochim. Acta, 2014, 133, 254–261 CrossRef CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894 CrossRef CAS PubMed.
- V. Soundharrajan, B. Sambandam, S. Kim, M. H. Alfaruqi, D. Y. Putro, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, Nano Lett., 2018, 18, 2402 CrossRef CAS PubMed.
- B. Y. Tang, J. Zhou, G. Z. Fang, S. Guo, X. Guo, L. T. Shan, Y. Tang and S. Q. Liang, J. Electrochem. Soc., 2019, 166, A480–A486 CrossRef CAS.
- Y. S. Cai, F. Liu, Z. G. Luo, G. Z. Fang, J. Zhou, A. Q. Pan and S. Q. Liang, Energy Storage Mater., 2018, 13, 168–174 CrossRef.
- B. Y. Tang, G. Z. Fang, J. Zhou, L. B. Wang, Y. P. Lei, C. Wang, T. Q. Lin, Y. Tang and S. Q. Liang, Nano Energy, 2018, 51, 579–587 CrossRef CAS.
- X. Guo, G. Z. Fang, W. Y. Zhang, J. Zhou, L. T. Shan, L. B. Wang, C. Wang, T. Q. Lin, Y. Tang and S. Q. Liang, Adv. Energy Mater., 2018, 1801819 CrossRef.
- P. He, G. B. Zhang, X. B. Liao, M. Y. Yan, X. Xu, Q. Y. An, J. Liu and L. Q. Mai, Adv. Energy Mater., 2018, 8, 1702463 CrossRef.
- S. Islam, M. H. Alfaruqi, B. Sambandam, D. Y. Putro, S. Kim, J. Jo, S. Kim, V. Mathew and J. Kim, Chem. Commun., 2019, 55, 3793–3796 RSC.
- F. Hu, W. Jiang, Y. D. Dong, X. Y. Lai, L. Xiao and X. Wu, RSC Adv., 2017, 7, 29481–29488 RSC.
- H. G. Bachmann and W. H. Barnes, Can. Mineral., 1962, 7, 219–235 CAS.
- H. M. Liu, Y. G. Wang, L. Liang, K. X. Wang, E. Hosono and H. S. Zhou, J. Mater. Chem., 2009, 19, 7885 RSC.
- Y. F. Dong, S. Li, K. N. Zhao, C. H. Han, W. Chen, B. L. Wang, L. Wang, B. A. Xu, Q. L. Wei, L. Zhang, X. Xu and L. Q. Mai, Energy Environ. Sci., 2015, 8, 1267–1275 RSC.
- H. N. He, G. H. Jin, H. Y. Wang, X. B. Huang, Z. H. Chen, D. Sun and Y. G. Tang, J. Mater. Chem. A, 2014, 2, 3563–3570 RSC.
- F. Hu, D. Xie, D. P. Zhao, G. H. Song and K. Zhu, J. Energy Chem., 2019, 38, 185–194 CrossRef.
- W. Y. Zhang, S. Q. Liang, G. Z. Fang, Y. Q. Yang and J. Zhou, Nano-Micro Lett., 2019, 11, 69 CrossRef CAS.
- T. Y. Wei, Q. Li, G. Z. Yang and C. X. Wang, Adv. Energy Mater., 2019, 1901480 CrossRef.
- R. Y. Wang, B. Shyam, K. H. Stone, J. N. Weker, M. Pasta, H. W. Lee, M. F. Toney and Y. Cui, Adv. Energy Mater., 2015, 5, 1401869 CrossRef.
- C. Xia, J. Guo, Y. J. Lei, H. F. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2017, 30, 1705580 CrossRef PubMed.
- P. Hu, T. Zhu, X. P. Wang, X. J. Wei, M. Y. Yan, J. T. Li, W. Luo, W. Yang, W. C. Zhang, L. Zhou, Z. Q. Zhou and L. Q. Mai, Nano Lett., 2018, 18, 1758–1763 CrossRef CAS PubMed.
- M. H. Alfaruqi, V. Mathew, J. Song, S. Kim, S. Islam, D. T. Pham, J. Jo, S. Kim, J. P. Baboo, Z. Xiu, K. S. Lee, Y. K. Sun and J. Kim, Chem. Mater., 2017, 29, 1684–1694 CrossRef CAS.
- Y. F. Zhang, C. X. Chen, W. B. Wu, F. Niu, X. H. Liu, X. H. Liu, Y. L. Zhong, Y. L. Cao, X. Liu and C. Huang, Ceram. Int., 2013, 39, 129–141 CrossRef CAS.
- W. D. Zhang, G. B. Xu, L. W. Yang and J. W. Ding, RSC Adv., 2016, 6, 5161–5168 RSC.
- Q. Q. Chen, Q. Xia, Y. X. Xu, P. F. Wang and Q. Q. Tan, Mater. Lett., 2019, 247, 178–181 CrossRef CAS.
- S. Li, M. H. Chen, G. Z. Fang, L. T. Shan, X. X. Cao, J. W. Huang, S. Q. Liang and J. Zhou, J. Alloys Compd., 2019, 801, 82–89 CrossRef CAS.
- B. Wu, G. B. Zhang, M. Y. Yan, T. F. Xiong, P. He, L. He, X. Xu and L. Q. Mai, Small, 2018, 14, 1703850 CrossRef PubMed.
- J. W. Hao, J. Mou, J. W. Zhang, L. B. Dong, W. B. Liu, C. J. Xu and F. Y. Kang, Electrochim. Acta, 2018, 259, 170–178 CrossRef CAS.
- J. Chen, S. Li, V. Kumar and P. S. Lee, Adv. Energy Mater., 2017, 7, 1700180 CrossRef.
- H. L. Pan, Y. Y. Shao, P. F. Yan, Y. W. Cheng, K. S. Han, Z. M. Nie, C. M. Wang, J. H. Yang, X. L. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- G. Z. Yang, T. Y. Wei and C. X. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 35079–35089 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |