
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadiation-induced curcumin release from curcumin–chitosan polymer films†
Rajat Chauhan,
Kelsey Kinney,
Archana Akalkotkar,
Betty M. Nunn,
Robert S. Keynton,
Patricia A. Soucy* and
Martin G. O'Toole *
*
Department of Bioengineering, University of Louisville, Louisville, KY 40292, USA. E-mail: tricia.soucy@louisville.edu; martin.otoole@louisville.edu
First published on 23rd April 2020
Abstract
The probability of human exposure to damaging radiation is increased in activities associated with long-term space flight, medical radiation therapies, and responses to nuclear accidents. However, the development of responsive countermeasures to combat radiation damage to biological tissue is lagging behind rates of human exposure. Herein, we report a radiation-responsive drug delivery system that releases doses of curcumin from a chitosan polymer/film in response to low level gamma radiation exposure. As a fibrous chitosan–curcumin polymer, 1 Gy gamma irradiation (137Cs) released 5 ± 1% of conjugated curcumin, while 6 Gy exposure releases 98 ± 1% of conjugated curcumin. The same polymer was formed into a film through solvent casting. The films showed similar, albeit attenuated behavior in water (100% released) and isopropyl alcohol (32% released) with statistically significant drug release following 2 Gy irradiation. ATR FT-IR studies confirmed glycosidic bond cleavage in the chitosan–curcumin polymer in response to gamma radiation exposure. Similar behavior was noted upon exposure of the polymer to 20 cGy (1 GeV amu−1, at 20 cGy min−1) high linear energy transfer (LET) 56Fe radiation based on FTIR studies. Density Functional Theory calculations indicate homolytic bond scission as the primary mechanism for polymer disintegration upon radiation exposure. Films did not change in thickness during the course of radiation exposure. The successful demonstration of radiation-triggered drug release may lead to new classes of radio-protective platforms for developing countermeasures to biological damage from ionizing radiation.
1. Introduction
Radiation exposure from medical radiation therapy (2–15 Gy), nuclear accidents (1–16 Gy), and space travel (1.8–5.5 mGy) results in a propensity for severe DNA damage, genomic instability, radiation sickness, tissue necrosis, cellular mutagenesis, and carcinogenesis in humans.1,3–6 Radiation levels as low as ∼0.1 Gy may cause DNA damage directly or through indirect damage via hydroxyl, superoxide or nitrogen free radicals generated in response to radiation exposure.1,7,8 Free radical-induced damage has been linked to severe metabolic dysfunctions, including loss of cell integrity, enzyme function, and genomic stability. This damage ultimately leads to the pathogenesis of many human diseases, such as inflammation, ischemia, atherosclerosis, arthritis, cancer, Parkinson's disease, and Alzheimer's disease, amongst others.8Recent advances in radioprotective agents include FDA approved amifostine,9 sulfhydryl group containing compounds (e.g.: cysteine, cysteamine),10 superoxide dismutase enzyme,11,12 nitroxides,13 selenium,9 vitamin (A, C, E),14–16 N-acetyl cysteine,17 soy-bean based Bowman–Birk protease inhibitor18 and herbal compounds (e.g. flavonoids, resveratrol, tea phenols, nutmeg).18,19 These compounds predominantly act as antioxidant-based free-radical scavengers, suppressing cell apoptosis and other aforementioned maladies during the radiation exposure through quenching of radiation-generated free radicals. However, the concentration of these radioprotective agents needed to counteract radiation exposure have demonstrated significant side effects and/or questionable efficacy against radiation-related pathologies, e.g. human carcinogenesis.9,12,15,20–22 Because of this shortcoming, there is a need for a non-toxic, cost effective radioprotective system that is highly sensitive to radiation stimuli to effectively release a potent agent at ambient temperatures. Ideally, the novel system will complement traditional radioprotective drugs, which require freezing or refrigeration to avoid loss of integrity and unpredictability in the radioprotectant's drug release profile.9,11,12,14–16,22
Curcumin (diferuloylmethane) is a small (MW 368.38 Da) polyphenol compound that displays an intense yellow color. The anticancer and radioprotective effects of curcumin have been demonstrated on many types of tissues, including skin, brain, colon, gastrointestinal, liver, lung, pancreas, mammary glands, prostate, breast, blood, and bone marrow.23–25 The potent antioxidant activity of curcumin is provided by its diketone and phenol moieties. It acts as a free radical scavenger, thus inhibiting reactive oxygen species (ROS) formation and subsequent damage to DNA caused by radiation exposure. Curcumin not only has the potential to prevent radiation damage with its antioxidant properties but can also initiate DNA repair processes in radiation damaged cells.26
This work builds upon our previous studies in conjugating curcumin to chitosan, an amine-bearing carbohydrate polymer derived from chitin. Covalent linkage of curcumin to chitosan to produce chitosan–curcumin (CC) polymer has previously shown (1) improved curcumin stability over ∼1 month in solutions at above-freezing and ambient temperatures, (2) improved solubility in aqueous media, (3) improved curcumin bioavailability, (4) controlled release of conjugated-curcumin over 19 days relative to the encapsulated-curcumin (doped), which had been reported to release completely from the spray-dried particles over a 2 h period, and (5) retention of nearly ∼100% recovery of curcumin's antioxidant potential upon cleavage from the polymer backbone via ester hydrolysis.27,28
Biomacromolecules have been found to degrade into fragments under radiation stress.8,29 Hydrolysis driven glycosidic bond (C–O–C) cleavage is well-known among carbohydrates and nucleic acids, rendering them an excellent drug-payload carrier in biological environments.29–33 Specially, cationic polysaccharides (i.e., chitosan) have a high sensitivity to controlled degradation of C–O–C bond by hydrolysis as well as under high-dose radiation exposures.26,34,35 Radiation induced C–O–C bond scission occurs more frequently among high molecular weight polymers and is described by homolysis or heterolysis mechanisms.34 Upon irradiation (i.e. γ-ray) of C–O–C bond, homolysis generates free radical (C˙; O˙) entities and heterolysis generates ionic pairs (C+/OH−) of the disintegrated polymer. Mechanistically, radical pairs react faster than ion-pairs.36 Therefore, faster radical-pair kinetics, with approaching solvent molecules, could lead to a facile polymeric dissolution in the aqueous or non-aqueous media.36,37 Additionally, homolytic rupture occurs at low energy as well as in the accidental radiation exposure range (1–6 Gy), favoring controlled drug release from radioprotective polymeric matrices.33,35 A controlled and targeted cleavage of C–O–C bond upon radiation, while maintaining the material properties (i.e., biological activity) of the polymer would be a hallmark in designing a stimuli-responsive drug delivery system.
Thus, the purpose of this work is to investigate the functionality of a novel radioprotective platform based on unique curcumin–chitosan conjugate polymer2 films for the targeted release of curcumin from exposure to radiation.
2. Experimental
2.1 Materials
Chitosan (low molecular weight), curcumin (85% pure), acetic acid, L-ascorbic acid, 2,2′-diphenylpicrylhydrazyl (DPPH), dichloromethane, glutaric anhydride, methanol (HPLC grade), 4-dimethylaminopyridine (DMAP), triethylamine, N-hydroxysulfosuccinimide (sulfo-NHS), 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC), diethylether, and 10× phosphate-buffered saline (PBS) were purchased from Sigma Aldrich (St. Louis, MO, USA) and used as received unless otherwise noted. Magnesium sulfate was purchased from Fisher Scientific (Pittsburgh, PA, USA).2.2 Chitosan–curcumin polymer fabrication
Chitosan–curcumin polymer (Fig. 1) was formulated using a previously published protocol.27 Briefly, glutaric anhydride modified curcumin was conjugated to chitosan 90/200 via EDC/NHS (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) chemistry in 1 wt/v% acetic acid solution.27
:1) chemistry in 1 wt/v% acetic acid solution.27
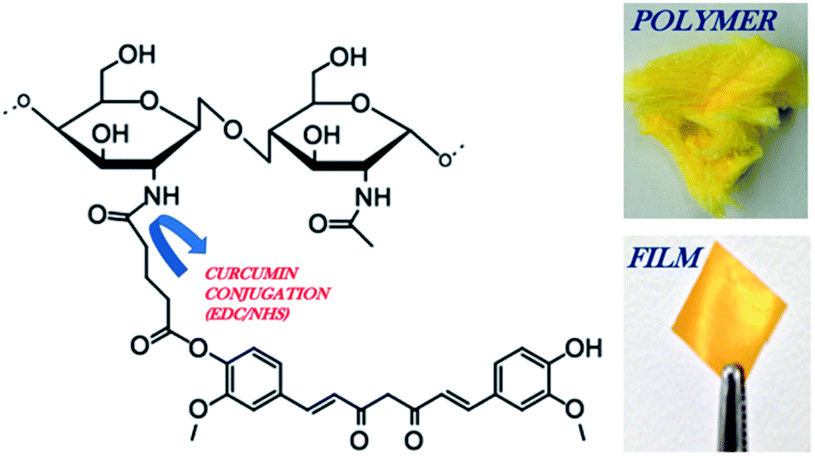 | ||
| Fig. 1 Schematic of chitosan 90/200 conjugated to glutaric anhydride modified curcumin via EDC/NHS chemistry. Image of the CC polymer and CC film. The reaction steps were previously reported, demonstrating the covalent conjugation of curcumin to chitosan through a pendant glutaric anhydride linker using amide bond coupling chemistry.26 | ||
2.3 Chitosan–curcumin film fabrication
Films were made by solvent-casting into a poly dimethyl siloxane (PDMS) mold on a Teflon coated pan to avoid film-tear during their removal from the surface. The mold was made by pouring PDMS (Sylgard®184 silicone elastomer kit: base and curing agent) around a p95 Petri dish placed inside a Teflon-coated pan. After vacuum-setting overnight at room-temperature, each Petri dish was removed, exposing the non-stick surface of the same surface area as the p95 dish (i.e., 55 cm2). 20.0 mL of 1.5 wt/v% curcumin–chitosan conjugate polymer in 1% acetic acid solution. Also, controls of chitosan doped with curcumin were prepared by adding 3 mL of ethanolic solution of curcumin (1 mg mL−1) to 20.0 mL of 2.0 wt/v% chitosan polymer in 1% acetic acid solution. This resulting mixture was stirred for 10 minutes for facile encapsulation of curcumin. The curcumin–chitosan conjugate as well as control solutions were poured onto the non-stick area created in the PDMS mold and desiccated in an aluminum-foiled covered vacuum chamber (at ∼20 mm Hg) for two days at room temperature. The film was then carefully removed from the mold and cut into 1.0 cm × 1.0 cm patches using a scalpel and ruler, unless noted otherwise. The film samples were stored at 4 °C and wrapped in aluminum foil until used.2.4 Radiation exposure
The chitosan–curcumin polymer was exposed to both low linear energy transfer (LET) radiation (γ) and high LET radiation exposure (56Fe) to assess the sensitivity of the polymer to different sources of radiation. All studies utilized 137Cs γ-radiation (Gammacell, Nordion) from 1–6 Gy in 1 Gy increments, except for the data in Fig. 3B, where the chitosan–curcumin polymer was exposed to 20 cGy of 56Fe radiation (1 GeV amu−1, at 20 cGy min−1) at Brookhaven National Lab's NASA Space Radiation Lab.2.5 ATR FT-IR analysis of chitosan–curcumin polymer
FT-IR spectra were measured with an FT-IR spectrometer (PerkinElmer Spectrum 100) with universal attenuated total reflectance (ATR) sample accessory. The chitosan–curcumin polymer was sandwiched between diamond/ZnSe crystal and pressure arm of universal ATR. The spectra were recorded over the wavenumber range 4000–500 cm−1.2.6 Density functional theory
Geometry optimization and frequency calculations for curcumin–chitosan conjugate were performed using the Gaussian 09 suite of programs. Density functional theory (DFT) calculations were completed using the B3LYP functional. For these calculations the 6-31G basis set was used for carbon, hydrogen, nitrogen and oxygen atoms. Input coordinates were taken from crystallographic data for curcumin and chitosan.38,392.7 UV-VIS analysis post-radiation exposure
UV-VIS was utilized to detect curcumin released into solution following γ-radiation exposure in the chitosan–curcumin polymer and the films. The chitosan–curcumin polymer (5 mg) was added to 3 mL of 1× PBS with 0.03 M L-ascorbic acid within a 15.0 mL centrifuge tube. The samples were radiated at 1 Gy intervals of γ-radiation for 1 minute, then centrifuged at 1000g for 1 minute and 1 mL of the supernatant was removed for UV-Vis characterization. A fresh 1 mL of 1× PBS with 0.03 M L-ascorbic acid was added to each sample to bring the total volume back to 3 mL and the cycle was repeated again until the sample had been exposed to a total of 6 Gy radiation. A total of three polymer samples were exposed to γ-radiation and three polymer samples were used as a control (0 Gy radiation). The control polymers immersed in 1× PBS with 0.03 M L-ascorbic acid in parallel with the radiated samples and provided a control for each period of time during the radiation cycles. UV absorption spectra of the supernatants were measured in 200–1000 nm wavelength range using a UV Visible Spectrometer (Varian Cary 50 BIO UV, Agilent Technologies). The peak absorbance wavelength for curcumin was identified at 430 nm. The peak absorbance was correlated to a concentration of curcumin using a standard curve of 1–27 μM of curcumin dissolved in methanol:PBS (1:1) solution.
The chitosan–curcumin films exposed to γ-radiation in 1× PBS with 0.03 M L-ascorbic acid, followed the same procedure for radiation exposure outlined above with each 1 cm2 sample added to 3 mL 1× PBS and 0.03 M L-ascorbic acid in a centrifuge tube. Analysis of the curcumin content in the supernatant was carried out with the UV-VIS Spectrometer as described above. A total of three polymer samples were exposed to each γ-radiation level and three polymer samples were used as a control (0 Gy γ-radiation). The controls were films immersed in 1× PBS with 0.03 M L-ascorbic acid in parallel with the radiated samples and provided a control for each period of time during the radiation cycles.
The chitosan–curcumin films were also exposed to γ-radiation in pure isopropyl alcohol (IPA) to eliminate hydrolysis induced release of curcumin, which allowed characterization of the radiation-induced released. These studies were carried out in a similar format to the films described above. Films were submerged with 1 mL IPA and then exposed to 1 Gy radiation. Afterwards, the samples were placed on an orbital shaker for 1 minute and each sample was then moved to 1 mL of fresh IPA. The IPA in the original container was used to measure the curcumin content with the UV-Vis Spectrometer. The cycle was repeated until the samples had received a total of 6 Gy radiation. Four sample films were exposed to each radiation level as described above and three films were controls and were not radiated. The controls were immersed in IPA in parallel with the radiated samples and placed on the orbital shaker. They were moved to fresh IPA each time the radiated samples were moved. Thus, these controls provide a control for each time point during the full radiation experiment. All IPA samples were analyzed by UV-VIS at 430 nm to record absorbance values that were correlated to a concentration of curcumin in IPA by a standard curve from 0.5–16 μM.
After the samples were exposed to 6 Gy, the solution (1× PBS with 0.03 M L-ascorbic acid or IPA) was completely removed and the dry samples were later solubilized in 1 wt/v% acetic acid to analyze the amount of curcumin remaining in the sample after exposure to 6 Gy γ-radiation. The peak absorbance (λ = 420 nm) was correlated to a concentration of curcumin using a standard curve of 2–30 μM of curcumin dissolved in 1 wt/v% acetic acid solution. The amount of curcumin loaded into a sample was calculated as the sum of the amount of curcumin remaining in a sample after exposure to 6 Gy γ-radiation and the total amount of curcumin released into the supernatant after exposure to each γ-radiation exposure. Using this value of the amount of curcumin loaded per sample, the release study data was analyzed as a percentage of curcumin released for each dose of γ-radiation.
2.8 Free radical scavenging of released curcumin via DPPH assay
Multiple chitosan–curcumin films adding up to a total weight of 5.0 mg were exposed to γ-radiation at 0, 3, or 6 Gy with no solvent. These radiated and non-radiated films were further dissolved in 1% acetic acid and wrapped into aluminum foil to protect against photoactivation. After 5 minutes, 3.0 mL of ethyl acetate was added, vortexed and the sample was placed at 4 °C overnight. Next day, the yellowish layer of ethyl acetate in the solution was carefully separated using micropipette and evaporated via rotavap. The yellowish solid obtained via rotavap was therefore re-dissolved in 4.0 mL methanol and UV-VIS absorbance was measured at 420 nm. The average curcumin concentration for 0, 3, and 6 Gy samples (n = 3 for each radiation dose) was calculated using a standard curve of 1–14 μM of curcumin dissolved in methanol solution.A stock solution of 1,1-diphenyl-2-picrylhydrazyl (DPPH) was made in methanol equimolar to the solutions of extracted curcumin from rotovap (based on UV-VIS measurements). A 1 mL stock DPPH solution was added to 1 mL of a rotary-evaporator extracted curcumin in methanol in ratios of 0.2 to 1.0 equivalents. These solutions were incubated for 45 minutes at 37 °C and the UV-Vis absorbance at 517 nm of DPPH were recorded. The DPPH percent radical scavenging by extracted curcumin (% inhibition of DPPH) was calculated by the following equation.
| % inhibition = (control − sample/control) × 100 |
2.9 SEM analysis of chitosan–curcumin films
After radiation (0, 3, or 6 Gy) of films without solvent, the samples were sputter coated with gold/palladium alloy followed by SEM imaging using a SE2 detector at 2.0 keV accelerating voltage on a Zeiss Supra 35 (Carl Zeiss AG).2.10 Statistical analysis
Data was imported into GraphPad Prism® (Version 6.07). All reported data are of the mean with error bars representing the standard deviation. When appropriate, statistical analysis was conducted with an unpaired t-test and significance was considered to be present at p < 0.05 for all data.3. Results and discussions
3.1 Radiation-induced curcumin release for chitosan–curcumin fibrous polymer
UV-VIS spectroscopic analysis of the supernatant from the chitosan–curcumin fibrous polymer following γ-radiation detected curcumin dispersed in PBS solution (Fig. 2). The released curcumin increased with each 1 Gy increment of γ-radiation. In the 1–4 Gy radiation dose range, 5–32% of the curcumin loaded into the chitosan–curcumin fibrous polymer was released into the PBS/L-ascorbic acid solution. The curcumin release nearly doubled from 32 ± 7% to 61 ± 3% after exposure to 5 Gy of γ-radiation. After exposure to 6 Gy of γ-radiation, nearly all (98 ± 1%) of the curcumin was released from the chitosan–curcumin fibrous polymer. The curcumin release from the fibrous polymer was statistically significant at radiation doses of 2 Gy and above, compared to no radiation (p < 0.05).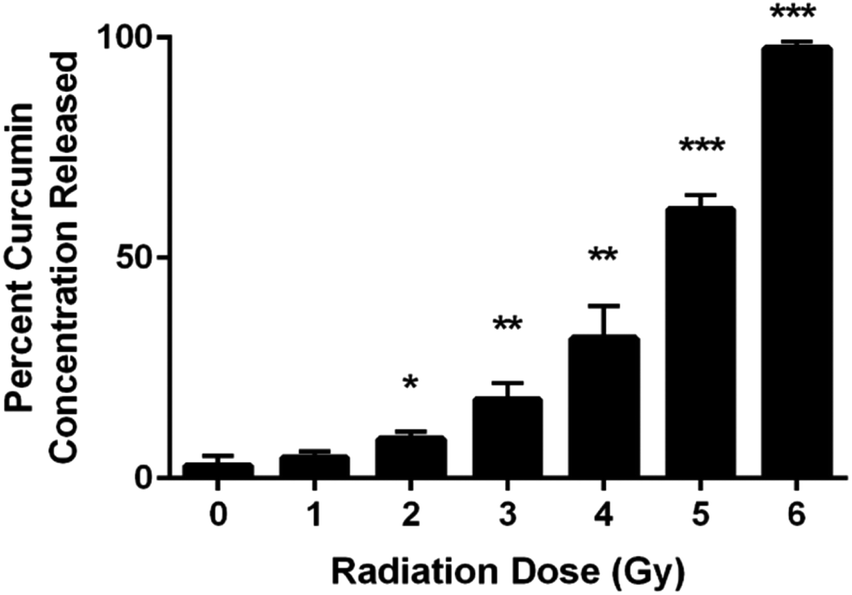 | ||
| Fig. 2 Incremental release of the percent cumulative curcumin concentrations from CC fibrous polymer at varying γ-radiation doses (0–6) Gy in PBS with L-ascorbic acid (unpaired t-test: 0 Gy vs. (1–6) Gy; *P < 0.05, **P < 0.005, ***P < 0.001). | ||
Chitosan–curcumin fibrous polymer samples exposed to either 5 or 6 Gy γ-radiation demonstrated higher levels of curcumin release (greater than 50%) than the lower γ-radiation doses which suggests a higher magnitude of homolytic rupture of the glycosidic bond at the 5 to 6 Gy doses.29–31,36,40,41 In the non-irradiated control samples (0 Gy in Fig. 2), only 3 ± 2% of the curcumin in the chitosan–curcumin fibrous polymer mixtures released over the time course of the experiment. This curcumin release at 0 Gy supports previously published results for the release of curcumin from chitosan–curcumin polymer, which concluded that it took 19 days to reach 100% curcumin release via ester-hydrolysis.27 However, this ester-hydrolysis release profile is not an ideal curcumin release mechanism for situations such as radiation exposure that require rapid drug response after the harmful stimuli.27,42 For comparison, the 6 Gy release of 98% curcumin occurred in 30 minutes. The radiation sensitivity for releasing curcumin from the chitosan–curcumin polymer makes this a unique system for radiation exposure situations.
3.2 ATR FT-IR analysis of chitosan–curcumin fibrous polymer
ATR FT-IR spectroscopic analysis of chitosan–curcumin fibrous polymer was used to monitor radiation-induced carbohydrate backbone disintegration under γ-radiation exposure (Fig. 3A). The ATR FT-IR transmission bands of chitosan–curcumin polymer/film at 3290 cm−1 (m), 1763 cm−1 (w), 1540 cm−1 (m) and 1025 cm−1 (s) were attributed to hydroxyl (–OH), curcumin, chitosan amine (–NH2) group and glycosidic (C–O–C) groups respectively.27,41,43 Per literature, the amine (–NH2) group is not altered after γ-radiation exposure, and so the ATR FT-IR analysis of radiation-induced cleavage was referenced to this band.29,44,45 The data showed radiation-induced scission of glycosidic bonds leading to the formation of hydroxyl groups. Subsequently, an increase in the hydroxyl band [% T(OH/–NH2) = 1.08 (non-radiated) vs. 1.19 (radiated)] and simultaneous decrease in glycosidic band (C–O–C) [% T(C–O–C/–NH2) = 0.79 (non-radiated) vs. 0.61 (radiated)] intensities were observed upon 6 Gy γ-radiation exposure (Fig. 3A). Gamma, ultraviolet and ultrasonic irradiation have been previously shown to have significant impact on the intrinsic properties of polysaccharides via bond scission.45–49 Wasikiewicz, et al. observed similar γ-radiation-induced polymeric degradation of chitosan and sodium alginate chains via cleavage of glycosidic bonds, albeit at much higher radiation exposure levels (5.0 kGy) than were needed to see the cleavage of the glycosidic bonds in a chitosan–curcumin polymer.45 They studied the effects of γ-radiation on the material properties of chitosan and sodium alginate in the kGy (0.5–200) range, an irradiation dose used for sterilization of long-term clinical products (i.e., implants). However, we investigated the variations in material properties of chitosan–curcumin polymer for an incidental or accidental exposure (1–6) Gy range, using low-energy γ-radiation exposure.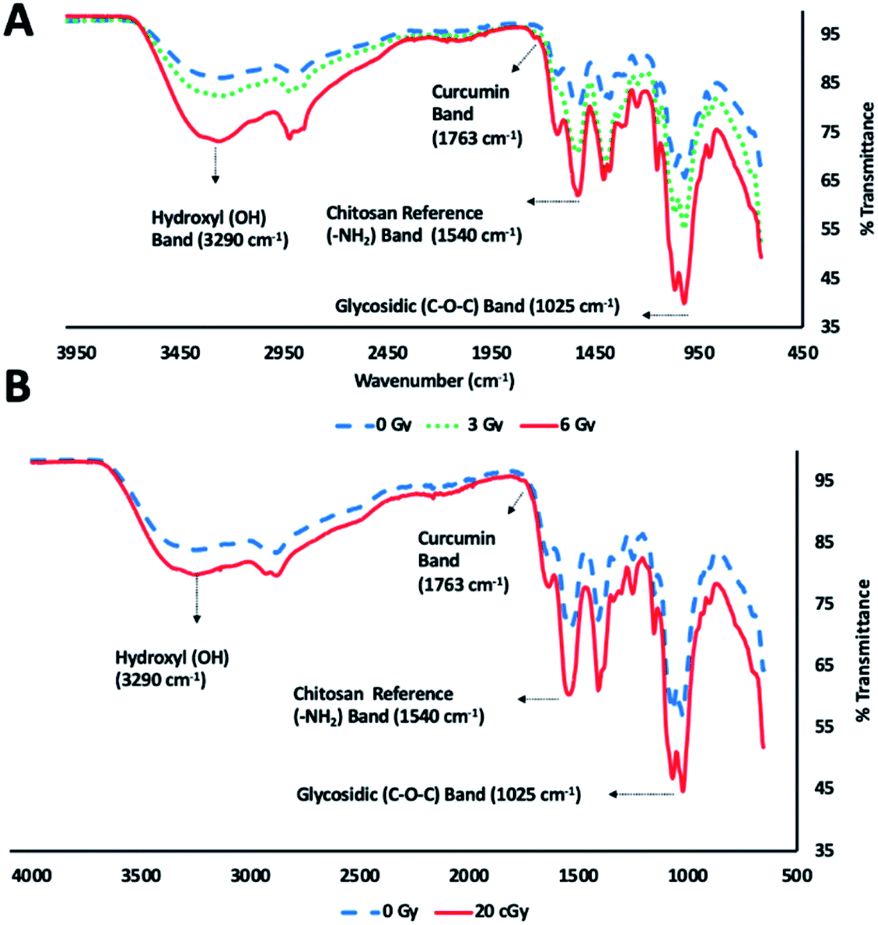 | ||
| Fig. 3 Glycosidic bond (C–O–C) disintegration in curcumin–chitosan conjugate fibrous polymer as displayed by FT-IR (ATR) spectra at (A) γ-radiation doses (0, 3 and 6), (B) 56Fe ionizing radiation dose (20 cGy) compared to non-irradiated conditions. | ||
Similar to the gamma radiation exposure, 56Fe radiation exposure at 20 cGy to the chitosan–curcumin fibrous polymer exhibited radiation dependent backbone disintegration of glycosidic linkages (Fig. 3B). The glycosidic scission by the heavy ion irradiation were demonstrated with increases in the hydroxyl (–OH) [% T(OH/–NH2) = 1.17 (non-radiated)–1.39 (radiated)] and simultaneous decrease in glycosidic (C–O–C) [% T(C–O–C/–NH2) = 0.84 (non-radiated)–0.69 (radiated)] transmission band intensities. Therefore, the chitosan–curcumin fibrous polymer displayed similar bond cleavage trends in response to both 20 cGy radiation flux of 56Fe ions and cesium (137Cs) emitted γ-radiation, respectively.
3.3 DFT analysis
To examine the relative bond stability within the chitosan–curcumin fibrous polymer, the thermodynamic stability and solubility of chitosan–curcumin conjugate [C38O17N2H49 (106 atoms)] in aqueous and in less polar solvents such as ethanol and isopropanol were analyzed with DFT (Fig. 4). The absorption, distribution, metabolism, and excretion (ADME) based pharmacokinetics and pharmacodynamics of pure chitosan in polar solvents has been previously reported using density functional theory (DFT) and molecular dynamics studies.50–53 Homolytic cleavage of the glycosidic bonds would produce two radical cations, whereas a heterolytic bond scission would produce two neutral species. The Gibbs free energy of solvation (ΔGsolvation) for both uncharged [0, singlet] [ΔGsolvation,water = −47.77; ΔGsolvation,ethanol = −46.08; ΔGsolvation,IPA = −34.55 kcal mol−1] and radical cationic [+1; doublet] [ΔGsolvation,water = −68.50; ΔGsolvation,ethanol = −64.35; ΔGsolvation,IPA = −62.22 kcal mol−1] optimized structures, indicates that the radical cation would be a more stable structure in water than ethanol and isopropanol solutions, thus favoring a hemolytic bond scission pathway.54 This trend is in agreement with previous reports of radiation-induced scission of cationic polysaccharides through homolysis in solution, resulting in the formation of two stable macromolecular radicals.36,40,41,54 These stable and mobile macromolecular radicals may later recombine, disproportionate or repolymerize in their aqueous or non-aqueous (e.g. ethanol, Isopropanol) environments.55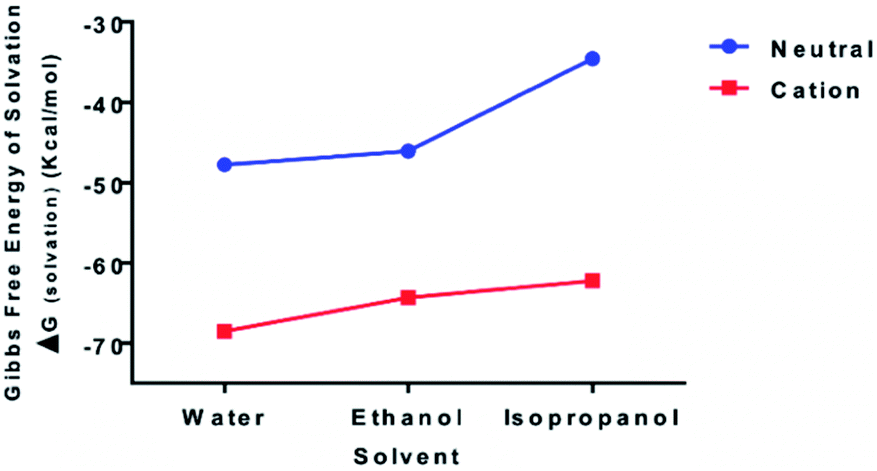 | ||
| Fig. 4 Increasing thermodynamic stability of curcumin–chitosan conjugate [C38O17N2H49 (106 atoms)] in water compared to lesser polar ethanol and isopropanol media as displayed by calculated Gibbs free energy of solvation (kcal mol−1) of neutral [0, singlet] and radical cationic [+1; doublet] states using B3LYP/6-31G and polarizable continuum model (PCM) level of theory. | ||
3.4 Chitosan–curcumin film thickness and morphology
The chitosan–curcumin polymer was utilized to create films (Fig. 5). The average thickness of the non-irradiated films was 12.77 ± 2.92 μm. Films exposed to 3 Gy and 6 Gy γ-radiation had an average thickness of 12.84 ± 0.43 μm and 13.02 ± 2.04 μm, respectively. The similar film thicknesses for these conditions implies that while γ-radiation induced bond scission as shown in the ATR FT-IR spectra of chitosan–curcumin fibrous polymer, no statistical changes of the bulk thickness of the films (whether through swelling or polymer degradation) were observed. | ||
| Fig. 5 Similar CC film thickness displayed by SEM images of cross-section of film for irradiated (6 Gy) and non-radiated (0 Gy) samples [inset (right-corner): SEM stub set-up with film on the carbon tape]. *White-line demonstrated with 2 Xs and 2 circles is a distance-measurement tool and the film-thickness reported herein was measured across 2 circles. | ||
The surface morphology of the chitosan–curcumin film samples (n = 3) was observed using SEM at 0 Gy and 6 Gy, respectively (Fig. 6). The films displayed a solid non-porous surface texture with no major changes in the surface morphology upon radiation exposure.
 | ||
| Fig. 6 Similar CC film morphology displayed by SEM images of films for irradiated (6 Gy) and non-radiated (0 Gy) samples. | ||
3.5 Radiation-induced curcumin release for chitosan–curcumin film
Curcumin released from chitosan–curcumin films were investigated in PBS with L-ascorbic acid (Fig. 7A). The non-radiated samples (black bars) represent the hydrolysis mediated curcumin release from the chitosan–curcumin films, while the films exposed to γ-radiation (gray bars) induced curcumin release from both hydrolysis and radiation. Films exposed to 1 Gy γ-radiation released twice as much curcumin as the control samples without radiation, but the increase was not statistically significant (p = 0.07). At 2 Gy γ-radiation, the curcumin released was still twice as high as the control and was statistically significant (p = 0.04). The 3 and 4 Gy γ-radiated films each had a statistically significant 47% increase in curcumin released compared to their respective controls (p < 0.02, and p < 0.002, respectively). Similarly, films exposed to 5 Gy γ-radiation had a 45% increase in curcumin release compared to its control and 6 Gy had a 42% increase compared to its control (P < 0.001 for both). This data is similar to the fibrous polymer release of curcumin following γ-radiation where a statistically significant increase was observed initially at the 2 Gy dose of γ-radiation and above. These results indicate that 2 Gy could be a threshold dose for inducing a statistically significant drug release in PBS. The films also demonstrated 100 ± 0% release of the loaded curcumin following exposure to 6 Gy of γ-radiation.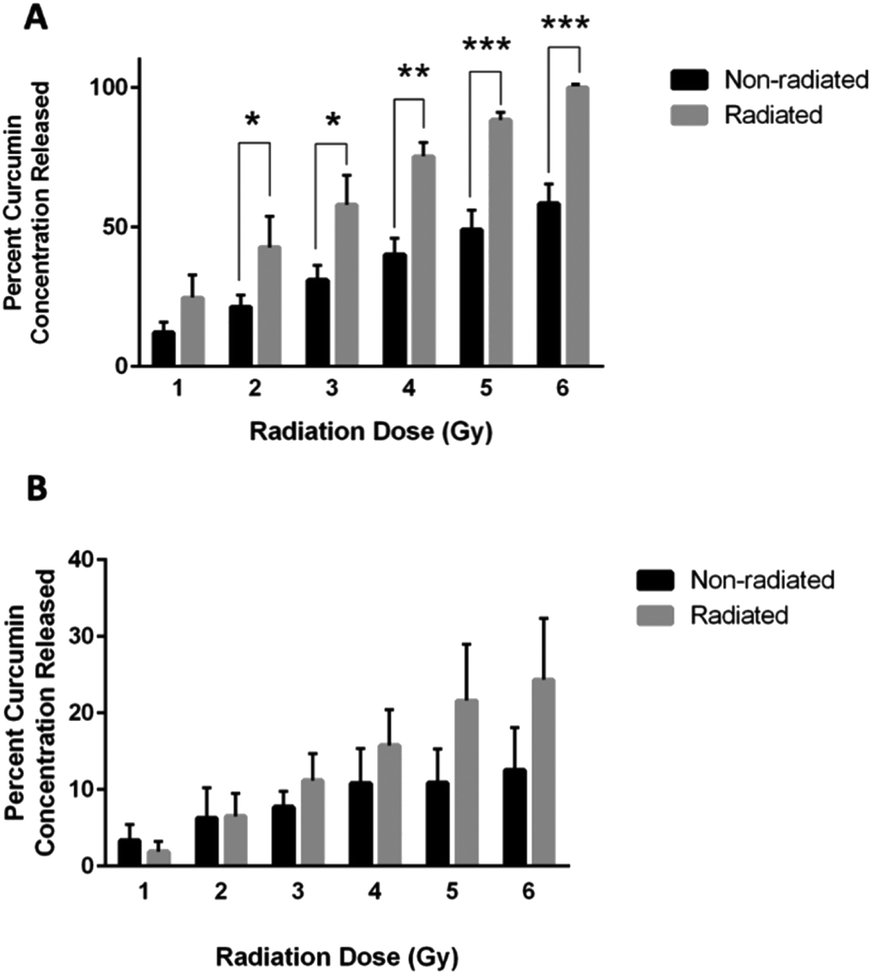 | ||
| Fig. 7 (A) Plot of percent curcumin concentrations released from CC films exposed to varying γ-radiation doses of 1–6 Gy (grey bars) and non-radiated controls (black bars) kept for each radiation time interval in PBS with L-ascorbic acid (unpaired t-test, *P < 0.05, **P < 0.005, ***P < 0.001). (B) Plot of percent curcumin concentrations released from CC films at varying γ-radiation doses of 1–6 Gy (grey bars) and non-radiated controls (black bars) kept for each radiation interval in isopropanol solutions. | ||
To remove ester hydrolysis as a mechanism for curcumin release from the chitosan–curcumin films, γ-radiation-induced release was studied in chitosan–curcumin films immersed in anhydrous IPA. Unlike in aqueous media, γ-radiated films in IPA did not have a statistically significant difference in the amount of curcumin released compared to control chitosan–curcumin films with no radiation (Fig. 7B). Although, films exposed to doses of 3–6 Gy γ-radiation did show a trend of increased curcumin release from films exposed to γ-radiation compared to their controls: 11 ± 4% for 3 Gy, 16 ± 5% for 4 Gy, 22 ± 7% for 5 Gy and 24 ± 8% for 6 Gy, respectively. Clearly, the films exposed to 6 Gy released approximately twice as much curcumin compared to non-radiated films. The similar amounts of curcumin released from control chitosan–curcumin films compared to radiated chitosan–curcumin films in isopropanol could be attributed to the inherently superior curcumin solubility in alcohols favoring ester-cleavage.37 The lower release of curcumin from films in IPA (24 ± 8%) compared to release in 1× PBS with L-ascorbic acid (100 ± 0%) at 6 Gy can be attributed to the following potential factors. First, there is less radical generation in non-polar IPA solutions, consistent with IPA's low dielectric constant (ε = 17.9) as compared to strongly hydrogen-bonding and polar aqueous (ε = 80.1) environments. Additionally, the greater amount of curcumin release in water under γ-radiation suggests water-derived radical species (e.g. hydroxy radicals) are major participators in glycosidic bond cleavage.31
In addition, as a control, curcumin released from chitosan films doped with curcumin were also investigated in IPA (Fig. S1†). The γ-radiated chitosan–curcumin doped films did not have a statistically significant difference in the amount of curcumin released compared to their control films, i.e. no radiation, at any radiation dose (Fig. S1†).
3.6 Free radical scavenging of released curcumin via DPPH assay
The antioxidant capacity of curcumin released from the chitosan–curcumin (conjugated) films were assessed using the DPPH assay (Fig. 8). Similar percent inhibitions (76 ± 7% for 0 Gy, 75 ± 7% for 3 Gy and 80 ± 3% for 6 Gy) were obtained for both radiated and non-radiated chitosan–curcumin films at 1:1 ratio with DPPH assay (p = 0.87 for 0 Gy compared to 3 Gy and p = 0.33 for 0 Gy compared to 6 Gy). The percent inhibition reported here was higher compared to previously published 52.4% inhibition with chitosan–curcumin polymers in an acetic acid/Tween-20 solution.27 The similar radical scavenging percentages of curcumin from films with and without radiation implies that the radiation induced glycosidic bond cleavage that released curcumin did not alter curcumin's free radical scavenger activity with DPPH at 1:1 molar ratio. This is important to note, as the gamma irradiation stimulus does not appear to alter the potential therapeutic effects of curcumin.
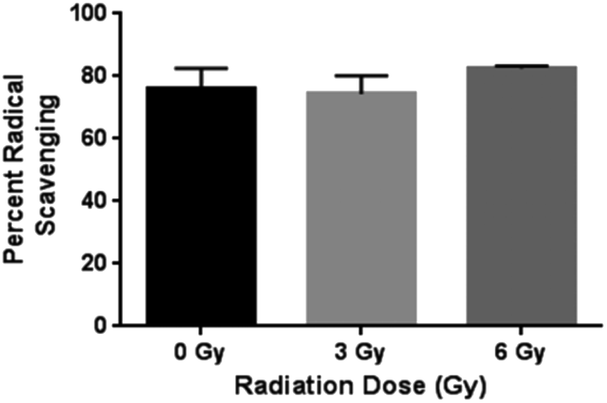 | ||
| Fig. 8 Statistically similar percent radical scavenging of DPPH demonstrated by CC films at γ-radiation doses 0, 3 and 6 Gy. | ||
4. Conclusions
Chitosan–curcumin conjugated polymer's sensitivity to radiation was studied. The chitosan–curcumin polymer were used to form films, which shows promise as a potential radioprotectant system since it is easy to construct and highly functional. Curcumin's chemical stability was displayed with its similar free radical scavenging activity with and without radiation exposure. Solubility of curcumin following radiation exposure was demonstrated with ∼100% recovery in aqueous and ∼32% recovery of concentration levels in non-aqueous (IPA) environments. The radiation exposure leads to glycosidic bond scission among the chitosan units of the chitosan–curcumin polymer. The advantage of current work is the controlled and statistically significant drug release from the chitosan–curcumin polymer under stimuli of γ-radiation in aqueous and polar media. Further studies to extend the observed triggered-burst release in thin films to other chemical radioprotectants, wound-healing, pulsatile drug delivery and additional triggered-burst induced targeted delivery applications are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this work was in part provided by NASA Grants NNX10AJ36G and NNX13AD33A and the University of Louisville Department of Bioengineering.References
- W. Sudprasert, P. Navasumrit and M. Ruchirawat, Int. J. Hyg. Environ. Health, 2006, 209, 503–511 CrossRef CAS PubMed.
- J. Chatterjee, R. K. Nairy, J. Langhnoja, A. Tripathi, R. K. Patil, P. P. Pillai and M. S. Mustak, Metab. Brain Dis., 2018, 33, 855–868 CrossRef CAS PubMed.
- F. El Ghissassi, R. Baan, K. Straif, Y. Grosse, B. Secretan, V. Bouvard, L. Benbrahim-Tallaa, N. Guha, C. Freeman, L. Galichet and V. Cogliano, Lancet Oncol., 2009, 10, 751–752 CrossRef PubMed.
- I. Furuno-Fukushi, Int. J. Radiat. Biol., 1996, 70, 209–217 CrossRef CAS PubMed.
- A. E. Metzger, E. C. Anderson, M. A. Van Dilla and J. R. Arnold, Nature, 1964, 204, 766 CrossRef.
- J. P. Christodouleas, R. D. Forrest, C. G. Ainsley, Z. Tochner, S. M. Hahn and E. Glatstein, N. Engl. J. Med., 2011, 364, 2334–2341 CrossRef CAS PubMed.
- E. I. Azzam, J.-P. Jay-Gerin and D. Pain, Cancer Lett., 2012, 327, 48–60 CrossRef CAS PubMed.
- J. A. Reisz, N. Bansal, J. Qian, W. Zhao and C. M. Furdui, Antioxid. Redox Signaling, 2014, 21, 260–292 CrossRef CAS PubMed.
- B. S. Margulies, T. A. Damron and M. J. Allen, J. Orthop. Res., 2008, 26, 1512–1519 CrossRef CAS PubMed.
- D. Citrin, A. P. Cotrim, F. Hyodo, B. J. Baum, M. C. Krishna and J. B. Mitchell, Oncologist, 2010, 15, 360–371 CrossRef PubMed.
- A. Petkau and C. A. Chuaqui, Radiat. Phys. Chem., 1984, 24, 307–319 CrossRef CAS.
- J. Pan, H. He, Y. Su, G. Zheng, J. Wu, S. Liu and P. Rao, Oxid. Med. Cell. Longevity, 2016, 2016, 5935080 Search PubMed.
- B. P. Soule, F. Hyodo, K.-i. Matsumoto, N. L. Simone, J. A. Cook, M. C. Krishna and J. B. Mitchell, Free Radical Biol. Med., 2007, 42, 1632–1650 CrossRef CAS PubMed.
- N. L. Gregory, Br. J. Radiol., 1978, 51, 473–474 CrossRef CAS PubMed.
- R. M. Niles, Nutrition, 2000, 16, 1084–1089 CrossRef CAS PubMed.
- V. Srinivasan and J. F. Weiss, Int. J. Radiat. Oncol., Biol., Phys., 1992, 23, 841–845 CrossRef CAS.
- F. Allaveisi, B. Hashemi and S. M. J. Mortazavi, Cell Tissue Banking, 2015, 16, 97–108 CrossRef CAS.
- K. Dittmann, C. Mayer, R. Kehlbach and H. P. Rodemann, Radiother. Oncol., 2008, 86, 375–382 CrossRef CAS PubMed.
- P. Okunieff, S. Swarts, P. Keng, W. Sun, W. Wang, J. Kim, S. Yang, H. Zhang, C. Liu, J. P. Williams, A. K. Huser and L. Zhang, Adv. Exp. Med. Biol., 2008, 614, 165–178 CrossRef CAS PubMed.
- K. Sideras, C. L. Hallemeier and C. L. Loprinzi, in Abeloff's Clinical Oncology, ed. J. O. Armitage, J. H. Doroshow, M. B. Kastan and J. E. Tepper, Content Repository Only!, Philadelphia, 5th edn, 2014, pp. 635–647, DOI:10.1016/B978-1-4557-2865-7.00043-6.
- J. A. Gangoiti, M. Fidler, B. L. Cabrera, J. A. Schneider, B. A. Barshop and R. Dohil, Br. J. Clin. Pharmacol., 2010, 70, 376–382 CrossRef CAS PubMed.
- M. I. Koukourakis, Br. J. Radiol., 2012, 85, 313–330 CrossRef CAS PubMed.
- F. A. Mettler, D. Brenner, C. N. Coleman, J. M. Kaminski, A. R. Kennedy and L. K. Wagner, Am. J. Roentgenol., 2011, 196, 616–618 CrossRef PubMed.
- Y. J. Surh and K. S. Chun, Adv. Exp. Med. Biol., 2007, 595, 149–172 CrossRef PubMed.
- S. Singh and B. B. Aggarwal, J. Biol. Chem., 1995, 270, 24995–25000 CrossRef CAS PubMed.
- H. Xu, T. Wang, C. Yang, X. Li, G. Liu, Z. Yang, P. K. Singh, S. Krishnan and D. Ding, Adv. Funct. Mater., 2018, 28, 1707140 CrossRef.
- M. G. O'Toole, P. A. Soucy, R. Chauhan, M. V. R. Raju, D. N. Patel, B. M. Nunn, M. A. Keynton, W. D. Ehringer, M. H. Nantz, R. S. Keynton and A. S. Gobin, Biomacromolecules, 2016, 17, 1253–1260 CrossRef PubMed.
- M. G. O'Toole, R. M. Henderson, P. A. Soucy, B. H. Fasciotto, P. J. Hoblitzell, R. S. Keynton, W. D. Ehringer and A. S. Gobin, Biomacromolecules, 2012, 13, 2309–2314 CrossRef PubMed.
- A. L. Millen, L. A. B. Archibald, K. C. Hunter and S. D. Wetmore, J. Phys. Chem. B, 2007, 111, 3800–3812 CrossRef CAS PubMed.
- D. S. H. Chu, J. G. Schellinger, J. Shi, A. J. Convertine, P. S. Stayton and S. H. Pun, Acc. Chem. Res., 2012, 45, 1089–1099 CrossRef CAS PubMed.
- I. P. Edimecheva, R. M. Kisel, O. I. Shadyro, K. Kazem, H. Murase and T. Kagiya, J. Radiat. Res., 2005, 46, 319–324 CrossRef CAS PubMed.
- C. J. Biermann, in Advances in Carbohydrate Chemistry and Biochemistry, ed. R. S. Tipson and D. Horton, Academic Press, 1988, vol. 46, pp. 251–271 Search PubMed.
- L. Li, S. Maiti, N. A. Thompson, I. J. Milligan and W. Du, ChemSusChem, 2017, 10, 4829–4832 CrossRef CAS PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- M. A. García, N. de la Paz, C. Castro, J. L. Rodríguez, M. Rapado, R. Zuluaga, P. Gañán and A. Casariego, J. Radiat. Res. Appl. Sci., 2015, 8, 190–200 CrossRef.
- W. K. Czerwinski, Macromolecules, 1995, 28, 5405–5410 CrossRef CAS.
- R. Pagni, J. Chem. Educ., 2005, 82, 382 CrossRef CAS.
- S. P. Parimita, Y. V. Ramshankar, S. Suresh and T. N. Guru Row, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o860–o862 CrossRef CAS.
- P.-K. Naito, Y. Ogawa, D. Sawada, Y. Nishiyama, T. Iwata and M. Wada, Biopolymers, 2016, 105, 361–368 CrossRef CAS PubMed.
- H. Zegota and C. Von, Z. Naturforsch., B: J. Chem. Sci., 1977, 32, 1060 Search PubMed.
- C. von Sonntag, in Advances in Carbohydrate Chemistry and Biochemistry, ed. R. S. Tipson and D. Horton, Academic Press, 1980, vol. 37, pp. 7–77 Search PubMed.
- A. Safavy, K. P. Raisch, S. Mantena, L. L. Sanford, S. W. Sham, N. R. Krishna and J. A. Bonner, J. Med. Chem., 2007, 50, 6284–6288 CrossRef CAS PubMed.
- C. Peniche-Covas, W. Argüelles-Monal and J. San Román, Polym. Degrad. Stab., 1993, 39, 21–28 CrossRef CAS.
- N. Duy, D. Phu, N. Tue Anh and N. Hien, Synergistic degradation to prepare oligochitosan by γ-irradiation of chitosan solution in the presence of hydrogen peroxide, 2011 Search PubMed.
- J. M. Wasikiewicz, F. Yoshii, N. Nagasawa, R. A. Wach and H. Mitomo, Radiat. Phys. Chem., 2005, 73, 287–295 CrossRef CAS.
- C. Zhou and H. Ma, J. Agric. Food Chem., 2006, 54, 2223–2228 CrossRef CAS PubMed.
- F. El-Ashhab, L. Sheha, M. Abdalkhalek and H. A. Khalaf, J. Assoc. Arab Univ. Basic Appl. Sci., 2013, 14, 46–50 Search PubMed.
- W. Yue, R. He, P. Yao and Y. Wei, Carbohydr. Polym., 2009, 77, 639–642 CrossRef CAS.
- K. Datta, S. Suman, B. V. S. Kallakury and A. J. Fornace Jr, PLoS One, 2012, 7, e42224 CrossRef CAS PubMed.
- B. Hassan, A. Shireen, K. Muraleedharan and V. M. A. Mujeeb, Int. J. Biol. Macromol., 2015, 74, 392–396 CrossRef CAS PubMed.
- A. E. Silviya, G. Kavitha, K. N. Kutty and P. K. K. Namboori, Int. J. Drug Delivery, 2015, 7, 5 Search PubMed.
- M. Sano, O. Hosoya, S. Taoka, T. Seki, T. Kawaguchi, K. Sugibayashi, K. Juni and Y. Morimoto, Chem. Pharm. Bull., 1999, 47, 1044–1046 CrossRef CAS.
- B. C. Deka and P. K. Bhattacharyya, J. Chem. Sci., 2016, 128, 589–598 CrossRef CAS.
- I. A. E. Agency, The Radiation Chemistry of Polysaccharides, International Atomic Energy Agency, Vienna, 2016 Search PubMed.
- Y. Sun, Applications of Ionizing Radiation in Materials Processing, Institute of Nuclear Chemistry and Technology, 2017, vol. 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00144a |
| This journal is © The Royal Society of Chemistry 2020 |