
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular dynamics simulations of loading and unloading of drug molecule bortezomib on graphene nanosheets†
Songwei Zenga,
Yu Jib,
Yue Shenb,
Ruiyao Zhub,
Xiaogang Wangb,
Liang Chen b and
Junlang Chen*ab
b and
Junlang Chen*ab
aSchool of Information and Industry, Zhejiang A&F University, Lin'an 311300, China. E-mail: chenjunlang7955@sina.com
bDepartment of Optical Engineering, Zhejiang A&F University, Lin'an 311300, China
First published on 28th February 2020
Abstract
Graphene has been regarded as one of the most hopeful candidates for transporting drugs to target cells because of its huge surface area and high cellular uptake. In this work, we performed molecular dynamics simulations to investigate the potential application of graphene as a substrate to carry and deliver drug molecules. Bortezomib (BOR) was selected as a model drug, as its atomic structure and polarity are suitable to be adsorbed on pristine graphene (PG) and graphene oxide (GO). First, BOR molecules are loaded on graphene surface to form graphene–BOR complexes, then these complexes readily enter the lipid bilayer and finally BOR releases from graphene surface into the membrane. The entry of graphene–BOR complexes into the membrane is mainly driven by the hydrophobic interactions between lipid tails and the basal plane of nanosheets, while the electrostatic interaction between the polar groups of BOR and lipid headgroups contributes to the release of BOR from graphene into the membrane. Different from PG, BOR molecules are hard to remove from GO surface after the complex enters the lipid bilayer. The electrostatic attraction from the oxygen-containing groups enhances the binding of BOR on GO. Potential of mean force calculations confirm that BOR on GO has lower free energy than it adsorbed on PG surface. The results indicate that the adsorption intensity and release rate of graphene nanosheets can be tuned by oxidation and electrification, and graphene served as substrate to transport and release particular drug molecules is feasible.
Introduction
Graphene and its derivatives have shown increasing potential in the fields of biomedicine and biotechnology because of the unique atomic structure, physical and chemical properties.1–3 For biomedical applications, graphene and its derivatives have been found as potential carriers for drug and gene delivery due to their large specific surface area available for efficient drug loading and abundant functional groups for easy surface modification.4,5 The graphene-based composites are often water-soluble and show high cellular uptake. The significance of loading drugs on nanocarriers is avoiding enzymatic degradation or hydrolization before reaching the target cells.6,7 In early 2008, Dai et al. first introduced graphene-based nanomaterials into the field of biomedical sciences.8 They successfully synthesized PEGylated graphene oxide (GO) sheets to adsorb SN38, a camptothecin analogue. The formed GO–PEG–SN38 composite can access the cell interior via passive penetration or endocytosis.Since then, interest in its biomedical applications has gained great momentum. Functionalized nano-graphene has been extensively explored for applications in biosensors,9–12 cellular imaging13–15 and drug delivery.16–18 For example, the same group used GO–PEG composite to load another anticancer drug (doxorubicin, DOX) and the loaded drug exhibited a pH responsive release property.19 Zhang et al. further investigated the targeted delivery of multiple anticancer drugs by using functionalized GO.20 In their work, two types of anticancer drugs, DOX and camptothecin (CPT), were loaded together onto folic acid (FA) conjugated GO (GO–FA) to specifically target MCF-7 human breast cancer cells. The GO–FA complex possesses a selective cytotoxicity to those positive cells. In the delivery of paclitaxel (PTX), Xu et al. developed GO–PEG/PTX composites, which exhibit a relatively high loading capacity. Compared with free PTX, GO–PEG/PTX of same amount led to lower cell viability, with an approximately 30% and 10% relative cell viability of A549 and MCF-7, respectively.21 Except the delivery of small therapeutic molecules, graphene-based nanomaterials have also been explored for gene delivery due to their unique physiochemical properties. Plasmid DNA or small interfering RNA loaded on GO–PEI complex is delivered into cells to knockdown the expression of its target gene.22–24
Complementary to experimental studies, density functional theory (DFT) and molecular dynamics (MD) simulations have been extensively employed to study graphene loading and delivering drug molecules.25–30 The theoretical calculations can provide the detailed interactions between drugs and their nanovectors, such as π–π stacking, electrostatic interaction and hydrogen bonding. For example, Mahdavi et al. performed MD simulations to investigate the drug DOX loading and release on pristine graphene (PG) and GO.28 They found that the adsorption and desorption of drug on graphene are pH-dependent. The neutral pH is beneficial to the drug loading on GO, while the release occurs in an acidic condition (pH = 5). Using DFT and MD simulations, Hasanzade et al. investigated the adsorption of anticancer drug thioguanine (TG) on GO sheets.29 DFT calculations show that the hydrogen bond interactions between TG molecules and the functional groups of GO play the main role in the adsorption. MD results demonstrate that the drug takes longer time to bind with GO and the system becomes unstable when water and ethanol exist. Similarly, Safdari et al. performed DFT and MD simulations to study the interaction of another anticancer drug 5-fluorouracil (5-FU) with GO.30 The results illustrate that the adsorption of 5-FU on GO is exothermic and increasing the simulation temperature is beneficial to form hydrogen bond between their polar groups.
However, the above calculations are only performed at gas phase or in the water solution without cell membranes. The passive penetration of graphene–drug complex into the membrane and the subsequent drug release have yet to be studied theoretically. In this work, we use classic MD simulations to investigate the loading of small drug molecule bortezomib (BOR) on PG and GO in the water solution and the release of this drug into the lipid membrane. We find that the graphene–BOR complexes can readily penetrate the lipid bilayer and then BOR molecules begin to release from PG into the bilayer but are difficult to leave from GO surface. Potential of mean force calculations confirm that the binding affinity of BOR to GO is much stronger than that to PG. The hydrogen bonding enhances the adsorption of BOR on GO surface.
