DOI:
10.1039/C9RA10280A
(Paper)
RSC Adv., 2020,
10, 4860-4868
pH-responsive polymeric nanoparticles with tunable sizes for targeted drug delivery†
Received
7th December 2019
, Accepted 10th January 2020
First published on 29th January 2020
Abstract
Biodegradable nanoparticles (NPs) have shown great promise as intracellular imaging probes, nanocarriers and drug delivery vehicles. In this study, we designed and prepared amphiphilic cellulose derivatives via Schiff base reactions between 2,3-dialdehyde cellulose (DAC) and amino compounds. Polymeric NPs were facilely fabricated via the self-assembly of the as-synthesized amphiphilic macromolecules. The size distribution of the obtained NPs can be tuned by changing the amount and length of the grafted hydrophobic side-chains. Anticancer drugs (DOX) were encapsulated in the NPs and the drug-loaded NPs based on cellulose derivatives were stable in neutral and alkaline environments for at least a month. They rapidly decomposed with the efficient release of the drug in acidic tumor microenvironments. These drug-loaded NPs have the potential for application in cancer treatment.
Introduction
Biodegradable NPs with suitable dimensions, smooth surface and increased water solubility have gained considerable interest due to their environmental friendliness and biocompatibility.1–3 Various biodegradable NPs have been widely used in drug delivery,4–9 biosensors, and medical imaging.10–13 In particular, biodegradable NPs have potential advantages in improving the therapeutic value of various medicinal drugs and bioactive molecules by improving the bioavailability, solubility and retention time.14,15 Biodegradable NPs derived from nontoxic and green materials are expected to be in high demand by consumers and industries. Current research for biodegradable NPs is mainly focused on developing biodegradable polymers for the self-assembly of NPs.16–22
In recent years, numerous studies have been carried out on polysaccharides and their derivatives due to their tremendous advantages including non-toxicity, biocompatibility, hydrophilicity and stability.23–25 It is well known that cellulose is the most abundant renewable polysaccharide produced by plants. Cellulose also provides a reactive platform with numerous OH groups that can be functionalized to achieve different properties.26,27
Biocompatible and biodegradable NPs for drug delivery can be administered via different routes including oral, intravenous, intraperitoneal, and transdermal.13 The NPs from cellulose derivatives have functional groups on the cellulose backbone, which provide the NPs with attached drugs the ability to be used via physical adsorption.
In our recent study, biocompatible functional cellulose derivatives were prepared by the reaction of DAC with oleylamine and aminoethyl rhodamine (AERhB) via Schiff base bonds.27 AERhB was used as a model compound in place of cancer therapeutic drugs containing amine groups. The release of this model compound AERhB was due to the pH change. However, many applied cancer therapeutic drugs do not contain amine groups and cannot be integrated via Schiff base formation. In comparison, the encapsulation of such drugs into NPs should be another applicable approach for subsequent drug delivery following cellular uptake through endocytosis and cell cytotoxicity assay.
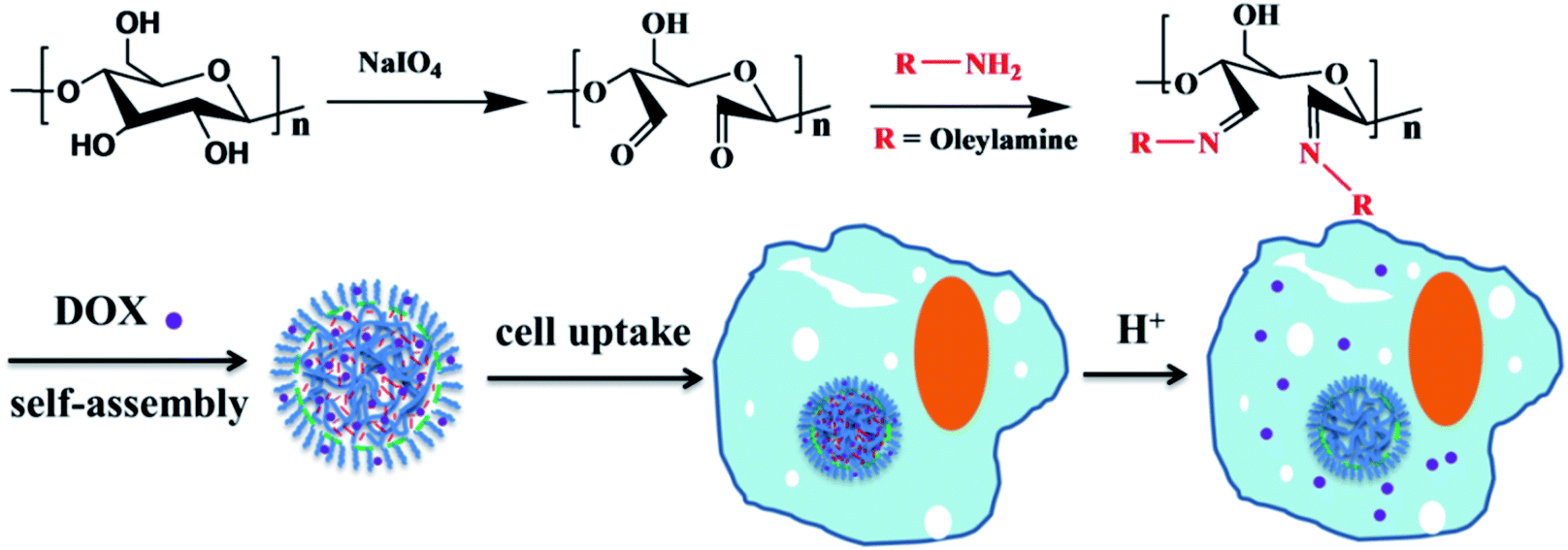
Herein, dialdehyde cellulose (DAC) was synthesized by the periodate oxidation of cellulose under mild aqueous conditions, resulting in the formation of dialdehyde groups from the hydroxyl groups at the C2 and C3 positions of anhydroglucose units.28–30 Then, pH-responsive NPs were facilely fabricated via the self-assembly of a synthetic amphiphilic functional cellulose derivative. To achieve this, biocompatible functional cellulose was constructed based on DAC by conjugating with a variety of amines via Schiff base bonds, followed by a nanoprecipitation technique in aqueous solution. A predetermined amount of DOX was added to the DAC derivative solution with stirring at room temperature for 2 h; the DOX-loaded NPs were obtained by using a nanoprecipitation technique. The NPs containing encapsulated anticancer drugs (DOX) were stable in neutral and alkaline environments. They rapidly decomposed and efficiently released the drugs due to the cleavage of the Schiff base linkages under endosomal/lysosomal conditions after the internalization of the NPs by tumor cells. Thus, these NPs based on cellulose derivatives are promising carriers for biomedicine.
Experimental
Materials
Microcrystalline cellulose (MCC) with a granule size of 50 μm, hydroxylamine hydrochloride, oleylamine, and rhodamine B base (97%) were purchased from Sigma-Aldrich. NaIO4, ethylene glycol, aqueous hydrochloric acid (HCl, 37% w/w), sodium hydroxide (NaOH), ethanol, and N,N-dimethylformamide (DMF) were received from TH. Geyer (Germany). All chemicals were of analytical grade or higher. Deionized water with the conductivity of <0.05 μS cm−1 was used throughout the experiment. Aminoethyl rhodamine (AERhB) was prepared in two steps from rhodamine B as reported before.31 The dialysis membrane with a molecular weight cut-off of 3500 Da was bought from Thermo Fisher Scientific. Human lung cancer cell line (A549) cells were obtained from the Cell Bank of the Chinese Academy of Sciences, Shanghai, China. DMEM medium containing 10% (v/v) fetal bovine serum was obtained from Gibco, USA, along with 100 g mL−1 streptomycin and 100 U mL−1 penicillin.
Fabrication of dialdehyde cellulose (DAC)
DAC was prepared through periodate oxidation of MCC as previously described.27,32,33 MCC (1 g), NaIO4 (1.65 g) and 50 mL water were stirred at 250 rpm for 72 h at room temperature (RT) in the dark. Ethylene glycol was added to this mixture to quench the residual periodate. Thereafter, the oxidized cellulose was purified by dialysis against water. The purified DAC was heated at 80 °C for 4 h, and then the residual solid was separated from the sample by centrifuging at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. The supernatant was collected and stored at 4 °C.
000 rpm for 30 min. The supernatant was collected and stored at 4 °C.
Preparation of NPs from DAC derivatives
The preparation methods were based on a previous report with slight modifications.27 Firstly, 0.5 g of samples with various molar ratios of DAC and hexylamine (1:0.5, 1:0.75, 1:1, 1:2, 1:5) were respectively added to ethanol in a flask and the system was stirred for 1 h at room temperature until completely dissolved in ethanol. Solutions of DAC–hexylamine at the concentration of 10 mg mL−1 were obtained, and 1 mL of each solution was added dropwise into 10 mL water by using a syringe pump at a rate of 0.2 mL min−1. The NPs-H (NPs-H1:0.5, NPs-H1:0.75, NPs-H1:1, NPs-H1:2, NPs-H1:5) with various molar ratios of DAC:hexylamine were obtained using the same method.27 The obtained NPs suspensions were loaded in dialysis bags and dialyzed against deionized water for three days to eliminate water-soluble impurities and DMF. Five kinds of NPs-O (NPs-O1:0.5, NPs-O1:0.75, NPs-O1:1, NPs-O1:2, NPs-O1:5) of DAC–oleylamine and the NPs-R (NPs-R1:0.5, NPs-R1:0.75, NPs-R1:1, NPs-R1:2, NPs-R1:5) suspensions were obtained via the same approach.
Preparation of DOX-loaded NPs
The DAC derivative from DAC and oleylamine with a molar ratio of 1:1 was added to DMF, then stirred and heated to 50 °C, until completely dissolved in DMF. A predetermined amount of DOX·HCl was added and the solution was stirred at room temperature for 2 h. Then, 1 mL of derivative and DOX·HCl solution was dropped into 10 mL water at a rate of 0.2 mL min−1 via a syringe pump. Subsequently, the obtained DOX-loaded NPs suspension was dialyzed against deionized water for 24 h to remove free DOX and byproducts.
Cell culture condition
Human lung cancer cell line (A549) cells were cultured in DMEM medium containing 10% (v/v) fetal bovine serum, 100 g mL−1 streptomycin and 100 U mL−1 penicillin. Cells were maintained at 37 °C and 5% CO2 humidified atmosphere.
Cytotoxicity assay
The cell viabilities of free DOX and DOX-loaded NPs on A549 cells were evaluated by using the CCK8 method. The A549 cells (4 × 103 cells per well) were seeded in a 96-well plate (four replicates for each treatment). After 24 h, free DOX (final concentration 20 μg mL−1) and DOX released from NPs under pH 4.0, pH 5.0 and pH 7.0 conditions, where the pH of the DOX solution was adjusted to 7.0, were added to the cell medium and incubated for 24 h. The 10 μL CCK8 solution was then added to each well and incubated for 3 h in a humidified incubator (5% CO2, 37 °C). The absorbance was determined at 450 nm using a SpectraMax M5 multi-mode microplate reader. Cell images after the different treatments were captured using a Nikon microscope.
Evaluation of the pH-triggered destabilization of NPs
In order to evaluate the pH sensitivity of DAC-derived NPs, various kinds of NPs suspensions (1 mg mL−1) were transferred to dialysis bags and immersed in 100 mL acetate buffer solutions (pH 4.0) and 100 mL phosphate buffer solution (PBS) (pH 7.0), respectively. Each sample was maintained at 37 °C with continuous stirring at 200 rpm. After the predetermined time intervals, 1 mL of the NPs suspensions were extracted from the dialysis bags and the size distribution of NPs was measured via dynamic light scattering. A portion of the NPs suspensions (1 mL) was added to dialysis bags so that the entire suspensions were not influenced for each measurement.
In vitro DOX release from DOX-loaded NPs
DOX-loaded NPs suspensions (10 mL, 1 mg mL−1) were added to a dialysis membrane, which was then incubated in 200 mL acetate buffer solution (pH 4.0), 200 mL PBS of pH 5.0, and 200 mL PBS of pH 7.0 at 37 °C in a shaking water bath at a rate of 200 rpm. At the predetermined times (6 h, 12 h, 24 h, 48 h), aliquots of 200 μL of samples from outside the dialysis bags were measured by the validated HPLC method (Fig. S4–S7†). DOX-release experiments were conducted in triplicate and the results were expressed as the average with standard deviations.
The cumulative amount of released DOX was calculated according to the following equation:
| Cumulative drug release (%) = (Mt/M0) × 100, |
where
Mt denotes the amount of drug released at time
t and
M0 represents the initial amount of drug in NPs.
Measurements
FTIR spectra were recorded on a PerkinElmer SP one FTIR spectrometer. Elemental analysis was performed on an elemental analyzer Vario EL III CHN instrument from Elementar (Hanau, Germany). The sizes of the NPs were determined on a Zetasizer Nano ZS instrument (Malvern Ltd, UK). Three measurements were performed for each sample at 25 °C, and the average values were taken as the final result. Scanning electron microscopy was performed on a Leo SUPRA 35 instrument (Carl Zeiss SMT GmbH, Oberkochen, Germany). Diluted NP dispersions were dropped on silicon wafers and then dried at room temperature. Prior to the observation, the surfaces of the samples were coated with a layer of carbon of about 10 nm. High-Performance Liquid Chromatography (HPLC) (Shimadzu Corporation, Japan) was conducted using a Shim-pack VP-ODS column (150 mm × 4.6 mm, 5 μm), with a mobile phase of 0.002 mol L−1 sodium acetate buffer solution (pH 4.3) at a flow rate of 0.7 mL min−1 and a mobile phase of methanol at a flow rate of 0.3 mL min−1. The peak at 248 nm was detected with a UV detector. A SpectraMax M5 multi-mode microplate reader was used (Molecular Devices, USA). Cell images were captured using a Nikon microscope (ECLIPSE-Ti, Japan).
Results and discussion
Preparation and characterization of DAC derivatives
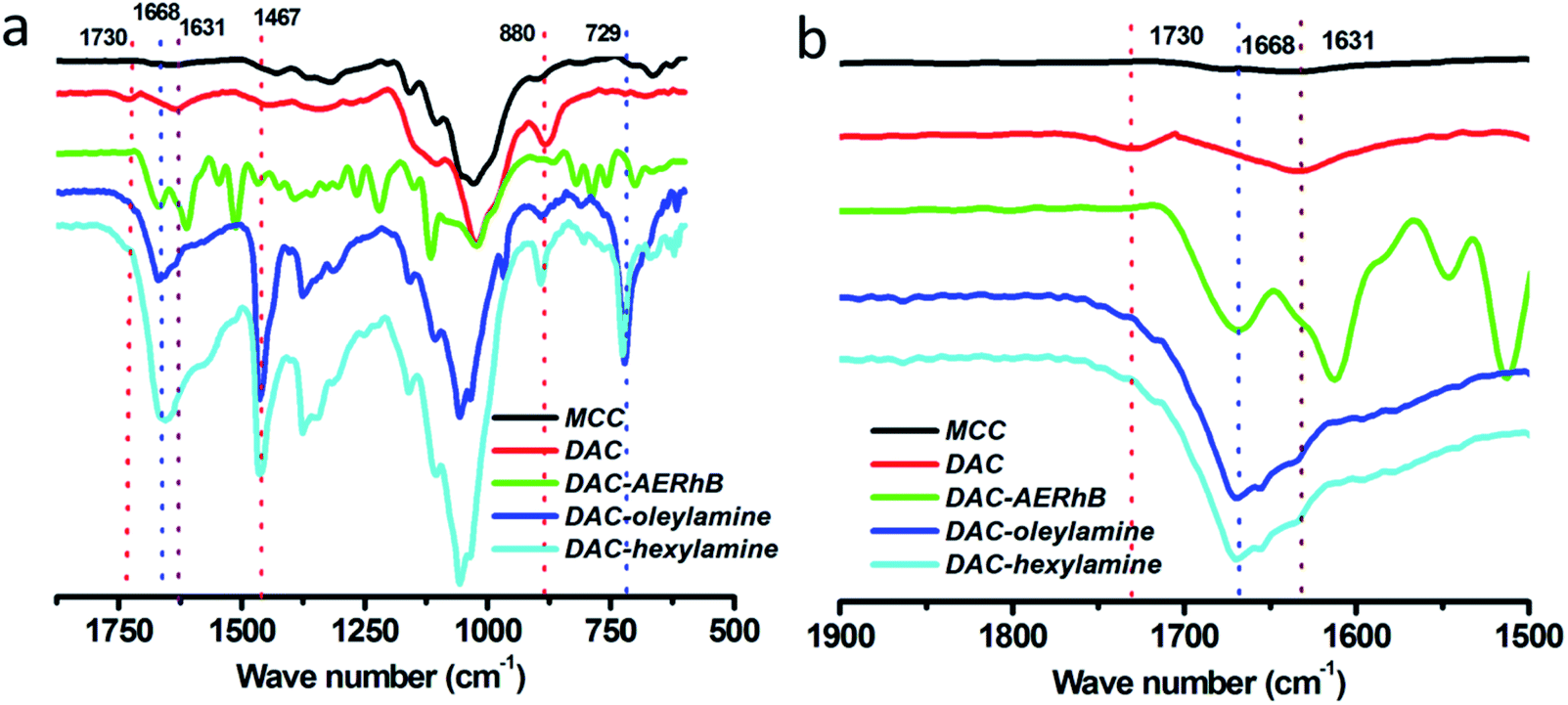
As shown in Scheme 1, 2,3-dialdehyde cellulose was obtained after the periodate oxidation of cellulose at room temperature in the dark. Using this DAC, three derivatives were further prepared via the Schiff base reaction using hexylamine, oleylamine and AERhB. As shown in Fig. 1, the structures of DAC, DAC–hexylamine, DAC–oleylamine and DAC–AERhB were confirmed by FTIR spectra. The characteristic absorption bands of the aldehyde carbonyl groups appeared at 1730 and 880 cm−1, which were due to carbonyl group stretching and the hemiacetal linkages formed from the dialdehyde groups, respectively.32,34 The characteristic peak at 1730 cm−1 disappeared in DAC derivatives, which further confirmed the effective Schiff base coupling where the dialdehyde groups were quantitatively converted into the imine groups (Fig. 1b). New absorption peaks at about 1668 cm−1 for DAC–hexylamine, DAC–oleylamine and DAC–AERhB were ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations.35 The absorption peaks at 1467 and 729 cm−1 correspond to the C–H stretching vibrations of multiple methylene moieties of DAC–hexylamine and DAC–oleylamine.
N stretching vibrations.35 The absorption peaks at 1467 and 729 cm−1 correspond to the C–H stretching vibrations of multiple methylene moieties of DAC–hexylamine and DAC–oleylamine.
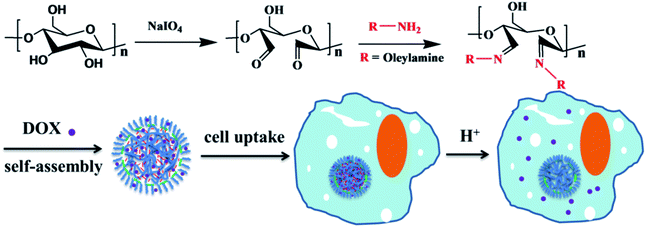 |
| | Scheme 1 Schematic representation of the formation of nanoparticles. | |
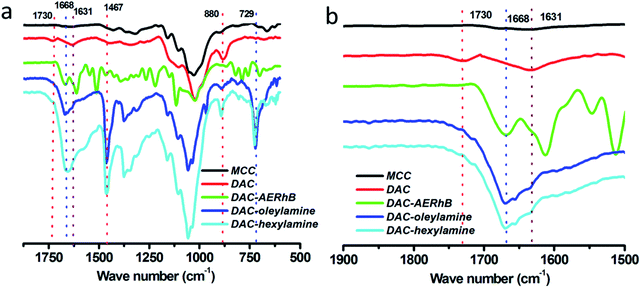 |
| | Fig. 1 (a) FTIR spectra (2000–500 cm−1) and (b) the extended view between 2000–1500 cm−1 of MCC, DAC, DAC–hexylamine, DAC–oleylamine and DAC–AERhB, respectively. | |
Synthesis and characterization of nanoparticles
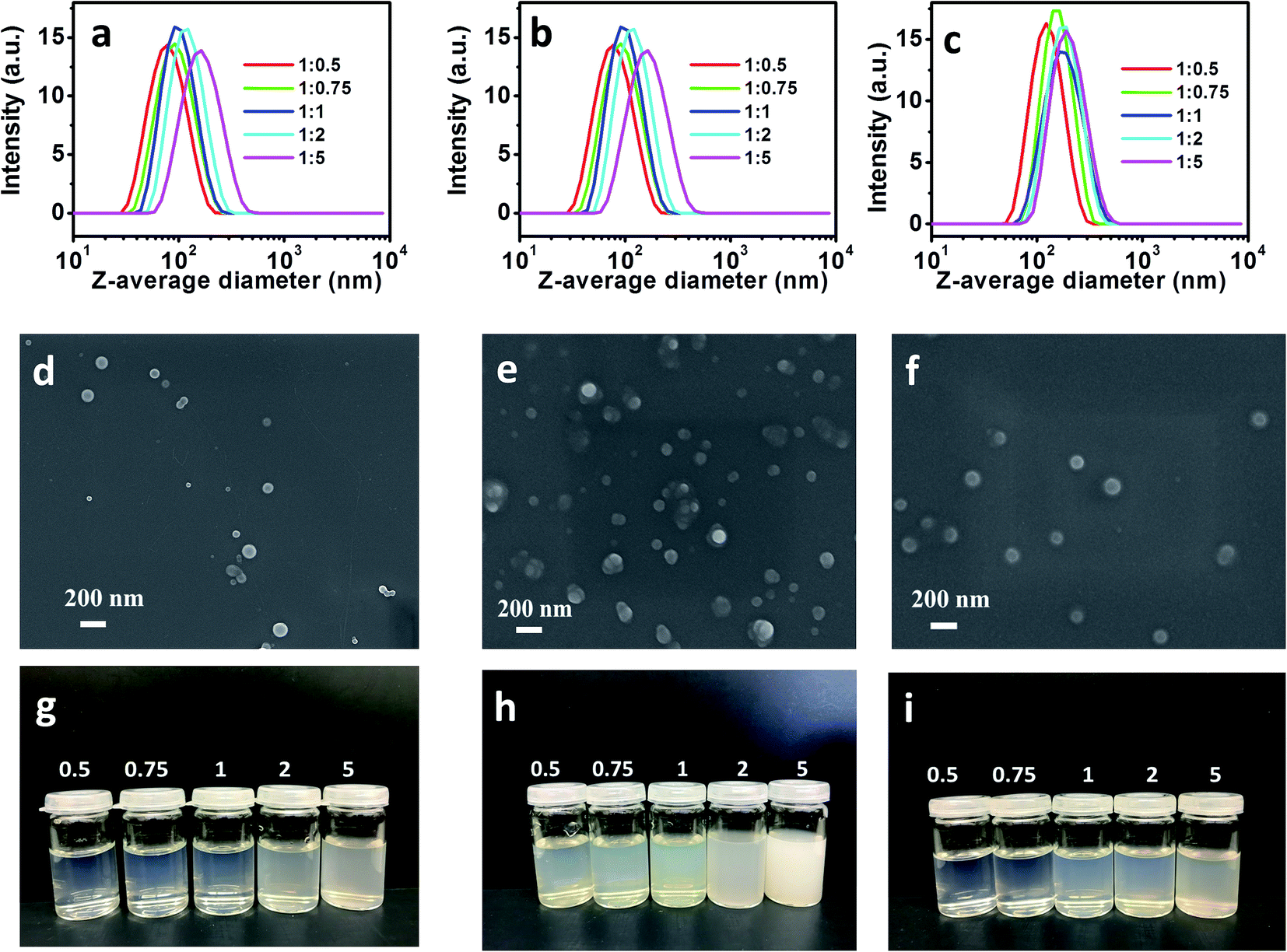
NPs were further prepared via the nanoprecipitation dropping technique using these DAC derivatives.36 Generally, 1 mL DMF solution of each DAC derivative (10 mg mL−1) was added to 10 mL of water by using a syringe pump at a rate of 0.2 mL min−1 under agitation at 800 rpm. The opalescent color of the resulting dispersions indicated the formation of NPs from DAC derivatives. These DAC derivatives are amphiphilic macromolecules that are composed of a hydrophilic DAC backbone and hydrophobic imine side groups. During the phase transition in the poor solvents, hydrophobic cores were formed by the aggregated hydrophobic groups, which were further surrounded by coiled DAC chains. In order to maintain a stable structure with minimum energy, sufficient amounts of polymer chains were promoted to form NPs.37,38 The samples prepared from DAC–hexylamine with five molar ratios of DAC and hexylamine (1:0.5, 1:0.75, 1:1, 1:2, 1:5) self-assembled into NPs with distinct sizes. Various kinds of NPs using DAC–oleylamine and DAC–AERhB were fabricated according to the same procedure. Z-average diameters and PDI of various kinds of NPs are shown in Table 1 and Fig. 2. Furthermore, colloidal stability evaluated by DLS revealed that the NPs dispersions were sufficiently stable during storage for more than 30 days or during strong dilutions. Moreover, the NPs exhibited a smooth surface according to their SEM images (Fig. 2).
