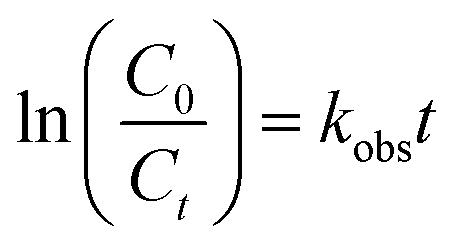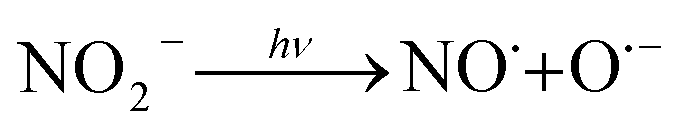DOI:
10.1039/D0RA00363H
(Paper)
RSC Adv., 2020,
10, 9907-9916
Degradation kinetics and mechanism of diclofenac by UV/peracetic acid
Received
13th January 2020
, Accepted 29th February 2020
First published on 9th March 2020
Abstract
In this work, the degradation kinetics and mechanism of diclofenac (DCF) by UV/peracetic acid (PAA) was investigated. The effects of pH, PAA dose and common water components such as inorganic ions and dissolved organic matter (DOM) on DCF degradation by UV/PAA were also evaluated. It was observed that the addition of PAA promoted the photodegradation of DCF due to the generation of reactive radicals in the photolysis of PAA, which was also confirmed by the radical scavenging experiment. The best degradation efficiency of DCF was obtained at pH 8.5. The removal of DCF was enhanced gradually with increasing PAA dose. Since NO3− is a photosensitive substance which can generate HO˙ under UV irradiation, its existence promoted the degradation of DCF. The presence of CO32− could slightly improve DCF degradation, which might be due to the role of generated carbonate radicals. Cl−, SO42− and Fe3+ had little effect on DCF removal, while Cu2+ could enhance DCF degradation because of its catalytic ability for PAA decomposition. An inhibition effect on DCF removal was observed in the presence of DOM, and it was more obvious in higher concentration of DOM. The elimination of total organic carbon (TOC) was low. According to the twelve reaction products detected in the UV/PAA system, the probable transformation mechanism of DCF was proposed exhibiting eight reaction pathways, i.e., hydroxylation, decarboxylation, formylation, dehydrogenation, dechlorination–hydrogenation, dechlorination–cyclization, dechlorination–hydroxylation and amidation. This study indicates that UV/PAA is a promising method for DCF removal from contaminated water.
1. Introduction
Pharmaceuticals and personal care products (PPCPs) as emerging contaminants have received more and more attention in recent years. Due to their widespread use and low human metabolic capability, they have been detected frequently in wastewater treatment plants (WWTPs) and natural water environment with concentrations of ng L−1 to μg L−1.1–4 Studies have shown that WWTPs are important sources of PPCPs in natural water, because most of them cannot be completely removed by traditional wastewater treatment processes.5–7 Diclofenac (DCF), a non-steroidal anti-inflammatory drug (NSAID), is extensively utilized to treat rheumatism or arthritis, relieve fever and ease pain.8,9 Since DCF cannot be effectively removed in WWTPs due to its stable chemical structure, it is detected widely in surface water, groundwater and even drinking water.10–12 Although DCF has a low acute toxicity, its occurrence can not only induce the production of drug-resistant bacteria but also have adverse effects on aquatic organisms, thus threatening human health and the ecosystem.13,14 Therefore, developing a few new effective technologies to degrade DCF from the contaminated water is very essential.
Advanced oxidation processes (AOPs) are considered to be promising methods to remove refractory organic pollutants, which are mainly attributed to the role of highly active radicals like hydroxyl radical (HO˙). Until now, AOPs have been widely used in the treatment of refractory organic wastewater with high concentration and emerging contaminants, which can improve the biodegradability of pollutants or even completely mineralize them. A number of AOPs such as UV/H2O2,15 photocatalysis,16,17 photo-Fenton reaction,18 Fenton-like reaction,19 electrochemical oxidation,20 transition metal catalyzed persulfate or peroxymonosulfate,21,22 etc. have been successfully applied to degrade DCF or other pharmaceuticals in wastewater.
The oxidation potential of PAA is 1.96 V, higher than H2O2 (1.78 V) and aqueous chlorine (1.48 V) but slightly lower than that of persulfate (2.01 V).23–25 More importantly, PAA exhibits an excellent disinfection activity without mutagenic and carcinogenic disinfection byproducts during its use, and thus it is widely used for medical, food and wastewater disinfection.26,27 Additionally, PAA can be activated to generate many kinds of radicals in a wide range of pH.28 Therefore, PAA may be a promising oxidant in AOPs. However, PAA has not been extensively studied for the removal of PPCPs in wastewater so far. Similar to H2O2, PAA can be decomposed to produce HO˙ through different activation methods like UV irradiation, transition metal catalysis and so on.29,30 Among them, UV activated PAA (UV/PAA) is considered to be a promising technology for the removal of organic pollutants based on two facts that: (1) UV is now widely used for wastewater disinfection in WWTPs, and (2) it will not bring secondary pollution. Currently, the information on the destruction of organic contaminants by UV/PAA is very limited. Daswat et al.31 found that the addition of PAA could improve the degradation of chlorophenol compared with UV alone. Cai et al.32 reported that six PPCPs could hardly be oxidized by PAA but be removed quickly by UV/PAA. Chen et al.33 studied the influence of water components on naproxen degradation in UV/PAA system. Rizzo et al.34 found that UV/PAA was more efficient than sunlight/PAA for the degradation of emerging contaminants in the tertiary treatment of urban wastewater. To date, very limited literature is available on DCF removal by UV/PAA.
In this work, the degradation of DCF by UV/PAA was systematically studied. Firstly, the contribution of UV photolysis and reactive radicals to DCF removal in UV/PAA system was investigated. Then, the effects of various factors including initial pH, PAA dose and common water components like inorganic ions and dissolved organic matter (DOM) on DCF degradation were explored. Finally, the degradation products of DCF were detected and identified, and probable transformation mechanism was subsequently proposed.
2. Materials and methods
2.1. Materials
Peracetic acid (15%, w/w) was purchased from Sinopharm Chemical Reagent Co. Ltd. (China). Diclofenac sodium (>99%) and fulvic acid (FA) were obtained from Aladdin (China). Methanol and acetic acid were HPLC grade. Sodium chloride (NaCl), sodium sulfate (Na2SO4), potassium nitrate (KNO3), sodium carbonate (Na2CO3), ferric chloride hexahydrate (FeCl3·6H2O), cupric sulfate pentahydrate (CuSO4·5H2O), monosodium phosphate dihydrate (NaH2PO4·2H2O), disodium hydrogen phosphate heptahydrate (Na2HPO4·7H2O), sodium hydroxide (NaOH), sulphuric acid (H2SO4), potassium iodide (KI), manganese sulfate monohydrate (MnSO4·H2O), ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O), soluble starch, potassium permanganate (KMnO4) and sodium hyposulfite pentahydrate (Na2S2O3·5H2O) were all analytical reagent grade and purchased from Chengdu Kelong Chemical Reagent Co. Ltd. (China). All the chemicals were used as received without further purification. Ultrapure water (18 MΩ cm) was used to prepare aqueous solutions.
2.2. Analysis
The quantification of DCF was measured by a high performance liquid chromatograph (HPLC, Waters 2695, USA), and its degradation products were determined by an ultra performance liquid chromatograph coupled with a quadrupole time-of-flight mass spectrometer (UPLC-QTOF/MS, Waters Xevo G2-XS QT, USA). The detailed analytical processes can be obtained in our previous researches.35,36 The UV light intensity was measured by an ultraviolet radiometer (UVC-254, Lutron, Taiwan). The total organic carbon (TOC) value was measured by a TOC analyzer (VCSH-ASI, Shimadzu, Japan). The absorption spectrum of DCF was measured by a UV-Vis spectrometer (8452A, Hewlett Packard). The concentration of PAA was determined using a titration method and the detailed procedures referred to the national standard of peracetic acid water solution (GB/T 19104-2008, China). The pH of solution was measured by a pH meter (PHS-3C, Leici, China). The degradation of DCF by UV/PAA followed pseudo first-order kinetic model based on data fitting degree and the observed rate constants (kobs) for DCF were calculated by the eqn (1):| |
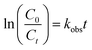 | (1) |
where C0 and Ct were the concentration of DCF at initial time and time t, respectively.
2.3. Photochemical experiments
Experiments were performed in a self-made collimated beam device equipped with two low pressure UV lamps (15 W) which emitted nearly monochromatic light at 254 nm. The average UV intensity was measured to be 2.1 mW cm−2. A glass Petri dish was used as the reactor that was placed at a magnetic stirrer for better mixing. The pH of solution was adjusted by 5 mM phosphate buffer to the desired value. For DCF degradation experiments, the initial concentration of DCF and PAA were 1 and 50 μM, respectively. At each given time, 0.5 mL sample was taken out and quenched immediately with 0.5 mL 5 mM Na2S2O3. For DCF transformation mechanism investigation, the initial concentration of DCF was increased to 10 μM. All experiments were repeated three times to minimize error.
3. Results and discussion
3.1. Degradation of DCF by UV/PAA
DCF could hardly be oxidized by PAA alone within 15 min, while it could be degraded by direct UV photolysis and its degradation rate increased when PAA was added into the reaction solution, as depicted in Fig. 1a. The promotion might be attributed to the production of HO˙ which came from the photolysis of PAA, as shown in eqn (2).37,38 In order to prove the effect of HO˙, methanol (MeOH) and tert-butyl alcohol (TBA), two widely used scavengers for HO˙ (kMeOH/HO˙ = 9.7 × 108 M−1 s−1, kTBA/HO˙ = 6 × 108 M−1 s−1),39,40 were added into the UV/PAA system, respectively. The radical oxidation of DCF in UV/PAA system was almost inhibited by adding MeOH, while it was partly inhibited by adding TBA. This result showed that in addition to HO˙, other radicals such as CH3COO˙ probably enhanced the removal of DCF and MeOH might be also a scavenger for CH3COO˙. The degradation of DCF in UV, UV/PAA and UV/PAA/TBA systems all followed the pseudo first-order kinetic model, and their observed rate constants (kobs) were 0.0655, 0.1161, and 0.0789 min−1, respectively. The degradation of DCF by UV/PAA was probably attributed to direct photolysis, HO˙ oxidation and other radicals oxidation. The contribution of each process was estimated based on the kobs obtained from DCF degradation by different systems (UV, UV/PAA, and UV/PAA/TBA), as shown in eqn (3)–(6). The result of the above calculation showed that the kdirect photolysis, kHO˙ oxidation and kother radicals oxidation of DCF were 0.0655, 0.0372, and 0.0134 min−1, respectively. Therefore, the contribution of UV photolysis, HO˙ oxidation and other radicals oxidation to DCF removal in UV/PAA system was 56%, 32% and 12%, respectively. To assess the superior performance of UV/PAA for DCF degradation, two control experiments were carried out, including UV/persulfate and UV/H2O2, to remove DCF at the same conditions, as shown in Fig. 1b. The degradation of DCF by UV/persulfate and UV/H2O2 also followed the pseudo first-order kinetic model. The result showed that the kobs of different systems followed the order kUV/PAA (0.1161 min−1) > kUV/persulfate (0.1071 min−1) > kUV/H2O2 (0.0857 min−1). Therefore, UV/PAA could be regarded as a promising process to degrade DCF.| |
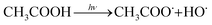 | (2) |
| | |
kUV/PAA = kdirect photolysis + kHO˙ oxidation + kother radicals oxidation
| (3) |
| | |
kdirect photolysis = kUV
| (4) |
| | |
kHO˙ oxidation = kUV/PAA − kUV/PAA/TBA
| (5) |
| | |
kother radicals oxidation = kUV/PAA/TBA − kUV
| (6) |
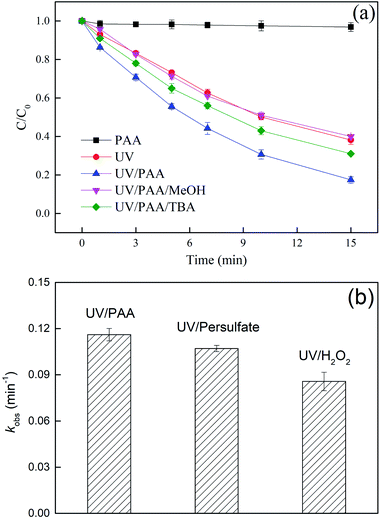 |
| | Fig. 1 Removal of DCF in different reaction systems (a) and the observed rate constants of DCF in UV/PAA, UV/persulfate and UV/H2O2 system (b). Experimental conditions: [DCF]0 = 1 μM, [PAA]0 = 50 μM, [persulfate]0 = 50 μM, [H2O2]0 = 50 μM, [MeOH]0 = 5 mM, [TBA]0 = 5 mM, 5 mM phosphate buffer at pH 7.0. | |
3.2. Effect of initial pH
As shown in Fig. 2a, no significant effect of pH was observed in the only UV system, indicating that although the deprotonation of DCF could occur when pH changed, the degradation of DCF by UV had nothing to do with whether DCF was deprotonation, which was due to the same UV absorbance for DCF under 254 nm at pH 3–11, as shown in Fig. 2b. However, the kobs of DCF in UV system was only 0.065 min−1. The optimal pH for DCF degradation in UV/PAA system was at 8.5, as shown in Fig. 2c. The kobs increased gradually from 0.096 to 0.1902 min−1 in the range of pH 3.0–8.5, but it decreased to 0.1536 min−1 at pH 11.0. Since the pKa value of PAA is 8.2, it was existed in two different forms at different pH, i.e., protonated form (PAA0) at acidic or neutral condition and deprotonated form (PAA−) at alkaline pH. Furthermore the molar absorption coefficients (ε) of PAA0 and PAA− at 254 nm were 8.0 M−1 cm−1 and 41.6 M−1 cm−1, respectively.32 PAA− had a faster photolysis rate than PAA0 because of its higher UV absorption at 254 nm, leading to more formation of HO˙ at alkaline pH condition. The effect of pH on photolysis of PAA within 15 min was shown in Fig. 2d. The photolysis of PAA in alkaline condition was higher than acidic or neutral condition, which was consist with the study of Cai et al.32 At pH 11, possible explanations for the decrease of DCF removal rate included that: (1) the concentration of OH− was 1000 times higher than that of DCF, which could compete with DCF for HO˙ (kOH−/HO˙ = 1.2 × 1010 M−1 s−1),41 and (2) the redox potential of HO˙ is lower at basic pH than acid or neutral pH according to Nernst equation.42 Combined with the above reasons, the kobs was found to be the highest at pH 8.5.
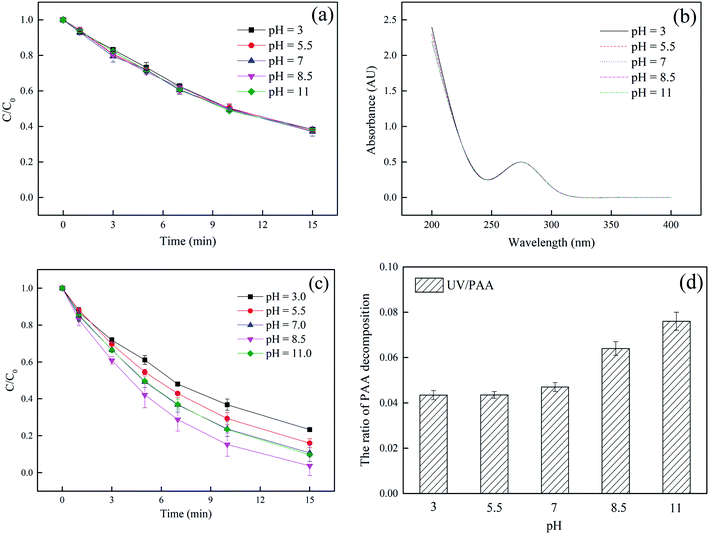 |
| | Fig. 2 Effect of initial pH on DCF removal by UV (a), absorption spectrum of DCF (b), DCF removal by UV/PAA (c) and photolysis of PAA (d). Experimental conditions: [DCF]0 = 1 μM, [PAA]0 = 50 μM, 5 mM phosphate buffer. | |
3.3. Effect of PAA concentration
The oxidant dose is a significant parameter to evaluate whether the UV/PAA is economical and efficient for DCF degradation. The effect of initial PAA concentration on kobs is shown in Fig. 3. The kobs enhanced gradually from 0.0967 to 0.182 min−1 with increasing PAA concentration from 10 to 150 μM. When PAA concentration was below 50 μM, the kobs increased linearly with the increase of PAA dose, which was probably attributed to the generation of more HO˙ and CH3COO˙. Although kobs was still increased in higher PAA concentration, its growth rate slowed down, which probably resulted from the scavenging effect of PAA on HO˙, because excessive PAA could compete with DCF for HO˙ (kPAA/HO˙ = (9.33 ± 0.3) × 108 M−1 s−1).32 In addition, more PAA molecules in system probably absorbed more photons which inhibited the direct photolysis of DCF.
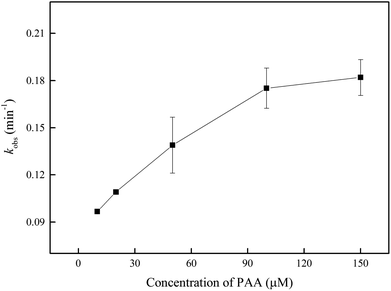 |
| | Fig. 3 Effect of PAA concentration on kobs by UV/PAA. Experimental conditions: [DCF]0 = 1 μM, 5 mM phosphate buffer at pH 7.0. | |
3.4. Effect of common water components
The composition of natural water is complex, which consists of various components such as inorganic ions and DOM. In order to apply UV/PAA to remove DCF from natural water, it is necessary to investigate their effects on DCF removal.
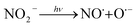
3.4.1 Effect of Cl−, SO42− and NO3−. The effect of Cl− on DCF degradation is shown in Fig. 4a. Its existence had almost no influence on DCF removal with the changed concentration from 1 to 10 mM. Although Cl− could react with HO˙ to form ClOH˙− which could further transform to Cl˙ and Cl2˙−, as presented in eqn (7)–(9), eqn (8) was difficult to occur under neutral condition. Therefore, the formed ClOH˙− might mainly undergo self-decomposition to regenerate HO˙ in current condition, as shown in eqn (10).43 The presence of SO42− showed no apparent influence on the degradation of DCF as shown in Fig. 4b, because it could not react with HO˙.44 The NO3−, as a well-known photosensitizer, probably had two effects on DCF removal in UV/PAA system. On one hand, NO3− could be excited to generate HO˙ under UV, which promoted DCF removal, as presented in eqn (11)–(14).45 On the other hand, NO3− also probably absorbed UV photons which inhibited the direct photolysis of DCF. Moreover, the NO2− produced during the NO3− photolysis process was a strong HO˙ scavenger (kNO2−/HO˙ = 1.0 × 1010 M−1 s−1),39 thus it could also compete with DCF for HO˙. As shown in Fig. 4c, the removal efficiency of DCF was slightly improved by NO3−, especially at higher NO3− concentration, indicating that the positive effect of NO3− on DCF removal by UV/PPA was stronger than negative effects. Furthermore, further study was needed to estimate if mutagenic and carcinogenic nitrated byproducts were formed in the presence of nitrate.| | |
Cl− + HO˙ → ClOH˙− k = (4.3 ± 0.4) × 109 M−1 s−1
| (7) |
| | |
ClOH˙− + H+ → Cl˙ + H2O k = (2.1 ± 0.7) × 1010 M−1 s−1
| (8) |
| | |
Cl˙ + Cl− → Cl2˙− k = 2.1 × 1010 M−1 s−1
| (9) |
| | |
ClOH˙− → Cl− + HO˙ k = (6.1 ± 0.8) × 109 M−1 s−1
| (10) |
| |
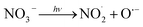 | (11) |
| |
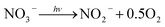 | (12) |
| |
 | (13) |
| | |
O˙− + H2O → HO˙ + OH−
| (14) |
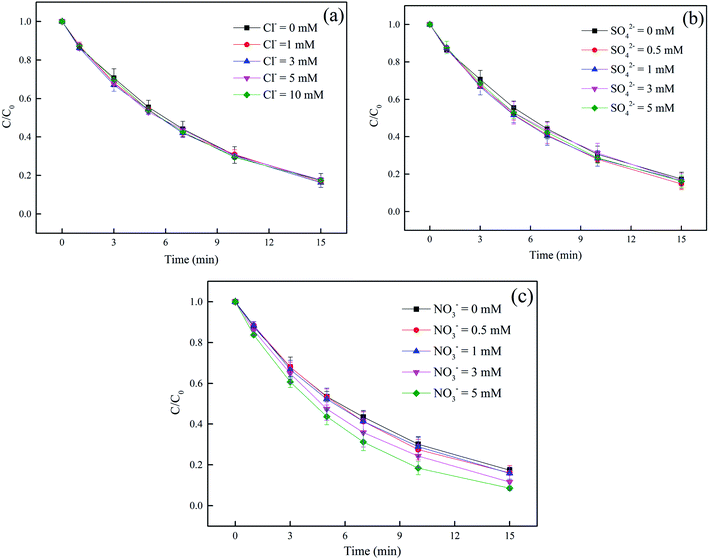 |
| | Fig. 4 Effect of Cl− (a), SO42− (b) and NO3− (c) on DCF removal by UV/PAA. Experimental conditions: [DCF]0 = 1 μM, [PAA]0 = 50 μM, 5 mM phosphate buffer at pH 7.0. | |
3.4.2 Effect of CO32−. Some studies reported that CO32− could quench HO˙ (kCO32−/HO˙ = 3.9 × 108 M−1 s−1) as presented in eqn (15), leading to the inhibition on the degradation of organic pollutants in HO˙ oxidation system.46,47 However, no such inhibition was observed in our study. Conversely, the presence of CO32− could promote the degradation of DCF by UV/PAA, as shown in Fig. 5. When the concentration of CO32− increased from 0.5 to 5 mM, the pH of the solution enhanced from 9.9 to 10.8. To eliminate the influence of pH on DCF degradation, its removal by UV and UV/PAA in phosphate buffer at pH 9.9, 10.3, 10.6 and 10.8 was used as a control, respectively. A similar degradation trend of DCF in UV system under different pH conditions (pH 9.9, 10.3, 10.6 and 10.8) was observed, which was consist with the previous results. Compared with the UV/PAA/buffer system, the degradation of DCF was faster in UV/PAA/CO32− system, which was probably due to the generation of carbonate radical (CO3˙−) through the reaction of CO32− with HO˙. Since CO3˙− is an electrophilic single-electron oxidant with the oxidation potential of 1.78 V, it can react with electron-rich compounds, such as phenols, anilines, and sulfur-containing organic compounds, through electron transfer or hydrogen extraction.48–50 Due to the presence of aniline group in DCF structure, it might react with CO3˙− with the second-order rate constant of 2.7 × 107 M−1 s−1,51 which could result in the enhancement on DCF degradation in the presence of CO32−. Liu et al.52 also found CO3˙− played a significant role on oxytetracycline removal in UV/H2O2/CO32− system.| | |
HO˙ + CO32− → CO3˙− + OH−
| (15) |
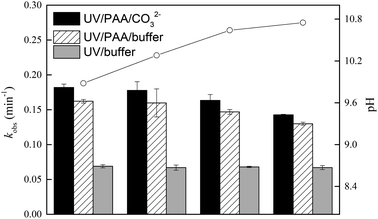 |
| | Fig. 5 The observed rate constants of DCF in UV/PAA system with the addition of CO32−. Experimental conditions: [DCF]0 = 1 μM, [PAA]0 = 50 μM, no phosphate buffer. Degradation of DCF by UV and UV/PAA in phosphate buffer (5 mM) at pH 9.9, 10.3, 10.6 and 10.8 was used as a control, respectively. | |
3.4.3 Effect of Fe3+ and Cu2+. It was found that the removal of DCF was hardly affected by Fe3+ in this study, as shown in Fig. 6a. Although Fe3+ is a catalyst for Fenton-like reaction and FeOH2+ can also produce HO˙ under UV radiation, these reactions usually occur in acid condition.53,54 In current condition (5 mM phosphate buffer at pH 7.0), Fe3+ was mainly existed in the form of Fe(OH)3 or FePO4,55 and consequently extra HO˙ could hardly be generated by the above pathways. Unlike Fe3+, the presence of Cu2+ significantly promoted DCF degradation and the higher the concentration of Cu2+ was, the better the promotion effect was, as shown in Fig. 6b. Cu2+ is also a kind of catalyst for Fenton-like reaction, which is adaptable to a wide range of pH, especially in near-neutral condition.56,57 As a result, the improvement effect might be attributed to the formation of extra HO˙ and CH3COO˙ in the decomposition of PAA catalyzed by Cu2+, as shown in eqn (16).57| |
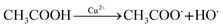 | (16) |
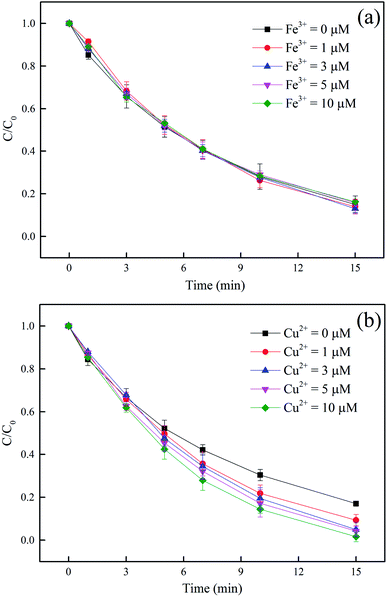 |
| | Fig. 6 Effect of Cu2+ (a) and Fe3+ (b) on DCF removal by UV/PAA. Experimental conditions: [DCF]0 = 1 μM, [PAA]0 = 50 μM, 5 mM phosphate buffer at pH 7.0. | |
3.4.4 Effect of DOM. In this work, fulvic acid was used as the representative of DOM. As shown in Fig. 7, the removal of DCF was inhibited in the presence of DOM and it was more obvious in higher concentration of DOM. There are two probable reasons to explain this phenomenon. Firstly, DOM is a kind of photosensitive substance that could compete with DCF and PAA for UV radiation, which would affect the direct photolysis of DCF and the formation of reactive radicals produced from the decomposition of PAA.58 In addition, DOM is a well-known HO˙ scavenger (kDOM/HO˙ = 2.23 × 108 L (mol C)−1 s−1),59 and it might compete with DCF for HO˙. Zhang et al.60 also found that the existence of DOM had a negative effect on azathioprine removal in UV and UV/H2O2 systems.
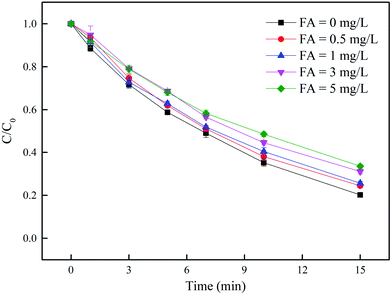 |
| | Fig. 7 Effect of DOM on DCF removal by UV/PAA. Experimental conditions: [DCF]0 = 1 μM, [PAA]0 = 50 μM, 5 mM phosphate buffer at pH 7.0. | |
3.5. The elimination of TOC
As shown in Fig. 8, DCF was completely removed within 50 min. However, only 8.6% TOC was eliminated. There were two possible reasons for the low removal of TOC: (1) the aromatic ring of DCF might not be easily destroyed by UV/PAA, so that it could not be split into smaller molecular weight hydrocarbons or finally be mineralized to CO2 and H2O; (2) PAA itself was an organic peroxide and its addition also partially contributed to TOC of the reaction system. PAA could not be completely mineralized during DCF removal in UV/PPA system. Although DCF and PAA were not completely mineralized to CO2 and H2O during DCF treatment by UV/PAA, they were probably split to smaller or non-toxic molecules, which could be further treated by the combination of biodegradation process. This process was mainly used as a pro-treatment process. Further study is needed to calculate the respective contribution of the transformation products of DCF and PAA to TOC in UV/PAA system.
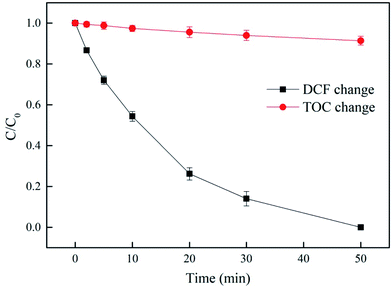 |
| | Fig. 8 The removal of TOC in UV/PAA system. Experimental conditions: [DCF]0 = 10 μM, [PAA]0 = 0.5 mM, no phosphate buffer. | |
3.6. Identification of reaction products and degradation pathways
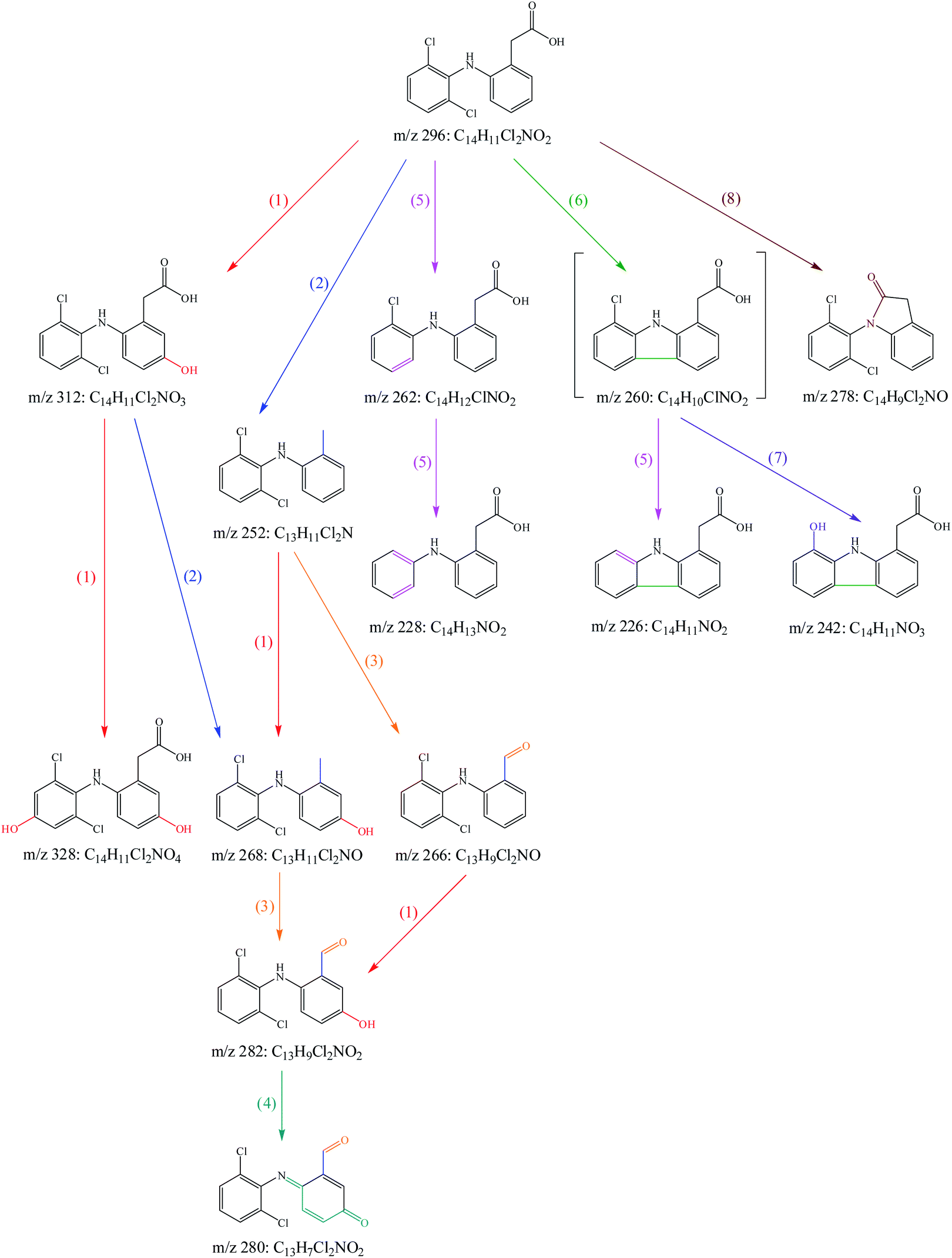
To investigate the transformation mechanism of DCF in UV/PAA system, its degradation products were detected by UPLC-QTOF/MS. Twelve transformation products of DCF were detected and identified in this system. Since direct photolysis might contribute significantly to DCF degradation in UV/PAA system, some products were probably derived from direct UV photolysis of DCF. Among these twelve transformation products, part of them were also detected by other researchers61–65 in UV photolysis of DCF, as shown in Table 1. According to these detected degradation products, the potential transformation mechanism of DCF by UV/PAA was proposed exhibiting eight different reaction pathways, i.e., hydroxylation, decarboxylation, formylation, dehydrogenation, dechlorination–hydrogenation, dechlorination–cyclization, dechlorination–hydroxylation, and amidation, as presented in Fig. 9.
Table 1 Transformation products of DCF by UV/PAA or UV
| Serial number |
Mass to charge ratio (m/z) |
The detected condition |
The detected condition61–65 |
| 1 |
328 |
UV/PAA |
— |
| 2 |
312 |
UV/PAA |
— |
| 3 |
282 |
UV/PAA |
— |
| 4 |
280 |
UV/PAA |
— |
| 5 |
278 |
UV/PAA |
— |
| 6 |
268 |
UV/PAA |
— |
| 7 |
266 |
UV/PAA |
UV |
| 8 |
262 |
UV/PAA |
UV |
| 9 |
252 |
UV/PAA |
UV |
| 10 |
242 |
UV/PAA |
UV |
| 11 |
228 |
UV/PAA |
UV |
| 12 |
226 |
UV/PAA |
UV |
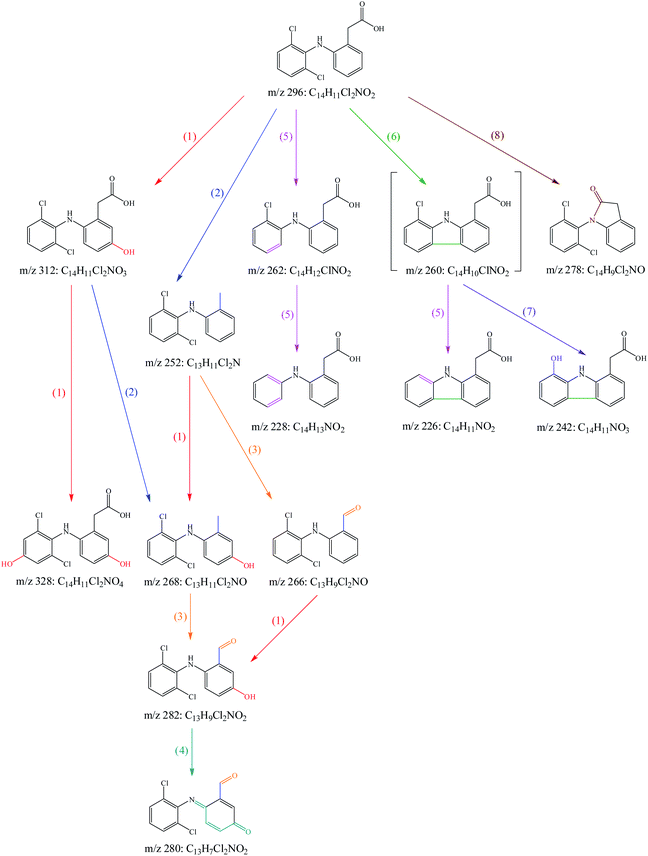 |
| | Fig. 9 Probable transformation pathways of DCF by UV/PAA: (1) hydroxylation, (2) decarboxylation, (3) formylation, (4) dehydrogenation, (5) dechlorination–hydrogenation, (6) dechlorination–cyclization, (7) dechlorination–hydroxylation, (8) amidation. Experimental conditions: [DCF]0 = 10 μM, [PAA]0 = 0.5 mM, no phosphate buffer. | |
(1) Hydroxylation. Hydroxylation was caused by the addition of HO˙ to aromatic ring of DCF through electron transfer due to its electrophilic nature, leading to the generation of mono-hydroxylation product m/z 312 and di-hydroxylation product m/z 328. Madhavan et al.66 also detected hydroxylation product m/z 312 in ultrasound assisted photocatalytic degradation of DCF in water.
(2) Decarboxylation. Decarboxylation referred to the removal of carboxyl group from acetic acid group in DCF structure, resulting in the formation of product m/z 252. It is well known that aromatic carboxylic acids are more prone to decarboxylate than saturated carboxylic acids. Under UV radiation, the molecule of DCF was in excitation state and then the C–C bond in acetic acid group was broken by HO˙ attack. The generated product m/z 252 could further be hydroxylated to produce the product m/z 268 which might also be generated from hydroxylation product m/z 312 through decarboxylation.
(3) Formylation. Formylation was occurred during the decomposition of the products of DCF. The methyl group in product m/z 252 could be oxidized to produce formyl group under HO˙ attack, leading to the production of formylation product m/z 266. It might undergo hydroxylation to generate product m/z 282 that could also be produced from the generated m/z 268 through this pathway. Similar formylation products were also detected in the degradation of DCF by ferrate.67
(4) Dehydrogenation. The structure of formed product m/z 282 contained not only phenol group but also aniline group, and both of them were electron-donating groups.68 Therefore, these sites were readily attacked by HO˙, yielding quinone-imine product m/z 280. Chong et al.8 detected similar product in DCF removal by FeCeOx catalyzed H2O2. Bedner and MacCrehan69 found that two quinone-imine byproducts in transformation of acetaminophen by chlorination were toxic compounds. However, further study was needed to estimate if the quinone-imine product (m/z 280) was also a toxic byproduct.
(5) Dechlorination–hydrogenation. Dechlorination–hydrogenation referred to the replacement a chlorine atom on the benzene ring with a hydrogen atom in DCF structure. During UV irradiation, the C–Cl bond was broken, resulting in the formation of carbocation through the transfer of an electron from carbon atom to chlorine radical, and subsequently it reacted with water molecule to generate dechlorination–hydrogenation products m/z 262 and 228.70 Yu et al.71 reported that dechlorination–hydrogenation was also an important pathway of DCF degradation by pulse radiolysis and γ-radiolysis.
(6) Dechlorination–cyclization. Dechlorination–cyclization was similar to dechlorination–hydrogenation except that the former underwent intramolecular electron transfer and subsequently cyclized producing the product m/z 260. Although this product was not detected in this work, it was found in others' studies where DCF was degraded by direct photolysis or photocatalysis.63,65,72,73 Product m/z 226 was detected in this study, which might be formed from m/z 260 through dechlorination–hydrogenation.
(7) Dechlorination–hydroxylation. Dechlorination–hydroxylation was similar to dechlorination–hydrogenation mechanism except that chlorine atom was replaced by hydroxyl group possibly from water molecule.70 It was probably occurred from the intermediate m/z 260 to form product m/z 242. Lekkerkerker-Teunissen et al.74 also detected this product in the transformation of DCF by UV/H2O2.
(8) Amidation. Amidation referred to the reaction of secondary amine with carboxylic acid to form carboxylic amine and then dehydrating to obtain the corresponding amide, which was probably occurred in the intramolecular structure of DCF, resulting in the production of amidation product m/z 278. Salaeh et al.75 also found this product in the photocatalytic degradation of DCF under simulated sunlight.
4. Conclusions
This study systematically investigated the degradation kinetics and transformation mechanism of DCF by UV/PAA. It was found that adding PAA into reaction solution could enhance DCF degradation in UV system, which was proved that the promotion effect was due to the role of HO˙ and CH3COO˙ produced by the photolysis of PAA. In the pH range of 3–11, DCF had the best degradation efficiency at pH 8.5. When PAA dose increased, the observed degradation rate constant was enhanced gradually. Presence of Cl−, SO42− and Fe3+ had almost no effect on DCF decomposition, while the existence of CO32− promoted DCF removal because of the contribution of CO3˙− formed through the reaction of CO32− with HO˙. Both NO3− and Cu2+ improved the degradation of DCF and the enhancement effect increased with the increase in their concentrations. An inhibition effect was observed in DCF removal in the presence of DOM, which was possibly attributed to its light screening effect and competition for reactive radical. The elimination of TOC was low at only 8.6%. Degradation mechanism of DCF in UV/PAA system was evaluated exhibiting eight different transformation pathways, i.e., hydroxylation, decarboxylation, formylation, dehydrogenation, dechlorination–hydrogenation, dechlorination–cyclization, dechlorination–hydroxylation and amidation based on twelve detected degradation products. This study showed that UV/PAA as an AOP could remove pharmaceutical pollutants such as DCF from wastewater and provided some valuable information on the degradation of DCF for the potential application.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Sichuan Science and Technology Programs (2017SZ0175 and 2018SZDZX0026). Yiqing Liu is also thankful to the financial support from the Fundamental Research Funds for the Central Universities (2682018CX32).