Computational methods
The simulation system consisted of a fully hydrated lipid bilayer, a graphene nanosheet and small drug molecules. The hydrated bilayer developed by Tieleman et al. was composed of 128 dipalmitoylphosphatidylcholine (DPPC) lipids and about 5000 water molecules.31 Bortezomib (abbreviated as BOR), an anticancer drug, was adopted as a model drug molecule, which has two aromatic rings and is prone to form π–π stacking with graphene. Two kinds of graphene nanosheets (PG and GO) were constructed. GO was developed based on the Lerf–Klinowski model with epoxy, hydroxyl groups attached on the basal plane and carboxyl groups bonded to the edge randomly.32 The ratio of carbon atoms to epoxy, hydroxyl and carboxyl groups was 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1, representing a typical oxidation of PG.33 The force field parameters for DPPC lipids and PG were taken from Berger et al. and Patra et al., respectively.34,35 The force field parameters for BOR were developed by Automated Topology Builder.36 Water was represented by the SPC model.37 The carbon atoms in PG were treated as uncharged Lennard-Jones (LJ) spheres with a cross section of σcc = 0.34 nm and a depth of the potential well of εcc = 0.36 kJ mol−1.38 All parameters are compatible with GROMOS 53a6 force field.39
:2:2:1, representing a typical oxidation of PG.33 The force field parameters for DPPC lipids and PG were taken from Berger et al. and Patra et al., respectively.34,35 The force field parameters for BOR were developed by Automated Topology Builder.36 Water was represented by the SPC model.37 The carbon atoms in PG were treated as uncharged Lennard-Jones (LJ) spheres with a cross section of σcc = 0.34 nm and a depth of the potential well of εcc = 0.36 kJ mol−1.38 All parameters are compatible with GROMOS 53a6 force field.39
All simulations were performed under the isothermal–isobaric (NPT) ensemble by using the Gromacs package 5.1.2 with periodic boundary conditions in all three directions.40–42 The vdW interactions were calculated with a smooth cutoff of 1.2 nm, whereas the particle-mesh Ewald (PME) method was used to calculate the long-range electrostatic interactions.43,44 The pressure was controlled semi-isotropically by a Berendsen barostat at 1 bar and the temperature was kept stable at 323 K using the V-rescale method.45,46 Bond lengths within water and non-water molecules were constrained by the SETTLE and LINCS algorithms, which allowed a time step of 2.0 fs.47,48
The adsorption energy of BOR on PG at aqueous phase was computed from the potential of mean force (PMF) using umbrella sampling.49 First, we conducted steered MD simulation to pull the molecule away from the PG surface along the normal to PG surface (z-axis).50 Then, 32 windows were generated, in which the z coordinates of COM distance between the molecule and PG in each window differed by about 0.1 nm to ensure sufficient sampling. Each window was run for 10 ns, and data in the last 5 ns was used for sampling. Finally, the PMF profile was obtained by the Weighted Histogram Analysis Method (WHAM), implanted in the GROMACS package as ‘gmx wham’.51 The free energy of BOR across the lipid membrane was calculated by the same approach.
Results and discussion
1. The loading of BOR on PG and GO
To evaluate the stability of graphene–BOR complexes at the aqueous phase, we first investigate the distribution of BOR in the water solution with a fixed PG or GO nanosheet. Graphene nanosheets as drug carriers are interesting because both sides of a single sheet could be accessible for drug binding. As shown in Fig. 1A, all BOR molecules are adsorbed on both sides of PG and GO nanosheets after equilibrium. Mass density of BOR demonstrates that they are distributed almost evenly on both sides of the basal plane (see Fig. 1C). The two symmetric peaks were localized at z = ±0.38 nm, close to the vdW radius of carbon atoms on PG. The adsorption intensity can be more quantitatively elucidated by the PMF of BOR at different positions along the normal to graphene surface (Fig. 1D). The PMF of BOR in the aqueous phase far away from the PG/GO surface, where the interactions between BOR and graphene can be neglected, is set as zero. We find that the minimum value of PMF is at z = 0.39 nm. Such position is in good agreement with the mass density profile, further confirming the energetically favorable location of BOR on both PG and GO surface. The binding energies at this point reach highly −60.1 ± 1.3 kJ mol−1 and −81.9 ± 2.2 kJ mol−1, implying that the adsorption of BOR on PG/GO surface is thermodynamically stable.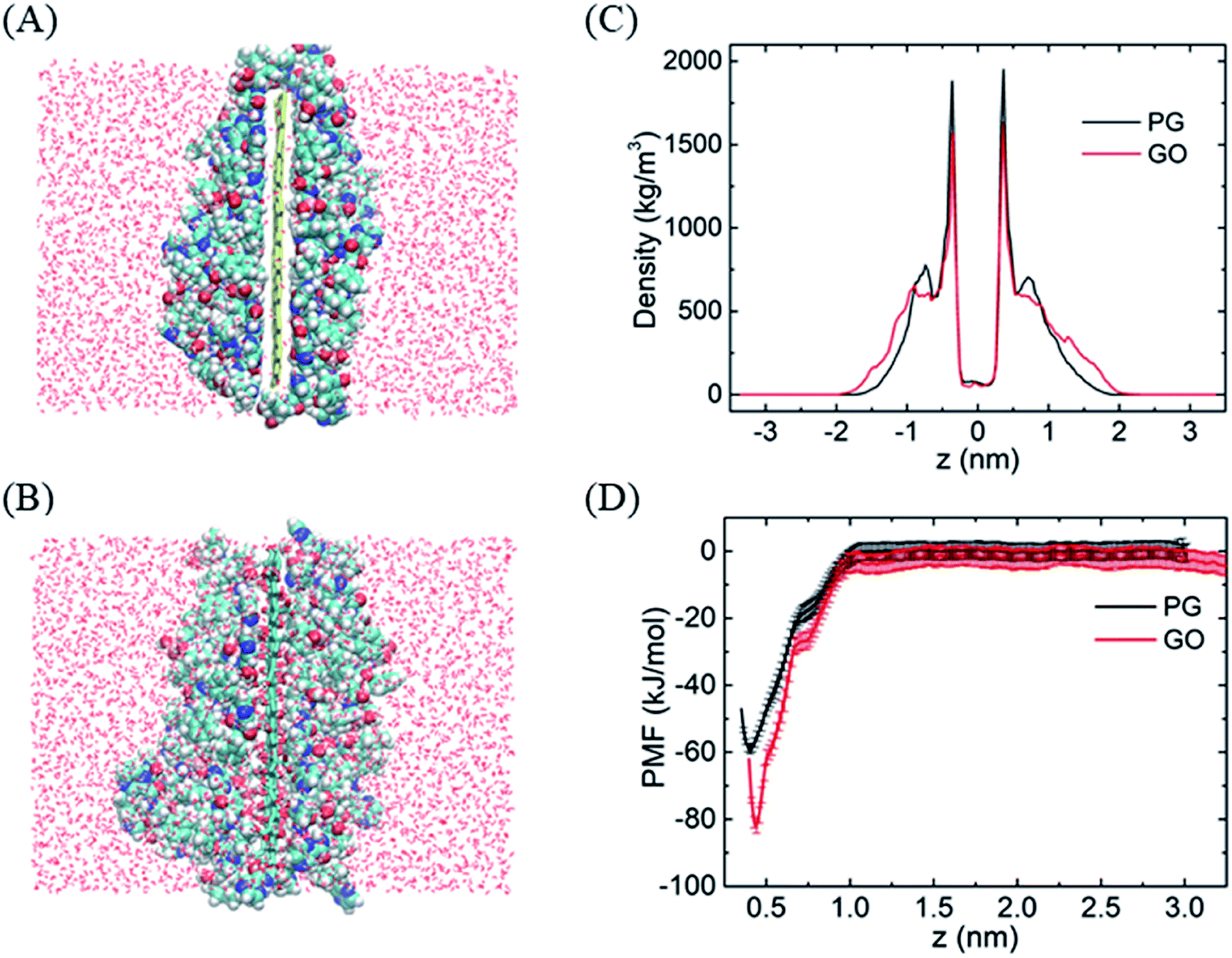 | ||
| Fig. 1 The adsorption of BORs on PG and GO nanosheets. (A and B) The structures of BOR molecules on PG/GO surface, (C) mass density profiles of BOR. The center of PG/GO is set as z = 0. (D) PMF profiles of BOR on PG/GO surface. Error bars denote the standard deviation as calculated using the bootstrap error analysis.51 | ||