Table 1 Z-average diameters (d) and PDI of three groups of NPs. NPs of DAC–hexylamine, DAC–oleylamine and DAC–AERhB are referred to as NPs-H, NPs-O and NPs-R, respectively
| NPs |
Dispersant |
Z-average (d) (nm) |
PDI |
| 1:0.5 (NPs-H) |
Water |
72.9 ± 0.5 |
0.118 ± 0.007 |
| 1:0.75 (NPs-H) |
Water |
86.3 ± 0.7 |
0.117 ± 0.007 |
| 1:1 (NPs-H) |
Water |
94.2 ± 0.8 |
0.104 ± 0.009 |
| 1:2 (NPs-H) |
Water |
113.2 ± 1.0 |
0.116 ± 0.011 |
| 1:5 (NPs-H) |
Water |
150.7 ± 1.4 |
0.126 ± 0.013 |
| 1:0.5 (NPs-O) |
Water |
126.8 ± 0.6 |
0.109 ± 0.005 |
| 1:0.75 (NPs-O) |
Water |
145.5 ± 0.5 |
0.091 ± 0.003 |
| 1:1 (NPs-O) |
Water |
158.6 ± 0.8 |
0.081 ± 0.009 |
| 1:2 (NPs-O) |
Water |
180.7 ± 0.7 |
0.096 ± 0.01 |
| 1:5 (NPs-O) |
Water |
208.1 ± 2.2 |
0.138 ± 0.012 |
| 1:0.5 (NPs-R) |
Ethanol |
117.6 ± 1.1 |
0.092 ± 0.003 |
| 1:0.75 (NPs-R) |
Ethanol |
148.1 ± 1.0 |
0.078 ± 0.004 |
| 1:1 (NPs-R) |
Ethanol |
163.6 ± 1.1 |
0.119 ± 0.005 |
| 1:2 (NPs-R) |
Ethanol |
172.8 ± 1.3 |
0.114 ± 0.008 |
| 1:5 (NPs-R) |
Ethanol |
187.8 ± 2.7 |
0.122 ± 0.010 |
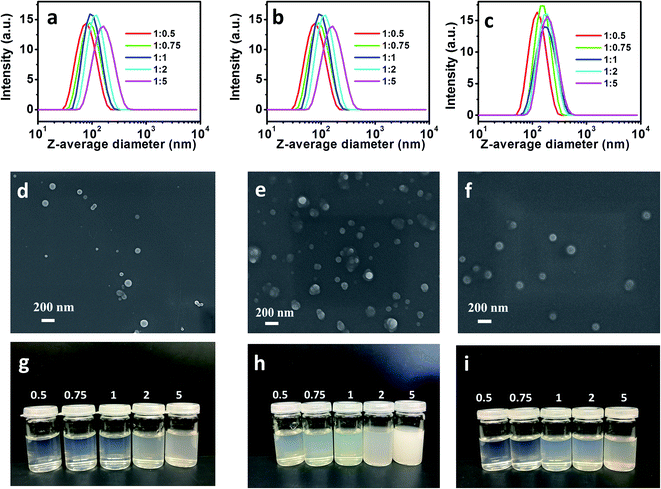 |
| | Fig. 2 DLS curves of (a) NPs-H suspensions, (b) NPs-O suspensions, and (c) NPs-R suspensions. SEM image of (d) original NPs-H1:1, (e) original NPs-O1:1, and (f) original NPs-R1:1. (g), (h) and (i) Optical images of NPs suspensions obtained from various (g) DAC–hexylamine, (h) DAC–oleylamine and (i) DAC–AERhB, respectively. | |
As shown in Fig. 2, SEM and DLS were performed to evaluate the aggregation behaviors of NPs as well as their sizes and morphologies. As shown in Fig. 2d–f, most of the obtained NPs were spherical, while only a small number of them had irregular shapes. The average diameters were measured using DLS to be 72.9–150.7 nm for NPs of DAC–hexylamine, 126.8–208.1 nm for NPs of DAC–oleylamine and 117.6–187.8 nm for NPs of DAC–AERhB. The size distributions of NPs are quite narrow, with PDIs less than 0.13. It can be seen from Table 1 that the mean sizes of NPs from five molar ratios of DAC and hexylamine (1:0.5, 1:0.75, 1:1, 1:2, 1:5) are 72.9 nm, 86.3 nm, 94.2 nm, 113.2 nm and 150.7 nm, respectively. These results indicate that the size of self-aggregated DAC–hexylamine increased with the increasing amount of attached side chains along the DAC backbone.39–41 It is well known that the hydrophobic groups aggregate together to form a hydrophobic core. Increasing the amount of hydrophobic groups will enlarge the diameters of NPs. This tendency was also observed for NPs of DAC–oleylamine and NPs of DAC–AERhB. Moreover, the mean sizes (at the same molar ratios) increased in the following order: DAC–hexylamine < DAC–AERhB < DAC–oleylamine. This fact further indicates that the diameters of NPs were strongly affected by the length of the hydrophobic side-chains.42 In particular, longer chain segments as hydrophobic groups along the polymeric backbone could result in larger aggregates due to the formation of larger hydrophobic cores and thus promote the formation of larger NPs.
pH-induced size changes in the nanoparticles
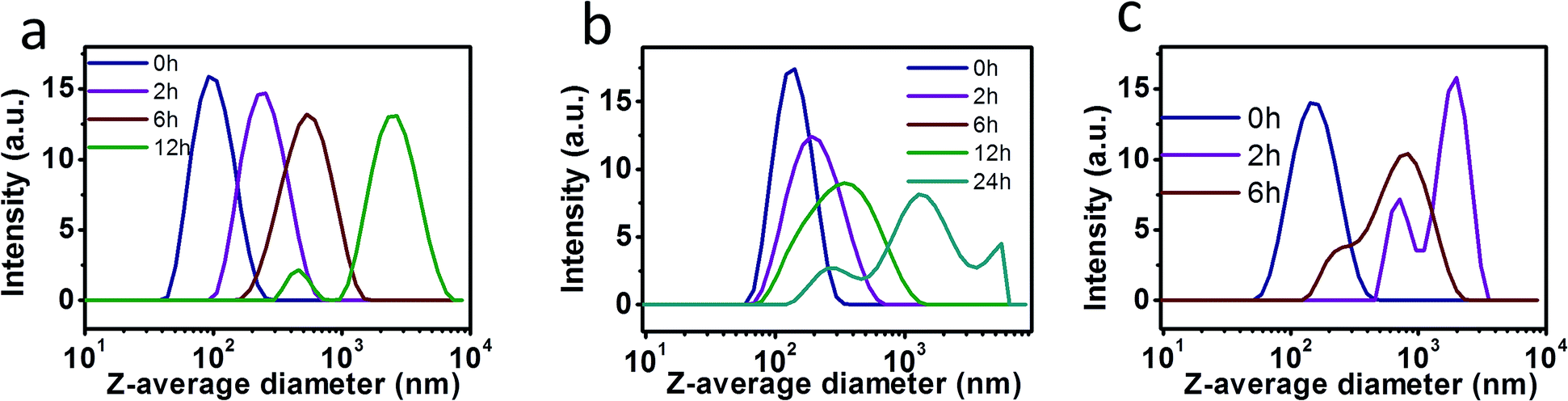
To investigate the pH-triggered size changes in the NPs, the size variations of NPs under environments of pH 7.0 and pH 4.0 were monitored using DLS measurement. Each kind of NPs was put into dialysis bags and immersed in acetate buffer solution at pH 4.0 or phosphate buffer solution (PBS) at pH 7.0, while the temperature was maintained at 37 °C. DLS results indicated that the suspensions were sufficiently stable during the 30 day storage in PBS at pH 7.0. The pH value of the solution was then decreased to 4.0. As shown in Table 2 and Fig. 3, the mean size of NPs-H1:1 (with the molar ratio of DAC to hexylamine of 1:1) was 94.2 nm immediately after adjusting the pH value to 4.0. The average size of NPs-H1:1 increased after 2 h and 6 h to 207.3 and 443.2 nm, respectively. Moreover, the size further increased to 2300 nm after 12 h and then the NPs began to decompose. With respect to NPs-O1:1 (with the molar ratio of DAC to oleylamine of 1:1), the size of the NPs increased from 158.6 to 188.4, 273.1, 650.6, 979.7 nm after incubation for 2 h, 6 h, 12 h, 24 h, respectively. However, the long storage time of 12 h and 24 h led to a broad distribution of NPs sizes, as shown by wide DLS curves of the NPs. In a similar manner, NPs-R1:1 (with the molar ratio of DAC to AERhB of 1:1) in solutions at pH 4.0 exhibited a dramatically increased average diameter of 540.5 nm within the first 2 h. After 6 h of incubation, most of the swollen NPs were formed with the average diameter of 2006 nm, while a small part of NPs had diameters of around 712 nm. In contrast to the original dimensions, the size increase was attributed to the decomposition of the side chains from the DAC backbone and the continuously increasing hydrophilicity led to the vesicular swelling.
Table 2 Z-average diameters of three kinds of NPs in water at pH 4 after various times
| NPs |
Z-average diameters (nm) |
| 0 h |
2 h |
6 h |
12 h |
24 h |
| NPs-H1:1 |
94.2 ± 0.8 |
207.3 ± 1.2 |
443.2 ± 3.5 |
1697 ± 11.6 |
|
| NPs-O1:1 |
158.6 ± 0.6 |
188.4 ± 2.0 |
273.1 ± 3.1 |
650.6 ± 5.7 |
979.7 ± 10.5 |
| NPs-R1:1 |
163.6 ± 1.1 |
540.5 ± 4.6 |
2006 ± 12.1 |
|
|
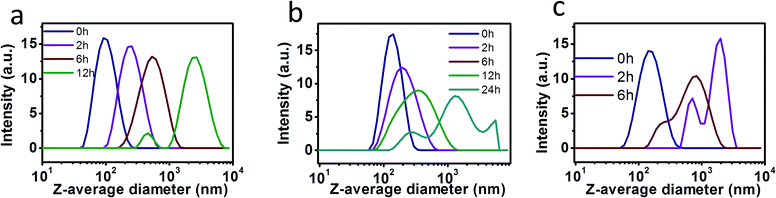 |
| | Fig. 3 Stability of diverse NPs at pH 4.0 using DLS measurements of their aqueous suspensions. (a) NPs-H1:1 in water of pH 4.0, (b) NPs-O1:1 in water of pH 4.0, and (c) NPs-R1:1 in ethanol of pH 4.0 after diverse treatment times. | |
pH-induced size change in nanoparticles
To investigate pH-triggered size changes in NPs, the size variations in NPs at pH 7.0 and pH 4.0 were monitored using DLS measurements. Each kind of NPs was put into dialysis bags and immersed in acetate buffer solution at pH 4.0 or phosphate buffer solution (PBS) at pH 7.0, while the temperature was maintained at 37 °C. DLS results indicated that the suspensions were sufficiently stable during the 30 days storage in PBS at pH 7.0. The pH value of the solution was then decreased to 4.0. As shown in Table 2 and Fig. 3, the mean size of NPs-H1:1 (with the molar ratio of DAC to hexylamine of 1:1) was 94.2 nm immediately after adjusting the pH value to 4.0. The average size of NPs-H1:1 increased after 2 h and 6 h to 207.3 and 443.2 nm, respectively. Moreover, the size further increased to 2300 nm after 12 h and then the NPs began to decompose. With respect to NPs-O1:1 (with the molar ratio of DAC to oleylamine of 1:1), the size of the NPs increased from 158.6 to 188.4, 273.1, 650.6, 979.7 nm after incubation for 2 h, 6 h, 12 h, 24 h, respectively. However, the long storage times of 12 h and 24 h led to broad distributions of NPs sizes, as shown by wide DLS curves of the NPs. In a similar manner, NPs-R1:1 (with the molar ratio of DAC to AERhB of 1:1) in solutions at pH 4.0 exhibited a dramatically increased average diameter of 540.5 nm within the first 2 h. After incubation for 6 h, most of the swollen NPs were formed with an average diameter of 2006 nm, while a small amount of NPs had diameters of around 712 nm. In contrast to the original dimensions, the size increase was attributed to the decomposition of side chains from the DAC backbone and the continuously increasing hydrophilicity led to vesicular swelling.
NPs containing Schiff base bonds were highly stable under conditions of pH 7.0 or in alkaline solutions, but they were easily cleavable in acidic conditions. The hydrolysis of Schiff base linkages can only start from the NPs surface in weakly acid conditions and slowly penetrate the interior of the NPs, leading to the shrinkage of the core, while the shell becomes swollen and loose.43–46 Among the three kinds of NPs in aqueous solutions at pH 4.0, the stability of the NPs changed in the following order: NPs-O1:1 > NPs-H1:1 > NPs-R1:1. Therefore, the larger the side chains are for the formation of NPs, the more difficult it is for H+ to penetrate the interior of the NPs to disassemble hydrophobic cores. Compared to NPs fabricated with shorter hydrophobic chains, the NPs from longer hydrophobic side chains possess improved stability in acidic conditions.
Loading and in vitro drug release of DOX
Based on the above results, more stable NPs-O1:1 were chosen for the encapsulation of drugs. During the co-nanoprecipitation process, anticancer drugs (DOX) were encapsulated NPs via intermolecular interactions including hydrogen bonding, π–π stacking and electrostatic interactions between DOX and NPs-O1:1.47,48 In detail, the DAC derivatives from DAC and oleylamine with the molar ratio of 1:1 were completely dissolved in DMF at 50 °C, then DOX·HCl, with a weight of 30% of that of the DAC derivatives, was added to the DAC derivatives solution under stirring at room temperature for 2 h. The DOX-loaded NPs were obtained by using the nanoprecipitation technique in aqueous solution. The obtained DOX-loaded NPs suspension was dialyzed against DI water for 24 h in a dialysis bag to remove free DOX and byproducts. The loading content in the vesicles was 10.2% with the loading efficiency of 29.5%. The average diameters of DOX-loaded NPs were similar to the corresponding NPs.
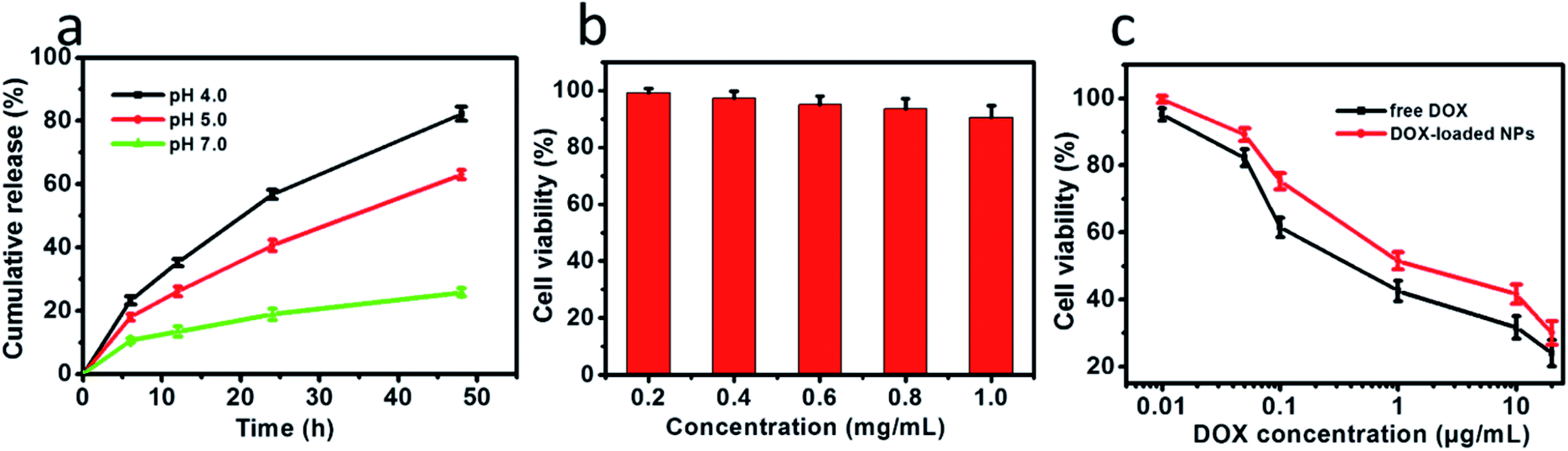
The in vitro drug release profiles by the drug-loaded NPs were explored at pH 7.0 and in acidic environments (pH 4.0 and 5.0) to simulate the pH of the endosomal or lysosomal microenvironments. The step-wise pH-responsive release of DOX from DOX-loaded NPs was further measured using HPLC (Fig. S1–S4†). After 48 h release at 37 °C, 25.8% at pH 7.0, 63.0% at pH 5.0 and 82.2% at pH 4.0 were determined, respectively. As shown in Fig. 4a, the relatively cumulative release of DOX from NPs was slow at pH 7.0. In comparison, DOX-loaded NPs exhibited a higher cumulative release in an acidic tumor microenvironment of pH 5.0 and pH 4.0. These results revealed the selective release of drugs in the cancer microenvironment. The controlled release of DOX was predominantly driven by the acid-induced degradation of the Schiff base linkages and, therefore, the disassembly of NPs.
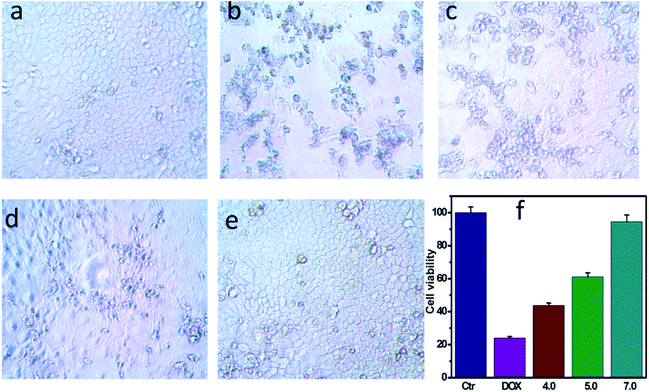 |
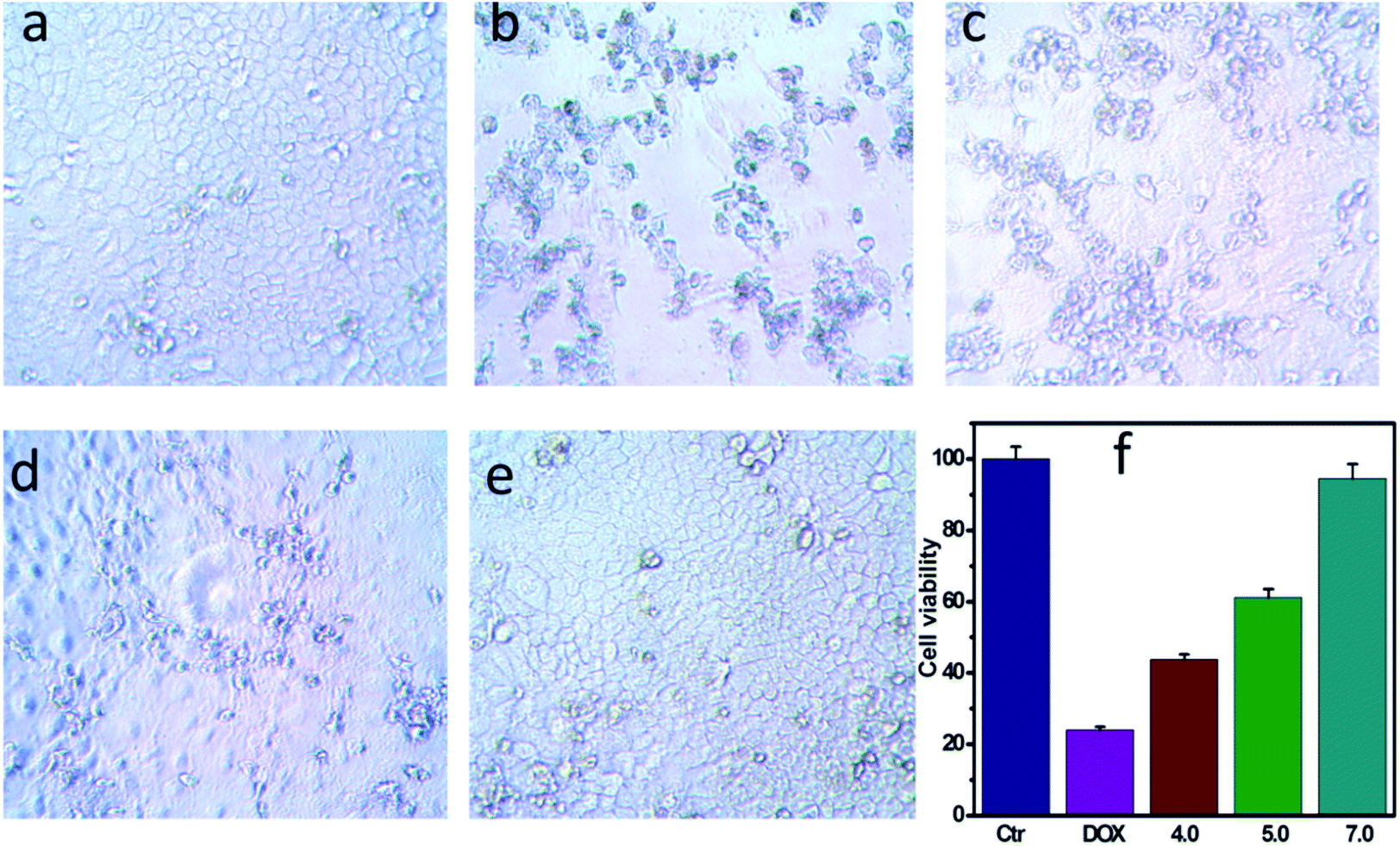
| | Fig. 4 Changes in cell viabilities. (a) A549 cells incubated with NPs for 24 h at pH 7.0. (b) A549 cells incubated with NPs and DOX together (final concentration 20 μg mL−1) for 24 h at pH 7.0. (c) A549 cells incubated with DOX-loaded NPs for 24 h at pH 4.0. (d) A549 cells incubated with DOX-loaded NPs for 24 h at pH 5.0. (e) A549 cells incubated with DOX-loaded NPs for 24 h at pH 7.0. (f) Cell viability of A549 cells incubated with DOX-loaded NPs at various pH for 24 h. | |
Cell uptake and drug release
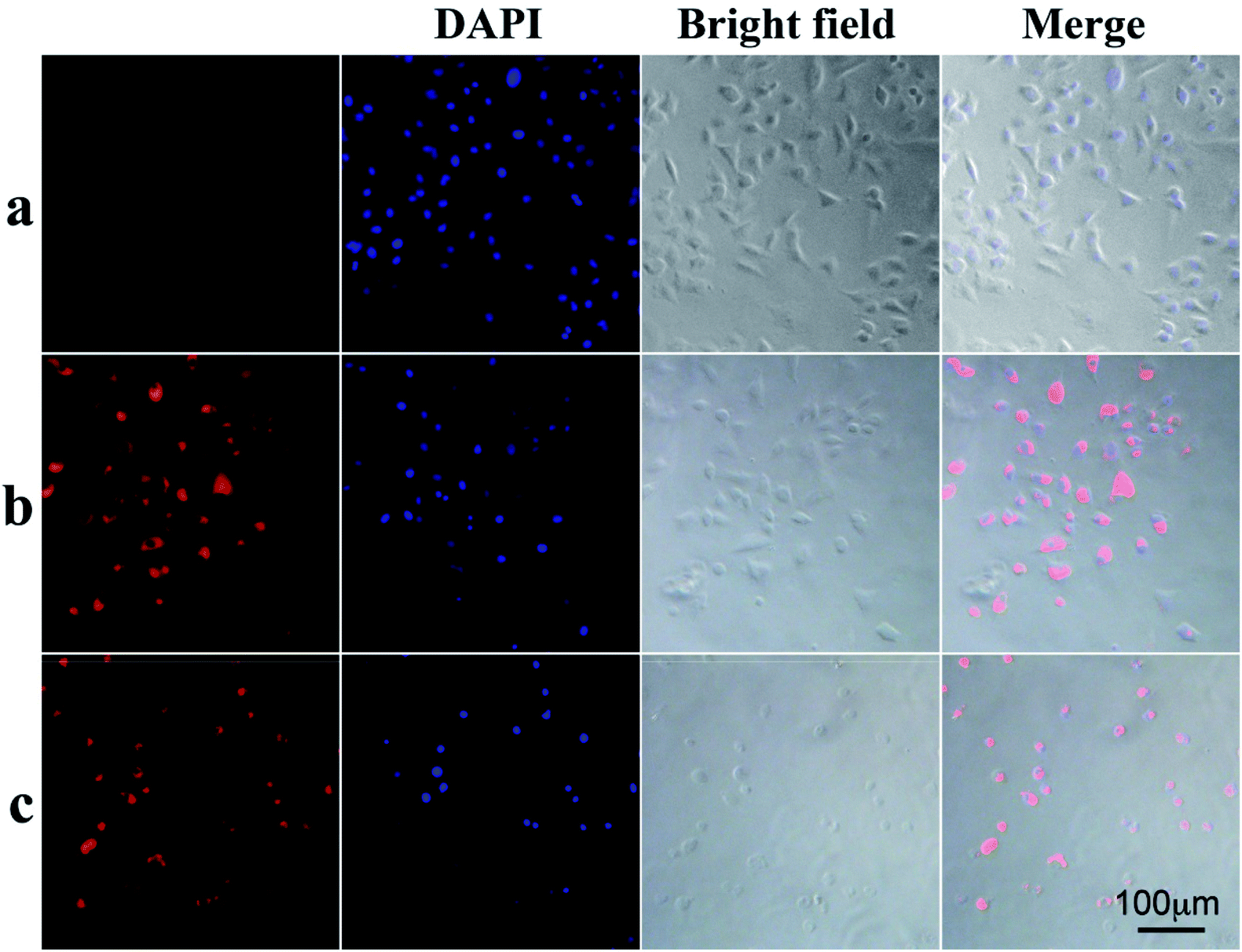
To demonstrate that the drug-loaded NPs internalized by cancer cells have effective therapeutic effects against cancer, the cellular uptake by A549 cells and intracellular drug release behavior of DOX-loaded NPs were examined. DOX was released from NPs under conditions of pH 4.0, pH 5.0 and pH 7.0 after 48 h at 37 °C, and then these NPs suspensions were adjusted to pH 7.4, added to the cell medium and incubated for 24 h. As shown in Fig. 4, cell viabilities of A549 cells incubated at pH 4.0, pH 5.0 and pH 7.0 lay between those after the treatment solely with NPs or free DOX. Moreover, cell viabilities of A549 cells changed in the following order: pH 4.0 < pH 5.0 < pH 7.0. To determine whether NPs and DOX-loaded NPs were endocytosed by tumor cells, fluorescence microscopy was used to trace the cellular uptake. Representative images of A549 incubated with blank NPs and DOX-loaded NPs for 24 h are shown in Fig. 6(a–c). Compared with the images of the blank sample in Fig. 6c, the strong red fluorescence at the periphery of the cell nucleus was observed, indicating the effective cellular uptake of the DOX-loaded NPs from 0.1 mL mL−1 to 0.2 mL mL−1 (Fig. 6b and c), and the cytosolic release of the DOX in the acidic tumor cells. These results indicated that DOX-loaded NPs can enter cancer cells and release drugs in response to intracellular pH values. In particular, the drug-loaded NPs were internalized by A549 cells with the efficient release of DOX from the NPs, and further escaped from the endo/lysosomes to the nucleus. The results were also in accordance with the cell viability (Fig. 4 and 5).
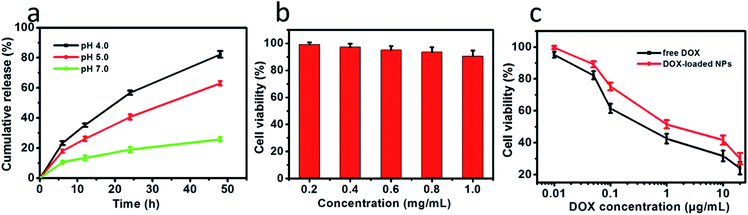 |
| | Fig. 5 (a) pH-triggered release of DOX from NPs. (b) The cells were incubated for 24 h with DOX-loaded NPs at concentrations ranging from 0.2 to 1.0 mg mL−1. (c) Viabilities of A549 cells after 24 h incubation with DOX-loaded NPs and free DOX as a function of DOX dosages. All the data are presented as the average ± standard deviation. | |
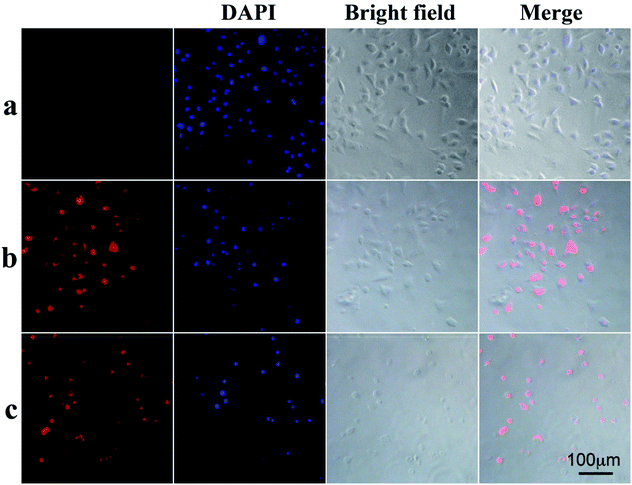 |
| | Fig. 6 Cellular uptake and release of the drug: (a) A549 cells incubated with 0.1 mg mL−1 NPs for 24 h. (b) A549 cells incubated with 0.1 mg mL−1 DOX-loaded NPs for 24 h. (c) A549 cells incubated with 0.2 mg mL−1 DOX-loaded NPs for 24 h. | |
In vitro cell viability assay
The biocompatibility of NPs is a key issue for the drug delivery system. The in vitro cytotoxicity of NPs to A549 cells was evaluated using the CCK-8 assay. As shown in Fig. 5b, the NPs was no toxicity toward A549 cells, owing to cellulose derivatives possessing non-toxicity, biocompatibility, hydrophilicity and stability. The cell viabilities were more than 90% even at concentrations of NPs up to 1.0 mg mL−1, indicating the excellent biocompatibility of these biodegradable NPs. Moreover, the DOX-loaded NPs showed high efficiency of antitumor activity toward A549 cells after incubation for 24 h. The results demonstrated that the viability of A549 cells depended on the DOX concentration (Fig. 5c). Furthermore, DOX-loaded NPs and free DOX exhibited different uptake pathways. Compared with DOX molecules that rapidly diffused into cells, the DOX-loaded NPs had to be endocytosed into the cells. Free DOX molecules are quicker during the internalization process than DOX-loaded NPs, and DOX-loaded NPs with an equal amount of DOX exhibited high efficiency in cancer cell inhibition. These results imply that the drug delivery system could effectively inhibit the growth of cancer cells.
Conclusion
In this report, a novel group of amphiphilic biocompatible macromolecules was designed and facilely fabricated via the formation of Schiff base linkages between dialdehyde cellulose (DAC) and amino-containing compounds. The size of polymeric NPs from 72.9 to 208 nm were altered by tuning the amount and length of the hydrophobic side chains. The NPs remained stable in neutral and alkaline environments and rapidly decomposed in acidic solution due to the inherent properties of Schiff bases. The NPs are not toxic to cancer cells and NPs-based cellulose derivatives are suitable for drug delivery. More importantly, the drug-loaded NPs showed pH-sensitive behaviors that rapidly and thoroughly released the drug in the acid extracellular microenvironments of cancer cells. Moreover, the drug-loaded NPs exhibited dose-dependent cytotoxicity in A549 cells. These results indicate that this drug delivery system can serve as a carrier for pH-regulated drug delivery to cancer cells.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
X. P. thanks the National Natural Science Foundation of China (51563012, 21865013), Sailing Project of 100 People in Jiang Xi, China and Science and Technology Project of Jiang Xi provincial department of education. P. L. and B. P. thank the China Scholarship Council (CSC) for the financial support.
References
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 24–36 CrossRef
 .
. - M. D. Joshi, V. Patravale and R. Prabhu, Int. J. Nanomed., 2015, 1001, DOI:10.2147/ijn.s56932 .
- U. Capasso Palmiero, L. Morosi, E. Bello, M. Ponzo, R. Frapolli, C. Matteo, M. Ferrari, M. Zucchetti, L. Minoli, M. De Maglie, P. Romanelli, M. Morbidelli, M. D'Incalci and D. Moscatelli, J. Controlled Release, 2018, 276, 140–149 CrossRef CAS PubMed .
- M. Kovaliov, S. Li, E. Korkmaz, D. Cohen-Karni, N. Tomycz, O. B. Ozdoganlar and S. Averick, RSC Adv., 2017, 7, 47904–47912 RSC .
- Y. Tu, F. Peng, A. A. M. André, Y. Men, M. Srinivas and D. A. Wilson, ACS Nano, 2017, 11, 1957–1963 CrossRef CAS PubMed .
- D. Chen, G. Zhang, R. Li, M. Guan, X. Wang, T. Zou, Y. Zhang, C. Wang, C. Shu, H. Hong and L.-J. Wan, J. Am. Chem. Soc., 2018, 140, 7373–7376 CrossRef CAS PubMed .
- P. Yuan, H. Zhang, L. Qian, X. Mao, S. Du, C. Yu, B. Peng and S. Q. Yao, Angew. Chem., Int. Ed., 2017, 56, 12481–12485 CrossRef CAS PubMed .
- C. Li, Y. Zhang, Z. Li, E. Mei, J. Lin, F. Li, C. Chen, X. Qing, L. Hou, L. Xiong, H. Hao, Y. Yang and P. Huang, Adv. Mater., 2018, 30, 1706150 CrossRef PubMed .
- N. K. Garg, B. Singh, G. Sharma, V. Kushwah, R. K. Tyagi, S. Jain and O. P. Katare, RSC Adv., 2015, 5, 62989–62999 RSC .
- W. Kim, S. H. Lee, Y. J. Ahn, S. H. Lee, J. Ryu, S. K. Choi and S. Choi, Biosens. Bioelectron., 2018, 111, 59–65 CrossRef CAS PubMed .
- Y. Lyu, J. Zeng, Y. Jiang, X. Zhen, T. Wang, S. Qiu, X. Lou, M. Gao and K. Pu, ACS Nano, 2018, 12, 1801–1810 CrossRef CAS PubMed .
- M. J. Sailor and J.-H. Park, Adv. Mater., 2012, 24, 3779–3802 CrossRef CAS PubMed .
- A. S. Wadajkar, T. Kadapure, Y. Zhang, W. Cui, K. T. Nguyen and J. Yang, Adv. Healthcare Mater., 2012, 1, 450–456 CrossRef CAS PubMed .
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed .
- D. B. Shenoy and M. M. Amiji, Int. J. Pharm., 2005, 293, 261–270 CrossRef CAS PubMed .
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS .
- M. Sponchioni, L. Morosi, M. Lupib and U. Capasso Palmiero, RSC Adv., 2017, 7, 50981–50992 RSC .
- K. Sevim and J. Pan, Acta Biomater., 2018, 66, 192–199 CrossRef CAS PubMed .
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed .
- C. M. Thomas and J.-F. Lutz, Angew. Chem., Int. Ed., 2011, 50, 9244–9246 CrossRef CAS PubMed .
- H. Kargarzadeh, J. Huang, N. Lin, I. Ahmad, M. Mariano, A. Dufresne, S. Thomas and A. Gałęski, Prog. Polym. Sci., 2018, 87, 197–227 CrossRef CAS .
- S. F. Chin, F. B. Jimmy and S. C. Pang, J. Drug Delivery Sci. Technol., 2018, 43, 262–266 CrossRef CAS .
- Y. Wu, X. Zhang, H. Li, P. Deng, H. Li, T. He, J. Rong, J. Zhao and Z. Liu, J. Mater. Chem. B, 2018, 6, 6646–6659 RSC .
- M. Swierczewska, H. S. Han, K. Kim, J. H. Park and S. Lee, Adv. Drug Delivery Rev., 2016, 99, 70–84 CrossRef CAS PubMed .
- D. Das and S. Pal, RSC Adv., 2015, 5, 25014–25050 RSC .
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC .
- X. W. Peng, P. W. Liu, B. Pang, Y. W. Yao, J. X. Wang and K. Zhang, Carbohydr. Polym., 2019, 216, 113–118 CrossRef CAS PubMed .
- J. Lindh, D. O. Carlsson, M. Strømme and A. Mihranyan, Biomacromolecules, 2014, 15, 1928–1932 CrossRef CAS PubMed .
- A. J. Varma and M. P. Kulkarni, Polym. Degrad. Stab., 2002, 77, 25–27 CrossRef CAS .
- U. J. Kim, S. Kuga, M. Wada, T. Okano and T. Kondo, Biomacromolecules, 2000, 1, 488–492 CrossRef CAS PubMed .
- W. Li, W. Wang, Y. Yang and K. Zhang, J. Mater. Chem. A, 2014, 2, 13675–13681 RSC .
- P. Liu, C. Mai and K. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 5313–5319 CrossRef CAS .
- H. Yang, D. Z. Chen and T. G. M. van de Yen, Cellulose, 2015, 22, 1743–1752 CrossRef CAS .
- X. Tian and X. Jiang, Cellulose, 2017, 25, 987–998 CrossRef .
- S. M. A. S. Keshk, A. M. Ramadan and S. Bondock, Carbohydr. Polym., 2015, 127, 246–251 CrossRef CAS PubMed .
- J. Wang and K. Zhang, J. Phys. Chem. C, 2018, 122, 7474–7483 CrossRef CAS .
- H. Wang, G. Liu, S. Dong, J. Xiong, Z. Du and X. Cheng, J. Mater. Chem. B, 2015, 3, 7401–7407 RSC .
- K. Zhang, A. Geissler and T. Heinze, Part. Part. Syst. Charact., 2015, 32, 258–266 CrossRef CAS .
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed .
- S. Y. Chae, S. Son, M. Lee, M.-K. Jang and J.-W. Nah, J. Controlled Release, 2005, 109, 330–344 CrossRef CAS PubMed .
- L. Choisnard, A. Ge`ze, J. Putaux, Y. Wong and D. Wouessidjewe, Biomacromolecules, 2006, 7, 515–520 CrossRef CAS PubMed .
- K. Y. Lee, J.-H. Kim, I. C. Kwon and S. Y. Jeong, Colloid Polym. Sci., 2000, 278, 1216–1219 CrossRef CAS .
- M. Huang, K. Zhao, L. Wang, S. Lin, J. Li, J. Chen, C. Zhao and Z. Ge, ACS Appl. Mater. Interfaces, 2016, 8, 11226–11236 CrossRef CAS PubMed .
- H. Feng, C. Wang, J. Zhou, J. Liu, J. Zhang, R. Guo, J. Liu, A. Dong and L. Deng, Macromol. Chem. Phys., 2018, 219, 1800062 CrossRef .
- C. Wu, L. Xu, L. Shi, X. Gao, J. Li, X. Zhu and C. Zhang, Biomater. Sci., 2018, 6, 2261–2269 RSC .
- Y. Wan, Y. Bu, J. Liu, J. Yang, W. Cai, Y. Yin, W. Xu, P. Xu, J. Zhang and M. He, Polym. Chem., 2018, 9, 3415–3424 RSC .
- X. Wang, Y. Yang, Y. Zhuang, P. Gao, F. Yang, H. Shen, H. Guo and D. Wu, Biomacromolecules, 2016, 17, 2920–2929 CrossRef CAS PubMed .
- C. Wu, L. Xu, L. Shi, X. Gao, J. Li, X. Zhu and C. Zhang, Biomater. Sci., 2018, 6, 2261–2269 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10280a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Hao Cui
*ab,
Hao Cui