
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc–iron silicate for heterogeneous catalytic ozonation of acrylic acid: efficiency and mechanism
Yue Liu *ab,
Jimin Shenb,
Laiqun Zhaoa,
Weiqiang Wangb,
Weijin Gonga and
Fanfan Zhenga
*ab,
Jimin Shenb,
Laiqun Zhaoa,
Weiqiang Wangb,
Weijin Gonga and
Fanfan Zhenga
aSchool of Energy & Environment Engineering, Zhongyuan University of Technology, Zhongyuan Road 47, Zhengzhou, China. E-mail: yue5757@sina.com
bState Key Laboratory of Urban Water Resources and Environment, School of Municipal & Environmental Engineering, Harbin Institute of Technology, Harbin 150090, China
First published on 4th March 2020
Abstract
This research aimed at researching the degradation of acrylic acid (AA) in aqueous solution, by catalytic and non-catalytic ozonation processes performed in a semi-continuous reactor. Zinc–iron silicate was synthesized and characterized using X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) analysis, Fourier transformation infrared (FT-IR) and energy dispersive spectrometry (EDS). The characterization studies showed that Fe–Si binary oxide, Zn–Si binary oxide, ZnO and Fe2O3 deposits were formed on the surface of poor crystallinity zinc–iron silicate which contained abundant functional groups. Catalytic ozonation test results revealed that zinc–iron silicate exhibited high catalytic activity and stability in catalytic ozonation of AA in aqueous solution. The inclusion of zinc–iron silicate in the ozonation process enhanced AA decomposition by 28.7% and TOC removal by 20%, compared to the ozonation alone. The main AA removal mechanisms involved direct oxidation by ozone and indirect oxidation by hydroxyl radicals generated by the ozone chain reaction accelerated by zinc–iron silicate. The surface characteristics and chemical composition are significant factors determining the catalytic activity of zinc–iron silicate.
Introduction
Acrylic acid (C3H4O2, AA) is an unsaturated fatty acid, consisting of a vinyl group and a carboxyl group. AA is an important organic chemical synthetic raw material, mainly used in many industrial fields such as textile, synthetic fibre, printing, papermaking, leather chemicals, and flocculants and so on. AA has a strong stimulating effect on skin, eyes and the respiratory tract. AA wastewater is biologically toxic and poses a great hazard to ecology and human health. At present, the main method for treating AA wastewater is incineration, but the incineration method is costly and easily causes secondary pollution. In recent years, researchers at home and abroad have continuously developed some new methods for the treatment of AA wastewater, including biological methods, Fenton oxidation, supercritical water oxidation and catalytic ozonation.1–3Catalytic ozonation is the use of catalyst to accelerate the decomposition of ozone to produce strong oxidative active radicals (e.g., hydroxyl radicals, ˙OH).4–6 ˙OH has high oxidation rate constants with almost all organic compounds in aqueous solution.7 According to the morphology of the catalyst, the catalytic ozonation is divided into homogeneous catalytic ozonation, and heterogeneous catalytic ozonation. In the homogeneous catalytic ozonation process, ozone is catalyzed by soluble metal ions. However, after reaction, the soluble metal ion catalyst is difficult to separate from aqueous solution. So it is easy to produce secondary pollution. In the heterogeneous catalytic ozonation process, ozone is catalyzed by a solid catalyst (such as ZnO, ZnOOH, FeOOH, activated carbon) without any additional thermal, light energy or second pollution involving ironic catalyst separation.8 In recent years, heterogeneous catalytic ozonation has received wide attention in practical applications.9
At present, research on heterogeneous catalysts mainly focuses on three aspects: metal oxides, supported metals, and some porous materials (activated carbon and microporous and mesoporous silicate).10,11 In several cases, zinc oxide and iron oxide which are chemically stable and non-toxic have shown high activity for the destruction of pollutants with ozone.12–15 However, the micro- and nano-sized zinc oxide or iron oxide powers are difficult to be separated from treated water, and this disadvantage restrict the spreading use of them in the actual water treatment process.
Silicates materials, with the uniform mesoporous channel structure, high surface area, relatively wide pore size distribution, massive hydroxyl groups, good chemical stability and low cost, have received great attention as catalysts.16 The unique characteristics of silicates make it an appropriate matrix for the development of novel heterogeneous catalysts by introducing metal oxides and metal complexes into silicate with a controlled manner to develop well-defined active sites. With this in mind, zinc–iron silicate was synthesized in the laboratory.
The purpose of the present paper is to investigate the characteristic of the synthesized novel material zinc–iron silicate, to explore its catalytic performance in ozonation of AA in aqueous solution, and to study the reaction mechanisms at the solid–water interface.
Results and discussion
Characterization
The morphologies of the as-prepared zinc–iron silicate were observed using SEM images. The SEM images (Fig. 1 (a) ×10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, (b) ×20000) revealed that the zinc–iron silicate had irregular structure and consisted of agglomerated particles. The EDS results (Fig. 1(c)) revealed that the atomic % ratio of Zn to Fe was close to 1.0 which was consistent with the designed value 1:1, implying the full utilization of both metal ions. Further component analysis by XRD (Fig. 1(d)) showed that there was no single, sharp, intense and easily recognizable peak, but only two low intensity and vaguely recognizable peaks at 34.4° and 62.0°. Because of the lack of a relationship between crystallinity and a specific Miller index, the peaks were excessively wide. Therefore, zinc–iron silicate was believed to be amorphous, which was consistent with the results of the SEM.
000, (b) ×20000) revealed that the zinc–iron silicate had irregular structure and consisted of agglomerated particles. The EDS results (Fig. 1(c)) revealed that the atomic % ratio of Zn to Fe was close to 1.0 which was consistent with the designed value 1:1, implying the full utilization of both metal ions. Further component analysis by XRD (Fig. 1(d)) showed that there was no single, sharp, intense and easily recognizable peak, but only two low intensity and vaguely recognizable peaks at 34.4° and 62.0°. Because of the lack of a relationship between crystallinity and a specific Miller index, the peaks were excessively wide. Therefore, zinc–iron silicate was believed to be amorphous, which was consistent with the results of the SEM.
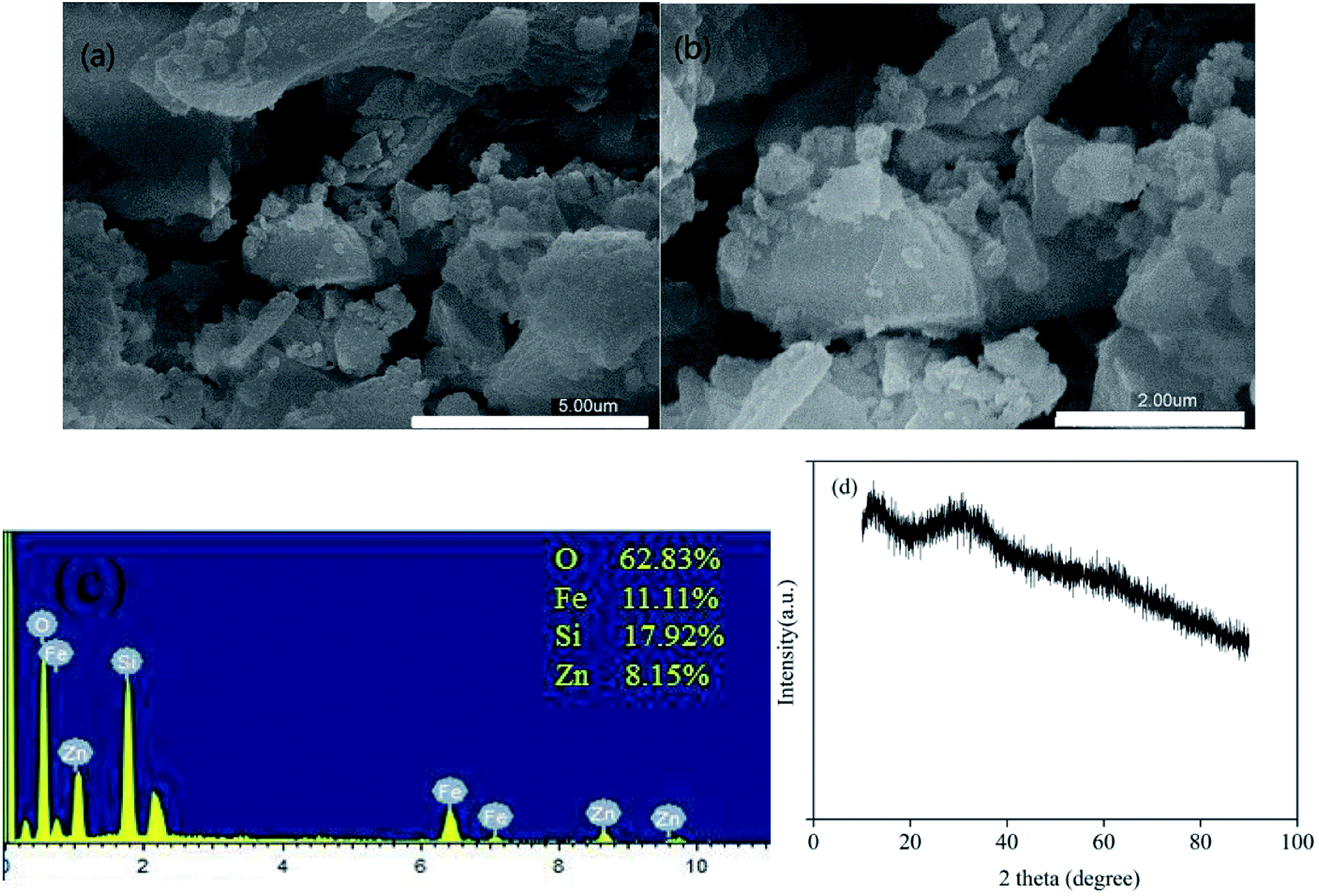 | ||
| Fig. 1 SEM images (a) ×10000, (b) ×20000, (c) EDS results, (d) XRD pattern of zinc–iron silicate. | ||
The structure and surface functional groups of zinc–iron silicate was recorded by FTIR spectrum. As shown in Fig. 2. The peaks at 1080 and 490 cm−1 represent Si–O–Si bonds and Zn–O bonds. The peaks at 3540 and 1680 cm−1 indicate the presence of hydroxyls and coordinated water molecules on the surface of zinc–iron silicate.17 The peaks at 1124 and 662 cm−1 represent the bending vibration of Fe–O–Si and Zn–O–Si bonds peaks.18,19 The presence of Fe–O–Si and Zn–O–Si bonds indicated that the iron and zinc were dispersed onto the silica matric. This result was confirmed by XPS analysis.
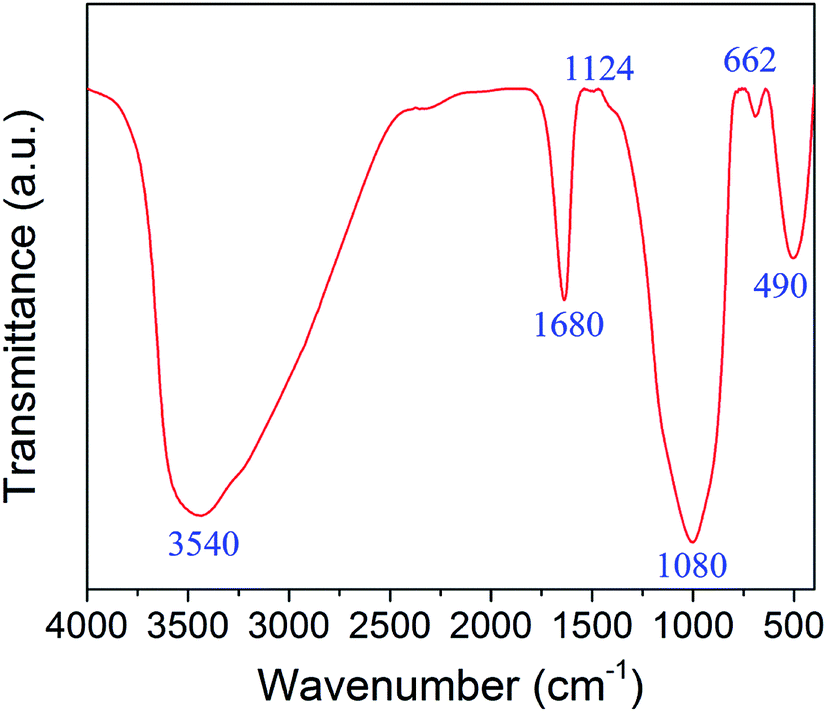 | ||
| Fig. 2 FTIR spectrum of the zinc–iron silicate. | ||
The surface chemistry of zinc–iron silicate was studied by XPS analysis. As shown in the Fig. 3(a), the spectrum contains Fe, Zn, O, Si, and C. The peaks of C 1s can be attributed to contamination caused by sample preparation or by the pumping oil used in the XPS instrument itself. As shown in Fig. 3(b), the XPS spectra of Zn 2p3/2 showed an unsymmetrical Zn 2p3/2 peak. The binding energy (BE) of Zn 2p3/2 in the zinc–iron silicate samples was found to be 1025.3 eV, which is higher than that of pure ZnO (1023.5 eV).20 The wide Zn 2p3/2 signals obtain for zinc–iron silicate could be fitted satisfactorily to two principal peaks after deconvolution. The peak at 1023.5 eV belongs to ZnO. Meanwhile, another peak appears at 1025.6 eV. The BE of Fe 2p3/2 obtain for zinc–iron silicate is 713 eV (Fig. 3(d)), and the signals could be fitted to two peaks. The peak at 710.5 eV belongs to Fe3+ of Fe2O3, and another peak appears at 709.9 eV belong to Fe–Si binary oxides.21 As shown in Fig. 3(c), the O 1s XPS spectra exhibited asymmetric peaks, indicating that different degrees of oxygen contribution existed on the zinc–iron silicate surface. The O 1s peaks could be fitted satisfactorily into seven peaks after deconvolution. They corresponded to Fe–O (529.9 eV), –OH (532.3 eV), Fe–Si binary oxides (532.1 eV), Zn–Si binary oxides (531.8 eV), Zn–O (530.3 eV), Si–O–Si (533.3 eV) and adsorbed water (535.8 eV), respectively.21 The BE of Si 2p was found to be 103.1 eV (Fig. 3(e)), which was higher than that of pure SiO2 (100.2 eV). The electronegativities (Pauling term), the ionicities (Madelung term), and the final states (relaxation term) in the environment of the photoionized atom could result in the core-level shifts.22 These analyses indicated that the core-level shifts of Zn, Fe and Si were due to the formation of Si–O–Zn bond and Si–O–Fe.
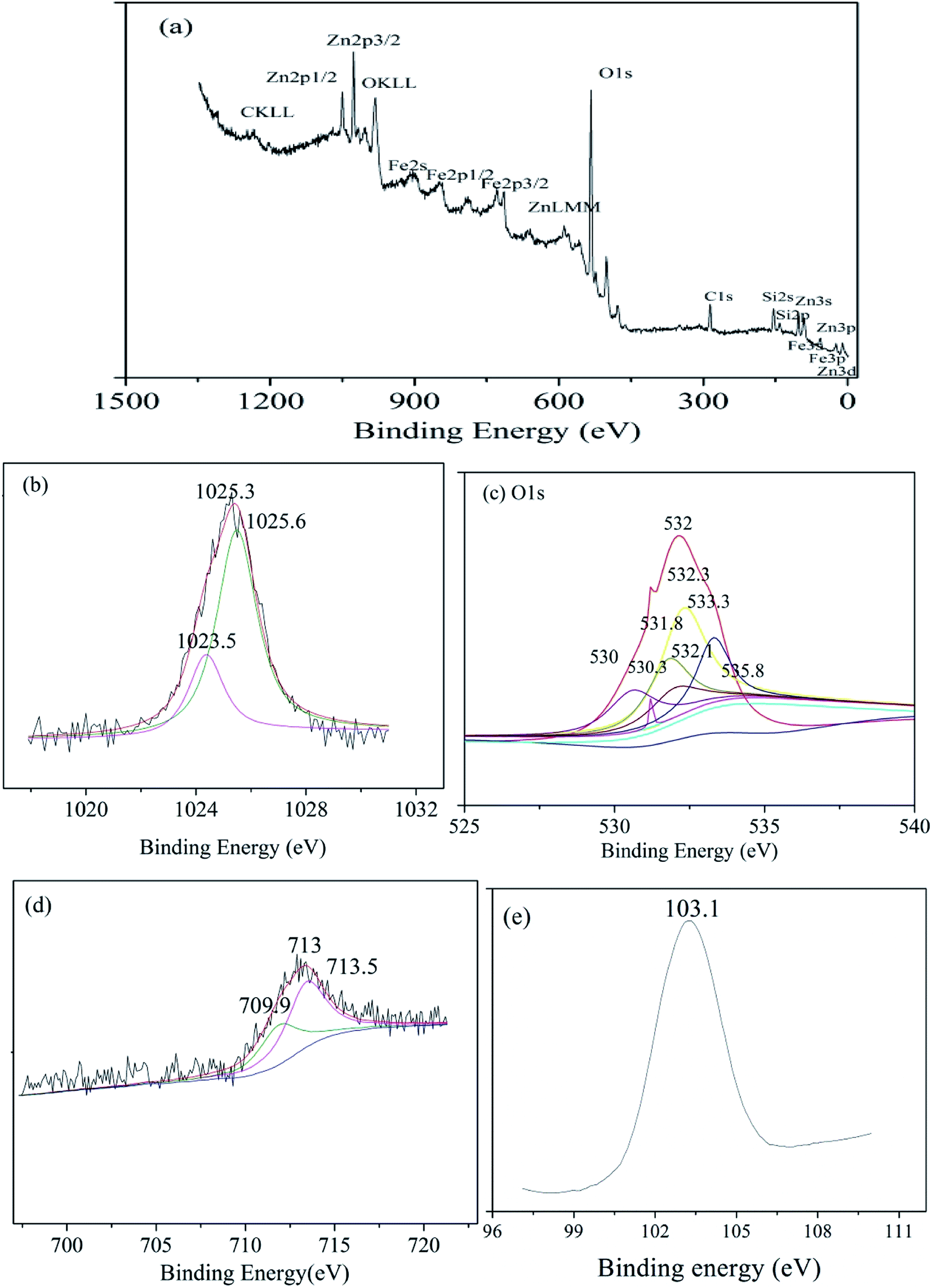 | ||
| Fig. 3 XPS spectra of (a) zinc–iron silicate, (b) Zn 2p3/2, (c) O 1s curves fitted, (d) Fe 2p3/2, (e) Si 2p. | ||
On the basis of SEM, EDS, XRD, FTIR, and XPS analysis, zinc–iron silicate was mainly composed by Fe–Si binary, Zn–Si binary, ZnO and Fe2O3.
Activity and stability of zinc–iron silicate
The experiments of ozonation alone, catalytic ozonation and adsorption on catalysts were conducted to study the catalytic activity of as-synthesized zinc–iron silicate. In addition, we investigated the leaching of Zn2+ and Fe3+ after catalytic ozonation experiment. Fig. 4(a) shows the removal curves of AA along with reaction time. Less than 3% AA was adsorbed on the surface of the zinc–iron silicate, zinc silicate and iron silicate, respectively. These results suggested that during the three catalytic ozonation processes self-adsorption had slightly contribution on the removal of AA. Moreover, Fig. 4(a) shows that the removal of AA was 69.3%, 93%, 91% and 98% by O3 alone, O3/iron silicate, O3/zinc silicate and O3/zinc–iron silicate after 20 min, respectively. The best result was obtained with zinc–iron silicate. Meanwhile, as shown in Fig. 4(b), the presence of three catalysts all increased TOC removal compared with ozonation alone. Catalytic ozonation with zinc–iron silicate removed 42.7% of TOC compared to 36.1% with zinc silicate and 37.9% with iron silicate after 20 min, respectively. An increase in TOC degradation of approximately 20% was observed for O3/zinc–iron silicate relative to the effect of ozonation alone. After reaction, Zn2+ and Fe3+ concentrations in treatment water were all below the detection limit. This finding suggested that the presence of the different silicate could generate positive influence on the ozone molecules in the removal and mineralization of AA. Furthermore, the results also suggested that the mixture of zinc and iron silicate could increase the catalytic activity compared with the sole metal silicate. This phenomenon was consistent with the results described by Pang et al.23 The use of different metal ion mixing could effectively improve the electron transport capacity of the catalyst surface interface, and further improve the catalytic performance.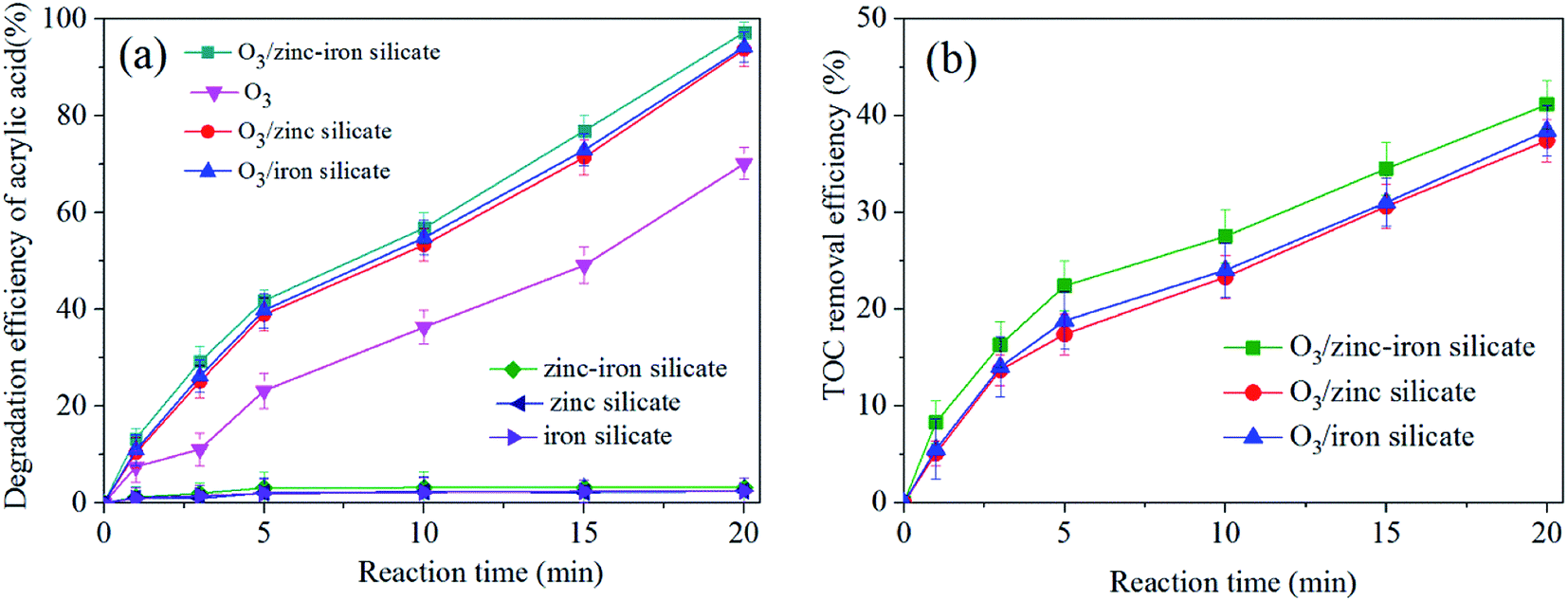 | ||
| Fig. 4 (a) Degradation of AA versus reaction time in different processes. (b) Removal of TOC versus reaction time in different processes. Ozone dose: 18 mg min−1, catalyst dose: 500 mg, initial concentration of AA solution: 1000 mg L−1, pH = 5.4, flow rate of oxygen 0.6 L min−1, reaction temperature 20 °C. | ||
A good catalyst, not only maintain a high catalytic activity but also have good stability. To study the recyclability of the zinc–iron silicate, we carried out five continuous cycles of the catalytic ozonation of AA under the same conditions. At the end of one cycle (20 min), the catalyst was collected, rinsed gently with ultra-pure water and drying at 60 °C, ready for use in the next cycle. Fig. 5 shows that after five continuous cycles, the catalytic activity of zinc–iron silicate did not decrease significantly, in which 97%, 96%, 95.7%, 95.2% and 95% of the AA was removed, whereas 41%, 39.9%, 39.7%, 39.3%, and 39% of the TOC was removed, respectively. These results showed that zinc-silicate was an effective, high activity and fine stability catalyst in ozonation.
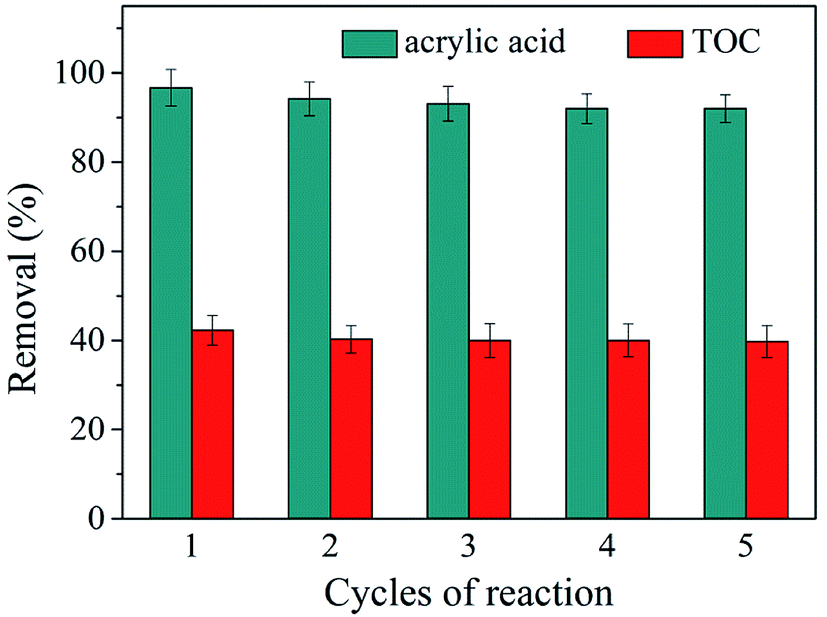 | ||
| Fig. 5 Recycling of zinc–iron silicate in catalyzed ozonation. Ozone dose: 18 mg min−1, catalyst dose: 500 mg, initial concentration of AA solution: 1000 mg L−1, pH = 5.4, flow rate of oxygen 0.6 L min−1, reaction temperature 20 °C. | ||
Effect of ozone dosage
The effect of catalyst dosage was also investigated and each dataset was modelled using a pseudo-first-order expression (eqn (1)):
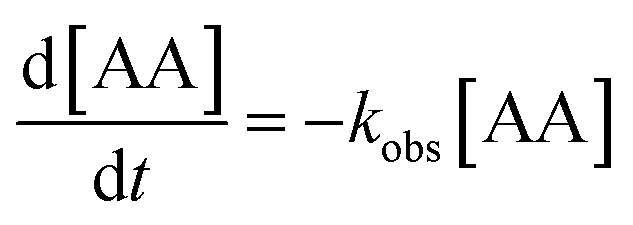 | (1) |
According to Table 1 and Fig. 6, AA degradation kinetics was obviously catalyst dosage dependent. The kobs was increased along with the catalyst dosage, particularly, the kobs of 500 mg L−1 was nearly 2 times more than that 100 mg L−1. This high degradation kinetics may be ascribed the sufficient surface active sites provide by zinc–iron silicate for catalysing the decomposition of O3. When the dosage increased to 800 mg L−1, the kobs constant was 0.128 which was close to the 500 mg L−1. According to the He et al.24 reported, this phenomenon may be because that further increase of the catalyst dosage lead to more ˙HO, and ˙HO react with molecular O3 resulting in ineffective ozone consumption.
| Dosage (mg L−1) | Initial pH | kobs (min−1) | R2 |
|---|---|---|---|
| 100 | 5.4 | 0.061 | 0.988 |
| 300 | 5.4 | 0.072 | 0.997 |
| 500 | 5.4 | 0.113 | 0.995 |
| 800 | 5.4 | 0.128 | 0.989 |
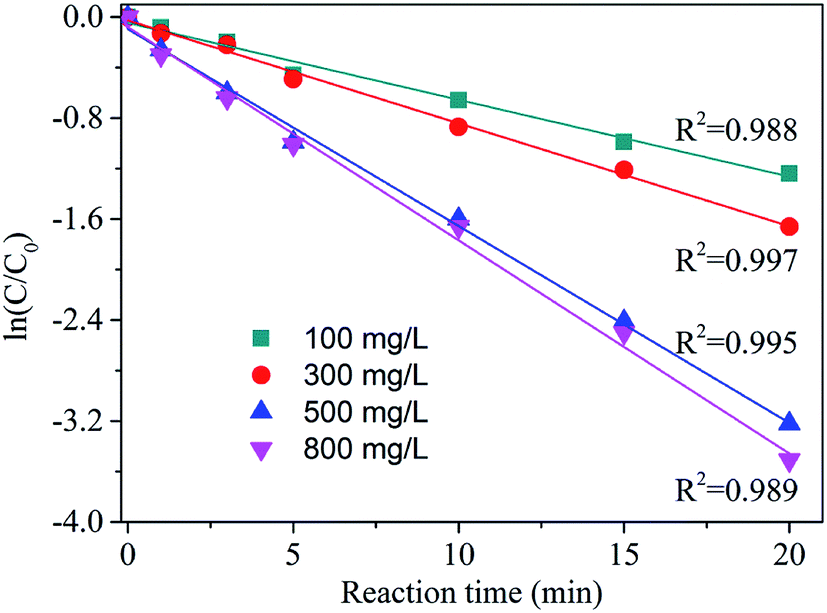 | ||
| Fig. 6 AA degradation kinetics by catalytic ozonation under selected processes. Ozone dose: 18 mg min−1, initial concentration of AA solution: 1000 mg L−1, pH = 5.4, flow rate of oxygen 0.6 L min−1, reaction temperature 20 °C. | ||
Effect of Zn/Fe molar ratio
The effect of the Zn/Fe molar ration of zinc–iron silicate on AA degradation was also investigated in catalytic ozonation. As shown in Fig. 7, the results clearly demonstrated that the five different Zn/Fe molar ration catalysts significantly enhanced AA removal relative to ozonation alone. However, with different Zn/Fe molar ratios exhibited different catalytic activities in catalytic ozonation. The removal efficiency of AA increased along with the increase of the molar ratio of Zn/Fe from 0.1 to 1. However, further increase in molar ratio of Zn/Fe to 2.0, the removal efficiency has no significant change. These results indicated that optimal critical molar ratio of Zn/Fe was 1.0 under the present experimental conditions. The reason may be because that the surface properties of zinc–iron silicate were affected by the different ration of Zn/Fe, and the surface properties may influence the catalytic activity of the zinc–iron silicate.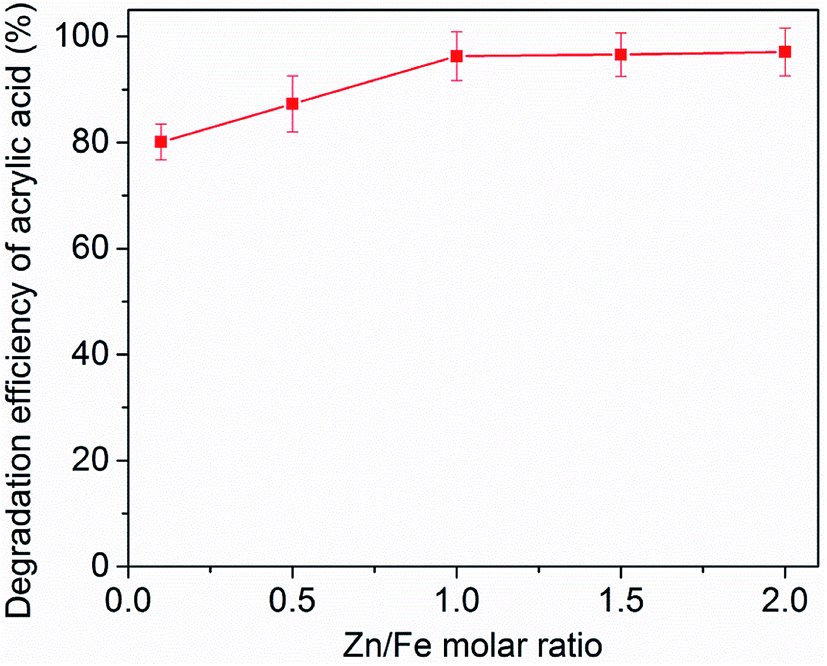 | ||
| Fig. 7 Effect of Zn/Fe molar ratio on AA removal. Ozone dose: 18 mg min−1, catalyst dose: 500 mg, initial concentration of AA solution: 1000 mg L−1, pH = 5.4, flow rate of oxygen 0.6 L min−1, reaction temperature 20 °C. | ||
Discussion the possible catalytic ozonation mechanisms
Experimental observations show that the presence of zinc–iron silicate catalyst improved the ozonation of AA. Therefore, it is deduced that in the process of zinc–iron silicate catalytic ozonation, some active species were formed. In general, in the heterogeneous catalytic ozonation process, ˙OH is the main reactive species.25 In order to determine whether ozone was transformed into ˙OH in the zinc–iron silicate catalytic ozoantion process, tert-butanol (TBA) a well-known ˙OH scavenger was used. As shown in Fig. 8, for the catalyzed ozonation process, the presence of TBA in aqueous solution had a negative effect on the efficiency of AA degradation even at a very low concentration (0.1 mmol L−1). Obviously, as the concentration of TBA increased from 0 to 0.3 mmol L−1, the efficiency of AA degradation decreased from 96.7% to 55%. However, the removal rate of AA did not decrease further when the concentration of TBA was increased to 0.5 mmol L−1. This phenomenon was consistent with the free radical characteristics described by our previous research.26 The results of this experiment indirectly proved that ˙OH is formed in the process of catalytic ozonation of AA by zinc–iron silicate, and ˙OH act as oxidants to participate in the oxidation of AA. However, the excess TBA in this experiment did not completely inhibit the removal efficiency of AA, which indicates that AA was removed following two possible mechanisms: (1) indirect reaction with ˙OH formed by the decomposition of ozone by zinc–iron silicate in aqueous solution; (2) direct reaction by ozonation alone.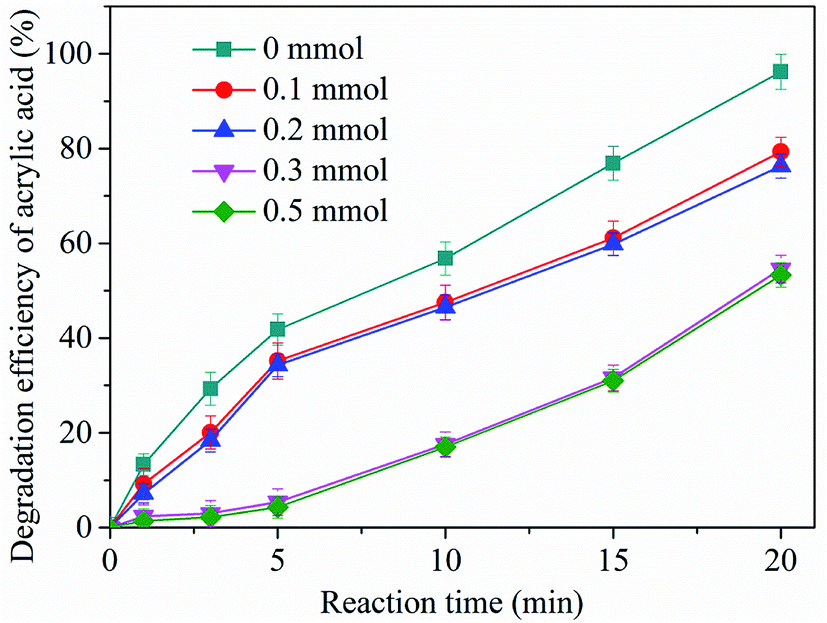 | ||
| Fig. 8 Effects of TBA on the degradation of AA. Ozone dose: 18 mg min−1, catalyst dose: 500 mg, initial concentration of AA solution: 1000 mg L−1, pH = 5.4, flow rate of oxygen 0.6 L min−1, reaction temperature 20 °C. | ||
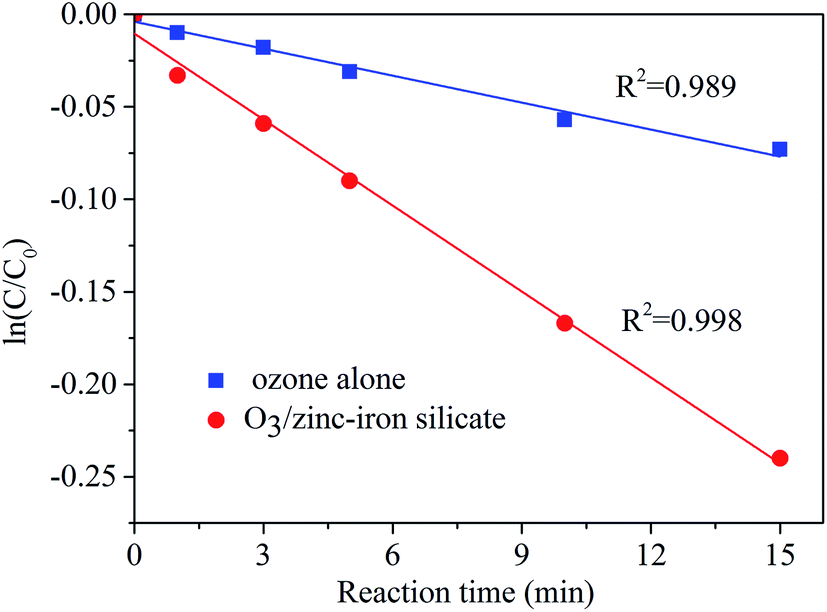
In heterogeneous catalytic ozonation processes, in order to exert the activity of the solid catalyst, adsorption of ozone and/or organic molecule on the surface of the catalyst has to take place. As state in the section of activity and stability of zinc–iron silicate, in zinc–iron silicate catalytic ozonation process, a rather low adsorption of AA was observed (Fig. 4). To study the catalytic mechanism, zinc–iron silicate was used to investigate the decomposition of ozone. As demonstrated in Fig. 9, it is obvious that the decomposition of ozone followed first-order kinetics with and without the zinc–iron silicate, and the presence of zinc–iron silicate accelerates the decomposition of ozone. Ozone decomposition rate increased by 3.13 times compared to ozonation alone process.
 | ||
| Fig. 9 The decomposition of ozone in the process of O3/zinc–iron silicate process. Ozone dosage: 1.0 mg, catalyst dose: 500 mg, initial pH of the ozone solution was adjusted to pH = 7.0 with NaOH, reaction temperature 20 °C. | ||
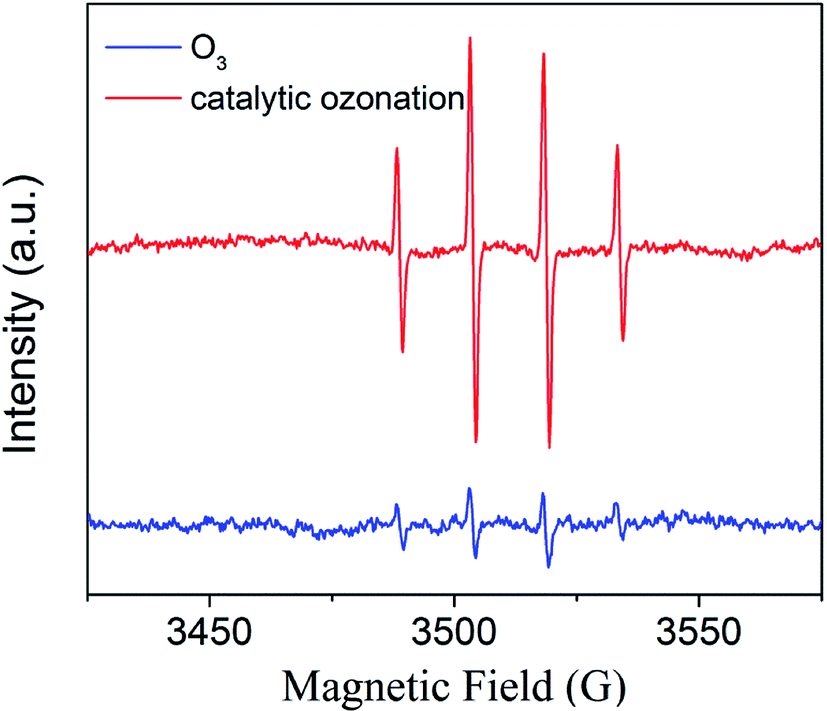
The spin-trapping/EPR technique is suitable for investigating ˙OH generation. Fig. 10 shows typical EPR spectra obtained during ozonation alone and zinc–iron silicate catalytic ozonation process. According to the EPR spectra results, ˙OH was produced in the two processes mentioned above. In the zinc–iron silicate catalyzed ozonation process the DMPO-OH signals were much stronger than those in the ozonation alone. In other word, the presence of the zinc–iron silicate accelerated the generation of ˙OH in the ozonation system.
 | ||
| Fig. 10 Spectra of DMPO-OH signals. Ozone dosage: 2.0 mg L−1, catalyst dose: 1000 mg, initial pH of the ozone solution was adjusted to pH = 7.0 with NaOH, reaction temperature 20 °C, DMPO = 100 mmol L−1. | ||
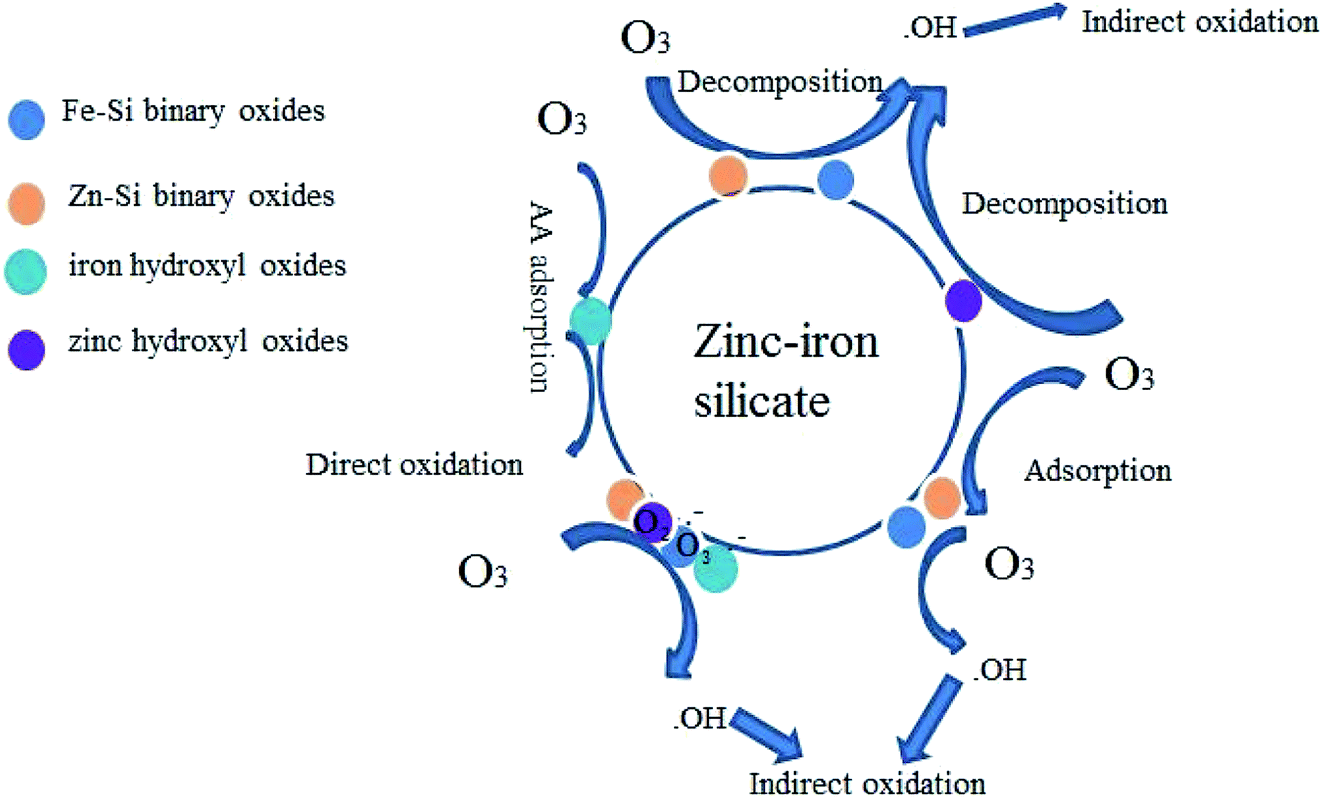
The results shown in Fig. 4 and 8–10 confirmed that ˙OH participates in the zinc–iron silicate catalyzed ozonation process. There are generally three possible mechanisms expatiating on heterogeneous catalytic ozonation: the enhancement of catalytic ozonation of involves the adsorption of ozone or pollutant or both of them on the catalyst surface leading to the formation of free radicals which react with non-adsorbed species in the aqueous solution. Comparison the results of adsorption experiment and Fig. 9, the zinc–iron silicate catalytic ozonation process follows the first case. Moreover, it is reasonable to explain that ˙OH was not the main oxidative species responsible for the high degradation effectiveness, because the ˙OH if exit in homogeneous system, it must be scavenged by TBA. Therefore the surface characteristics and chemical composition of zinc–iron silicate play an important role in the oxidant mechanism. According to the XPS results, the Zn and Fe in the zinc–iron silicate sample substituted the silicon ion in the silicic acid and linked with the frame work via oxygen ions and formed Fe–Si binary oxides, Zn–Si binary oxides, ZnO and Fe2O3. Because of the desaturation of electrons on the surface of Zn and Fe, it was easily developing negative centers or Brønested acid sites.27 The coordinative unsaturation of Fe–Si binary oxides, Zn–Si binary oxides, ZnO and Fe2O3 encourage water molecules adsorbed on the surface of the zinc–iron silicate, resulting in the formation of surface hydroxyl groups.28 The FTIR spectrum (Fig. 2) has proven that there is an abundance of hydroxyl groups on the surface of zinc–iron silicate. Saturated deprotonating experiments results showed that the density of surface hydroxyl groups on zinc–iron silicate was 2.58 × 10−2 mol g−1, which was much more than that of iron silicate and zinc silicate as reported by Liu et al.29 For zinc–iron silicate catalytic ozonation system, a mechanism (depicted in Fig. 11) of zinc–iron silicate catalyzed ozonation delineated as below.
 | ||
| Fig. 11 Scheme of mechanism proposed in the process of catalytic ozonation. | ||
The surface hydroxyl groups on the surface of the zinc–iron silicate initiate ozone decomposition (eqn (2) and (3)):
| O3 + OH− → HO2− + O2 | (2) |
| O3 + HO2− → OH + O2 + O2˙− | (3) |
Due to the electrophilic character of the zinc–iron silicate, ozone could be adsorbed to the negative centers or Brønsted acid sites generation of superoxide radicals. As shown in the Fig. 11, zinc–iron silicate can accelerate the ozone decomposition (eqn (3), (4) and (6)).
| O3 + O2˙− → O3˙− + O2 | (4) |
| O3˙− + H+ → HO3˙ | (5) |
| HO3˙ → ˙OH + O2 | (6) |
Whether on the surface of the catalyst or in the bulk of the aqueous phase, free radicals can set off a radical chain reaction, and precede oxidation step-wisely. Therefore, the surface characteristics and chemical composition are significant factors determining its activity.
Experimental
Materials and reagents
The model water was prepared by spiking 1000 mg L−1 AA (Tianjin Chemical Factory, China, 99.5%) in Milli-Q water. Synthetic 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was supplied by Sigma (USA). Methyl alcohol was purchased from Sigma-Aldrich (U.S.). Zinc nitrate, ferric nitrate, sodium silicate, sodium thiosulfate, sodium hydroxide, t-butanol, indigo, and borate were all of analytical grade and used without further purification in the experiment processes. The used water was supplied by an ultrapure water system (Millipore Q Biocel system).Synthesis of catalysts
The zinc–iron silicate was prepared using Zn(NO3)2, Fe(NO3)3 and Na2SiO3 as the starting materials. The Zn/Fe molar ratios = 1:1 were dissolved into 1 L ultra-pure water. Then, 0.2 mol L−1 Na2SiO3 solution was slowly titrated into the mixed solution under magnetic stirring at 100 rpm to pH 7. The pH of the suspension was adjusted to 12 by NaOH solution at 25 °C and then incubated further at 60 °C for 24 h. The precipitate was then collected and washed several times with ultra-pure water until the pH and conductivity of the supernatant remained constant and baked at 90 °C for 6 h. Finally, the dry solid catalyst was ground up, sieved and stored in a vacuum desiccator. This catalyst (zinc–iron silicate, Zn/Fe molar ratio = 1:1) exhibited the highest activity and was used in the experiments. As references, different Zn/Fe molar ratio (0.5:1, 1.5:1 and 2:1) zinc–iron silicates, zinc silicate and iron silicate were synthesized via the same procedure.
Ozonation procedure
All the catalytic ozonation experiments in this study were conducted in semi-continuous flow mode at ambient temperature 20 °C. The glass reactor was a cylindrical reactor (h = 600 mm, r = 60 mm) with a volume of 1.2 L. In each experiment, the reactor was filled with 1000 mL ultrapure water. Ozone was produced in situ from pure oxygen using the ozone generator (CF-G-3-010 g, Qingdao Guolin, China) and continuously fed to the solution through a silica bubble diffuser at the bottom of the reactor. The initial aqueous ozone concentration was controlled by changing the ozone gas intake and electrical current of the ozone generator. The desired amounts of catalysts and AA stock solution were dosed into the reactor by taking off its base. Then, the ozone generator was turned on to initiate the experiment. Water samples (each being 15 mL) were taken from the ozonation reactor at a predetermined time intervals (0, 1, 3, 5, 10, 15, 20 min) to analyse the residual concentration of the AA. The oxidation reaction was quenched by addition of Na2S2O3 solution (0.1 mol L−1). All the experiments were conducted three times and the average date and error bars were shown in the figures. For comparative purpose, adsorption on zinc–iron silicate, zinc silicate and iron silicate catalytic ozonation experiment, reusability experiments and the ozonation alone experiments were performed with the same system under identical experimental conditions.Ozone decomposition experiment was conducted in batch mode. The glass reactor was a flat-bottomed flask with a volume of 1.2 L. In the experiment, the reactor was filled with 1000 mL ultra-pure water. The initial aqueous ozone concentration was controlled 1 mg L−1 and catalyst dosage was 500 mg. The reactor was sealed and the magnetic stirrer was turned on to initiate the experiment. After a designated time interval, 25 mL of the solution was sampled from the reactor and to measure the residual dissolved ozone.
Analytical methods
The concentration of ozone in the gas was measured using the indigo method.30 The concentration of AA was analyzed by a 1200 high performance liquid chromatography (HPLC, Agilent, USA) equipped with an UV-Vis detector. A reverse-phase 4.6 mm × 250 mm, 5 μm Sepax GP-C18 column was used at 40 °C. Samples were analyzed with an eluent (borate buffer solution and methyl alcohol in the ratio 3:7 v/v) at a flow rate of 0.5 mL min−1 and detected at 230 nm. The injection volume was 1 mL. TOC analyzer (TOC-VCPH, Shimadzu, Japan) was used to analyze the total organic carbon (TOC). The crystalline structure of the catalyst was conducted by X-ray diffraction (XRD, D8 Advance diffractometer, Germany). The elemental concentration of the catalyst was analyzed by energy dispersive spectrometry (EDS, Genesis, USA). The surface morphology of the catalyst was conducted by scanning electron microscopy (SEM, Quanta 200, Netherlands). The surface chemical composition of the catalyst was analyzed by X-ray photoelectron spectroscopy (XPS, PHI 5700, USA). The surface properties of the samples were measured by Fourier transform infrared spectroscopy (FT-IR, Spectrum One, USA) with a spectral range of 4000 to 400 cm−1. The concentrations of Zn2+ and Fe3+ from the catalyst into solution were determined using an inductively coupled plasma atomic emission spectrometer (Optima 5300DV, USA). The density of surface hydroxyl groups of the zinc–iron silicate was measured by saturated deprotonation method described by Laiti et al.31
Conclusions
Zinc–iron silicate was synthesized in the laboratory. SEM, EDS, XRD, FTIR, and XPS analyses confirmed that zinc–iron silicate was amorphous and mainly composed by Fe–Si, Zn–Si binary oxides, ZnO and Fe2O3. The catalytic activity of zinc–iron silicate was investigated in heterogeneous catalytic ozonation process using AA as a target pollutant under semi-continuous flow mode. The results showed that zinc–iron silicate had good stability and could greatly improve the removal efficiency of AA relative to ozonation alone. Adsorption of AA on the surface of zinc–iron silicate exhibited no remarkable influence on the degradation efficiency of AA. The catalyst with 1:1 Zn/Fe molar ratios achieved the highest AA removal among all the tried conditions. The formation of ˙OH in the zinc–iron silicate catalytic ozonation process was confirmed by spin-trapping/EPR technology and TBA experiment. The coordinative unsaturation of Fe–Si binary oxides, Zn–Si binary oxides, ZnO and Fe2O3 composite can act as Lewis acids and Brønsted bases to form surface hydroxyl groups. These were the active sites triggering a chain reaction of ozone to form of the ˙OH. The AA removal mechanisms involved direct oxidation by ozone in the bulk of aqueous solution and the indirect oxidation by ˙OH generated by the ozone chain reaction accelerated by zinc–iron silicate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the National Natural Science Foundation of China (No. 51308561), Key Scientific Program of Higher Education Institutions (19B560012), Open Project of State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology (QA201941), Henan innovation team (19IRTSTHN010), and Zhongyuan University Special funds for scientific research (K2019YY011).Notes and references
- W. Li, J. Huang, H. Wang, A. Qi and J. Xie, Catalytic wet oxidation treatment of acrylic acid wastewater, J. Jilin Inst. Chem. Technol., 2007, 24(3), 3–6 CAS.