The strong adsorption of BOR on graphene surface mainly originates from the hydrophobic π–π stacking interactions, which are determined based upon two geometrical criteria: the distance between two adjacent aromatic planes (d < 0.48 nm) and the angles between axes normal to the two adjacent planes (α < 30°) of the aromatic structures.52,53 It is found that one or two aromatic rings on BOR stick to the PG/GO surface in the first adsorbed layer. The typical structures of π–π stacking are presented in Fig. 2A and B, in which the two aromatic rings are close to the PG/GO surface with almost parallel orientations. As to GO, it should be pointed out that π–π interactions are established between aromatic rings and unoxided regions of GO. Two parameters, namely, the vertical distance and included angle between two aromatic rings and PG/GO, are employed to characterize these interactions, as shown in Fig. 2C and D. The most probable vertical distance between aromatic rings and PG/GO surface is located at z = 0.36 nm, which is consistent with the vdW radius of carbon atoms. Meanwhile, the most probable included angles between BOR and PG/GO are approximately 10° and 22°, indicating that the aromatic rings of BOR are almost parallel to the PG surface. These results further confirm that the loading of BOR on PG/GO is highly stable and π–π stacking is the main force in such adsorption. Besides π–π interactions, there are averaged 1.4 hydrogen bonds (Fig. 2E) formed between BOR and GO because of the oxygen-containing groups, which can explain the higher adsorption energy (∼20 kJ mol−1) of BOR on GO than that on PG surface.
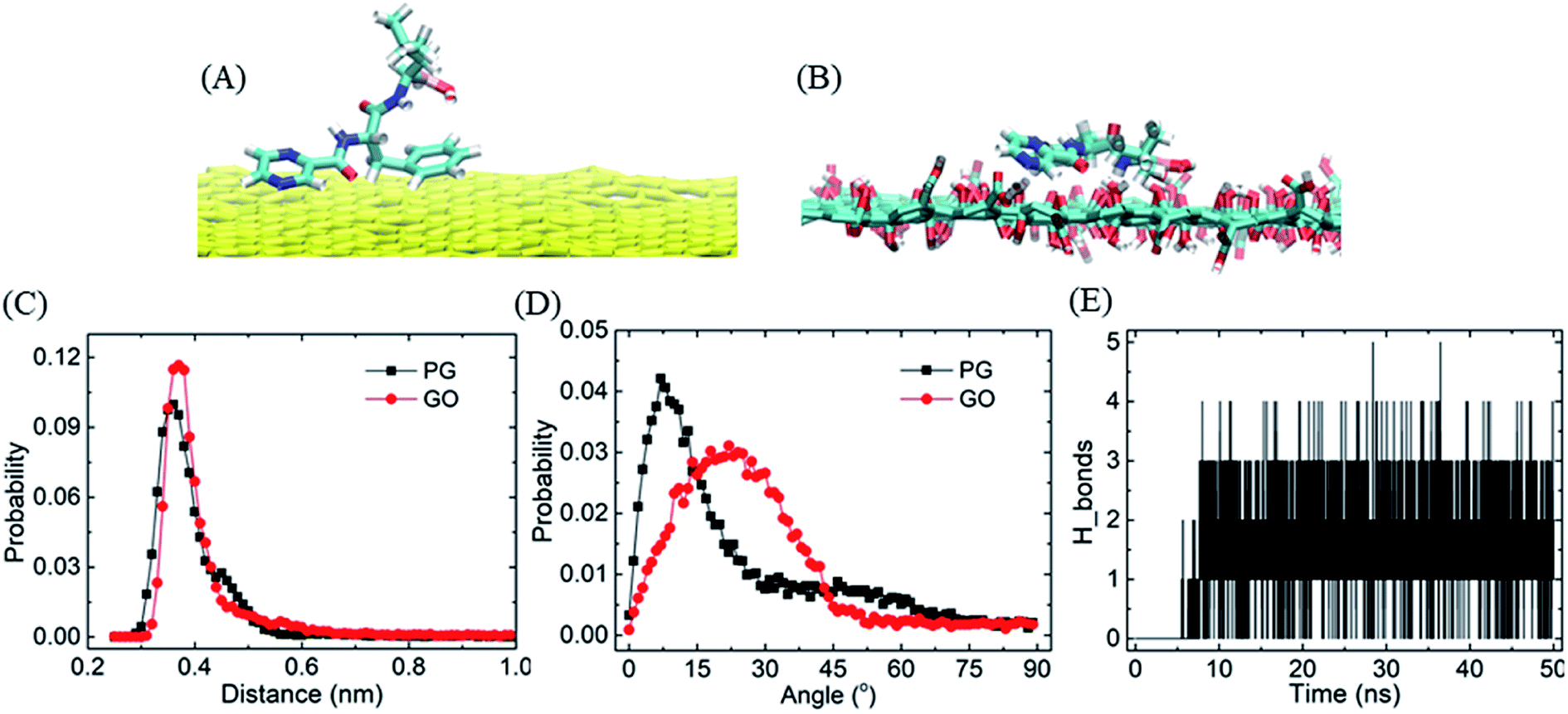 | ||
| Fig. 2 The interactions between BOR and PG/GO. (A and B) The structures of BOR on PG and GO, (C and D) the probability of the vertical distance and included angle between two aromatic rings and PG/GO, (E) the number of hydrogen bonds formed between BOR and GO. | ||
Similarly, because of the hydrophobic π–π stacking interactions, BOR molecules are prone to form cluster in the water solution. We therefore calculate the cohesive energy of BOR at aqueous phase, which is described by the PMF of BOR at the different position, as shown in Fig. 3. The cohesive energy of BOR cluster is approximately −48.5 ± 0.8 kJ mol−1, slightly weaker than the adsorption energy of BOR on PG (−60.1 kJ mol−1, Fig. 1C), indicating that BOR can be preferably adsorbed on PG/GO surface. On the other hand, this strong cohesive energy makes BOR molecules stacked tightly. As a result, the complexes of PG/GO surrounded by BOR molecules are highly stable in the water solution.
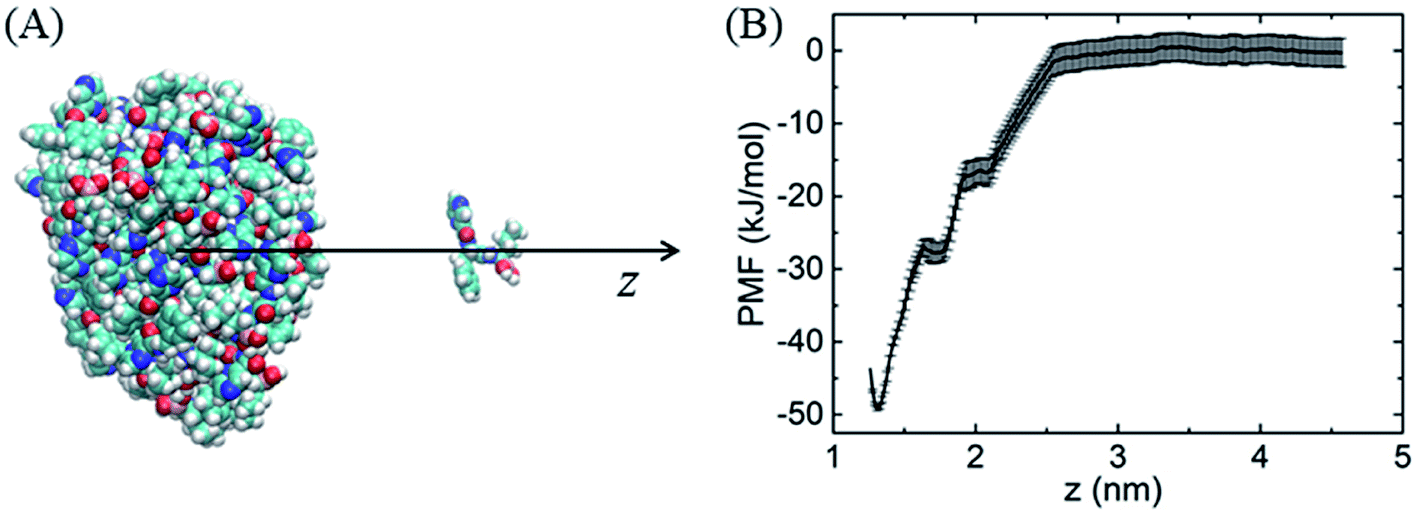 | ||
| Fig. 3 (A) Schematic illustration of sampling structure, (B) PMF profile of BOR cluster at aqueous phase. | ||