References
- T. Alvarino, S. Suarez, E. Katsou, J. Vazquez-Padin, J. M. Lema and F. Omil, Water Res., 2015, 68, 701–709 CrossRef CAS PubMed.
- L. Arpin-Pont, M. J. M. Bueno, E. Gomez and H. Fenet, Environ. Sci. Pollut. Res., 2016, 23, 4978–4991 CrossRef PubMed.
- J. Wang and L. Chu, Radiat. Phys. Chem., 2016, 125, 56–64 CrossRef CAS.
- Y. Yang, Y. S. Ok, K. H. Kim, E. E. Kwon and Y. F. Tsang, Sci. Total Environ., 2017, 596–597, 303–320 CrossRef CAS PubMed.
- W. Chen, J. Xu, S. Lu, W. Jiao, L. Wu and A. C. Chang, Chemosphere, 2013, 93, 2621–2630 CrossRef CAS PubMed.
- A. J. Ebele, M. A. E. Abdallah and S. Harrad, Emerg. Contam., 2017, 3, 1–16 CrossRef.
- J. Wang and S. Wang, J. Environ. Manage., 2016, 182, 620–640 CrossRef CAS PubMed.
- S. Chong, G. Zhang, N. Zhang, Y. Liu, T. Huang and H. Chang, J. Hazard. Mater., 2017, 334, 150–159 CrossRef CAS PubMed.
- D. Vogna, R. Marotta, A. Napolitano, R. Andreozzi and M. Ischia, Water Res., 2004, 38, 414–422 CrossRef CAS PubMed.
- D. Stulten, S. Zuhlke, M. Lamshoft and M. Spiteller, Sci. Total Environ., 2008, 405, 310–316 CrossRef PubMed.
- S. Suarez, M. Carballa, F. Omil and J. M. Lema, Rev. Environ. Sci. Bio/Technol., 2008, 7, 125–138 CrossRef CAS.
- Y. Zhang, S. U. Geiben and C. Gal, Chemosphere, 2008, 73, 1151–1161 CrossRef CAS PubMed.
- M. Letzel, G. Metzner and T. Letzel, Environ. Int., 2009, 35, 363–368 CrossRef CAS PubMed.
- J. Schwaiger, H. Ferling, U. Mallow, H. Wintermayr and R. D. Negele, Aquat. Toxicol., 2004, 68, 141–150 CrossRef CAS PubMed.
- O. Rozas, C. Vidal, C. Baeza, W. F. Jardim, A. Rossner and H. D. Mansilla, Water Res., 2016, 98, 109–118 CrossRef CAS PubMed.
- Y. Lin, H. Liu, C. Yang, X. Wu, C. Du, L. Jiang and Y. Zhong, Appl. Catal., B, 2020, 264, 118479 CrossRef.
- Y. Lin, X. Wu, Y. Han, C. Yang, Y. Ma, C. Du, Q. Teng, H. Liu and Y. Zhong, Appl. Catal., B, 2019, 258, 117969 CrossRef CAS.
- M. G. Alalm, A. Tawfik and S. Ookawara, J. Environ. Chem. Eng., 2015, 3, 46–51 CrossRef CAS.
- Y. Su, D. Jassby, S. Song, X. Zhou, H. Zhao, J. Filip, E. Petala and Y. Zhang, Environ. Sci. Technol., 2018, 52, 6466–6475 CrossRef CAS PubMed.
- M. M. Cid-Ceron, D. S. Guzman-Hernandez, M. T. Ramirez-Silva, A. Galano, M. Romero-Romo and M. Palomar-Pardave, Electrochim. Acta, 2016, 199, 92–98 CrossRef CAS.
- S. Wu, H. He, X. Li, C. Yang, G. Zeng, B. Wu, S. He and L. Lu, Chem. Eng. J., 2018, 341, 126–136 CrossRef CAS.
- S. Wu, H. Li, X. Li, H. He and C. Yang, Chem. Eng. J., 2018, 353, 533–541 CrossRef CAS.
- M. Y. Hua, H. C. Chen, R. Y. Tsai and Y. C. Lin, Electrochim. Acta, 2011, 56, 4618–4623 CrossRef CAS.
- T. Luukkonen and S. O. Pehkonen, Crit. Rev. Environ. Sci. Technol., 2017, 47, 1–39 CrossRef CAS.
- C. Liang, C. F. Huang and Y. J. Chen, Water Res., 2008, 42, 4091–4100 CrossRef CAS PubMed.
- K. Zhang, X. Zhou, P. Du, T. Zhang, M. Cai, P. Sun and C. H. Huang, Water Res., 2017, 123, 153–161 CrossRef CAS PubMed.
- M. Kitis, Environ. Int., 2004, 30, 47–55 CrossRef CAS PubMed.
- F. Zhou, C. Lu, Y. Yao, L. Sun, F. Gong, D. Li, K. Pei, W. Lu and W. Chen, Chem. Eng. J., 2015, 281, 953–960 CrossRef CAS.
- S. Rothbart, E. E. Ember and R. van Eldik, New J. Chem., 2012, 36, 732–748 RSC.
- P. Sun, T. Zhang, B. Mejia-Tickner, R. Zhang, M. Cai and C. H. Huang, Environ. Sci. Technol., 2018, 5, 400–404 CAS.
- D. P. Daswat and M. Mukhopadhyay, Chem. Eng. J., 2012, 209, 1–6 CrossRef CAS.
- M. Cai, P. Sun, L. Zhang and C. H. Huang, Environ. Sci. Technol., 2017, 51, 14217–14224 CrossRef CAS PubMed.
- S. Chen, M. Cai, Y. Liu, L. Zhang and L. Feng, Water Res., 2019, 150, 153–161 CrossRef CAS PubMed.
- L. Rizzo, T. Agovino, S. Nahim-Granados, M. Castro-Alferez, P. Fernandez-Ibanez and M. I. Polo-Lopez, Water Res., 2019, 149, 272–281 CrossRef CAS PubMed.
- H. Wang, S. Wang, Y. Liu, Y. Fu, P. Wu and G. Zhou, Chemosphere, 2019, 237, 124518 CrossRef PubMed.
- H. Shi, G. Zhou, Y. Liu, Y. Fu, H. Wang and P. Wu, RSC Adv., 2019, 9, 31370–31377 RSC.
- C. Caretti and C. Lubello, Water Res., 2003, 37, 2365–2371 CrossRef CAS PubMed.
- E. V. Rokhina, K. Makarova, E. A. Golovina, H. V. As and J. Virkutyte, Environ. Sci. Technol., 2010, 44, 6815–6821 CrossRef CAS PubMed.
- G. V. Buxton, C. L. Greenstock and W. P. Helman, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- S. Zhang, X. Quan, J. F. Zheng and D. Wang, Water Res., 2017, 122, 86–95 CrossRef CAS PubMed.
- N. S. Shah, X. He, H. M. Khan, J. A. Khan, K. E. O'Shea, D. L. Boccelli and D. D. Dionysiou, J. Hazard. Mater., 2013, 263, 584–592 CrossRef CAS PubMed.
- P. S. Rao and E. Hayon, J. Am. Chem. Soc., 1974, 5, 1287–1294 CrossRef.
- Y. Liu, X. He, Y. Fu and D. D. Dionysiou, Chem. Eng. J., 2016, 284, 1317–1327 CrossRef CAS.
- N. Gao, Y. Deng and D. Zhao, J. Hazard. Mater., 2009, 164, 640–645 CrossRef CAS PubMed.
- O. S. Keen, N. G. Love and K. G. Linden, Water Res., 2012, 46, 5224–5234 CrossRef CAS PubMed.
- X. He, M. Pelaez, J. A. Westrick, K. E. O'Shea, A. Hiskia, T. Triantis, T. Kaloudis, M. I. Stefan, A. A. Cruz and D. D. Dionysiou, Water Res., 2012, 46, 1501–1510 CrossRef CAS PubMed.
- C. Wu and K. G. Linden, Water Res., 2010, 44, 3585–3594 CrossRef CAS PubMed.
- S. Canonica, T. Kohn, M. Mac, F. J. Real, J. Wirz and U. von Gunten, Environ. Sci. Technol., 2005, 39, 9182–9188 CrossRef CAS PubMed.
- M. L. Dell'Arciprete, J. M. Soler, L. Santos-Juanes, A. Arques, D. O. Martire, J. P. Furlong and M. C. Gonzalez, Water Res., 2012, 46, 3479–3489 CrossRef PubMed.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- Y. Huang, M. Kong, D. Westerman, E. G. Xu, S. Coffin, K. H. Cochran, Y. Liu, S. D. Richardson, D. Schlenk and D. D. Dionysiou, Environ. Sci. Technol., 2018, 52, 12697–12707 CrossRef CAS PubMed.
- Y. Liu, X. He, X. Duan, Y. Fu and D. D. Dionysiou, Chem. Eng. J., 2015, 276, 113–121 CrossRef CAS.
- Y. Rao, D. Xue, H. Pan, J. Feng and Y. Li, Chem. Eng. J., 2016, 283, 65–75 CrossRef CAS.
- B. C. Faust and J. Hoigne, Atmos. Environ., Part A, 1990, 24, 79–89 CrossRef.
- Y. Liu, X. He, Y. Fu and D. D. Dionysiou, J. Hazard. Mater., 2016, 305, 229–239 CrossRef CAS PubMed.
- L. Li, X. Fu, J. Ai, H. Zhao, W. Zhang, D. Wang and Z. Liu, Sep. Purif. Technol., 2019, 211, 972–982 CrossRef CAS.
- D. A. Nichela, A. M. Berkovic, M. R. Costante, M. P. Juliarena and F. S. G. Einschlag, Chem. Eng. J., 2013, 228, 1148–1157 CrossRef CAS.
- Y. Liu, X. He, X. Duan, Y. Fu, D. Fatta-Kassinos and D. D. Dionysiou, Water Res., 2016, 95, 195–204 CrossRef CAS PubMed.
- P. Westerhoff, S. P. Mezyk, W. J. Cooper and D. Minakata, Environ. Sci. Technol., 2007, 41, 4640–4646 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang, Y. Xiao, V. W. C. Chang and T. T. Lim, Chem. Eng. J., 2016, 302, 526–534 CrossRef CAS.
- A. Aguera, L. A. Perez Estrada, I. Ferrer, E. M. Thurman, S. Malato and A. R. Fernandez-Alba, J. Mass Spectrom., 2005, 40, 908–915 CrossRef CAS PubMed.
- T. Poiger, H. R. Buser and M. D. Muller, Environ. Toxicol. Chem., 2001, 20, 256–263 CrossRef CAS PubMed.
- M. Kovacic, D. J. Perisic, M. Biosic, H. Kusic, S. Babic and A. L. Bozic, Environ. Sci. Pollut. Res., 2016, 23, 14908–14917 CrossRef CAS PubMed.
- R. Salgado, V.
J. Pereira, G. Carvalho, R. Soeiro, V. Gaffney, C. Almeida, V. Vale Cardoso, E. Ferreira, M. J. Benoliel, T. A. Ternes, A. Oehmen, M. A. M. Reis and J. P. Noronha, J. Hazard. Mater., 2013, 244–245, 516–527 CrossRef CAS PubMed.
- O. S. Keen, E. M. Thurman, I. Ferrer, A. D. Dotson and K. G. Linden, Chemosphere, 2013, 93, 1948–1956 CrossRef CAS PubMed.
- J. Madhavan, P. S. S. Kumar, S. Anandan, M. Zhou, F. Grieser and M. Ashokkumar, Chemosphere, 2010, 80, 742–752 CrossRef PubMed.
- J. Zhao, Y. Liu, Q. Wang, Y. Fu, X. Lu and X. Bai, Sep. Purif. Technol., 2018, 192, 412–418 CrossRef CAS.
- Z. Zhou and J. Jiang, J. Pharm. Biomed. Anal., 2015, 106, 37–45 CrossRef CAS PubMed.
- M. Bedner and W. A. MacCrehan, Environ. Sci. Technol., 2006, 40, 516–522 CrossRef CAS PubMed.
- J. A. Khan, X. He, N. S. Shah, H. M. Khan, E. Hapeshi, D. Fatta-Kassinos and D. D. Dionysiou, Chem. Eng. J., 2014, 252, 393–403 CrossRef CAS.
- H. Yu, E. Nie, J. Xu, S. Yan, W. J. Cooper and W. Song, Water Res., 2013, 47, 1909–1918 CrossRef CAS PubMed.
- J. Eriksson, J. Svanfelt and L. Kronberg, Photochem. Photobiol., 2010, 86, 528–532 CrossRef CAS PubMed.
- C. Martinez, M. Canle, M. I. Fernandez, J. A. Santaballa and J. Faria, Appl. Catal., B, 2011, 107, 110–118 CrossRef CAS.
- K. Lekkerkerker-Teunissen, M. J. Benotti, S. A. Snyder and H. C. van Dijk, Sep. Purif. Technol., 2012, 96, 33–43 CrossRef CAS.
- S. Salaeh, D. J. Perisic, M. Biosic, H. Kusic, S. Babic, U. L. Stangar, D. D. Dionysiou and A. L. Bozic, Chem. Eng. J., 2016, 304, 289–302 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Yongsheng Fu*
* and
Yongsheng Fu*