
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral mono- and dicarbamates derived from ethyl (S)-lactate: convenient chiral solvating agents for the direct and efficient enantiodiscrimination of amino acid derivatives by 1H NMR spectroscopy†
Federica Balzano * and
Gloria Uccello-Barretta
* and
Gloria Uccello-Barretta
Department of Chemistry and Industrial Chemistry, University of Pisa, via Moruzzi 13, 56124 Pisa, Italy. E-mail: federica.balzano@unipi.it
First published on 29th January 2020
Abstract
New chiral solvating agents (CSAs) for NMR spectroscopy have been obtained from ethyl (S)-lactate, a very cheap commercially available product. By a sequence of simple chemical modifications of its functional groups, monocarbamoylated and dicarbamoylated derivatives were obtained, the potentialities of which as CSAs for NMR spectroscopy have been explored. Their ability to differentiate the resonances of enantiomeric mixtures of amino acids bearing a 3,5-dinitrobenzoyl moiety at the amino group and with the carboxyl function derivatized as methyl ester or amide has been probed. Almost every CSA was able to originate enantiodiscrimination in the 1H NMR spectra, with (2S)-1-(3,5-dimethylphenylcarbamoyloxy)-2-(3,5-dinitrophenylcarbamoyloxy)propane (4) standing out for efficiency and versatility.
Introduction
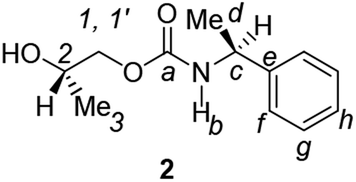
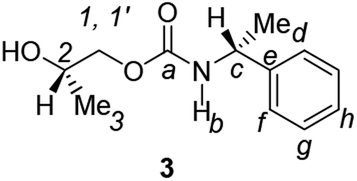
Divergent pharmacological behaviour of several enantiomers of chiral active ingredients still represents a critical aspect in biomedical and pharmaceutical research,1,2 which has given great impulse to the development of more and more sophisticated stereoselective synthetic procedures,3–6 as well as methods for assessing the stereoisomeric purities of chiral materials.7–10 The development of validated procedures of quantification of enantiomers requires suitable analytical or preparative methods of separation: several approaches are available, chromatographic and spectroscopic mainly.11–16 Among spectroscopic methods, nuclear magnetic resonance (NMR) spectroscopy15,16 has been playing a leading role since the 60s/70s, when the possibility to detect different NMR signals of enantiomers, transferred into a diastereomeric environment by use of suitable chiral auxiliaries, had been suggested. Under the impulse of these earliest experiments, three main classes of chiral auxiliaries for NMR spectroscopy were proposed, i.e. chiral derivatizing agents (CDAs), chiral solvating agents (CSAs) and chiral lanthanide shift reagents (CLSRs). CSAs have emerged for their best practicality of use and favourable spectroscopic features: any preliminary chemical derivatization procedure is not required as for CDAs, since CSAs are simply mixed to the enantiomeric substrates into the NMR tube, and severe linewidth broadenings are not caused as in the case of paramagnetic CLSRs, which could be detrimental in terms of accuracy and reproducibility of enantiomers quantification.Proposed chiral solvating agents span from very simple low molecular weight compounds, to natural products or systems with highly preorganized structures able to enhance the enantioselectivity of the interaction with the chiral substrates.15,16 However, in the everyday practice of NMR detection and quantification of enantiomers, mainly for trial separations, chiral auxiliaries coming from commercially available inexpensive products should be privileged. With this idea in mind, we took into consideration ethyl lactate, a very cheap product, as an attractive building block for the development of new CSAs for NMR spectroscopy, obtained by simple derivatization procedures of its functional groups.17–19 In particular, we focused on the ethyl lactate derivative (2S)-2-(tetrahydro-2-pyranoxy)-1-propanol (I), in which the primary hydroxyl was carbamoylated by preserving the secondary one as in the compounds 1, 2, and 3 (Fig. 1). Compound 1 was endowed with an electron rich 3,5-dimethylphenyl ring, 2 and 3 were diastereomeric derivatives in which an additional chiral centre was introduced. CSAs 1–3 were furtherly carbamoylated by introducing the π-acid 3,5-dinitrophenyl moiety on the secondary hydroxyl, in the perspective of having in hands new polyamidic CSAs (compounds 4–6, Fig. 1), in which hydrogen bond donor/acceptor interaction could be reinforced by attractive π–π interactions.
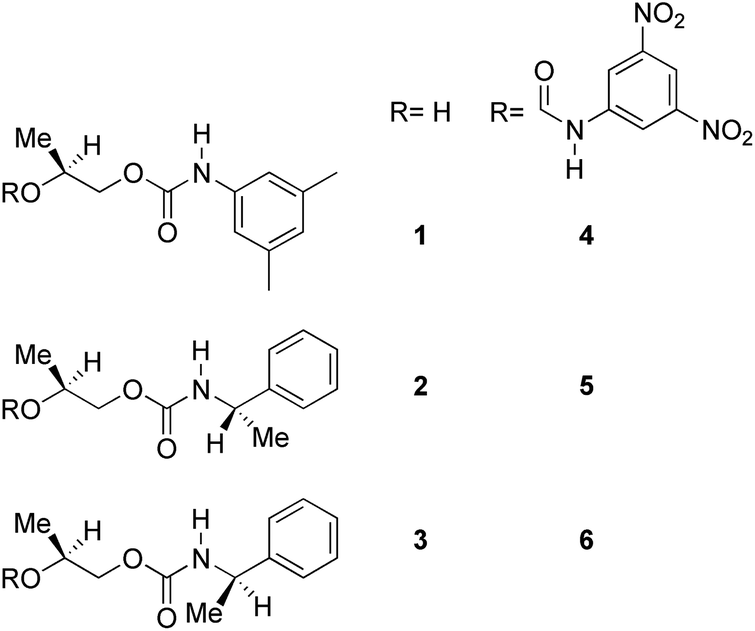 | ||
| Fig. 1 Chemical structures of CSAs 1–6. | ||
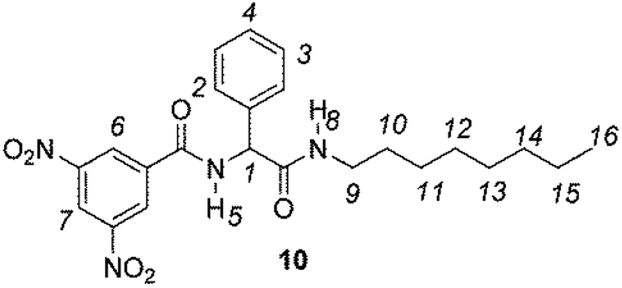
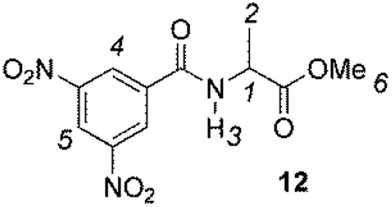
The enantiodiscriminating efficiency of above said new chiral auxiliaries was probed towards N-3,5-dinitrobenzoyl derivatives of amino acids 7–12 (Fig. 2) in which the carboxyl function was derivatized as amide function (7–10) or as methyl ester (11–12). Compound 13 was a simple derivative of an amine (Fig. 2).
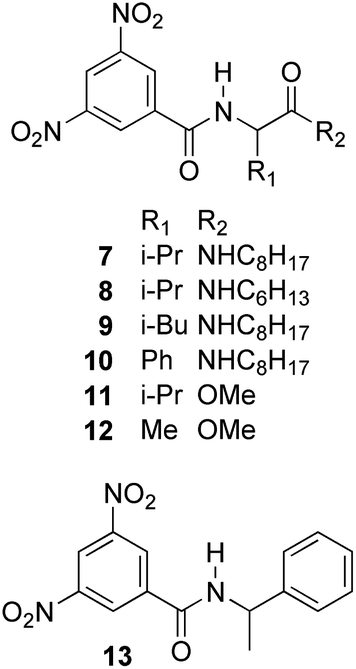 | ||
| Fig. 2 Chemical structures of derivatives 7–13. | ||
Results and discussion
Synthesis of CSAs 1–6
Ethyl (S)-lactate is a chiral pool reagent20 used for the preparation of enantiomerically pure analogs via straightforward steps including alcohol protection and direct reduction of the ester to a primary alcohol as in the case of I (ESI, Scheme S1†).21 Derivative I has been used as a chiral scaffold to prepare the CSAs 1–3 in high yields (84–100%) in two steps: reaction of the primary alcohol function with selected isocyanates in toluene at reflux for 24 hours and their subsequent deprotection in the presence of Amberlyst-15H in methanol at room temperature for 3 hours. The reaction of the secondary alcohol group of the lactate-derived chiral carbamates 1–3 with 3,5-dinitrophenyl isocyanate in toluene at reflux afforded the CSAs 4–6 in essentially quantitative yield (Scheme 1).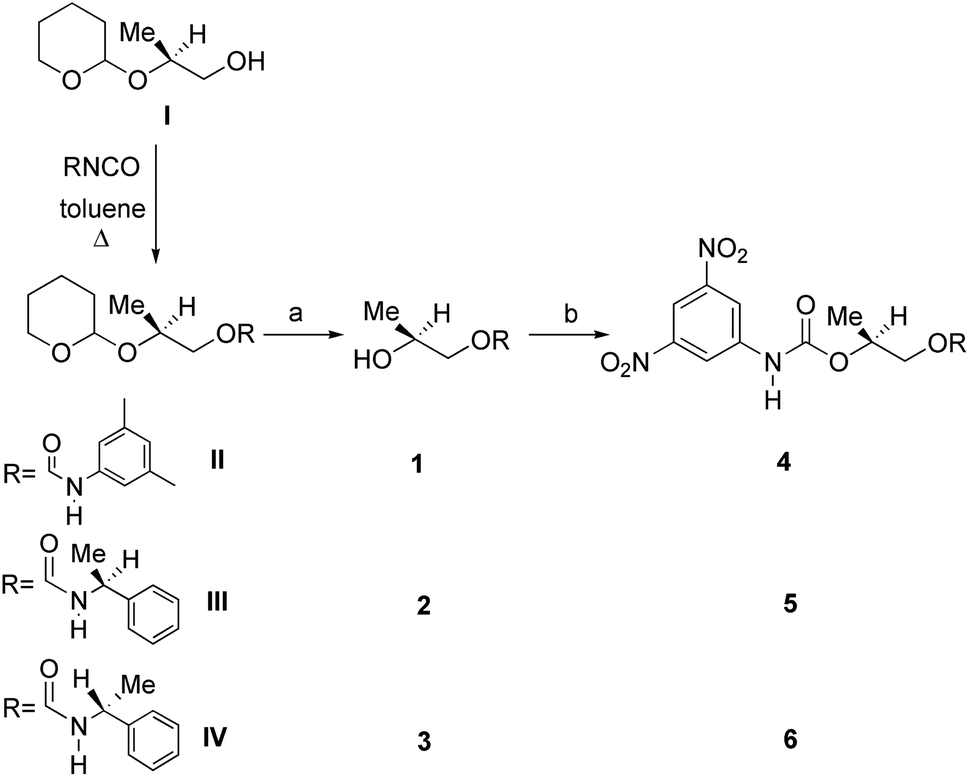 | ||
| Scheme 1 Synthesis of CSAs 1–6: (a) Amberlyst-15H, MeOH; (b) 3,5-dinitrophenyl isocyanate, toluene at reflux. | ||
All chiral solvating agents were fully characterized in CDCl3 solution by 1H and 13C NMR spectroscopy by comparing homonuclear and heteronuclear scalar or dipolar correlations in COSY (COrrelation SpectroscopY), ROESY (Rotating-frame Overhauser Enhancement SpectroscopY), HSQC (Heteronuclear Single Quantum Correlation) and HMBC (Heteronuclear Multiple Bond Correlation) maps.
1H-NMR enantiodiscrimination experiments
Chiral auxiliaries 1–6 were employed as 1H NMR chiral solvating agents for the enantiodiscrimination of chiral compounds 7–13 (Fig. 2) in CDCl3 solution.CSAs efficiency was evaluated by comparing nonequivalences data (Table 1) obtained from 1H NMR spectra of equimolar CSA/substrate mixtures, recorded in the same experimental conditions (total concentration 20 mM, 25 °C). Nonequivalence (ΔΔδ = |ΔδS − ΔδR|, ppm; where ΔδS = δS − δfree and ΔδR = δR − δfree) is defined as the difference between the chemical shifts of corresponding enantiotopic nuclei in the presence of the CSA (δS and δR) and it describes the enantiodifferentiation capability of a chiral solvating agent towards a selected class of compounds.
| Substrate proton | CSA-1 | CSA-2 | CSA-3 | CSA-4 | CSA-5 | CSA-6 |
|---|---|---|---|---|---|---|
| 7-NHα | — | 0.018 | 0.014 | 0.202 | 0.061 | 0.014 |
| 7-NHβ | — | — | — | 0.095 | 0.032 | — |
| 7-Hortho | — | 0.004 | 0.004 | 0.019 | 0.003 | — |
| 7-Hpara | 0.003 | — | 0.003 | 0.055 | 0.015 | — |
| 8-NHα | 0.007 | 0.024 | 0.013 | 0.210 | 0.068 | 0.011 |
| 8-NHβ | — | 0.004 | — | 0.101 | 0.037 | — |
| 8-Hortho | — | 0.006 | 0.003 | 0.019 | 0.003 | — |
| 8-Hpara | 0.003 | 0.003 | 0.002 | 0.107 | 0.015 | — |
| 9-NHα | — | 0.023 | 0.031 | 0.105 | 0.030 | 0.024 |
| 9-NHβ | — | — | 0.008 | 0.052 | 0.015 | 0.010 |
| 9-Hortho | — | 0.005 | 0.006 | 0.022 | 0.002 | 0.004 |
| 9-Hpara | 0.001 | 0.003 | 0.003 | 0.041 | 0.011 | 0.001 |
| 10-NHα | — | 0.006 | 0.009 | 0.009 | 0.011 | 0.018 |
| 10-NHβ | — | — | — | 0.023 | 0.032 | 0.017 |
| 10-Hortho | — | 0.003 | 0.003 | 0.008 | — | 0.006 |
| 10-Hpara | — | 0.001 | 0.001 | 0.012 | — | 0.006 |
| 11-NH | — | 0.011 | 0.012 | 0.015 | — | 0.016 |
| 11-Hortho | — | 0.002 | — | 0.003 | — | 0.001 |
| 11-Hpara | — | — | — | 0.006 | — | — |
| 12-NH | — | 0.013 | 0.011 | 0.013 | — | 0.015 |
| 12-Hortho | — | 0.003 | — | 0.003 | — | 0.003 |
| 12-Hpara | — | — | — | 0.005 | — | — |
| 13-NH | — | 0.021 | 0.016 | — | 0.013 | 0.026 |
In regard to the amino acid derivatives 7–12, complexation shifts and doublings of NMR signals were observed for NH (Fig. 3, Table 1), 3,5-dinitrophenyl (Fig. 3, Table 1), and aliphatic protons in the presence of one equivalent of almost all the chiral solvating agents (ESI, Fig. S1†).
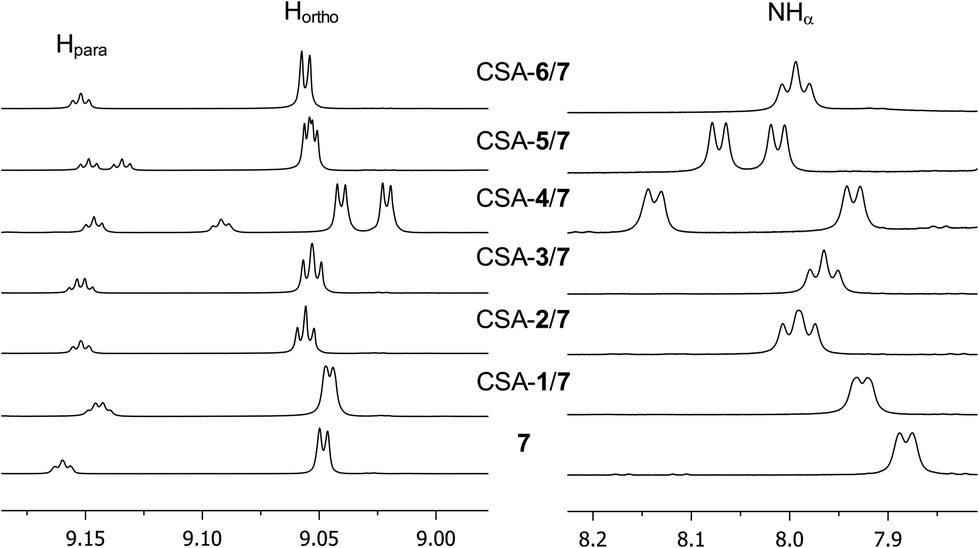 | ||
| Fig. 3 1H NMR (600 MHz, CDCl3, 25 °C) spectral regions corresponding to 3,5-dinitrobenzoyl and NHα protons of 7 (10 mM) in the presence of one equivalent of CSA. | ||
Among CSAs 1–3, the presence of a further stereogenic centre, as in 2 and 3, makes them more effective in chiral recognition processes with respect to the compound 1 with an achiral π-basic moiety (Table 1). Indeed, negligible nonequivalences (0–0.007 ppm) were measured only on some protons of the derivatives 7–12 in the presence of CSA 1, whereas doublings of the enantiomeric signals were detected in the presence of one equivalent (10 mM) of 2 or 3, leading to the best enantiodifferentiation for the amino acid NHα proton (0.006–0.031 ppm, Table 1). The diastereomeric relationship of 2 and 3 does not seem affect the enantiodiscriminating efficiency, thus suggesting that the lactate moiety is mainly involved in the enantioselective interactions that contribute to the differentiation of NMR signals of the two enantiomers.
The presence of two carbamoyl functions as in CSAs 4–6, led to better enantiodiscriminations. Interestingly, CSA 4, derived from the less efficient monocarbamoylated CSA 1, produced the best results in the enantiodifferentiation experiments (Table 1). In particular, in the 1H NMR spectrum of the equimolar mixture CSA-4/7 (total concentration 20 mM) two well-separated triplets (0.095 ppm) and doublets (0.202 ppm) were detected for the NHβ (ESI, Fig. S1†) and NHα (Fig. 3) protons, respectively, allowing a very accurate integration of enantiomeric signals. The enantioseparations of signals due to 3,5-dinitrophenyl moiety were relevant too (0.019 ppm and 0.055 ppm for ortho and para protons, respectively, Fig. 3, Table 1). The effect of the change of absolute configuration in 5 with respect to 6 is not clear.
The increasing of the total concentration, from 20 mM to 60 mM, produced a slight enhancement of the nonequivalences measured on protons of derivative 7, more significant for the CSAs 2 and 3 with respect to dicarbamates 4–6 (ESI, Table S1 and Fig. S2†).
Similar trend of the nonequivalences was observed for every diamide derivative of amino acids (8–10). The presence of the phenyl moiety in 10 produced a lowering of the nonequivalences with respect to amino acid derivatives with alkyl groups, as in 7, 8 and 9 (Table 1).
The length of the alkyl chain bound to the NHβ of the amino acid derivative did not influence the nonequivalences, as confirmed by comparing 7 and 8, respectively bearing C8H17 and C6H13 groups (Table 1).
The role of derivatization at the carboxyl function emerged in the comparison between valine derivatives 8 and 11, in the presence of CSA 4. Nonequivalence detected at the NHα proton spectrum of diamide 8 was 14-fold superior in comparison with 11, the carboxyl function of which was derivatized as methyl ester (Table 1).
The less extent of enantiodiscrimination towards methyl esters derivatives was confirmed for the alanine derivative 12 too. It is noteworthy that nonequivalence was very responsive to temperature: lowering temperature from 25 °C till to −40 °C remarkably and progressively increased nonequivalences of 3,5-dinitrophenyl and NH protons of 12 (Table 2), reasonably as the consequence both of increase of association constants of diastereomeric solvates and enhanced conformational restrictions.
| Substrate | Proton | 25 °C | 0 °C | −20 °C | −40 °C |
|---|---|---|---|---|---|
| 12 | NH | 0.013 | 0.028 | 0.052 | 0.089 |
| Hortho | 0.003 | 0.005 | 0.011 | 0.019 | |
| Hpara | 0.005 | 0.012 | 0.024 | 0.048 |
Finally, CSAs 2–3 and their corresponding dicarbamate derivatives 5–6 were able to discriminate the enantiomers of 3,5-dinitrobenzoyl derivative of α-phenylethylamine (13) in a comparable way (Table 1). It is interesting to note that no chiral recognition of 13 was observed not only in the presence of CSA 1, which showed the lowest enantiodiscriminating efficiency towards compounds 7–12, but also in the presence of 4, which showed the best efficiency.
Conclusions
Ethyl (S)-lactate represents a cheap commercially available chiral reagent which can be subjected to straightforward synthetic transformations, in view of the design of several classes of new chiral auxiliaries.Its derivative (2S)-2-(tetrahydro-2-pyranoxy)-1-propanol can be exploited to prepare new monocarbamoyl and dicarbamoyl derivatives which are promising chiral solvating agents for the NMR differentiation of amino acid derivatives.
In particular, (2S)-1-(3,5-dimethylphenylcarbamoyloxy)-2-(3,5-dinitrophenylcarbamoyloxy)propane (4) constitutes a polyamidic platform in which hydrogen bond and π–π interactions are potentially able to cooperate in order to generate privileged attractive interactions with N-3,5-dinitrobenzoyl derivatives of amino acids, both aliphatic and aromatic, with the carboxyl functions derivatized as amide or methyl ester. Equimolar amounts of 4 at moderately low concentration lead to remarkable differentiation of several NMR resonances of the enantiomeric mixtures, which is the basis for the development of any accurate and reliable methods of determination of enantiomeric composition.
Experimental
General experimental methods
NMR measurements were mainly performed in CDCl3 on a spectrometer operating at 600 MHz and 150 MHz for 1H and 13C nuclei, respectively. The temperature was controlled to ±0.1 °C. All 1H chemical shifts are referenced to TMS as external standard. The multiplicity of NMR signals is referred as singlet (s), broad singlet (br s), doublet (d), double doublet (dd), triplet (t), quartet (q), and multiplet (m). The 2D NMR spectra were obtained by using standard sequences. The spectral width used was the minimum required in both dimensions. The COSY (COrrelation SpectroscopY) maps were acquired by using a relaxation delay of 3 s, 256 increments of 2–4 scans and 2k data points. The ROESY (Rotating-frame Overhauser Enhancement SpectroscopY) spectra were recorded in the phase-sensitive mode, by employing a mixing time of 0.6 s. The pulse delay was maintained at 5 s; 256 increments of 4 scans and 2k data points each were collected. The data matrix was zero-filled to 2k × 1k and a Gaussian function was applied for processing in both dimensions. The gradient 1H–13C HSQC (Heteronuclear Single Quantum Correlation) and HMBC (Heteronuclear Multiple Bond Correlation) spectra were obtained in 4–16 scans per each of 256 increments and a relaxation delay of 2 s. The HMBC experiments were optimized for a long-range 1H–13C coupling constant of 8 Hz. Thin layer chromatography (TLC) was carried out on silica gel plates (Merck, Silica G-60).Materials
3,5-Dimethylphenyl isocyanate, 3,5-dinitrophenyl isocyanate, (S)-α-phenylethyl isocyanate, (R)-α-phenylethyl isocyanate, valine, N-(3,5-dinitrobenzoyl)phenylglycine, N-(3,5-dinitrobenzoyl)leucine, 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinine (EEDQ), and Amberlyst-15H were purchased from Aldrich and used without purification.Anhydrous toluene, methanol (MeOH), and tetrahydrofuran (THF) were prepared according to standard procedures.22 (2S)-2-(Tetrahydro-2-pyranoxy)-1-propanol (I) was kindly gifted by Prof. Rita Menicagli.
Synthesis of derivatives II–IV
A solution of I (6.87 mmol for II and 13.74 mmol for III–IV) and the appropriate isocyanate (6.78 mmol) in anhydrous toluene (30 mL) was stirred, under nitrogen atmosphere, and heated at reflux for 18–24 h. The crude products were recovered by removing the solvent under reduced pressure. II–IV were used in the following reactions without further purification.Synthesis of CSAs 1–3
A mixture of II–IV (6.25 mmol) and Amberlyst-15H (280 mg) in MeOH (30 mL) was stirred at room temperature for 3 h. The resin was filtered off and the filtrate concentrated under reduced pressure to give CSAs 1–3. For 2 and 3, the crude residue was dissolved in CH2Cl2. The organic layer was washed with H2O to remove the degradation product of the precursor used in excess, 1,2-propandiol, and dried over anhydrous Na2SO4. Usual work-up afforded 2 and 3 in high yield (84–91%).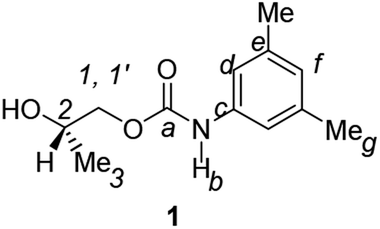
Synthesis of CSAs 4–6
A solution of 1–3 (2.23 mmol) and 3,5-dinitrophenyl isocyanate (2.23 mmol) in anhydrous toluene (30 mL) was stirred, under nitrogen atmosphere, and heated at reflux for 18 h. CSAs 4–6 were obtained with an almost quantitative yield by removing the solvent under reduced pressure.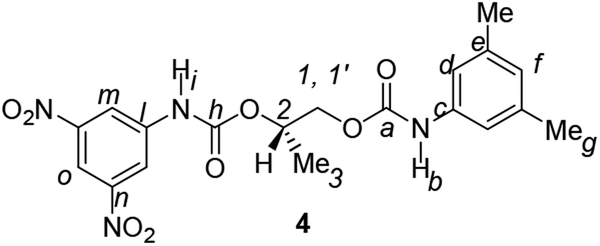
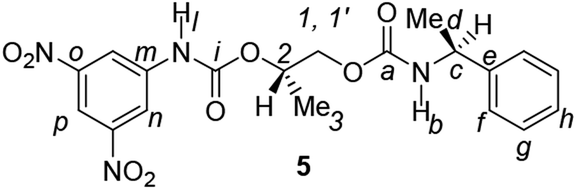
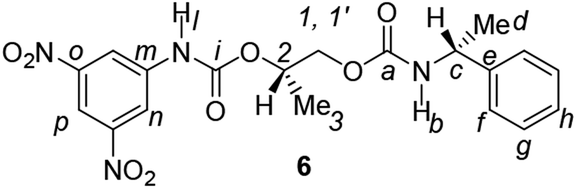
Synthesis of racemates 7–8
A mixture of valine (19.4 mmol), propylene oxide (56.2 mmol) and 3,5-dinitrobenzoyl chloride (19.4 mmol) in anhydrous THF (130 mL) was stirred, under nitrogen atmosphere, overnight at room temperature. After solvent evaporation under reduced pressure, the residue was dissolved in Et2O. The organic layer was washed with NaHCO3 solution (10%), dried over anhydrous Na2SO4, and concentrated to afford N-(3,5-dinitrobenzoyl)valine, which was used without purification. To a mixture of N-(3,5-dinitrobenzoyl)valine (16 mmol) and EEDQ in anhydrous THF (120 mL), stirred, under nitrogen atmosphere, at room temperature for 3 h, was added the appropriate amine (8.03 mmol), and stirred at room temperature for further 15 h. After solvent evaporation under reduced pressure, the crude products 7–8 were purified by recrystallization (THF/hexane).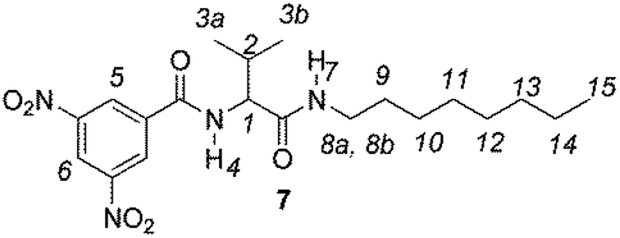
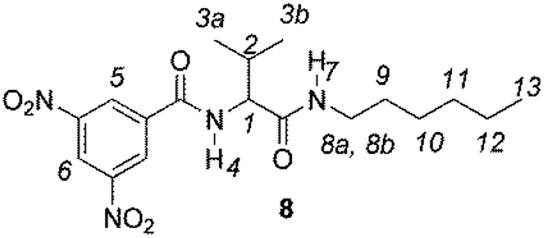
Synthesis of racemates 9–10
To a mixture of N-(3,5-dinitrobenzoyl)amino acid (16 mmol) and EEDQ in anhydrous THF (120 mL), stirred, under nitrogen atmosphere, at room temperature for 3 h, was added the appropriate amine (8.03 mmol). The reaction mixture was stirred at room temperature for further 15 h. After solvent evaporation under reduced pressure, the crude products 9–10 were purified by recrystallization (THF/hexane).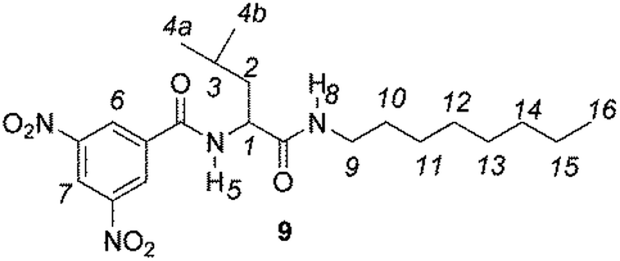
Synthesis of racemates 11–12
A solution of N-(3,5-dinitrobenzoyl)valine or N-(3,5-dinitrobenzoyl)alanine (3 mmol) in anhydrous MeOH (30 mL) was stirred, under nitrogen atmosphere, at 0 °C, and added of HCl gas for 2 h. The mixture was heated at reflux for 1 h. The crude product, obtained by removing the solvent under reduced pressure, was dissolved in CH2Cl2 and washed with saturated NaHCO3 solution, H2O, and dried over anhydrous Na2SO4. 11 and 12 were obtained chemically pure (TLC: SiO2, CHCl3; 1H NMR), after the usual work-up.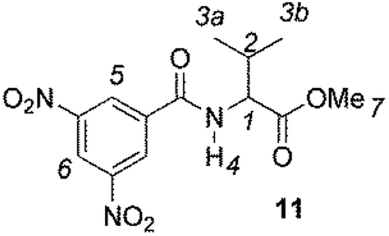
Synthesis of 13
To a solution of α-phenylethylamine (8.3 mmol) and triethylamine (12.5 mL, 9.0 mmol) in anhydrous THF (50 mL) was added dropwise at 0 °C a solution of 3,5-dinitrobenzoyl chloride in THF. The mixture was stirred at room temperature for 16 h. By monitoring the total conversion of the precursor (TLC, CHCl3/hexane 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the reaction was quenched by adding H2O. By concentration under reduced pressure, the crude product was obtained, and dissolved in CH2Cl2; the organic layer was washed with HCl (10%), Na2CO3 (10%), H2O, and dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded 13.
:1), the reaction was quenched by adding H2O. By concentration under reduced pressure, the crude product was obtained, and dissolved in CH2Cl2; the organic layer was washed with HCl (10%), Na2CO3 (10%), H2O, and dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded 13.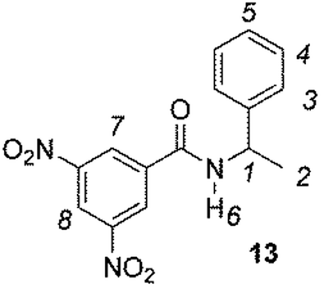
1H NMR (300 MHz, CDCl3, 25 °C) δ: 1.66 (H-2, d, J = 6.9 Hz, 3H); 5.34 (H-1, m, 1H); 6.58 (H-6, d, J = 7.3 Hz, 1H); 7.30–7.40 (H-3, H-4, and H-5, m, 5H); 8.91 (H-7, d, J = 2.3 Hz, 2H); 9.13 (H-8, t, J = 2.3 Hz, 1H).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by University of Pisa (PRA2018_23 “Functional Materials”).Notes and references
- H. Alkadi and R. Jbeily, Infect. Disord.: Drug Targets, 2018, 18, 88 CAS.
- E. Sanganyado, Z. Lu, Q. Fu, D. Schlenk and J. Gan, Water Res., 2017, 124, 527 CrossRef CAS PubMed.
- B. Patel, G. Grant, M. Zunk and S. Rudrawar, Eur. J. Org. Chem., 2019, 6005 CrossRef CAS.
- F. Tan and H.-G. Cheng, Chem. Commun., 2019, 55, 6151 RSC.
- M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2019, 17, 3670 RSC.
- L. Wei, C. Shen, Y.-Z. Hu, H.-Y. Tao and C.-J. Wang, Chem. Commun., 2019, 55, 6672 RSC.
- Differentiation of Enantiomers I, ed. V. Schurig, Springer-Verlag Berlin Heidelberg, 2013 Search PubMed.
- Differentiation of Enantiomers II, ed. V. Schurig, Springer-Verlag Berlin Heidelberg, 2013 Search PubMed.
- G. Uccello-Barretta, F. Balzano, F. Aiello and R. Settambolo, From Chiral Drugs to Chiral Metabolites: The NMR Approach, in Applications of NMR Spectroscopy, ed. A.-u. Rahman and M. Iqbal Choudhary, Bentham Science Publishers, 2015, vol. 1, ch. 6, p. 182 Search PubMed.
- A. Calcaterra and I. D'Acquarica, J. Pharm. Biomed. Anal., 2018, 147, 323 CrossRef CAS PubMed.
- R. A. Patil, C. A. Weatherly and D. W. Armstrong, Chiral Gas Chromatography, in Chiral Analysis: Advances in Spectroscopy, Chromatography and Emerging Methods, ed. P. L. Polavarapu, Elsevier, 2nd edn, 2018, ch. 11, p. 468 Search PubMed.
- M. F. Wahab, C. A. Weatherly, R. A. Patil and D. W. Armstrong, Chiral Liquid Chromatography, in Chiral Analysis: Advances in Spectroscopy, Chromatography and Emerging Methods, ed. P. L. Polavarapu, Elsevier, 2nd edn, 2018, ch. 12, p. 507 Search PubMed.
- B. Chankvetadze, Enantioseparations by Capillary Electromigration Techniques, in Chiral Analysis: Advances in Spectroscopy, Chromatography and Emerging Methods, ed. P. L. Polavarapu, Elsevier, 2nd edn, 2018, ch. 13, p. 565 Search PubMed.
- R. Franzini, A. Ciogli, F. Gasparrini, O. H. Ismail and C. Villani, Recent Developments in Chiral Separations by Supercritical Fluid Chromatography, in Chiral Analysis: Advances in Spectroscopy, Chromatography and Emerging Methods, ed. P. L. Polavarapu, Elsevier, 2nd edn, 2018, ch. 14, p. 607 Search PubMed.
- F. Balzano, G. Uccello-Barretta and F. Aiello, Chiral Analysis by NMR Spectroscopy: Chiral Solvating Agents, in Chiral Analysis: Advances in Spectroscopy, Chromatography and Emerging Methods, ed. P. L. Polavarapu, Elsevier, 2nd edn, 2018, ch. 9, p. 367 Search PubMed.
- T. J. Wenzel, Differentiation of Chiral Compounds Using NMR Spectroscopy, Wiley & Sons, Inc., Hoboken, USA, 2nd edn, 2018 Search PubMed.
- G. Uccello-Barretta, C. Rosini, D. Pini and P. Salvadori, J. Am. Chem. Soc., 1990, 112, 2707 CrossRef CAS.
- G. Uccello-Barretta, M.-G. Berni and F. Balzano, Tetrahedron: Asymmetry, 2007, 18, 2565 CrossRef CAS.
- K. D. Klika, M. Budovská and P. Kutschy, J. Fluorine Chem., 2010, 131, 467 CrossRef CAS.
- E. J. J. Grabowski, (S)-Ethyl Lactate, in e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, 2001, pp. 1–4. DOI:10.1002/047084289x.re095.
- R. G. Ghirardelli, J. Am. Chem. Soc., 1973, 95, 4987 CrossRef CAS.
- W. L. F. Armarego, Purification of Laboratory Chemicals, Butterworth-Heinemann Cambridge, UK, 8th edn, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme of the synthesis of derivative I. 1H NMR spectral regions corresponding to NHβ and methyl protons of 7 (10 mM) in the presence of one equivalent of CSA (1–6). Nonequivalences data (total concentration 60 mM) for 7 in the presence of one equivalent of CSA (1–6). 1H NMR spectral regions corresponding to DNB and amide (NHα and NHβ) protons of 7 (10 mM and 30 mM) in the presence of one equivalent of CSA (1–6). 1H and 13C NMR spectra of CSAs 1–6. See DOI: 10.1039/d0ra00200c |
| This journal is © The Royal Society of Chemistry 2020 |