
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedent formation of methylsilylcarbonates from iridium-catalyzed reduction of CO2 with hydrosilanes†
Jefferson Guzmán,
Pilar García-Orduña,
Fernando J. Lahoz and
Francisco J. Fernández-Alvarez*
and
Francisco J. Fernández-Alvarez*
Departamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), Universidad de Zaragoza, Facultad de Ciencias, 50009, Zaragoza, Spain. E-mail: paco@unizar.es
First published on 5th March 2020
Abstract
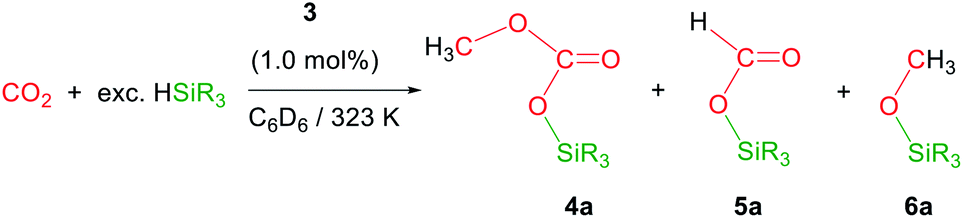
The iridium complex [Ir(μ-CF3SO3)(κ2-NSiMe2)2]2 (3) (NSiMe2 = {4-methylpyridine-2-yloxy}dimethylsilyl) has been prepared by reaction of [Ir(μ-Cl)(κ2-NSiMe2)2]2 (1) with two equivalents of AgCF3SO3. The solid structure of 3 evidenced its dinuclear nature, being a rare example of an iridium species with triflate groups acting as bridges. The 3-catalyzed reduction of CO2 with HSiMe(OSiMe3)2 affords a mixture of the corresponding silylformate and methoxysilane together with the silylcarbonate CH3OCO2SiMe(OSiMe3)2 (4a). This is the first time that the formation of silylcarbonates has been observed from the catalytic reduction of CO2 with silanes. Analogous behaviour has been observed when HSiMe2Ph and HSiMePh2 were used as reductants.
Introduction
The catalytic reaction of CO2 with silicon-hydrides has proven to be an effective and thermodynamically favoured methodology for its reduction to formate, formaldehyde, methanol or methane (Scheme 1).1,2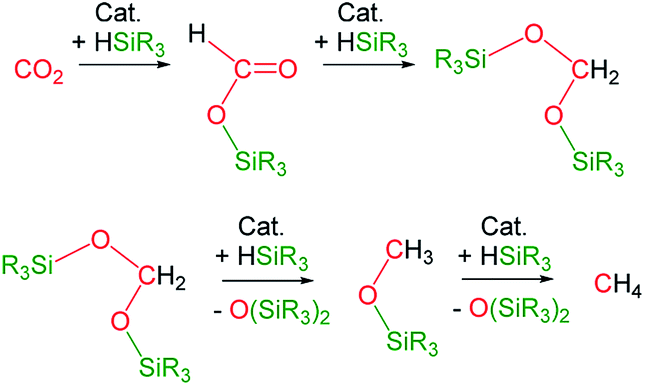 | ||
| Scheme 1 Products from the catalytic reduction of CO2 with silicon-hydrides reported so far. | ||
However, to the best of our knowledge the formation of methylsilylcarbonates, MeOCO2SiR3 (Fig. 1),3 as products of the catalytic reduction of CO2 with silicon-hydrides has not been reported so far.
