
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new spinel high-entropy oxide (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 with fast reaction kinetics and excellent stability as an anode material for lithium ion batteries†
Hong Chen ,
Nan Qiu*,
Baozhen Wu,
Zhaoming Yang,
Sen Sun and
Yuan Wang*
,
Nan Qiu*,
Baozhen Wu,
Zhaoming Yang,
Sen Sun and
Yuan Wang*
Key Laboratory of Radiation Physics and Technology, Ministry of Education, Institute of Nuclear Science and Technology, Sichuan University, Chengdu 610064, People's Republic of China. E-mail: qiun@scu.edu.cn; wyuan@scu.edu.cn
First published on 6th March 2020
Abstract
It is well known that transition metal oxides (TMOs) have attracted extensive attention as promising anodes for next-generation lithium ion batteries (LIBs) owing to their low cost and high theoretical capacities. However, the huge volume changes upon lithiation/delithiation cycling gradually cause drastic particle pulverization in the electrodes, thus leading to fast capacity fading and limiting their practical applications. High-entropy oxides with enhanced electronic conductivity and multiple electrochemically active elements display stepwise lithium storage behaviors, thus efficiently alleviating the volume change induced electrode pulverization problem. Herein, we report the synthesis of a new kind of spinel (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 material via a facile one-step solid state reaction method and subsequent high-energy ball-milling. When used as anodes for LIBs, the submicrometer-sized (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles exhibit superior lithium storage properties, delivering a large reversible capacity of 504 mA h g−1 at a current density of 100 mA g−1 after 300 cycles, and notably an exceptional rate capacity of 272 mA h g−1 at 2000 mA g−1. Our work highlights that rational design of high-entropy oxides with different electrochemically active elements and novel structures might be a useful strategy for exploring high-performance LIB anode materials in next-generation energy storage devices.
1. Introduction
Due to the ever-increasing energy demands and concern around global warming, advanced renewable energy technologies such as portable electronics, electric vehicles and green grid energy storage, have received much attention.1–5 To satisfy the growing demand of advanced rechargeable lithium-ion batteries (LIBs), tremendous efforts have been devoted to explore the electrode materials with long cycle lives, high rates and reversible specific capacities.6–8 Graphite is a commonly used anode material in current commercial LIBs.9 Nevertheless, its low theoretical capacity (372 mA h g−1) and the problem of Li dendrite formation limits the large-scale application in future LIBs system.10,11 Therefore, developing suitable anode materials with higher lithium storage capacities and superior cycle stability is urgently demanded.12–14 As promising anodes for LIBs, transition metal oxides (TMOs, such as Co3O4,15 ZnO,16 CuO,17–21 Fe3O4,22–25 Fe2O3 (ref. 25–27)) have attracted extensive attention owing to their low-cost and high theoretical capacities. However, the large volume changes upon the lithiation/delithiation process gradually causes drastic electrode pulverization, thus leading to fast capacity fading.3,28,29In order to solve the pulverization problem and to achieve enhanced cycling stability, it is reported that introducing foreign atoms into the single metal oxides could be a predominant effective method. A wide variety of ternary oxides have been studied as prospective anode materials for Li-ion batteries which undergo Li-cycling via the “conversion reaction,” and those have been recently reviewed by Reddy et al.12,25,30–37 In particular, iron-based spinel oxides, such as ferrites with general formula AFe2O4 (A = Zn, Ni, Co, Cu, Cd)38–40 and (Ni1−xZnx)Fe2O4 (x = 0, 0.2, 0.25, 0.4, 0.5, 0.6, 0.75, 0.8, 1)41,42 have been extensively studied for their Li-cyclability. Specifically, Zhao and Luo et al.43–45 suggested that the in situ formed Cu nanoclusters from CuFe2O4 not only promoted the reversible formation and decomposition of Li2O but also functioned as an excellent buffer matrix for the Fe2O3 electrode, preventing the aggregation of Fe nanograins and buffering volume changes and structural stress of the electrode during cycling. Thus, excellent electrochemical performance was achieved in the composite CuFe2O4 electrode. Meanwhile, another effective method was complexing with electrochemically inactive materials such as MgO and CaO, which can cause a “spectator effect” to restrain the agglomeration of active nanograins.46–52 Therefore, more efforts and rational design strategies must be done to exploit suitable TMO anode materials with good cycling stability and high specific capacity, which will satisfy practical applications.
High-entropy oxides (HEOs), as a new kind of multicomponent and single-phase solid solution TMOs, have greater electronic conductivity and multiple electrochemically active components as compared with single metal oxides.53–58 They display stepwise lithium storage behaviors, thus efficiently alleviating the volume change and then solving induced electrode pulverization problem to some extent. Herein, we have successfully synthesized a new spinel-structured (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 materials via a facile one-step solid state reaction method and subsequently high-energy ball-milling. When used as anodes for LIBs, the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode exhibits superior lithium storage properties, delivering a large reversible capacity of 504 mA h g−1 at a current density of 100 mA g−1 after 300 cycles, and notably an exceptional rate capacity of 272 mA h g−1 at 2000 mA g−1. Meanwhile, a high capacity retention of 96.2% after 800 cycles at 2000 mA g−1 was obtained.
2. Experimental section
2.1. Materials synthesis
All chemical reagents (purchased from Alfa Aesar Chemical Reagent Co., Ltd) were of analytical purity and used without any further purification. Equimolar amounts of the oxides, MgO (0.01 mol, 0.4030 g, 99.99%), TiO2 (0.01 mol, 0.7987 g, 99%), FeO (0.01 mol, 0.7184 g, 99%), ZnO (0.01 mol, 0.8139 g, 99.9%) and CuO (0.01 mol, 0.7954 g, 99.9%) were mixed as pre-alloyed powder by a planetary ball mill. To ensure adequate mixing, all pre-alloyed powder was milled for at least 2 hours. The mixed powder was then separated into several samples with a weight of 0.500 g per sample, and then pressed into pellets with 1.50 cm diameter using a uniaxial hydraulic press at 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 N. The pre-alloyed pellets were sintered at 1000 °C for 24 hours by using a Protherm PC442 tube furnace. Then checked via X-ray diffraction to ensure phase purity and that peaks remain narrow and intense. The geometrical density of the as-prepared pellets was in the 75–80% range. These pellets were then mixed into an isopropyl alcohol slurry and milled again into powder with yttrium-stabilized balls for 10 hours, then all the powder samples were dried in a fume hood at room temperature.
000 N. The pre-alloyed pellets were sintered at 1000 °C for 24 hours by using a Protherm PC442 tube furnace. Then checked via X-ray diffraction to ensure phase purity and that peaks remain narrow and intense. The geometrical density of the as-prepared pellets was in the 75–80% range. These pellets were then mixed into an isopropyl alcohol slurry and milled again into powder with yttrium-stabilized balls for 10 hours, then all the powder samples were dried in a fume hood at room temperature.