2. The entry of PG/GO–BOR complexes in the membrane
We then begin to study the passive lipid membrane permeation of the PG–BOR complex. According to the size of membrane, we cut an ultra-small graphene nanosheet (∼2 × 2 nm2). First, the PG sheet is covered by BOR molecules. Then the first layer of BOR molecules is retained to form a PG–BOR conjugate, which consists of a PG nanosheet and 6 BOR molecules. Each side of PG loads 3 drugs. The complex is initially placed out of the lipid bilayer (Fig. 4A, snapshot at t = 0 ns), and then released. It diffuses slowly towards the interface between lipid headgroups and water solution at the beginning. As shown in Fig. 3B, the COM distance between the complex and bilayer decreases gradually from 3.93 nm to 2.09 nm in the first 23 ns. BOR molecules remain adsorbed to the PG outside of the bilayer. Once reaching the interface, the complex is fast adsorbed into bilayer (Fig. 4A, snapshot at t = 21, 27 ns), as there is a sharp fall in COM distance curve. The COM distance continues to decline from 2.09 nm to 0.51 nm in the following 3 ns. In this stage, the insertion is driven mainly by the electrostatic interactions between polar groups on BOR and DPPC lipids. After insertion, the complex remains in the bilayer center until the end of simulation. The COM distance fluctuates slightly in the range of 0 to 0.5 nm.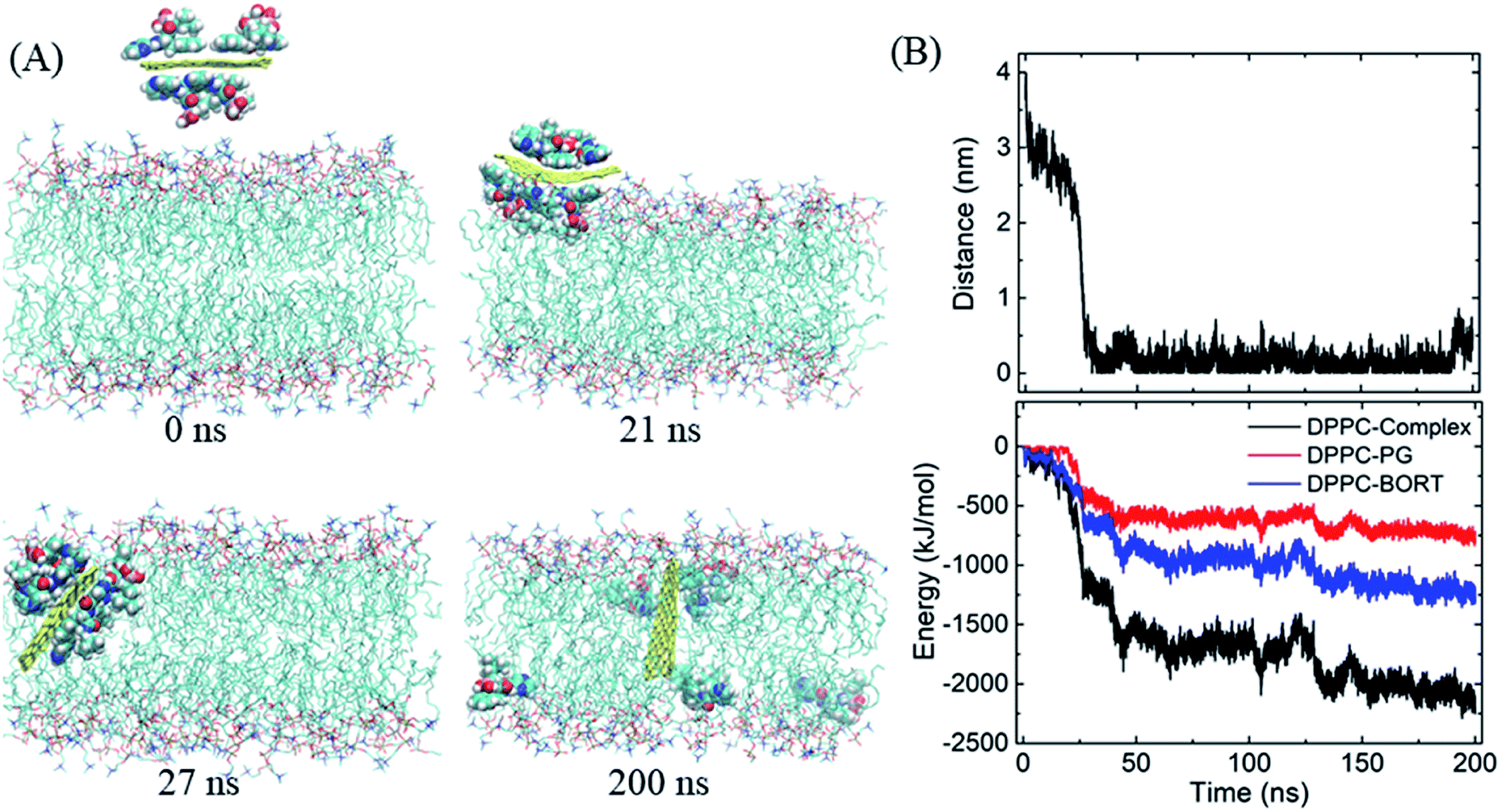 | ||
| Fig. 4 Insertion of the PG–BOR complex in the membrane. (A) Snapshots at critical time points. Water molecules are not shown for clarity. (B) COM distance and interaction energy between the complex and lipid bilayer as functions of simulation time. | ||
However, both PG and lipid tails as well as BOR are much hydrophobic, therefore, the hydrophobic interactions play the dominant role when the complex enters the membrane. To obtain a more quantitative picture of these interactions, we calculated the interaction energy between the complex and lipid bilayer. Here, the interaction energy was defined as the vdW interaction between the complex and membrane, as the carbon atoms on PG were uncharged. And the energy can be divided into two parts, since the complex consists of PG and BOR molecules. The energy curve presents the same trend as that of COM distance (Fig. 4B). The energy difference of PG–BOR complex in and out of the bilayer reaches approximately 2000 kJ mol−1 (PG contributes about 600 kJ mol−1, while BOR molecules provide the rest 1400 kJ mol−1). It is clear that the huge fall of the energy makes the complex fast adsorbed into the membrane.