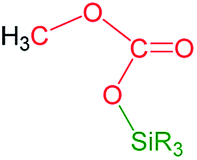 | ||
| Fig. 1 Methylsilylcarbonates. | ||
On the other hand, hydrosiloxanes are obtained in large scale as side products of the silicone industry and therefore, the development of catalytic processes effective for the selective reduction of CO2 based on hydrosiloxanes, instead of hydrosilanes, as reductants is of great interest.4 In this context, in recent years we have focused our research on the application of iridium species as catalysts for the reduction of CO2 with 1,1,1,3,5,5,5-heptamethyltrisiloxane, HSiMe(OSiMe3)2.5,6
Recently, we have described the synthesis and catalytic behavior of the iridium(III) complexes [Ir(μ-Cl)(κ2-NSiMe2)2]2 (1) and [Ir(CF3CO2)(κ2-NSiMe2)2] (2) (NSiMe2 = {4-methylpyridine-2-yloxy}dimethylsilyl) (Scheme 2).6 Species 2 catalyzed the selective reduction of CO2, depending on the reaction conditions, to silylformate or methoxysilane with HSiMe(OSiMe3)2.6 As a continuation of our studies on the chemistry of iridium complexes with pyridine-2-yloxyI-silyl ligands, we have found that the triflate derivative [Ir(μ-CF3SO3)(κ2-NSiMe2)2]2 (3) (Scheme 2) catalyzes the reaction of CO2 (3 bar) with HSiMe(OSiMe3)2 to give a mixture of the corresponding silylformate and methoxysilane along with a new compound that has been characterized by multinuclear NMR spectroscopy as CH3OCO2SiMe(OSiMe3)2 (4a). To the best of our knowledge, this is the first time that the formation of silylcarbonates from the catalytic reduction of CO2 with silanes has been observed. The results from these studies are described below.
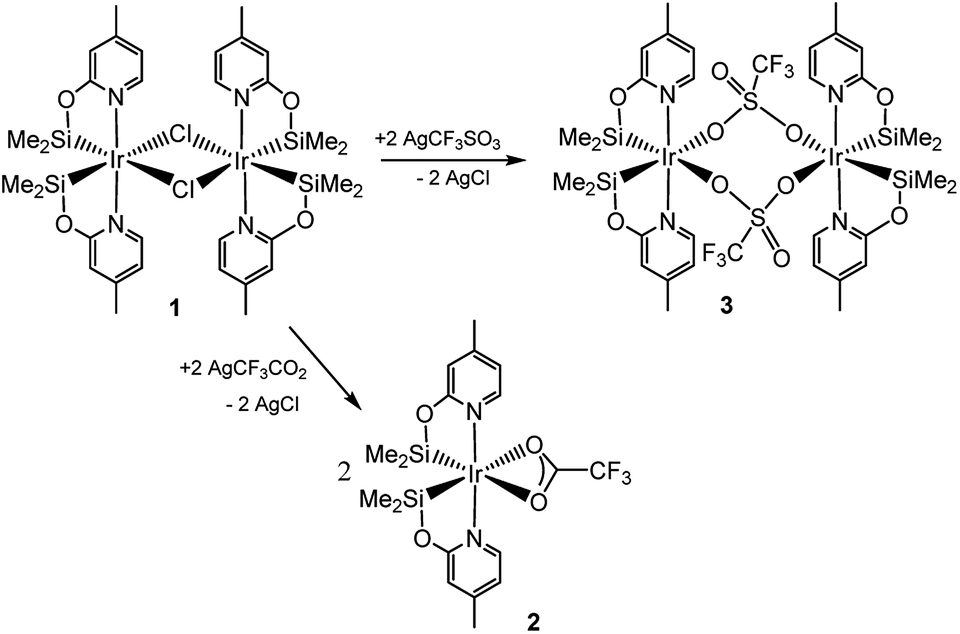 | ||
| Scheme 2 Preparation of the catalytic precursors 2 and 3. | ||
Results and discussion
Synthesis and characterization of the catalyst precursor
The reaction of complex 1 with two equivalents of AgCF3SO3 leads to the formation of [Ir(μ-CF3SO3)(κ2-NSiMe2)2]2 (3), which has been isolated as a white solid in 89% yield. The solid structure of complex 3 evidenced its dinuclear nature (Fig. 2). As far as we know, 3 is the first example of an iridium species with triflate groups acting as bridges. The molecule exhibits a crystallographic inversion center, with half the molecule as the independent part. Each metal atom shows an octahedral geometry like that found for 1.6 Main difference concerns the separation between the iridium atoms (6.0870(2) Å) in 3, significantly longer than that observed for 1 with chlorine bridges (4.0718(12) Å).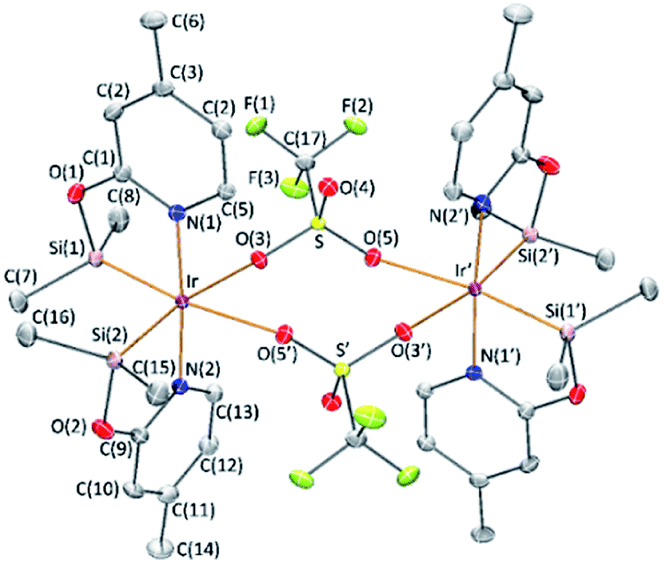 | ||
| Fig. 2 Molecular structure of compound 3. For clarity hydrogen atoms have been omitted. Primed atoms are related to unprimed ones through 1 − x, 1 − y,−z symmetry operation. Selected bond distances (Å) and angles (°): Ir–Si(1), 2.2570(5); Ir–Si(2), 2.2615(5); Ir–O(3), 2.3653(12); Ir–O(5′), 2.4331(13); Ir–N(1), 2.0590(15); Ir–N(2), 2.0583(15); Si(1)–Ir–Si(2), 91.68(5); Si(1)–Ir–O(3), 95.18(3); Si(1)–Ir–O(5′), 172.22(3); Si(1)–Ir–N(1), 81.57(4); Si(1)–Ir–N(2), 94.94(4); Si(2)–Ir–O(3), 170.27(3); Si(2)–Ir–O(5′), 93.26(3); Si(2)–Ir–N(1), 95.76(4); Si(2)–Ir–N(2), 82.22(4); O(3)–Ir–O(5′), 80.67(4); O(3)–Ir–N(1), 92.06(5); O(3)–Ir–N(2), 90.32(5); O(5′)–Ir–N(1), 91.95(5); O(5′)–Ir–N(2), N(1)–Ir–N(2), 175.94(6). | ||
1H and 13C{1H} NMR spectra of 3 in C6D6 show only one pattern of resonances for the four NSiMe2 ligands, which agrees with its high symmetry in solution. 29Si{1H} NMR spectra of 3 show a resonance at δ 38.2 ppm, corresponding to the four silicon atoms, which compares well to the value of δ 39.7 ppm reported for 2.6 In addition, a singlet resonance at δ −76.97 ppm in the 19F{1H} NMR spectra confirm the presence of the triflate ligand.
3-Catalyzed reduction of 13CO2 with silicon hydrides
Preliminary studies on the 3-catalyzed reaction of CO2 (3 bar) with HSiMe(OSiMe3)2 at 298 K showed that 24 hours are required to achieve the conversion of 94% of the starting hydrosiloxane into a mixture of the corresponding silylformate (53.0%) and methoxysilane (21.4%) along with a new compound (25.6%) (TOF = 3.9 h−1). This behavior differs from that previously reported for 2, which under the same reaction conditions catalyzed the full conversion of HSiMe(OSiMe3)2 into silylformate (93.0%) and methoxysilane (7.0%) in 3.5 h (TOF = 28.6 h−1).6 Therefore, complex 2 is not only more active but also more selective than 3.To determine the nature of the new species observed when 3 is used as catalyst for the hydrosilylation of CO2, we decided to study this reaction using isotopically labeled 13CO2. Thus, 1H and 13C NMR studies of the 3-catalyzed (1.0 mol%) reaction of 13CO2 (2.7 bar, 323 K, C6D6) with HSiMe(OSiMe3)2 were performed. These experiments evidenced, since early reaction stages, the presence of a mixture of 13CH3O–13CO2SiMe(OSiMe3)2 (4a), together with the corresponding silylformate, H13CO2SiMe(OSiMe3)2 (5a), and the methoxysilane, 13CH3OSiMe(OSiMe3)2 (6a) (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | T (K) | 4ab | 5ab | 6ab | Conversionc | Time (h) |
| a General conditions: 3 (1.0 mol%) in 0.5 mL of dry C6D6.b Calculated by 1H NMR and expressed in mol%.c Calculated by 1H NMR relative to the starting hydrosiloxane and expressed in mol%.d After 24 h at 323 K.e Control experiment without iridium catalyst (Fig. S24 see ESI). | ||||||
| 1 | 323 | 26.7 | 65.2 | 8.1 | 70 | 3 |
| 2 | 323 | 18.8 | 70.0 | 11.2 | 98 | 12 |
| 3 | 323 | 19.0 | 70.0 | 11.0 | 98 | 24 |
| 4 | 358 | 11.7 | 68.7 | 19.6 | 100 | 24d |
| 5 | 358 | 8.5 | 68.0 | 23.5 | 100 | 48d |
| 6e | 323 | 0 | 0 | 0 | 0 | 24 |
The most characteristic resonance in the 1H NMR spectra of 4a is a doublet of doublets centered at δ 3.33 ppm (1JC–H = 146.9 Hz; 3JC–H = 4.1 Hz) corresponding to the 13CH3O protons (Fig. 3). This resonance shows a direct C–H bond correlation in the 1H–13C HSQC spectra with a doublet resonance centered at δ 54.1 ppm (3JC–C = 1.7 Hz), assigned to the 13CH3O carbon in the 13C APT NMR spectra, and also a C–H bond correlation in the 1H–13C HMBC spectra with a doublet resonance that appeared centered at δ 153.0 ppm (3JC–C = 1.8 Hz), assigned to the 13CO3 carbon in the 13C APT NMR spectra. These data compare well with those reported for alkyl-silyl-carbonates3 and support the structure proposed for 4a in Table 1 (see ESI†).
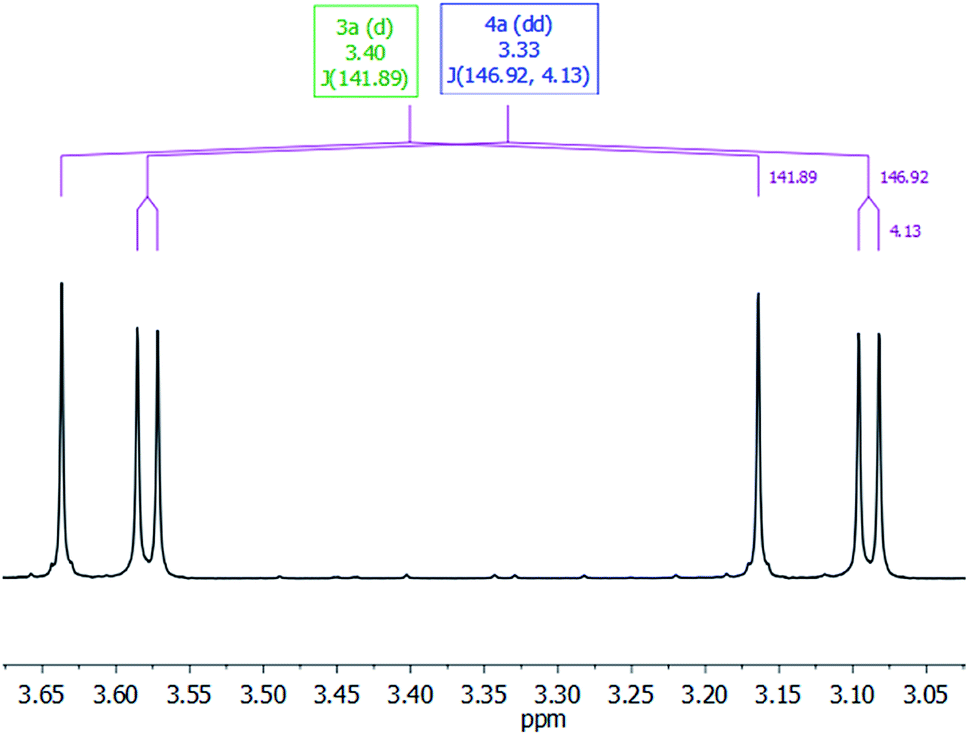 | ||
| Fig. 3 CH3O-region of the 1H NMR spectra in C6D6 of the 3-catalyzed reaction of 13CO2 (2.7 bar) with HSiMe(OSiMe3)2 at 323 K. | ||
Under the above described reaction conditions the slow consumption of HSiMe(OSiMe3)2 was observed (Table 1, entries 1 and 2). Thus, after 12 h the reaction is complete and 1H and 13C{1H} NMR spectra evidenced the consumption of all the starting 13CO2 and of the 98% of HSiMe(OSiMe3)2 to give a mixture of 4a (18.8 mol%), 5a (70.0 mol%) and 6a (11.2 mol%) (Table 1, entry 2). Heating this sample for another 12 h at 323 K did not evidence changes in the composition of the mixture (Table 1, entry 3). Interestingly, increasing the temperature at 358 K the slow transformation of 4a into 6a occurs (Table 1 entries 4 and 5), in addition, the formation of 13CO2 was observed. This outcome agrees with the known thermal behavior of alkyl-silyl-carbonates, ROCO2SiR3,3d,e which thermally decompose to give the corresponding methoxysilane and CO2 (Scheme 3, Path A).
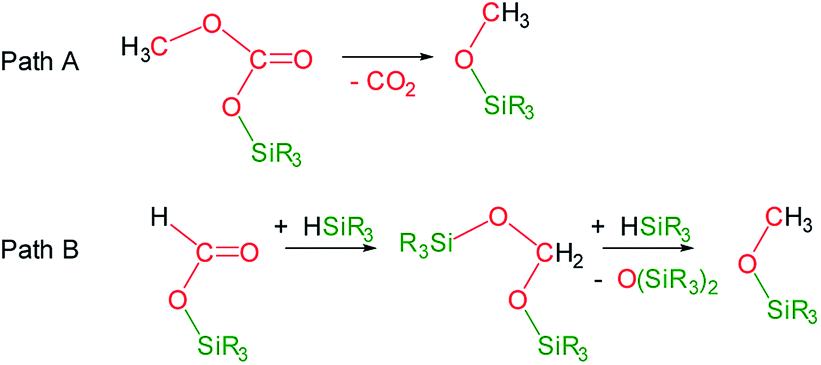 | ||
| Scheme 3 Reaction paths for the formation of methoxysilanes from the 3-catalyzed hydrosilylation of CO2. | ||
The formation of methoxy-silanes from the catalytic reduction of CO2 with hydrosilanes has been so far explained by reaction of bis(silyl)acetals with the corresponding hydrosilane (Scheme 3, Path B).6,7 Indeed, since it has been observed that at 323 K only traces of 4a are transformed into 6a (Table 1 entry 3) it is reasonable to assume that under these conditions 6a is formed by reactions of the bis(silyl)acetal 13CH2{OSiMe(OSiMe3)2}2 (7a) with HSiMe(OSiMe3)2. In agreement with this we have observed the presence of traces of 7a in the NMR spectra of this reaction (ESI†). However, after 24 h of reaction 98% of the starting hydrosiloxane was consumed and therefore, under these conditions, the formation of 6a at 358 K can only be explained by thermal decomposition of 4a (Scheme 3, Path B).
Analogous behavior has been observed when HSiMe2Ph and HSiMePh2 were used as reducing agents. Thus, the formation of the mixtures of methyl-silyl-carbonate 13CH3O–13CO2SiR3 (SiR3 = SiMe2Ph, 4b; SiMePh2, 4c) and the corresponding silylformate and methoxysilane from the 3-catalyzed reduction of 13CO2 with HSiR3 (SiR3 = SiMe2Ph, SiMePh2) was also observed (ESI†).
It should be mentioned, that the formation of the methyl-silyl-carbonates was not observed along the 2-catalyzed reduction of CO2 with HSiMe(OSiMe3)2.6 In this context, even though species 2 and 3 seem very similar, their reactivity against silanes is very different. Thus, while complex 3 is stable in presence of one equivalent of HSiMe(OSiMe3)2 at 323 K, the trifluoroacetate ligand in 2 is reduced to give the corresponding silylether CF3CH2OSiR3.5d,6 Therefore, it is reasonable to think that the stability of the triflate ligand could play a role stabilizing reaction intermediates, which could open new paths of reaction, no accessible in 2-catalyzed CO2 hydrosilylation reactions.
At this point the question arises, how methylsilylcarbonates are formed from the 3-catalyzed hydrosilylation of CO2. A plausible mechanism proposal is shown in Scheme 4. The formation of methylcarbonates has been explained by insertion of CO2 into the Ir–O bond of iridium–OCH3 species.8 Therefore, we propose that the reaction of an iridium-methoxy intermediate (A in Scheme 4) with CO2 could lead to the methylcarbonate species B, which by reaction with HSiR3 would afford the corresponding methylsilylcarbonate and the Ir–H species C. The next step, is the reaction of C with CO2 to give the iridium–formate intermediate D, which according to our previous results has been found to be a thermodynamically favoured process.5d,6 Finally, the reaction of the iridium–formate D with excess of silane could regenerate A closing the catalytic cycle.
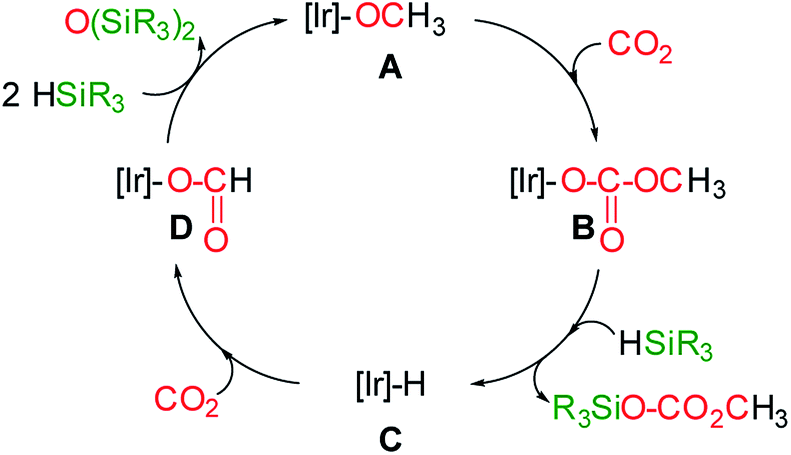 | ||
| Scheme 4 Plausible mechanism proposal for the formation of methyl-silyl-carbonates from the 3-catalyzed hydrosilylation of CO2. | ||
Experimental
General considerations
All manipulations were carried out under an argon atmosphere by Schlenk-type techniques or in a Glovebox MBraun Unilab. Organic solvents were dried by standard procedures and distilled under argon prior to use or obtained oxygen- and water-free from a solvent purification system (Innovative Technologies). 1H, 13C, 29Si and 19F NMR spectra were obtained on a Bruker AV-300, AV-400 or AV-500 spectrometer. Chemical shifts (δ), reported in ppm, are referenced to the residual solvent peaks and coupling constants (J) are reported in Hz.CCDC 1972218 contains the supplementary crystallographic data for this paper.
Crystal data compound 3. C34H48F6Ir2N4O10S2Si4·0.5(H2O); M = 1364.65; colourless prism 0.140 × 0.240 × 0.330 mm3; triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.0468(5), b = 12.0453(6), c = 12.0619(7) Å, α = 66.4720(10), β = 88.5460(10), γ = 88.7690(10)°, V = 1204.65(11) Å3; Z = 1; Dc = 1.881 g cm−3; μ = 5.783 mm−1; Tmin/Tmax: 0.1676/0.5996; 23
, a = 9.0468(5), b = 12.0453(6), c = 12.0619(7) Å, α = 66.4720(10), β = 88.5460(10), γ = 88.7690(10)°, V = 1204.65(11) Å3; Z = 1; Dc = 1.881 g cm−3; μ = 5.783 mm−1; Tmin/Tmax: 0.1676/0.5996; 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 425/5849 reflections measured/unique (Rint = 0.0179), number of data/restraint/parameters 5849/0/286, R1(F2) = 0.0131 (5771 reflections, I > 2σ(I)) and wR(F2) = 0.0324 (all data), final GoF = 1.034, largest difference peak: 0.988 e Å−3.
425/5849 reflections measured/unique (Rint = 0.0179), number of data/restraint/parameters 5849/0/286, R1(F2) = 0.0131 (5771 reflections, I > 2σ(I)) and wR(F2) = 0.0324 (all data), final GoF = 1.034, largest difference peak: 0.988 e Å−3.
Conclusions
In conclusion, the use of triflate, instead of trifluoroacetate, as ancillary ligand in the chemistry of Ir-(NSiMe2)2 species allows the preparation of 3, which is a rare example of a dinuclear iridium complex with triflate ligands acting as bridges.1H NMR studies of the 3-catalyzed reduction of CO2 with hydrosilanes evidenced the unprecedent formation of methylsilylcarbonates as reaction products, together with the corresponding silylformate and methoxysilane.
The results of this investigation show that the formation of methoxysilanes during the catalytic reduction of CO2 with silanes, which traditionally has been explained by the catalytic reaction of bis(silyl)acetals with silanes, could also be consequence of thermal decomposition of the corresponding methylsilylcarbonate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge MICINN/FEDER projects PGC2018-099383-B-100 and the Regional Government of Aragon/FSE 2014–2020 “Building Europe from Aragón” (group E42_17R) for funding.Notes and references
- (a) J. Chen, M. McGraw and E. Y.-X. Chen, ChemSusChem, 2019, 12, 4543–4569 CrossRef CAS PubMed; (b) F. J. Fernández-Alvarez and L. A. Oro, ChemCatChem, 2018, 10, 4783–4796 CrossRef; (c) C. Chauvier and T. Cantat, ACS Catal., 2017, 7, 2107–2115 CrossRef CAS; (d) F. J. Fernández-Alvarez, A. M. Aitani and L. A. Oro, Catal. Sci. Technol., 2014, 4, 611–624 RSC.
- For examples of transition-metal free catalyzed processes see: K. Motokura, C. Nakagawa, R. A. Pramudita and Y. Manaka, ACS Sustainable Chem. Eng., 2019, 7, 11056–11061 CrossRef CAS ; and references therein.
- (a) Y. Yamamoto and D. S. Tarbell, J. Org. Chem., 1971, 36, 2954–2956 CrossRef; (b) M. Paul, J. Dunoguès, R. Calas and E. Frainnet, J. Organomet. Chem., 1972, 38, 267–274 CrossRef CAS; (c) Y. Yamamoto, D. S. Tarbell, J. R. Fehlner and B. M. Pope, J. Org. Chem., 1973, 38, 2521–2525 CrossRef CAS; (d) R. Tacke, M. Link, A. Bentlage-Felten and H. Zilch, Z. Naturforsch., 1985, 40b, 942–947 CAS; (e) H. Yildirimyan and G. Gattow, Z. Anorg. Allg. Chem., 1985, 521, 135–144 CrossRef CAS.
- D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004–6011 CrossRef CAS PubMed.
- (a) R. Lalrempuia, M. Iglesias, V. Polo, P. J. Sanz Miguel, F. J. Fernández-Alvarez, J. J. Pérez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2012, 51, 12824–12827 CrossRef CAS PubMed; (b) E. A. Jaseer, M. N. Akhtar, M. Osman, A. Al-Shammari, H. B. Oladipo, K. Garcés, F. J. Fernández-Alvarez, S. Al-Khattaf and L. A. Oro, Catal. Sci. Technol., 2015, 5, 274–279 RSC; (c) A. Julián, E. A. Jaseer, K. Garcés, F. J. Fernández-Alvarez, P. García-Orduña, F. J. Lahoz and L. A. Oro, Catal. Sci. Technol., 2016, 6, 4410–4417 RSC; (d) A. Julián, J. Guzmán, E. A. Jaseer, F. J. Fernández-Alvarez, R. Royo, V. Polo, P. García-Orduña, F. J. Lahoz and L. A. Oro, Chem.–Eur. J., 2017, 23, 11898–11907 CrossRef PubMed; (e) A. I. Ojeda-Amador, J. Munarriz, P. Alamán-Valtierra, V. Polo, R. Puerta-Oteo, M. V. Jiménez, F. J. Fernández-Alvarez and J. J. Pérez-Torrente, ChemCatChem, 2019, 11, 5524–5535 CrossRef CAS.
- J. Guzmán, P. García-Orduña, V. Polo, F. J. Lahoz, L. A. Oro and F. J. Fernández-Alvarez, Catal. Sci. Technol., 2019, 9, 2858–2867 RSC.
- (a) T. C. Eisenschmid and R. Eisenberg, Organometallics, 1989, 8, 1822–1824 CrossRef CAS; (b) S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed; (c) S. N. Riduan, J. Y. Ying and Y. Zhang, ChemCatChem, 2013, 5, 1490–1496 CrossRef CAS; (d) D. S. Morris, C. Weetman, J. T. C. Wennmacher, M. Cokoja, M. Drees, F. E. Kühn and J. B. Love, Catal. Sci. Technol., 2017, 7, 2838–2845 RSC; (e) D. Specklin, F. Hild, C. Fliedel, C. Gourlaouen, L. F. Veiros and S. Dagorne, Chem.–Eur. J., 2017, 23, 15908–15912 CrossRef CAS PubMed; (f) M. Saleh, D. R. Powell and R. J. Wehmschulte, Organometallics, 2017, 36, 4810–4815 CrossRef CAS.
- N. Hazari and J. E. Heimann, Inorg. Chem., 2017, 56, 13655–13678 CrossRef CAS PubMed.
- SAINT+, version 6.01: Area-Detector Integration Software, Bruker AXS, Madison, 2001 Search PubMed.
- (a) SADABS (Version 2016/02), Bruker AXSMadison Search PubMed; (b) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and crystallographic information. CCDC 1972218. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra00204f |
| This journal is © The Royal Society of Chemistry 2020 |