2.2. Materials characterization
The phase structure was characterized by the X-ray diffraction (XRD, Bruker D8 Advance X-ray diffractometer) at voltage of 40 kV and current of 50 mA under Cu Kα radiation (λ = 1.54178 Å), with a 1.5° min−1 of scan speed and a range taken as 2θ = 10–90°. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images were acquired on JEM-1011 with the accelerating voltage of 200 kV and JEOL JSM-6700 M with the accelerating voltage of 10 kV, respectively. The sample for SEM characterization was sputtered a thin layer of gold prior to the measurements. X-ray photoemission spectroscopy (XPS) was performed to determine the compositions and chemical states by using a Thermo Fisher X-ray photoelectron spectrometer ESCALAB 250Xi (non-monochromated Mg Kα X-ray radiation as the excitation source). The high-resolution transmission electron microscopy (HRTEM) images, selected area electron diffraction (SAED) pattern, and EDS elemental mapping images were obtained by using a JEM-ARM200F (Schottky FEG Cs corrected TEM). EDX analysis was carried out in the STEM mode using a Tecnai G2 F20 transmission electron microscope. The sample for TEM measurements was prepared by dispersing the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 nanoparticles into petroleum ether followed by dropping onto a gold grid. A Tristar II 3020 physisorption system (Micromeritics Instrument Corporation, USA) was used to measure nitrogen adsorption–desorption isotherms at the liquid nitrogen temperature (77 K). The Brunauer–Emmett–Teller (BET) method was used to calculate the specific surface areas of the samples from the adsorption branches. The Barrett–Joyner–Halenda (BJH) model was employed to compute pore size distribution curves from the desorption branches.2.3. Electrochemical measurements
The working electrodes for LIBs were first fabricated by mixing active materials (submicrometer-sized (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles), Super P acetylene black (20 wt%) and poly(vinylidene fluoride) (Mw: 534000 g mol−1, Sigma-Aldrich) (10 wt%) using N-methyl pyrrolidone (NMP) as solvent to form a slurry with a weight ratio of 70:20:10. Then, the slurry was coated on Cu foil as current collector with a loading of 1.0 ± 0.2 mg cm−2, dried in a vacuum oven at 80 °C overnight, and then roll-pressed at 10 MPa for 2 minutes. After that, the electrochemical properties were investigated using coin-type cells (CR2025), and the assembly process was operated in the Ar-filled glovebox with H2O and O2 contents less than 1.0 ppm. The electrolyte was adopted by dissolving 1.0 M LiPF6 in ethylene carbonate/dimethyl carbonate (1/1 in volume), the counter electrode was lithium foil, and the separator was a Celgard 2400 microporous membrane. Cyclic voltammetry (CV) tests were carried out on the Autolab PGSTAT 302 N electrochemical station with a potential window of 0.01–3.0 V (vs. Li/Li+) at various scan rates from 0.1 to 1.0 mV s−1. Galvanostatic discharge/charge test (the mass loading of (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 is about 1.2 mg) was conducted on a LAND CT2001A multichannel battery test system (LAND Technology Co., Ltd., China) at room temperature. The electrochemical workstation was also used to measure the electrochemical impedance spectroscopy (EIS) with the frequency range from 106 Hz to 0.01 Hz with alternating current signal amplitude of 10 mV.
3. Results and discussion
3.1. Structure characterization of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles
Fig. 1(a) and (b) show the SEM and TEM images of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles, which display irregular spherical morphology with size in the ranges of 51–345 nm (average size 143 nm). The XRD patterns of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles are shown in Fig. 1(e) to investigate the crystal structure. The diffraction peaks at 2θ values of 18.21, 29.89, 35.26, 36.95, 42.79, 53.25, 56.64, 62.18, 73.56, and 89.09° correspond to the reflections of the (111), (220), (311), (222), (400), (422), (511), (440), (533), and (731) planes of a Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m spinel structure. Besides, no impurity peaks are detected, indicating the high purity of the spinel-structured materials. The polycrystalline diffraction rings of SAED pattern (as shown in Fig. 1(c)) present the microstructural characteristics of a typical face-centered-cubic (FCC) structure. The interplanar spacings (ring 1: d111 = 0.485 nm, ring 2: d220 = 0.297 nm, ring 3: d311 = 0.253 nm, ring 4: d400 = 0.209 nm, ring 5: d511 = 0.162 nm, and ring 6: d440 = 0.148 nm) are all assignable to the FCC structure. HRTEM image taken from an individual nanosheet (Fig. 1(d)) shows the well-defined lattice fringes with d spacing of 0.297 and 0.253 nm, which are in good agreement with the (220) and (311) planes of (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 phase, respectively. It is clear that the corresponding interplanar spacing values agreed well with the SAED data. In order to confirm the element distribution of (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles, EDS elemental mapping was performed (Fig. 1(f) and S1†), revealing that the Mg, Ti, Fe, Zn, Cu and O elements are homogeneously distributed within the region of a single particle with good overlapping.
m spinel structure. Besides, no impurity peaks are detected, indicating the high purity of the spinel-structured materials. The polycrystalline diffraction rings of SAED pattern (as shown in Fig. 1(c)) present the microstructural characteristics of a typical face-centered-cubic (FCC) structure. The interplanar spacings (ring 1: d111 = 0.485 nm, ring 2: d220 = 0.297 nm, ring 3: d311 = 0.253 nm, ring 4: d400 = 0.209 nm, ring 5: d511 = 0.162 nm, and ring 6: d440 = 0.148 nm) are all assignable to the FCC structure. HRTEM image taken from an individual nanosheet (Fig. 1(d)) shows the well-defined lattice fringes with d spacing of 0.297 and 0.253 nm, which are in good agreement with the (220) and (311) planes of (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 phase, respectively. It is clear that the corresponding interplanar spacing values agreed well with the SAED data. In order to confirm the element distribution of (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles, EDS elemental mapping was performed (Fig. 1(f) and S1†), revealing that the Mg, Ti, Fe, Zn, Cu and O elements are homogeneously distributed within the region of a single particle with good overlapping.
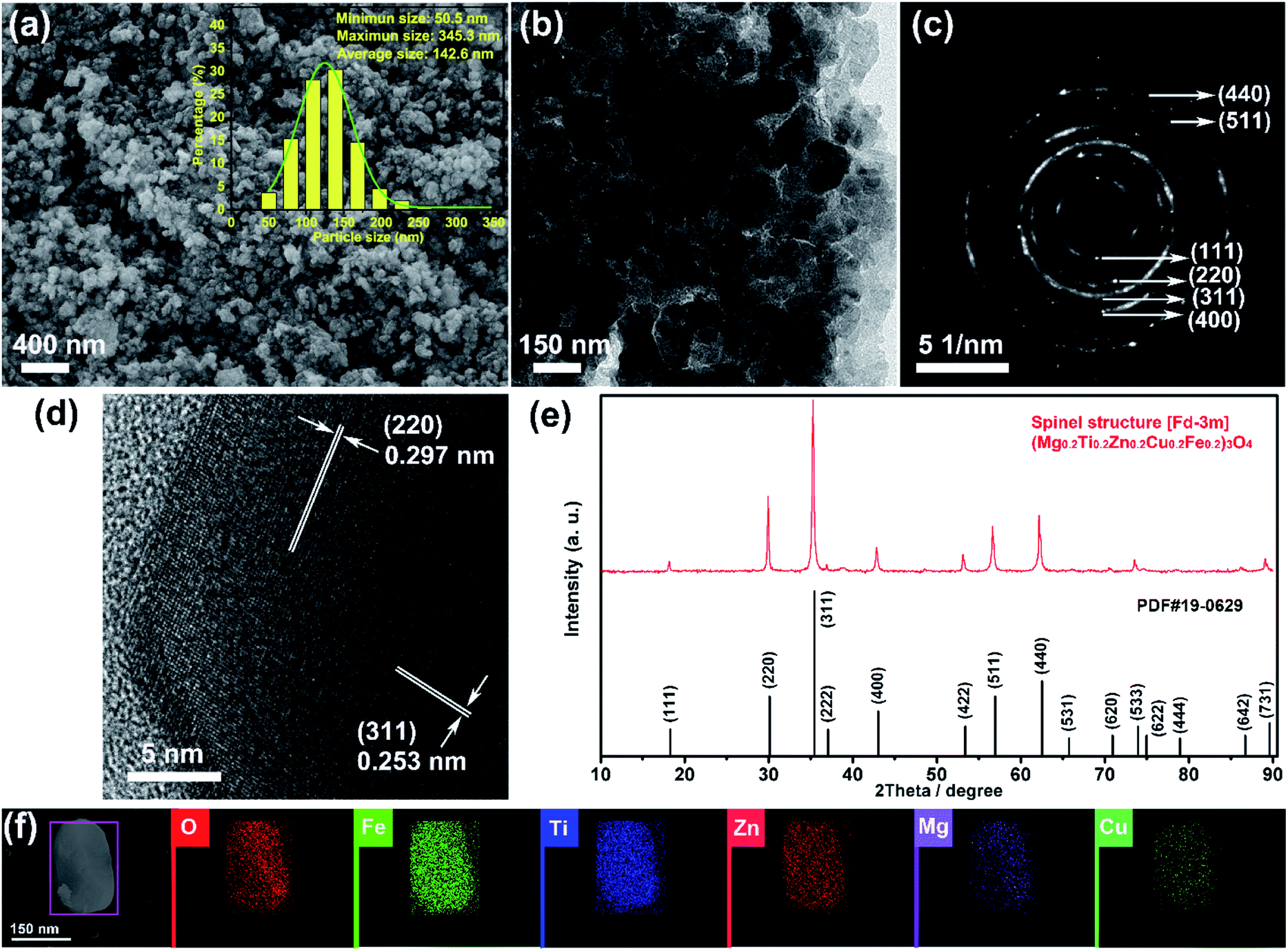 | ||
| Fig. 1 (a) SEM image and (b) TEM image of the synthesized irregular (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles; (c) SAED pattern, (d) HR-TEM image, (e) XRD pattern and (f) elemental mapping images of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles. | ||
It's well-known that the specific surface area, pore diameter, and pore volume all influence the electrochemical performance of the active electrode materials.58–60 The specific surface area and pore size distribution of the synthesized (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles were measured via nitrogen adsorption–desorption isotherm analysis (Fig. S2(a)†). The isotherm shows that the sample exhibits typical type-IV characteristics with a type-H1 hysteresis loop at high pressures, indicating the existence of open mesoporous characteristics.61 According to the Brunauer–Emmett–Teller model, it presents a BET specific surface area of 12.31 m2 g−1, a Langmuir surface area of 78.21 m2 g−1, and a total pore volume of 0.037 cm3 g−1. Pioneering research work of Fe2O3 by Reddy et al.35,42 suggested that anode materials with a high BET surface area would be ideal choice (Table S1†). Based on Barrett–Joyner–Halenda plots, the pore size of the four samples is similar and mainly in the range of 3–20 nm (Fig. S2(b and c)†), which is strong evidence that the sample contain a large number of mesoporous structure. Generally, the rich mesoporous configuration would benefit for the long cycle lives and better rate capabilities of LIBs,62 because such mesoporous structure possesses high specific surface area, which would facilitate charge transfer and reduce ion diffusion path lengths, and provide enough room for accommodating volume changes during cycling. To characterize the chemical compositions and oxidation state of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles, X-ray photoelectron spectroscopy (XPS) was performed. The XPS survey spectrum of the sample confirms the presence of Mg, Ti, Fe, Zn, Cu, and O elements in the material (Fig. S3†). Fig. S4(b)† shows that two main peaks are located at 231.9 and 235.1 eV in the Ti 2p XPS spectrum, corresponding to the Ti 2p3/2 and Ti 2p1/2 with oxidation state of Ti4+.63 In Fig. S4(c),† the O 1s peak is deconvoluted into two peaks. The peak around 529.5 eV arises from typical lattice O in (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4,64 while the other peak centered at 531.5 eV is attributed to defect sites with low O atom harmonization. As shown in the high-resolution Fe 2p XPS spectrum of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles (Fig. S4(d)†), two peaks centered at 710.8 and 724.1 eV are observed, corresponding to the electronic states of Fe 2p3/2 and Fe 2p1/2, respectively. Besides, the presences of two shake-up satellite peaks belong to the Fe3+.65,66 Two main peaks (Fig. S4(e)†) located at 933.3 and 953.5 eV in the Cu 2p XPS spectrum correspond to the Cu 2p3/2 and Cu 2p1/2 with oxidation state of Cu2+, respectively. In Fig. S4(f),† the existence of Zn2+ species is demonstrated by strong peaks of Zn 2p centered at the binding energies of 1021.3 and 1044.5 eV, which can be ascribed to Zn 2p3/2 and Zn 2p1/2, respectively.
3.2. Electrochemical performance studies of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particles as anode material for LIBs
Fig. 2(a) shows the CV curves of the first seven cycles. It represents the first cathodic scan with four reduction peaks for (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4, in which the first one is centered at 1.42 V, the second one at 0.65 V, the third one at 0.35 V, which can apparently corresponding to the lithiation of Fe3+/Fe0, Cu2+/Cu0, Zn2+/Zn0, the Li2O matrix formation and crystal structure destruction followed by Li-alloying of Zn, respectively.71–73 The fourth sharp peak at lower voltage (0.03 V) can be ascribed to the formation of a solid electrolyte interphase (SEI) layer on the electrode surface because of the reduction of solvents in the electrolyte.74,75
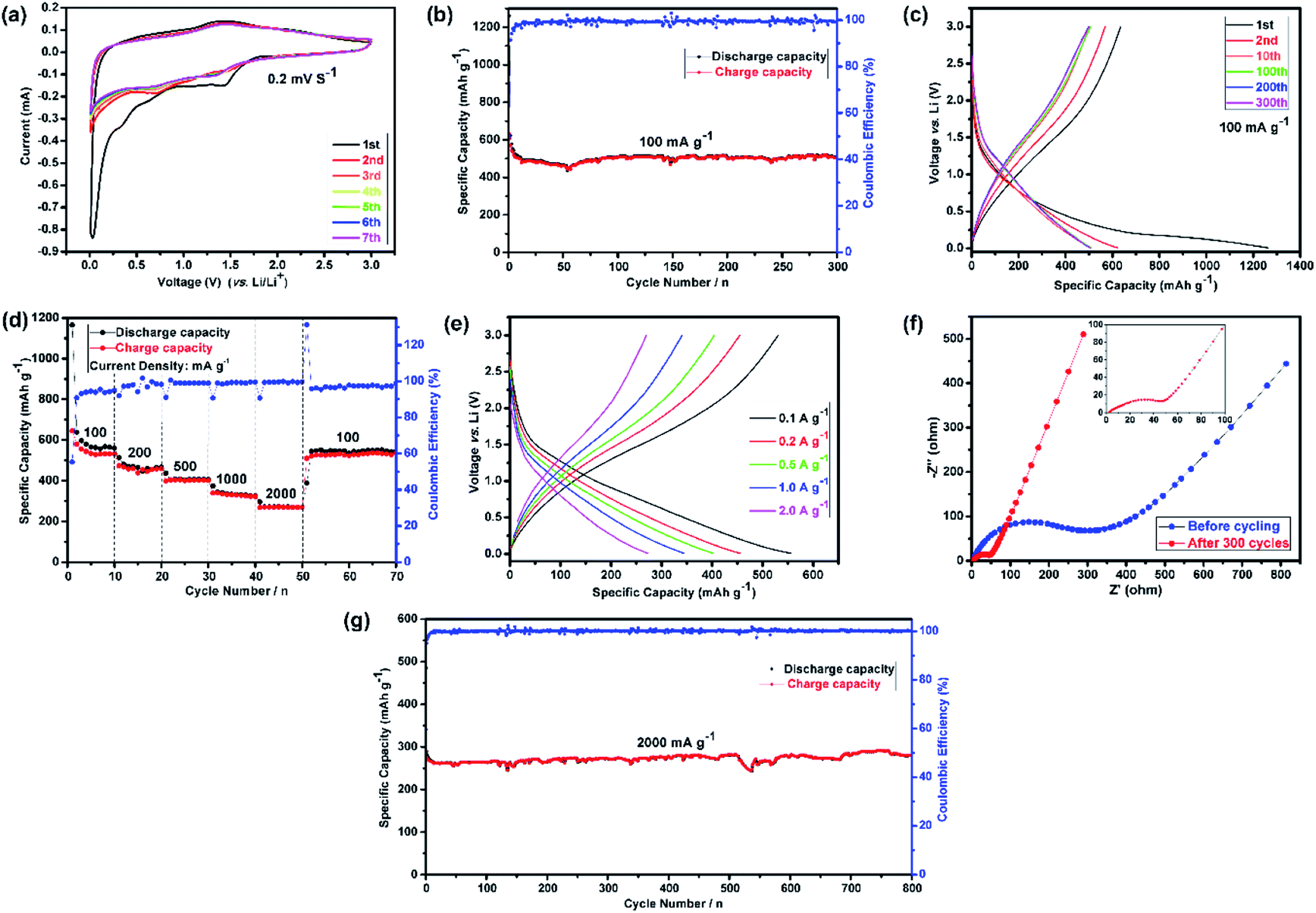 | ||
| Fig. 2 Half-cell electrochemical performances of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode for LIBs. (a) Representative cyclic voltammetry (CV) curves at a scanning rate of 0.2 mV s−1 in the voltage range of 0.01–3.00 V versus Li+/Li; (b) cycling performances and the corresponding coulombic efficiencies at 100 mA g−1 for 300 cycles; (c) discharge–charge voltage profiles for the 1st, 2nd, 10th, 100th, 200th and 300th cycles in the voltage range of 0.01–3.00 V at a current rate of 100 mA g−1; (d) rate performance at different current densities ranging from 100 to 2000 mA g−1; (e) the discharge–charge curves under different current densities; (f) Nyquist plots of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode: before cycling (at the open circuit voltage, 3.0 V) and after 300 cycles (at the specific charge voltage, 3.0 V) at a current density of 100 mA g−1; (g) cycling performances and the corresponding coulombic efficiencies at 2000 mA g−1 for 800 cycles. | ||
Subsequently, the broad peak centered at around 1.41 V in the anodic step is identified due to oxidation of metallic Fe, Cu and Zn to the corresponding metal oxides, followed by the decomposition of Li2O.13,76 According to Wang et al.,77 Li–Zn alloy formation occurs at 0.35 V and multistep dealloying process of Li–Zn alloy occurs in the range of 0.5–0.8 V vs. Li. As expected, three initial reduction peaks (1.42, 0.65 and 0.35 V) disappear, and the fourth one moves to 0.71 V in the second cycle. Commonly, this phenomenon can be observed in transition metal oxides owing to some side reactions and structural rearrangements only happen in the first cycle, which indicates the irreversibility of initial lithiation process and the SEI formation.13 In the following cycles, the CV curves are similar to the second cycle and almost are well-overlapped at 0.71 V and 1.41 V, suggesting the good electrochemical reversibility of the electrode.
Fig. 2(b) summarizes the cyclic performance with coulombic efficiency of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode at a current density of 100 mA g−1. As mentioned above, it is noticed that rapid capacity decay in the preliminary cycles because of complicated subsidiary reactions and irreversible structure transformation. Furthermore, the capacity values generally increase after specifically completing 50 cycles, which can be explained by the reversible formation/dissolution of polymeric gel-like film during the process of activation electrode materials for the transition metal oxide.78 It can be observed in Fig. 2(c) that the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode exhibit the first, second, 10th, 100th, 200th, and 300th charge/discharge curves at a current density of 100 mA g−1. During the first cycle, the voltage dropped to a plateau of 1.42 V from the open circuit potential and then slowly decreased to 0.01 V. The initial discharge and charge capacities were 1261 and 634 mA h g−1, corresponding to a low initial coulombic efficiency of 50.3%. However, the coulombic efficiency increased to about 99% after few initial cycles and maintained at this percentage for the entire test process. Finally, after 300 cycles, a high discharge specific capacity of 504 mA h g−1 was still maintained. The superior electrochemical cycling performance could be originated from the unique hierarchically assembled robust microspheres structure of the electrode after 1st discharge/charge cycle (Fig. S5†). On one hand, the well-connected microspheres can enhance the contact area between electrode and electrolyte, which facilitates to shorten the lithium ion diffusion length and accommodate the volume change during charge/discharge process. On the other hand, the spherical hierarchical morphology with low surface energy can alleviate the particle aggregation.79
Presently, to investigate the electrical conductivity and Li+ transfer in the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 anode, EIS plots of before cycling (at the open circuit voltage, 3.0 V) and after 300 cycles (at the specific charge voltage, 3.0 V) were evaluated. As shown in Fig. 2(f), the Nyquist plots show a semicircle in the high-to middle-frequency region (representing the ohmic resistance, SEI film resistance, and charge transfer resistance) and a straight sloping line in the low-frequency region (representing Li+ diffusion resistance).83,84 It is noted that the semicircle diameter of the fresh (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode is larger than that measured at the 300th cycle, indicating that the Li+ diffusion resistance is reduced after cycling. The semicircle diameter varies directly with impedance. The impedance data of all samples are analyzed by fitting the equivalent circuits inserted in Fig. 3. The equivalent circuits are composed of ohmic resistance (RS), SEI film resistance (RSEI), charge-transfer resistance (Rct), constant-phase element (CPE), and Warburg impedance (ZW). The parameters of the equivalent circuit are recorded in Table S3.† In this way, (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 exhibits excellent electrical conductivity. Fig. 3 shows the relationship between the real resistance (Z′) and the inverse square root of frequency (ω−1/2) of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode before and after 300 cycles. The diffusion coefficient values of Li+ into the electrodes can be calculated by the following equations:85,86
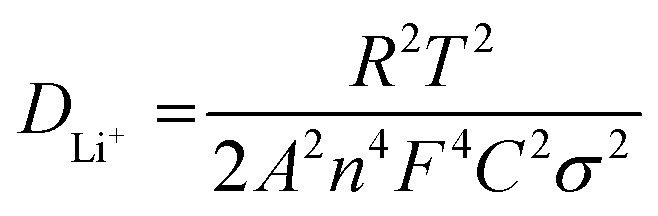
| Z′ = RS + Rct + σω−1/2 |
485.33 C mol−1), C is the concentration of lithium ion, and σ is the Warburg factor, which is related to Z′. The (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode after 300 cycles shows the Li+ diffusion coefficient of 4.716 × 10−15 cm2 s−1, 3.15 times higher than that of the fresh cell (1.497 × 10−15 cm2 s−1), explaining the good electrochemical performance. Hence, we believe that the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 material show higher electrical conductivity because of the five types of metals in the spinel structure, which reduce the activation energy for electron transfer relative to that in single-metal structures.
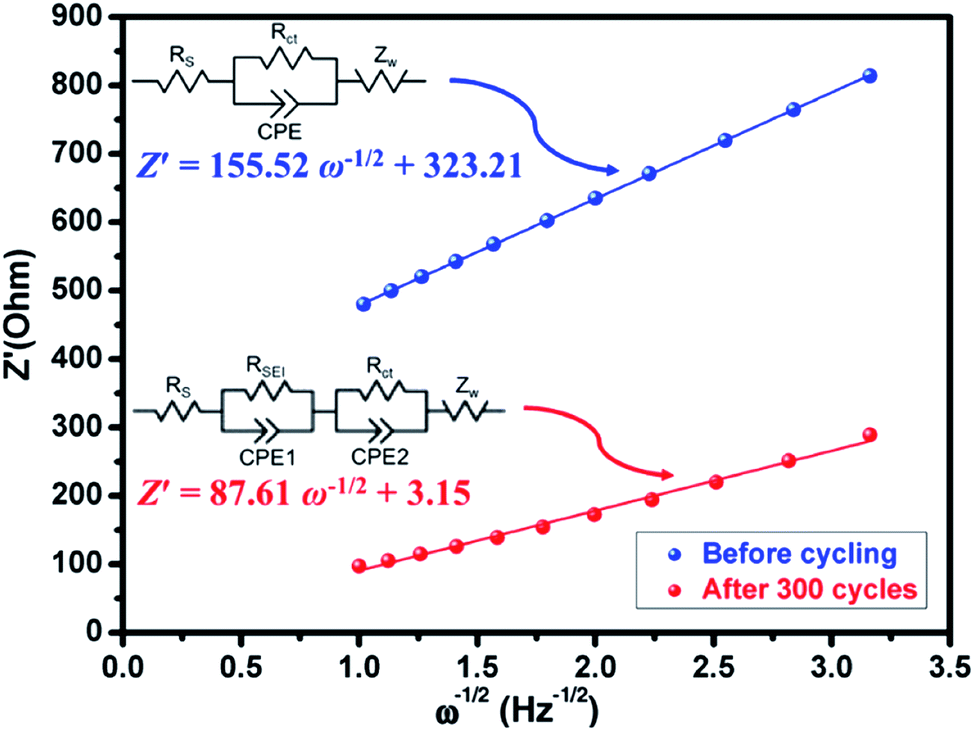 | ||
| Fig. 3 Plots of Z′ against ω−1/2 (inset: equivalent circuit) of the fresh (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 half-cell and the half-cell charged to 3.0 V after 300 cycles. | ||
| ip = avb |
| log(ip) = blog(v) + loga |
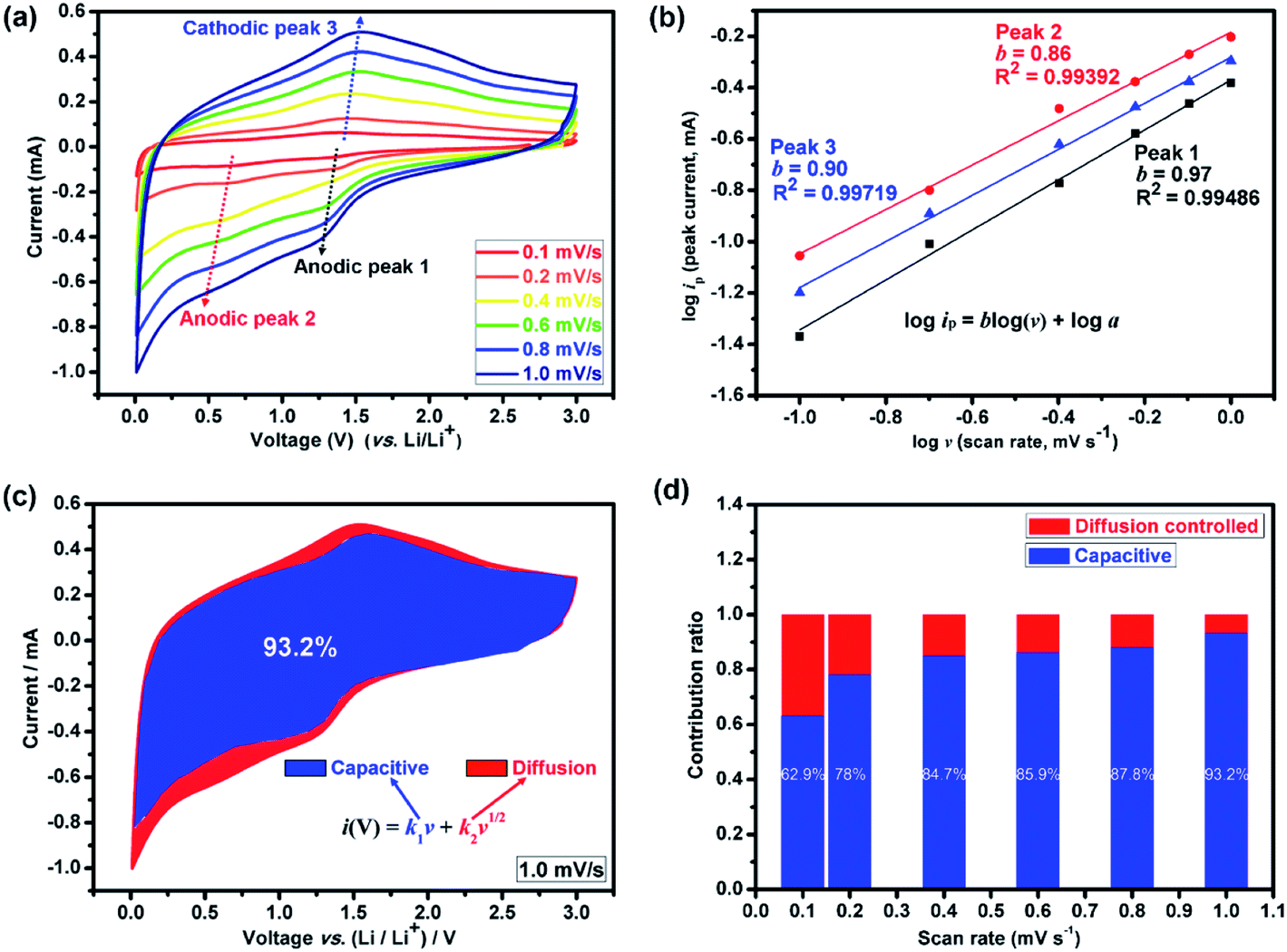 | ||
| Fig. 4 Kinetic analysis of the Li+ storage behavior. (a) Cyclic voltammetry (CV) curves of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode for LIBs at different scan rates of 0.1, 0.2, 0.4, 0.6, 0.8 and 1.0 mV s−1. (b) The corresponding logarithm peak current versus logarithm scan rate plots with inset showing the calculated b values. (c) Separation of capacitive (blue region) and diffusion (red region) controlled contribution at 1.0 mV s−1. (d) The normalized contribution ratio of pseudocapacitive capacities at different scan rates for the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode, which indicates that the electrode was mainly controlled by pseudocapacitive electrochemical behavior. | ||
The total capacitive contribution at a fixed scan rate can be quantitatively determined according to the following equation:
| i(V) = k1v + k2v1/2 |
| i(V)/v1/2 = k1v1/2 + k2 |
Thus, by determining k1 and k2 in Fig. S6,† we are able to quantify the fraction of the current due to each of these contributions at specific potentials. It presents that when increasing the scan rates, the capacitive contribution also increased (Fig. 4(d)). The blue areas in the CV curve shown in Fig. 4(c) represent the capacitive-controlled contribution at a scan rate of 1.0 mV s−1. Fig. S7† shows the CV curves with separation between the total current and surface capacitive current at different scan rates of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 electrode. At the scan rate of 0.1 mV s−1, the capacitive contribution is 62.9%; a high ratio of 93.2% can be obtained when the scan rate approaches 1.0 mV s−1. The dominant pseudocapacitive contribution indicates the superior rate capability, and associates with the structure of the (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 materials, in which the lithium ions can directly react with the active material without long distance diffusion pathways.
4. Conclusions
In this study, we have demonstrated the successful preparation of a new kind of spinel high-entropy oxide (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 material via a facile one-step solid state reaction method and subsequently high-energy ball-milling. When used as anode materials for LIBs, the electrode exhibited superior lithium storage properties, delivering a large reversible capacity of 504 mA h g−1 at a current density of 100 mA g−1 after 300 cycles, and notably an exceptional rate capacity of 272 mA h g−1 at 2000 mA g−1, respectively. Meanwhile, a high capacity retention of 96.2% after 800 cycles at 2000 mA g−1 was obtained. This superior electrochemical performance could be ascribed to the usage of novel (Mg0.2Ti0.2Zn0.2Cu0.2Fe0.2)3O4 particle structure, which can not only buffer the volumetric changes upon lithiation/delithiation processes, but also provide enlarged surface sites for lithium storage and facilitate the charge/electrolyte reaction. More importantly, our work highlights that the rational design and use of high-entropy oxides with different electrochemically active elements and novel structure as LIB anode materials might be promising for exploring next-generation energy storage devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (20826041C4208). This research was also partially supported by National Natural Science Foundation of China (Grant No. 11775150). We would like to thank the Analytical & Testing Center of Sichuan University for the X-ray diffraction work and we would be grateful to Dr Shan-ling Wang for her help of the transmission electron microscopy images.Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- N. J. Dudney and J. Li, Science, 2015, 347, 131 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- D. Xiong, X. Li, Z. Bai and S. Lu, Small, 2018, 14, 1703419 CrossRef PubMed.
- W. Liu, X. Li, D. Xiong, Y. Hao, J. Li, H. Kou, B. Yan, D. Li, S. Lu, A. Koo, K. Adair and X. Sun, Nano Energy, 2018, 44, 111–120 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- H. Wang, X. Yang, Q. Wu, Q. Zhang, H. Chen, H. Jing, J. Wang, S.-B. Mi, A. L. Rogach and C. Niu, ACS Nano, 2018, 12, 3406–3416 CrossRef CAS PubMed.
- S. Lim, J.-H. Kim, Y. Yamada, H. Munakata, Y.-S. Lee, S.-S. Kim and K. Kanamura, J. Power Sources, 2017, 355, 164–170 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- J. Wang, H. Wang, D. Cao, X. Lu, X. Han and C. Niu, Part. Part. Syst. Charact., 2017, 34, 1700185 CrossRef.
- M. V. Reddy, G. V. Subba Rao and B. V. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- K. Cao, T. Jin, L. Yang and L. Jiao, Mater. Chem. Front., 2017, 1, 2213–2242 RSC.
- L. Fan, X. Li, X. Song, N. Hu, D. Xiong, A. Koo and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 2637–2648 CrossRef CAS PubMed.
- M. V. Reddy, G. Prithvi, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2014, 6, 680–690 CrossRef CAS PubMed.
- L. Wang, G. Zhang, Q. Liu and H. Duan, Mater. Chem. Front., 2018, 2, 1414–1435 RSC.
- H. Zhang, G. Zhang, Z. Li, K. Qu, L. Wang, W. Zeng, Q. Zhang and H. Duan, J. Mater. Chem. A, 2016, 4, 10585–10592 RSC.
- Q. Zhang, J. Wang, D. Xu, Z. Wang, X. Li and K. Zhang, J. Mater. Chem. A, 2014, 2, 3865–3874 RSC.
- Q. Zhang, D. Xu, X. Zhou, X. Wu and K. Zhang, Small, 2014, 10, 935–943 CrossRef CAS PubMed.
- H. Huang, Y. Liu, J. Wang, M. Gao, X. Peng and Z. Ye, Nanoscale, 2013, 5, 1785–1788 RSC.
- M. V. Reddy, C. Yu, F. Jiahuan, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 4361–4366 CrossRef CAS PubMed.
- F.-X. Ma, H. Hu, H. B. Wu, C.-Y. Xu, Z. Xu, L. Zhen and X. W. Lou, Adv. Mater., 2015, 27, 4097–4101 CrossRef CAS PubMed.
- S. H. Lee, S.-H. Yu, J. E. Lee, A. Jin, D. J. Lee, N. Lee, H. Jo, K. Shin, T.-Y. Ahn, Y.-W. Kim, H. Choe, Y.-E. Sung and T. Hyeon, Nano Lett., 2013, 13, 4249–4256 CrossRef CAS PubMed.
- B. Wang, H. B. Wu, L. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 4165–4168 CrossRef CAS PubMed.
- S. Petnikota, S. K. Marka, A. Banerjee, M. V. Reddy, V. V. S. S. Srikanth and B. V. R. Chowdari, J. Power Sources, 2015, 293, 253–263 CrossRef CAS.
- J. Wang, H. Zhou, J. Nanda and P. V. Braun, Chem. Mater., 2015, 27, 2803–2811 CrossRef CAS.
- L. Zhang, H. B. Wu, R. Xu and X. W. Lou, CrystEngComm, 2013, 15, 9332–9335 RSC.
- J. Liang, Z. Fan, S. Chen, S. Ding and G. Yang, Chem. Mater., 2014, 26, 4354–4360 CrossRef CAS.
- Y. Chen, Y. Wang, X. Shen, R. Cai, H. Yang, K. Xu, A. Yuan and Z. Ji, J. Mater. Chem. A, 2018, 6, 1048–1056 RSC.
- M. V. Reddy, Z. Beichen, L. J. e. Nicholette, Z. Kaimeng and B. V. R. Chowdari, Electrochem. Solid-State Lett., 2011, 14, A79–A82 CrossRef CAS.
- M. V. Reddy, K. Y. H. Kenrick, T. Y. Wei, G. Y. Chong, G. H. Leong and B. V. R. Chowdari, J. Electrochem. Soc., 2011, 158, A1423–A1430 CrossRef CAS.
- M. V. Reddy, C. Yu, F. Jiahuan, K. P. Loh and B. V. R. Chowdari, RSC Adv., 2012, 2, 9619–9625 RSC.
- B. Das, M. V. Reddy and B. V. R. Chowdari, J. Alloys Compd., 2013, 565, 90–96 CrossRef CAS.
- M. V. Reddy, Y. Xu, V. Rajarajan, T. Ouyang and B. V. R. Chowdari, ACS Sustainable Chem. Eng., 2015, 3, 3035–3042 CrossRef CAS.
- M. V. Reddy, C. T. Cherian, K. Ramanathan, K. C. W. Jie, T. Y. W. Daryl, T. Y. Hao, S. Adams, K. P. Loh and B. V. R. Chowdari, Electrochim. Acta, 2014, 118, 75–80 CrossRef CAS.
- Y. Wu, P. Zhu, M. V. Reddy, B. V. R. Chowdari and S. Ramakrishna, ACS Appl. Mater. Interfaces, 2014, 6, 1951–1958 CrossRef CAS PubMed.
- D. Darbar, M. V. Reddy, S. Sundarrajan, R. Pattabiraman, S. Ramakrishna and B. V. R. Chowdari, Mater. Res. Bull., 2016, 73, 369–376 CrossRef CAS.
- C. T. Cherian, J. Sundaramurthy, M. V. Reddy, P. Suresh Kumar, K. Mani, D. Pliszka, C. H. Sow, S. Ramakrishna and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 9957–9963 CrossRef CAS PubMed.
- D. Darbar, M. R. Anilkumar, V. Rajagopalan, I. Bhattacharya, H. I. Elim, T. Ramakrishnappa, F. I. Ezema, R. Jose and M. V. Reddy, Ceram. Int., 2018, 44, 4630–4639 CrossRef CAS.
- M. V. Reddy, C. Yao Quan and S. Adams, Mater. Lett., 2018, 212, 186–192 CrossRef CAS.
- C. T. Cherian, M. V. Reddy, G. V. S. Rao, C. H. Sow and B. V. R. Chowdari, J. Solid State Electrochem., 2012, 16, 1823–1832 CrossRef CAS.
- M. V. Reddy, C. Y. Quan, K. W. Teo, L. J. Ho and B. V. R. Chowdari, J. Phys. Chem. C, 2015, 119, 4709–4718 CrossRef CAS.
- J. Zhao, Y. Cheng, X. Yan, D. Sun, F. Zhu and Q. Xue, CrystEngComm, 2012, 14, 5879–5885 RSC.
- L. Luo, R. Cui, H. Qiao, K. Chen, Y. Fei, D. Li, Z. Pang, K. Liu and Q. Wei, Electrochim. Acta, 2014, 144, 85–91 CrossRef CAS.
- L. Jin, Y. Qiu, H. Deng, W. Li, H. Li and S. Yang, Electrochim. Acta, 2011, 56, 9127–9132 CrossRef CAS.
- Y. Yin, N. Huo, W. Liu, Z. Shi, Q. Wang, Y. Ding, J. Zhang and S. Yang, Scr. Mater., 2016, 110, 92–95 CrossRef CAS.
- Y. Yin, W. Liu, R. Gao and S. Yang, ChemElectroChem, 2019, 6, 757–763 CrossRef CAS.
- A. K. Rai, T. V. Thi, J. Gim and J. Kim, Mater. Charact., 2014, 95, 259–265 CrossRef CAS.
- X. Xu, C. Niu, M. Duan, X. Wang, L. Huang, J. Wang, L. Pu, W. Ren, C. Shi, J. Meng, B. Song and L. Mai, Nat. Commun., 2017, 8, 460 CrossRef PubMed.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- D. W. Kim, Y. D. Ko, J. G. Park and B. K. Kim, Angew. Chem., Int. Ed., 2007, 46, 6654–6657 CrossRef CAS PubMed.
- N. Prasoetsopha, S. Pinitsoontorn, S. Fan, H. H. Hng and S. Maensiri, Ionics, 2017, 23, 395–403 CrossRef CAS.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef PubMed.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 1806236 CrossRef PubMed.
- Q. Wang, A. Sarkar, Z. Li, Y. Lu, L. Velasco, S. S. Bhattacharya, T. Brezesinski, H. Hahn and B. Breitung, Electrochem. Commun., 2019, 100, 121–125 CrossRef CAS.
- Q. Wang, A. Sarkar, D. Wang, L. Velasco, R. Azmi, S. S. Bhattacharya, T. Bergfeldt, A. Düvel, P. Heitjans, T. Brezesinski, H. Hahn and B. Breitung, Energy Environ. Sci., 2019, 12, 2433–2442 RSC.
- N. Qiu, H. Chen, Z. Yang, S. Sun, Y. Wang and Y. Cui, J. Alloys Compd., 2019, 777, 767–774 CrossRef CAS.
- H. Chen, N. Qiu, B. Wu, Z. Yang, S. Sun and Y. Wang, RSC Adv., 2019, 9, 28908–28915 RSC.
- W. Cheng, F. Rechberger, D. Primc and M. Niederberger, Nanoscale, 2015, 7, 13898–13906 RSC.
- H. Li, P. Zhou, F. Liu, H. Li, F. Cheng and J. Chen, Chem. Sci., 2019, 10, 1374–1379 RSC.
- K. Zhu, H. Gao, G. Hu, M. Liu and H. Wang, J. Power Sources, 2017, 340, 263–272 CrossRef CAS.
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26, 6622–6628 CrossRef CAS PubMed.
- P. Zhu, Y. Wu, M. V. Reddy, A. Sreekumaran Nair, B. V. R. Chowdari and S. Ramakrishna, RSC Adv., 2012, 2, 531–537 RSC.
- L. Qu, X. Hou, J. Mao, Q. Ru, S. Hu, X. Liu and K.-h. Lam, RSC Adv., 2016, 6, 96743–96751 RSC.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- I. Uhlig, R. Szargan, H. W. Nesbitt and K. Laajalehto, Appl. Surf. Sci., 2001, 179, 222–229 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Electrochim. Acta, 2005, 50, 3375–3382 CrossRef CAS.
- S. L. Tey, M. V. Reddy, G. V. Subba Rao, B. V. R. Chowdari, J. Yi, J. Ding and J. J. Vittal, Chem. Mater., 2006, 18, 1587–1594 CrossRef CAS.
- M. V. Reddy, B. Pecquenard, P. Vinatier and A. Levasseur, Electrochem. Commun., 2007, 9, 409–415 CrossRef CAS.
- B. Varghese, M. V. Reddy, Z. Yanwu, C. S. Lit, T. C. Hoong, G. V. Subba Rao, B. V. R. Chowdari, A. T. S. Wee, C. T. Lim and C.-H. Sow, Chem. Mater., 2008, 20, 3360–3367 CrossRef CAS.
- M. V. Reddy, B. L. Wei Wen, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 7777–7785 CrossRef CAS PubMed.
- C. T. Cherian, M. Zheng, M. V. Reddy, B. V. R. Chowdari and C. H. Sow, ACS Appl. Mater. Interfaces, 2013, 5, 6054–6060 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Mater. Chem., 2011, 21, 10003–10011 RSC.
- Y. Gao, L. Yin, S. J. Kim, H. Yang, I. Jeon, J.-P. Kim, S. Y. Jeong, H. W. Lee and C. R. Cho, Electrochim. Acta, 2019, 296, 565–574 CrossRef CAS.
- L. Hou, R. Bao, Y. Zhang, X. Sun, J. Zhang, H. Dou, X. Zhang and C. Yuan, J. Mater. Chem. A, 2018, 6, 17947–17958 RSC.
- Y. Deng, Q. Zhang, S. Tang, L. Zhang, S. Deng, Z. Shi and G. Chen, Chem. Commun., 2011, 47, 6828–6830 RSC.
- H. Wang, Q. Pan, Y. Cheng, J. Zhao and G. Yin, Electrochim. Acta, 2009, 54, 2851–2855 CrossRef CAS.
- P. F. Teh, Y. Sharma, S. S. Pramana and M. Srinivasan, J. Mater. Chem., 2011, 21, 14999–15008 RSC.
- L. Hu, H. Zhong, X. Zheng, Y. Huang, P. Zhang and Q. Chen, Sci. Rep., 2012, 2, 986 CrossRef PubMed.
- J. Wang, S. Xie, T. Liu, T. Yao, L. Zhu, A. M. Asiri, H. M. Marwani, X. Han and H. Wang, Mater. Today Commun., 2019, 20, 100578 CrossRef.
- P. Nithyadharseni, M. V. Reddy, B. Nalini, M. Kalpana and B. V. R. Chowdari, Electrochim. Acta, 2015, 161, 261–268 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Phys. Chem. C, 2007, 111, 11712–11720 CrossRef CAS.
- A. K. Rai, S. Kim, J. Gim, M. H. Alfaruqi, V. Mathew and J. Kim, RSC Adv., 2014, 4, 47087–47095 RSC.
- L. Hou, L. Lian, L. Zhang, G. Pang, C. Yuan and X. Zhang, Adv. Funct. Mater., 2015, 25, 238–246 CrossRef CAS.
- G. Li, X. Xu, R. Han and J. Ma, CrystEngComm, 2016, 18, 2949–2955 RSC.
- J. Zhu, L. Wei, J. Hu and C. Xue, J. Alloys Compd., 2017, 723, 729–735 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518 CrossRef CAS PubMed.
- L. Shen, H. Lv, S. Chen, P. Kopold, P. A. van Aken, X. Wu, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1700142 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00255k |
| This journal is © The Royal Society of Chemistry 2020 |