Similarly, the GO–BOR complex can also spontaneously enter the membrane and reach the bilayer center, as shown in Fig. 5A. Since GO has the same size as PG and is localized at bilayer center with similar orientation, the vdW interactions of GO–BOR complex and bilayer are close to those of PG–BOR. We therefore only calculated the electrostatic energy between GO and lipid membrane (Fig. 5B). It is found that the electrostatic energy is only −57.8 kJ mol−1 averaged over the last 100 ns, which can almost be neglected compared with the vdW energy of PG–BOR complex and bilayer (highly −2000 kJ mol−1). That is, the driving force of GO–BOR complex entering the membrane is still dominated by the hydrophobic interactions. However, the oxygen-containing groups on GO surface are unfavorable to drug release. Only one BOR molecule breaks away from GO surface after the complex entering the membrane (see snapshot at t = 200 ns).
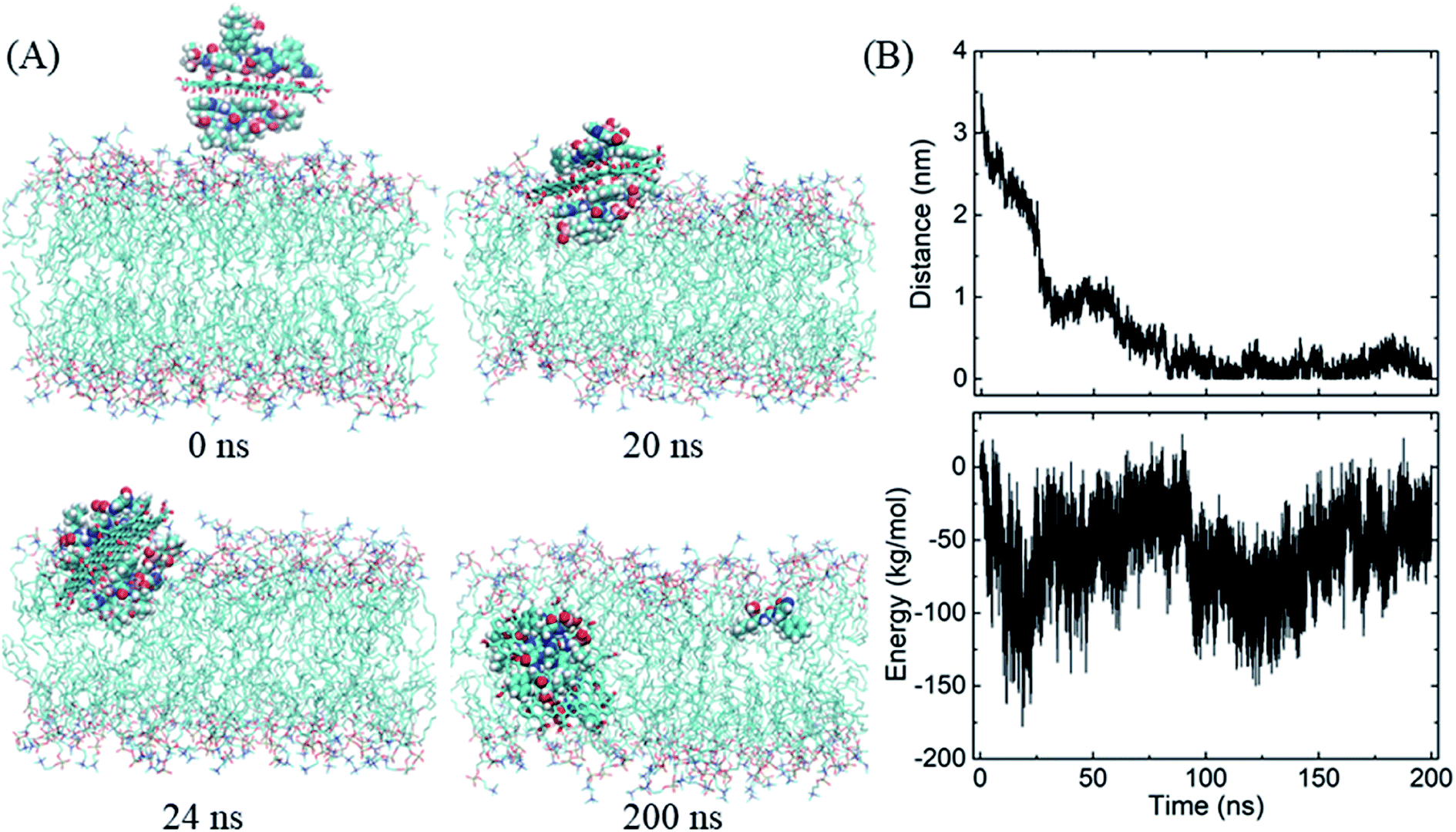 | ||
| Fig. 5 Insertion of the GO–BOR complex in the membrane. (A) Snapshots at critical time points. (B) COM distance and electrostatic energy between GO and lipid bilayer as functions of simulation time. | ||
3. BOR unloading from PG/GO into the membrane
After entering the membrane, BOR molecules gradually leave the PG surface and permeate into the bilayer (Fig. 4A, snapshot at t = 200 ns). Interestingly, it is found that if the complex is positioned out of but close to the membrane (in detail, the PG sheet is restrained while BOR molecules are free to move), BOR molecules still remain on the PG surface (see ESI, Fig. S1†). That is, only when the complex inserts the membrane, will the drug molecules release. To verify the robustness of the release of BOR from PG to membrane, we performed independent simulation that the complex was initially embedded in the bilayer center (Fig. 6A, t = 0 ns). At the end of simulation, all BOR molecules break away from the adsorption of PG and permeate into the lipid membrane (Fig. 6A, t = 200 ns). Different from PG, BOR on GO embedded in bilayer center (Fig. 6B, t = 0 ns) turns to be difficult to release. Only 4 of 6 BOR molecules leave GO surface and permeate into the membrane. This is because the adsorption of BOR on GO is much stronger than that on PG surface, due the electrostatic interactions of the oxygen-containing groups on GO and polar groups of BOR. Then, BOR molecules prefer