- W. Gong, F. Li and D. Xi, Supercritical Water Oxidation of Acrylic Acid Production Wastewater in Transpiring Wall Reactor, Environ. Eng. Sci., 2007, 26, 131–136 CrossRef.
- J. Xu, Discussion on treatment methods of high concentration acrylic acid and vinegar wastewater, Acry. Chem., 2006, 19(2), 14–18 Search PubMed.
- Y. Li, D. Li, J. Li, A. Hussain, H. Ji and Y. Zhai, Pretreatment of cyanided tailings by catalytic ozonation with Mn2+/O3, J. Environ. Sci., 2015, 28(1), 14–21 CrossRef CAS PubMed.
- O. Oputu, M. Chowdhury, K. Nyamayaro, O. Fatoki and V. Fester, Catalytic activities of ultra-small β-FeOOH nanorods in ozonation of 4-chlorophenol, J. Environ. Sci., 2015, 35(8), 83–90 CrossRef CAS PubMed.
- Y. Wang, W. Yang, X. Yin and Y. Liu, The role of Mn-doping for catalytic ozonation of phenol using Mn/γ-Al2O3 nanocatalyst: performance and mechanism, J. Environ. Chem. Eng., 2016, 4(3), 3415–3425 CrossRef CAS.
- Z. He, Q. lan, F. Cai, Z. Hong, J. Jiang and S. Chen, Effective Enhancement of the Degradation of Oxalic Acid by Catalytic Ozonation with TiO2 by Exposure of {001} Facets and Surface Fluorination, Ind. Eng. Chem. Res., 2012, 51(16), 5662–5668 CrossRef CAS.
- L. Yuan, J. Shen, P. Yan and Z. Chen, Interface Mechanisms of Catalytic Ozonation with Amorphous Iron Silicate for Removal of 4-Chloronitrobenzene in Aqueous Solution, Environ. Sci. Technol., 2018, 52, 1429–1434 CrossRef CAS PubMed.
- S. Zhang, L. Gutierrez, X. Niu, F. Qi and C. Jean-Philippe, The characteristics of organic matter influence its interfacial interactions with MnO2 and catalytic oxidation processes, Chemos, 2018, 209, 950–959 CrossRef CAS PubMed.
- J. Niu, W. Li and Z. Li, Preparation of Cu-Mn/Fe3O4@SiO2@KCC Nanocatalyst and Its Application in Catalytic Ozonation for Degradation of Terephthalic Acid, RSC Adv., 2007, 18(6), 354–360 Search PubMed.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Environmental implications of hydroxyl radicals (˙OH), Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- H. Jung and H. Choi, Catalytic decomposition of ozone and ara-Chlorobenzoic acid (pCBA) in the presence of nanosized ZnO, Appl. Catal., B, 2006, 66, 288–294 CrossRef CAS.
- Y. Liu, Q. Fan and J. Wang, Zn-Fe-CNTs catalytic in situ generation of H2O2 for Fenton-like degradation of sulfamethoxazole, J. Hazard. Mater., 2018, 342, 166–176 CrossRef CAS PubMed.
- M. Sui, L. Sheng, K. Lu and F. Tian, FeOOH catalytic ozonation of oxalic acid and the effect of phosphate binding on its catalytic activity, Appl. Catal., B, 2010, 96, 94–100 CrossRef CAS.
- S. Tang, D. Yuan, Q. Zhang, Y. Liu, Z. Liu and H. Huang, Fe–Mn bi-metallic oxides loaded on granular activated carbon to enhance dye removal by catalytic ozonation, Environ. Sci. Pollut. Res., 2016, 23, 18800–18808 CrossRef CAS PubMed.
- L. Yuan, J. Shen, Z. L. Chen and X. H Guan, Synergistic role of pumice surface composition in hydroxyl radical initiation in the catalytic ozonation process, Ozone: Sci. Eng., 2016, 38, 42–47 CrossRef CAS.
- G. L. Richmond, Molecular bonding and interactions at aqueous surfaces as probed by vibrational sum frequency spectroscopy, Chem. Rev., 2002, 102, 2693–2724 CrossRef CAS PubMed.
- Y. Zeng and J. Park, Characterization and coagulation performance of a novel inorganic polymer coagulant—poly-zinc-silicate-sulfate, Colloids Surf., A, 2009, 334, 147–154 CrossRef CAS.
- M. Selvaraj, B. R. Min, Y. G. Shul and T. G. Lee, Comparison of mesoporous solid acid catalysts in the production of DABCO by cyclization of ethanolamine: I. Synthesis and characterization of mesoporous solid acid catalysts, Microporous Mesoporous Mater., 2004, 74, 143–155 CAS.
- E. Dalchiele, P. Giorgi, R. Marotti, F. Martín, J. Ramos-Barrado, R. Ayouci and D. Leinen, Electrodeposition of ZnO thin films on n-Si(1 0 0), Sol. Energy Mater. Sol. Cells, 2001, 70, 245–254 CrossRef CAS.
- B. V. Crist, Handbook of the Elements and Native Oxides, XPS International: Mountain View, CA, 1999 Search PubMed.
- B. M. Reddy, B. Chowdhury and P. G. Smirniotis, An XPS study of the dispersion of MoO3 on TiO2–ZrO2, TiO2–SiO2, TiO2–Al2O3, SiO2–ZrO2, and SiO2–TiO2–ZrO2 mixed oxides, Appl. Catal., A, 2001, 211, 19–30 CrossRef CAS.
- X. Pang, Y. Guo, Y. Zhang, B. Xu and F. Qi, LaCoO3 perovskite oxide activation of peroxymonosulfate for aqueous 2-phenyl-5-sulfobenzimidazole degradation: effect of synthetic method and the reaction mechanism, Chem. Eng. J., 2016, 304, 897–907 CrossRef CAS.
- H. He, Y. Liu, W. Deli, X. Guan and Y. Zhang, Ozonation of dimethyl phthalate catalyzed by highly active CuxO-Fe3O4 nanoparticles prepared with zero-valent iron as the innovative precursor, Environ. Pollut., 2017, 227, 73–82 CrossRef CAS PubMed.
- O. Turkay, H. Inan and A. Dimoglo, Experimental and theoretical investigations of CuO-catalyzed ozonation of humic acid, Sep. Purif. Technol., 2014, 134, 110–116 CrossRef CAS.
- Y. Liu, Z. Chen, W. Gong, Y. Dou, S. Wang and W. Wang, Structural characterizations of zinc-copper silicate polymer (ZCSP) and its mechanisms of ozonation for removal of p-chloronitrobenzene in aqueous solution, Sep. Purif. Technol., 2017, 172, 251–257 CrossRef CAS.
- H. Valdes, V. Farfan, J. Manoli and C. A. Zaror, Catalytic ozone aqueous decomposition promoted by natural zeolite, J. Hazard. Mater., 2009, 165(1–3), 915–922 CrossRef CAS PubMed.
- F. Qi, B. Xu, Z. Chen, J. Ma, D. Suna, L. Zhang and F. Wu, Ozonation catalyzed by the raw bauxite for the degradation of 2,4,6-trichloroanisole in drinking water, J. Hazard. Mater., 2009, 168, 246–252 CrossRef CAS PubMed.
- Y. Liu, J. Shen, Z. Chen and Y. Liu, Degradation of p-chloronitrobenzene in drinking water by manganese silicate catalyzed ozonation, Desal, 2011, 279, 219–224 CrossRef CAS.
- H. Bader and J. Hoigné, Determination of ozone in water by the indigo method Original, Water Res., 1981, 15, 449–456 CrossRef CAS.
- E. Laiti, L. O. Ohman, J. Nordin and S. Sjöberg, Acid/base properties and phenylphosphonic acid complexation at the aged γ-Al2O3/water interface, J. Colloid Interface Sci., 1995, 175, 230–238 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |