
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering pH-responsive switching of donor–π–acceptor chromophore alignments along a peptide nanotube scaffold†
Yuki Tabata ,
Yusuke Kamano,
Shunsaku Kimura and
Hirotaka Uji*
,
Yusuke Kamano,
Shunsaku Kimura and
Hirotaka Uji*
Department of Material Chemistry, Graduate School of Engineering, Kyoto University, Kyoto-Daigaku-Katsura, Nishikyo-ku, Kyoto 615-8246, Japan. E-mail: uji.hirotaka.3w@kyoto-u.ac.jp
First published on 22nd January 2020
Abstract
A cyclic tri-β-peptide cyclo(β-Ala-β-Ala-β-Lys) having diethylaminonaphthalimide at the β-Lys side chain (CP3Npi) self-assembled into a peptide nanotube in a solution of HFIP and water. CD spectra of the CP3Npi nanotubes show a negative Cotton effect at 441 nm and a positive Cotton effect at 393 nm, indicating that D–π–A naphthalimide chromophores are aligned in a left-handed chiral way along the nanotube. The CP3Npi nanotubes bear positive charges under acidic conditions retaining the nanotube structure but pH-responsive switching of D–π–A naphthalimide alignments along the nanotube between a left-handed chiral and random arrangement was observed. The peptide nanotube is a stable scaffold for attaining pH-responsive alignment switching of side-chain chromophores.
1. Introduction
In the field of organic electronics, a wide range of organic molecules have been studied for their performance as electric wires, diodes, and transistors with expectations towards flexible and lightweight electronic devices. Further, the preparation cost will be diminished compared to the established silicon-based electronics when the solution process preparation method can be applied. The aromatic compounds in particular those having electron donor (D) and acceptor (A) substituents (D–π–A) have attracted common interests owing to their charge-conducting properties.1,2 The band gap energies of D–π–A molecules can be precisely modulated by adjusting the electronic properties of the electron-donating and withdrawing substituents. In addition, on the basis of the DFT calculations on the molecular orbital energies, p-type or n-type organic semiconductors can be designed.2 Another useful point of D–π–A molecules is the large dipole moment generated in the molecule, which can be used to facilitate a charge separation process upon molecular excitation by resisting against the large exciton binding energy.3,4 A p-type organic semiconductor showing a carrier mobility up to 20 cm2 V−1 s−1 has been attained by polymer carrying D and A substituents, and an n-type organic semiconductor of nearly 10 cm2 V−1 s−1 as well.5 The performances of those organic semiconductors are determined mainly by two factors of organic molecules themselves and their mutual arrangement in the nm-size devices. The latter point is generally resolved by crystallization of the organic molecules and chromophore alignment by using polymers taking extended conformation to keep regular and close π–π stacking.6,7 One of the hard challenges for the structural regulation of molecular assemblies of D–π–A chromophores is to align D–π–A chromophores into a parallel dipole–dipole arrangement despite of their electrostatic repulsions. We have proposed to use cyclic-peptide nanotubes as a scaffold for chromophores to align them linearly along the nanotube axis.8–10 Accordingly, the chromophores are obliged to take the parallel orientation within the distance of π–π interaction owing to the regular stacking of cyclic peptides with 5 Å interval.Another challenge for organic semiconductors is to switch the conduction state into the non-conductive state in response to stimuli from outside. The typical responsive system is the photosystem I, where solar energy is converted efficiently to chemical energy under the self-adjustment of the reaction activity by proton-potential generation across the thylakoid membranes to prevent over-production of the reactive species.11–13 Along this line, photochromic molecular switches were demonstrated to change the fluorescence yield resulting in the regulation of the photoinduced electron transfer.14–16 On the other hand, S. Ghosh et al. demonstrated solvent switchable tubular and vesicular nanostructures prepared from bola-shaped naphthalene imide amphiphiles, and also revealed morphology change influences severely on the conduction and recombination state of charge carriers through alteration in π–π stacking modes.17 Similarly, we propose here to switch nanostructures of D–π–A chromophores of n-type by external stimuli in the aim of control of the electron transfer ability of the nanostructure in a self-responsive manner to environment. But the novel point of the present paper is that the change in the chromophore alignment, spiral or random, is realized with keeping the nanotube structure. The responsive system is not accompanied by a drastic morphology change but chromophore-alignment change. This property provides robustness to the stimuli-responsive self-assemblies. Further the responsive system for n-type organic materials may be advantageous to avoid the concerns of their less stability and liable degradation.
Cyclic tripeptide composed of β-Ala-β-Ala-β-Lys(naphthalimide) was designed and synthesized (Fig. 1A). Naphthalimide (Npi) has been widely studied as a self-assembling key structure, whose electron-deficient imide group works as an electron acceptor moiety,18,19 leading to a high electron mobility.20,21 With introducing diethylamino group of an electron donating group at the 4th-position of the naphthalimide moiety, D–π–A chromophore (diethylamino group-naphthalene-imide group), which can sense pH-change, is attainable.22,23 The spatial arrangement of D–π–A Npi groups on the PNT and its pH-responsiveness are studied (Fig. 1B).
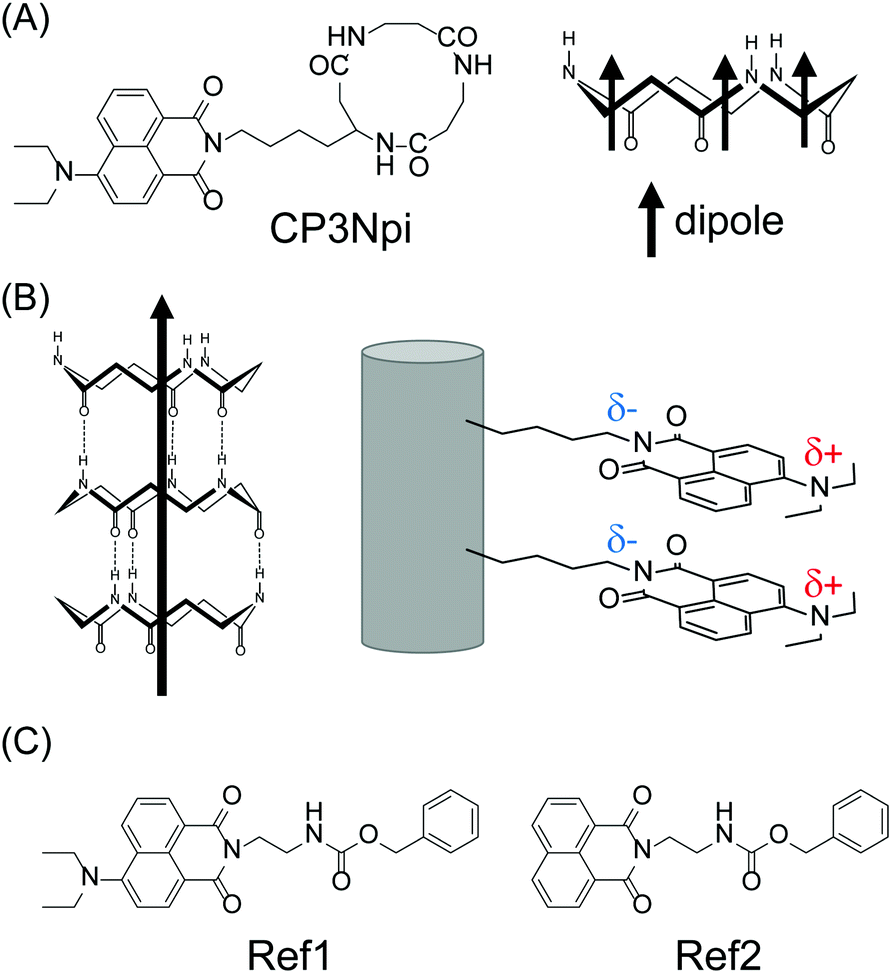 | ||
| Fig. 1 (A) Chemical structures of CP3Npi. (B) Schematic illustration of peptide nanotube composed of CP3Npi. (C) Chemical structures of reference compounds. | ||
2. Experimental section
2.1 Peptide synthesis
The cyclic peptide and reference compounds (Fig. 1) were synthesised by the conventional liquid phase method according to Scheme S1–S4 (Synthetic section in the ESI).† All chemicals were purchased from commercial suppliers and used without further purification. The synthetic compounds were identified by 1H NMR spectroscopy and ESI mass spectrometry. NMR spectra were recorded on a Bruker DPX-400 spectrometer. ESI mass spectra were obtained using Thermo Scientific Exactive Bench-Top LC-MS Spectrometer. The purity of the final compounds was checked by HPLC (Shimadzu HPLC Prominence 20 system) with ODS column (TSKgel ODS-120T).2.2 FTIR measurements
The FTIR spectra of the cyclic peptides were recorded on a Nicolet Magna 850 Fourier transform infrared spectrometer with a Ge-ATR attachment.2.3 Spectroscopies
UV, fluorescence, and CD spectra were recorded using a Shimadzu UV-2450 spectrophotometer, a JASCO FP-6600 spectrofluorometer, and a JASCO J-1500 circular dichroism spectrometer, respectively.2.4 Determination of pH
The pH measurements were performed with a pH meter (Horiba pH meter F-52, a glass electrode Horiba 9680-10D).2.5 Preparation of gold mica substrate
Gold mica substrate was prepared by following method. Gold (99.99%, 1000 Å) was vapor-deposited onto the mica substrate by a vacuum deposition system (Osaka Vacuum N-KS350). The gold mica substrate was thermally annealed before use.2.6 Atomic force microscopy (AFM)
AFM images were obtained by MultiMode 8-HR (Bruker) with the Peak Force QNM mode with a Bruker cantilever (SiN, 0.4 N m−1, 70 kHz) under ambient atmosphere at room temperature. A dispersion of crystals was cast and dried onto a gold mica substrate.3. Results and discussion
3.1 Peptide nanotube formation
CP3Npi is soluble in hexafluoroisopropanol (HFIP) or formic acid. Molecular assemblies were obtained by a solvent diffusion method which increased water content gradually into a HFIP solution of CP3Npi. Molecular assembling was confirmed by DLS measurement (Fig. 2A), and the molecular assemblies were also observed in HFIP/water (1/39 v/v).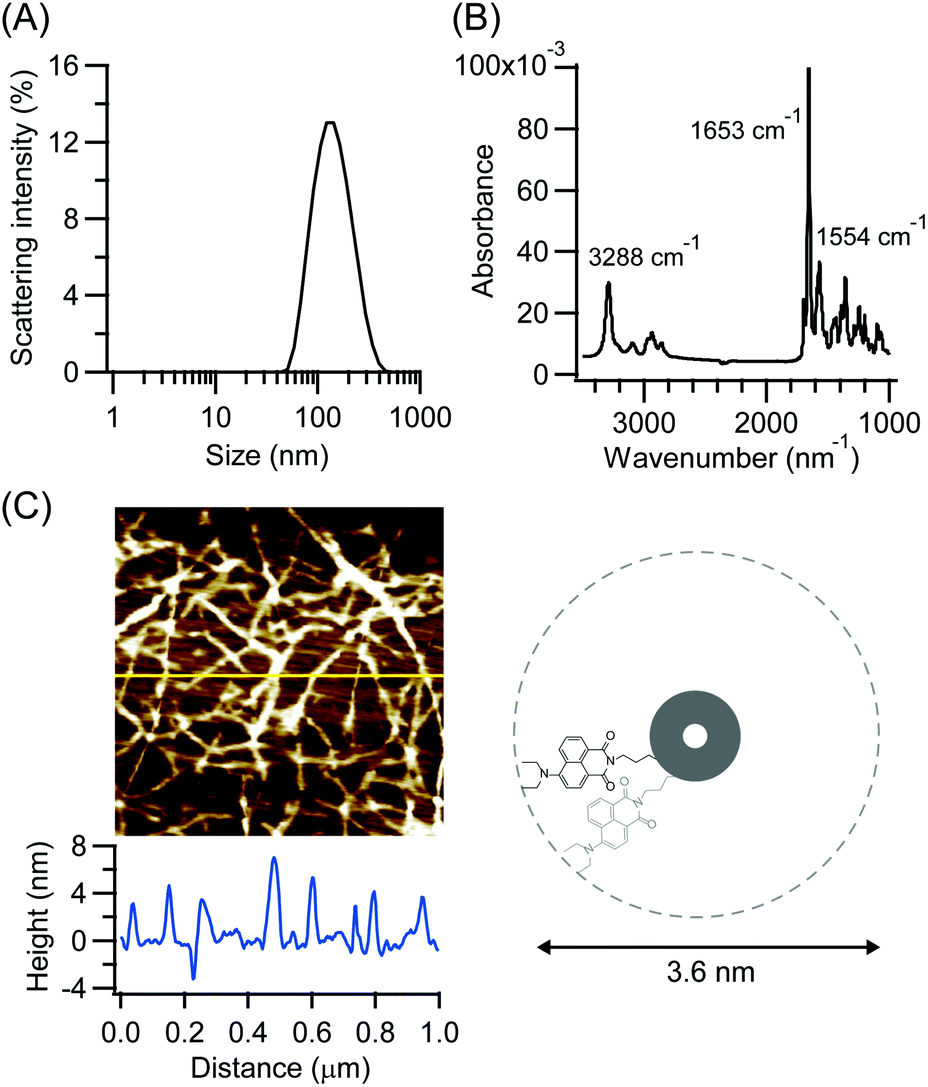 | ||
| Fig. 2 (A) The DLS profile of CP3Npi suspension prepared from CP3NPi in HFIP/water (1/39, v/v). (B) The ATR-FTIR spectrum of CP3Npi suspension prepared from CP3NPi in HFIP/water (1/39, v/v). (C) AFM image and height profile on silicon wafer of peptide nanotube prepared from CP3NPi in HFIP/water (1/39, v/v). | ||
Self-assembling structure via hydrogen bond formation was studied by FTIR spectroscopy. The CP3Npi suspension in HFIP/water was slowly dried on a Ge substrate for ATR-FTIR measurements. FTIR spectrum shows amide I and II bands at 1653 and 1554 cm−1, respectively (Fig. 2B). This profile suggests a parallel β-sheet-like structure similarly to the previous reports on cyclic tri-β-peptide nanotubes.10,24,25 Furthermore, the N–H stretching band was strongly observed at 3288 cm−1, suggesting homogeneous hydrogen bonds forming in CP3Npi molecular assemblies.26 Consequently, CP3Npi in HFIP/water is concluded to self-assemble into peptide nanotube owing to hydrogen bond formation.
Nanotube morphology was analyzed by AFM observations. The CP3Npi suspension in HFIP/water was placed on a silicon wafer, then the excess solvent was sucked up with a filter paper. AFM image was obtained using the Peak Force QNM mode under ambient atmosphere at room temperature with a Bruker cantilever (SiN, 0.4 N m−1, 70 kHz). Very thin fibril-like images of a few micrometers length were observed (Fig. 2C). Since the height profile indicates around 4 nm thickness which corresponds to the diameter of CP3Npi, single nanotube is considered to be generated without bundle formation, which makes a vivid contrast to the bundle formation of other PNTs due to anti-parallel dipole–dipole interaction between PNTs.10,24 It is considered that the bulky Npi moieties should hinder close contact of macrodipoles of PNTs, and the large D–π–A dipoles may be arranged to cancel out partly the macrodipole of the PNT.
3.2 Chromophore alignment
Alignment of the Npi moieties along PNT was analyzed by UV-vis spectroscopy. UV spectra show a hypsochromic shift from 460 nm in a HFIP solution to 432 nm in a HFIP/water suspension (Fig. 3A left), whilst Npi molecule without cyclic peptide moiety (Ref1, Fig. 1C left) did not show such a large extent of the shift (Fig. 3A right). The hypsochromic shift indicates that Npi moieties were arranged in a way of face to face (H-type) stacking in HFIP/water. Würthner studied self-assembly of push–pull chromophores, and revealed that dipolar merocyanine dye showed a hypsochromic shift upon dimer formation.27 In their system, dipolar molecules formed antiparallel dimer, but CP3Npi molecules should stack in a columnar arrangement to adopt one mode of nearly parallel stacking of Npi moieties out of three possible modes due to the constraint of stacking of cyclic tri-β-peptides.28,29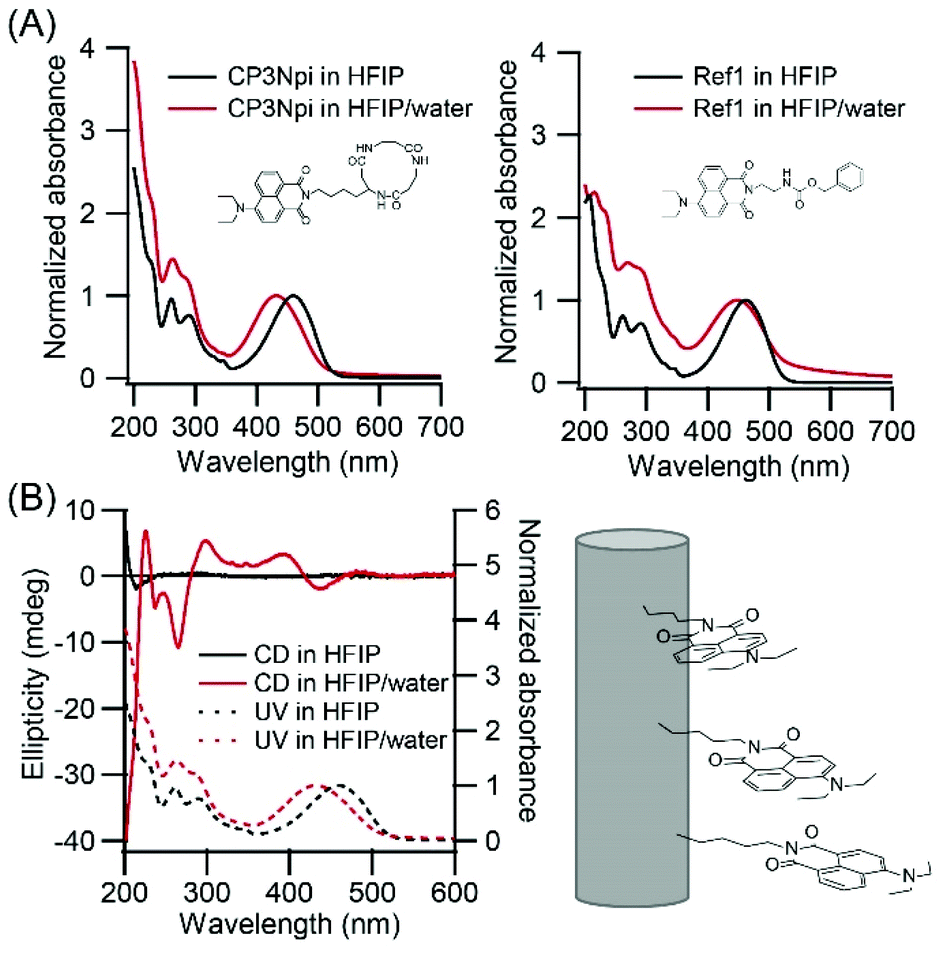 | ||
| Fig. 3 (A) Normalized UV-vis absorption spectra of CP3Npi and compound Ref1 in HFIP and HFIP/water (1/39, v/v). (B) CD spectra of CP3Npi in HFIP and HFIP/water (1/39 v/v). | ||
CD spectra also support a regular arrangement of Npi moieties in nanotubes. CP3Npi in HFIP solution shows no Cotton effect around 450 nm in the Npi absorption region, except a negative Cotton effect at 210 nm which is attributed cyclic peptide skeleton (Fig. 3B). On the other hand, CP3Npi in HFIP/water shows a negative Cotton effect at 441 nm and positive Cotton effect at 393 nm. According to the exciton chirality rule, Npi moieties were arranged by a left-handed chiral way upon PNT formation.
3.3 pH-Response of chromophores
It is well known that an amino group introduced at the 4th-position of the naphthalimide ring works as a pH responsive probe. UV-vis spectroscopy was carried out in HFIP/hydrochloric acid (HCl) aqueous solution (1/39 v/v) with varying HCl concentrations (Fig. 4A). Upon increase of HCl concentrations, the absorption near 435 nm decreased while the absorption near 335 nm increased. This behavior is explainable by protonation of diethylamino group at the 4th-position of naphthalimide, because the absorptions at 435 and 335 nm are related with conjugation of the diethylamino group with the naphthalimide ring as evidenced by comparison of UV spectra of compound Ref1 with the diethylamino group and Ref2 without it (Fig. 1C right, and Fig. S10†). Upon protonation of the diethylamino group, the induced Cotton effects in this region diminished and disappeared at 0.1 M HCl, suggesting that the Npi chiral arrangement was destroyed by electrostatic repulsion between protonated diethylamino groups.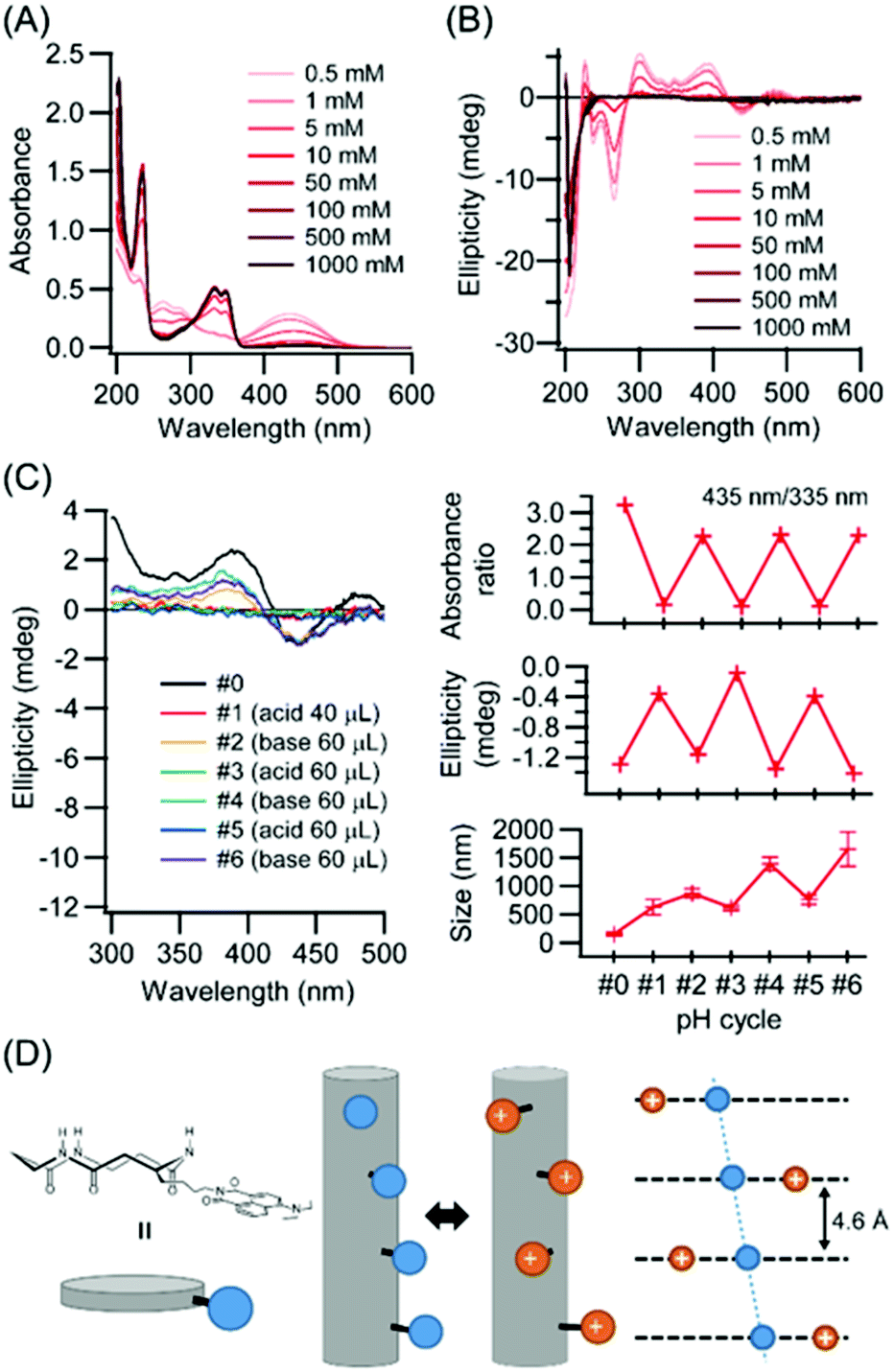 | ||
| Fig. 4 (A and B) Normalized UV-vis absorption and CD spectra of CP3Npi in HFIP/HCl aq. with various concentration of HCl (0.5, 1, 5, 10, 50, 100, 500, 1000 mM). (C) pH-Responsive UV, CD and DLS cycle profiles. (D) Schematic illustration of pH responsive behavior of CP3Npi peptide nanotube. | ||
The effect of protonation at the diethylamino group on molecular assembling morphology was investigated. Unexpectedly, under acidic conditions PNT molecular assemblies were retained as evidenced by DLS measurements (Fig. 4C). Electrostatic interactions between two charges in water decrease comparable to the thermal energy with the Bjerrum length lB (lB = 0.71 nm).30 The closest distance between two Npi moieties is 0.46 nm based on the cyclic peptide stacking distance for the neighboring pair,10,24 and the distance between first and third cyclic peptides along PNT exceeds over lB (Fig. 4D). There are three stacking modes with rotations by 0°, 120°, and 240° between first and second cyclic peptides along PNT.29 The rotation by 0° of the closest Npi moieties (blue spheres) illustrated in Fig. 4D causes severe electrostatic repulsions whilst other rotations (red spheres illustrated in Fig. 4D) can relieve it. Further, cations along PNT might exist as close as 0.46 nm without destroying the PNT formation owing to counterions, which is well known as the Manning–Oosawa counterion condensation.31,32 As a result, Npi moieties can rearrange the alignments with retaining PNT upon pH stimuli.
Repeatability of the pH responsive behavior was further analyzed by adding HCl and NaOH solutions. Table 1 shows the experimental conditions of the added volumes of 5 M HCl and 5 M NaOH aqueous solutions to a 2 mL CP3Npi suspension in HFIP/water (1/39 v/v). Under acidic conditions (pH cycle #1), protonation of the diethylamino group was confirmed by UV-vis absorbance ratio (435 nm/335 nm) and CD ellipticity of 441 nm. In addition, molecular assembly size was increased from 150 nm to 625 nm by DLS measurements. Therefore, even though Coulomb repulsion energy was expected to be increased, nanotubes kept their structures. On the other hand, under basic conditions (pH cycle #2), UV-vis absorption ratio and CD ellipticity were recovered again. This behavior was observed repeatedly from #1 to #6, suggesting the PNT structure is maintained under the pH cycle conditions, and the Npi groups are arranged alternately between the chiral and random alignments.
| Cycle | #0 | #1 | #2 | #3 | #4 | #5 | #6 |
|---|---|---|---|---|---|---|---|
| HCl aq. | — | 40 μL | — | 60 μL | — | 60 μL | — |
| NaOH aq. | — | — | 60 μL | — | 60 μL | — | 60 μL |
| pH | 5.4 | 1.0 | 8.5 | 1.0 | 8.5 | 1.0 | 8.5 |
4. Conclusions
We have successfully demonstrated the columnar alignment of D–π–A chromophores. The D–π–A Npi groups formed parallel H-type stacking along peptide nanotubes. The D–π–A chromophore alignment was shown to be switched between two states, the chiral and random alignments in response to pH changes. Cyclic β-peptide can be utilized, therefore, as a scaffold for developing functional materials using molecular dipole and switching properties, regarded as a new strategy for molecular devices. These D–π–A columns here are therefore expected to become an efficient charge separation system, which is under way.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported partially by JSPS KAKENHI Grant Number JP19K15375, The Kyoto University Foundation, Iketani Science and Technology Foundation (0311024-A), and The Kyoto Technoscience Center.Notes and references
- J. Zhang, J. Q. Jin, H. X. Xu, Q. C. Zhang and W. Huang, J. Mater. Chem. C, 2018, 6, 3485–3498 RSC.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2019, 1904545 CrossRef.
- A. Bolag, N. Sakai and S. Matile, Chem.–Eur. J., 2016, 22, 9006–9014 CrossRef CAS PubMed.
- E. Zojer, T. C. Taucher and O. T. Hofmann, Adv. Mater. Interfaces, 2019, 6, 1900581 CrossRef.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 1801079 CrossRef PubMed.
- S. Y. Son, Y. Kim, J. Lee, G. Y. Lee, W. T. Park, Y. Y. Noh, C. E. Park and T. Park, J. Am. Chem. Soc., 2016, 138, 8096–8103 CrossRef CAS PubMed.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. Mcree and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS PubMed.
- F. Fujimura, M. Fukuda, J. Sugiyama, T. Morita and S. Kimura, Org. Biomol. Chem., 2006, 4, 1896–1901 RSC.
- A. N. Tikhonov, Photosynth. Res., 2013, 116, 511–534 CrossRef CAS PubMed.
- T. Roach and A. Krieger-Liszkay, Curr. Protein Pept. Sci., 2014, 15, 351–362 CrossRef CAS PubMed.
- D. Takagi, K. Amako, M. Hashiguchi, H. Fukaki, K. Ishizaki, T. Goh, Y. Fukao, R. Sano, T. Kurata, T. Demura, S. Sawa and C. Miyake, Plant J., 2017, 91, 306–324 CrossRef CAS PubMed.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336 RSC.
- R. Berera, C. Herrero, I. H. M. van Stokkum, M. Vengris, G. Kodis, R. E. Palacios, H. van Amerongen, R. van Grondelle, D. Gust, T. A. Moore, A. L. Moore and J. T. M. Kennis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5343–5348 CrossRef CAS PubMed.
- S. D. Straight, G. Kodis, Y. Terazono, M. Hambourger, T. A. Moore, A. L. Moore and D. Gust, Nat. Nanotechnol., 2008, 3, 280–283 CrossRef CAS PubMed.
- A. Sikder, J. Sarkar, T. Sakurai, S. Seki and S. Ghosh, Nanoscale, 2018, 10, 3272–3280 RSC.
- M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef PubMed.
- F. Würthner, C. R. Saha-Moller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- A. T. Haedler, K. Kreger, A. Issac, B. Wittmann, M. Kivala, N. Hammer, J. Kohler, H. W. Schmidt and R. Hildner, Nature, 2015, 523, 196–199 CrossRef CAS PubMed.
- L. Zang, Acc. Chem. Res., 2015, 48, 2705–2714 CrossRef CAS PubMed.
- S. Ast, P. J. Rutledge and M. H. Todd, Phys. Chem. Chem. Phys., 2014, 16, 25255–25257 RSC.
- N. I. Georgiev, P. V. Krasteva and V. B. Bojinov, J. Lumin., 2019, 212, 271–278 CrossRef CAS.
- H. Uji, H. Kim, T. Imai, S. Mitani, J. Sugiyama and S. Kimura, Biopolymers, 2016, 106, 275–282 CrossRef CAS PubMed.
- Y. Tabata, Y. Kamano, H. Uji, T. Imai and S. Kimura, Chem. Lett., 2019, 48, 322–324 CrossRef CAS.
- G. P. Dado and S. H. Gellman, J. Am. Chem. Soc., 1994, 116, 1054–1062 CrossRef CAS.
- F. Würthner, Acc. Chem. Res., 2016, 49, 868–876 CrossRef PubMed.
- Y. Kamano, Y. Tabata, H. Uji and S. Kimura, RSC Adv., 2019, 9, 3618–3624 RSC.
- Y. Tabata, S. Mitani, H. Uji, T. Imai and S. Kimura, Polym. J., 2019, 51, 601–609 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic, New York, 3rd edn, 2011 Search PubMed.
- G. S. Manning, J. Chem. Phys., 1969, 51, 924–933 CrossRef CAS.
- B. O'Shaughnessy and Q. Yang, Phys. Rev. Lett., 2005, 94, 048302 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials synthetic section, NMR spectra, HPLC chromatograms, fluorescent spectra, UV spectra of reference compounds. See DOI: 10.1039/d0ra00231c |
| This journal is © The Royal Society of Chemistry 2020 |