to stay at the conjunctions between lipid headgroups and tails, as shown in the mass density profile (Fig. 6C and ESI, Fig. S2†), and such positions are in good agreement with PMF profile of BOR across the lipid bilayer. The minimum of PMF is about −72.3 ± 1.2 kJ mol−1 at the symmetric positions (z = ± 1.0 nm) from the bilayer center, indicating that BOR molecules are energetically favorable at these positions. The middle peak of red line corresponds to the BOR molecules that are not released. We repeated the entry and release of the complex with graphene loading more BOR molecules (see ESI, Fig. S3†), and obtained the similar results, indicating that using graphene to load and unload specific drug molecules is feasible.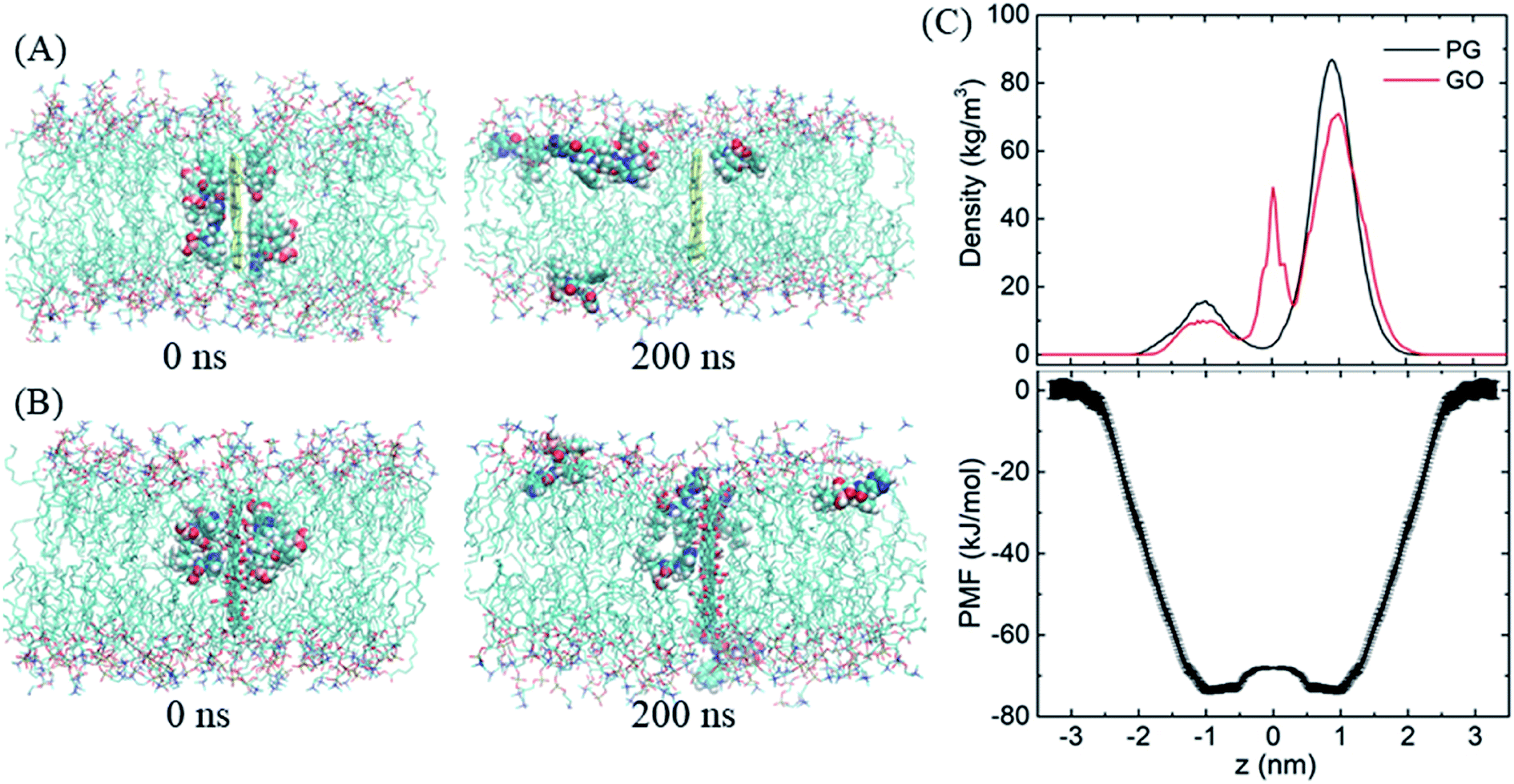 | ||
| Fig. 6 The release of BOR from graphene to membrane. (A and B) Initial and final snapshots of BOR release from PG/GO surface, (C) mass density and PMF profiles of BOR across the lipid bilayer. | ||
Conclusions
In summary, using MD simulations, we have investigated the adsorption of small anticancer drug BOR on graphene surface in the water solution, the insertion of graphene–BOR complexes in the lipid bilayer and the release of BOR from graphene surface into the membrane. The results show that the formed graphene–BOR complexes are highly stable in the water solution with or without lipid bilayer. The complexes can spontaneously permeate into the membrane and reach the bilayer center, mainly due to the hydrophobic interactions between the complex and lipid tails. Then, the drug BOR molecules release one by one from PG into the lipid membrane, which is driven mainly by the electrostatic interactions between lipid headgroups and BOR polar groups. As comparison, BOR molecules become difficult to leave from GO surface because of the electrostatic interactions between oxygen-containing groups and polar groups on BOR. Graphene exhibits prospective potential as nanovehicle to carry and deliver small drug molecules through oxidation or modification on its surface charge.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 11875236, U1832150, 61975185), the Zhejiang Provincial Natural Science Foundation of China (Grant No. LY18A040001), and Zhejiang Provincial Science and Technology Project (Grant No. LGN18C200017).References
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2011, 46, 2211–2224 CrossRef PubMed.
- H. Y. Mao, S. Laurent, W. Chen, O. Akhavan, M. Imani, A. A. Ashkarran and M. Mahmoudi, Chem. Rev., 2013, 113, 3407–3424 CrossRef CAS PubMed.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Chem. Res. Toxicol., 2012, 25, 15–34 Search PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS.
- Y. Wang, Z. Li, J. Wang, J. Li and Y. Lin, Trends Biotechnol., 2011, 29, 205–212 CrossRef CAS.
- Z. R. Yang, H. F. Wang, J. Zhao, Y. Y. Peng, J. Wang, B. A. Guinn and L. Q. Huang, Cancer Gene Ther., 2007, 14, 599–615 CrossRef CAS PubMed.
- S. Goenka, V. Sant and S. Sant, J. Controlled Release, 2014, 173, 75–88 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- S. He, B. Song, D. Li, C. Zhu, W. Qi, Y. Wen, L. Wang, S. Song, H. Fang and C. Fan, Adv. Funct. Mater., 2010, 20, 453–459 CrossRef CAS.
- C. Lu-Hua, H. Yang-Hao, C. Zhu-Ling, X. Chen and G. Chen-Nan, Angew. Chem., 2009, 121, 4879–4881 CrossRef.
- H. Dong, W. Gao, F. Yan, H. Ji and H. Ju, Anal. Chem., 2010, 82, 5511–5517 CrossRef CAS PubMed.
- S. S. Chou, M. De, J. Luo, V. M. Rotello, J. Huang and V. P. Dravid, J. Am. Chem. Soc., 2012, 134, 16725–16733 CrossRef CAS PubMed.
- Y. Wang, Z. Li, D. Hu, C.-T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS PubMed.
- J. Huang, C. Zong, H. Shen, M. Liu, B. Chen, B. Ren and Z. Zhang, Small, 2012, 8, 2577–2584 CrossRef CAS PubMed.
- H. Hong, K. Yang, Y. Zhang, J. W. Engle, L. Feng, Y. Yang, T. R. Nayak, S. Goel, J. Bean, C. P. Theuer, T. E. Barnhart, Z. Liu and W. Cai, ACS Nano, 2012, 6, 2361–2370 CrossRef CAS PubMed.
- S. Makharza, G. Cirillo, A. Bachmatiuk, I. Ibrahim, N. Ioannides, B. Trzebicka, S. Hampel and M. H. Rummeli, J. Nanopart. Res., 2013, 15, 2099 CrossRef.
- H. Zhao, R. Ding, X. Zhao, Y. Li, L. Qu, H. Pei, L. Yildirimer, Z. Wu and W. Zhang, Drug Discovery Today, 2017, 22, 1302–1317 CrossRef CAS PubMed.
- K. Yang, L. Feng and Z. Liu, Expert Opin. Drug Delivery, 2015, 12, 601–612 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- Z. Xu, S. Wang, Y. Li, M. Wang, P. Shi and X. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 17268–17276 CrossRef CAS PubMed.
- L. Feng, S. Zhang and Z. Liu, Nanoscale, 2011, 3, 1252–1257 RSC.
- H. Kim, R. Namgung, K. Singha, I. K. Oh and W. J. Kim, Bioconjugate Chem., 2011, 22, 2558–2567 CrossRef CAS PubMed.
- L. Zhang, Z. Lu, Q. Zhao, J. Huang, H. Shen and Z. Zhang, Small, 2011, 7, 460–464 CrossRef CAS PubMed.
- W. Qin, X. Li, W. W. Bian, X. J. Fan and J. Y. Qi, Biomaterials, 2010, 31, 1007–1016 CrossRef CAS PubMed.
- W. Zhao, A. Sugunan, Z.-B. Zhang and A. Ahniyaz, J. Phys. Chem. C, 2019, 123, 26282–26288 CrossRef CAS.
- M. H. Wang, Q. Wang, X. Lu, K. F. Wang, L. Fang, F. Ren, G. Lu and H. Zhang, J. Phys. Chem. B, 2017, 121, 7907–7915 CrossRef CAS PubMed.
- M. Mahdavi, F. Rahmani and S. Nouranian, J. Mater. Chem. B, 2016, 4, 7441–7451 RSC.
- Z. Hasanzade and H. Raissi, Appl. Surf. Sci., 2017, 422, 1030–1041 CrossRef CAS.
- F. Safdari, H. Raissi, M. Shahabi and M. Zaboli, J. Inorg. Organomet. Polym., 2017, 27, 805–817 CrossRef CAS.
- D. P. Tieleman and H. J. C. Berendsen, J. Chem. Phys., 1996, 105, 4871–4880 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- N. V. Medhekar, A. Ramasubramaniam, R. S. Ruoff and V. B. Shenoy, ACS Nano, 2010, 4, 2300–2306 CrossRef CAS PubMed.
- O. Berger, O. Edholm and F. Jahnig, Biophys. J., 1997, 72, 2002–2013 CrossRef CAS PubMed.
- N. Patra, B. Wang and P. Kral, Nano Lett., 2009, 9, 3766–3771 CrossRef CAS PubMed.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- G. Hummer, J. C. Rasaiah and J. P. Noworyta, Nature, 2001, 414, 188–190 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. v. d. Spoel and R. v. Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- B. Hess, C. Kutzner, D. v. d. Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. v. Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- J. A. Lemkul and D. R. Bevan, J. Phys. Chem. B, 2004, 114, 1652–1660 CrossRef PubMed.
- S. Park and K. Schulten, J. Chem. Phys., 2004, 120, 5946–5961 CrossRef CAS PubMed.
- J. S. Hub, B. L. d. Groot and D. v. d. Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS.
- C. A. Hunter, J. Mol. Biol., 1991, 218, 837–846 CrossRef CAS PubMed.
- G. B. McGaughey, M. Gagne and A. K. Rappe, J. Biophys. Chem., 1998, 273, 15458–15463 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00261e |
| This journal is © The Royal Society of Chemistry 2020 |