
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring the AlCl3/iPr2O/Et2O initiation system for highly reactive polyisobutylene synthesis in pure n-hexane
Dan Xie ,
Shan Zhu and
Yangcheng Lu*
,
Shan Zhu and
Yangcheng Lu*
State Key Laboratory of Chemical Engineering, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: luyc@tsinghua.edu.cn; Fax: +86 10 62773017; Tel: +86 10 62773017
First published on 31st January 2020
Abstract
This paper reports the flow synthesis of highly reactive polyisobutylenes (HRPIBs) in pure n-hexane using properly prepared AlCl3·Et2O crystals in conjunction with AlCl3·iPr2O solution as coinitiators. By preparing AlCl3·iPr2O solution and AlCl3·Et2O crystals separately, the cationic polymerization of isobutylene proceeded smoothly under a wide range of monomer concentrations (0.33–1.30 M) in the presence of H2O as an initiator, affording a high yield (∼89%) and a moderate exo-olefin terminal group content (60–75%) in 10 min. The various functions of iPr2O and Et2O in the initiator solution were comprehensively revealed from the polymerization results, attenuated total reflection-Fourier transform infrared and 27Al nuclear magnetic resonance spectra, and density functional theory simulations. AlCl3·iPr2O was confirmed to be the key component that stabilized carbenium ions. The AlCl3·Et2O complex was the key component to promote proton elimination. Free Et2O should be removed to inhibit its negative effect on isomerization. This new strategy may lead to high commercial interest in HRPIB synthesis in pure green solvent and could potentially be extended to other initiation systems containing solid Lewis acids.
Introduction
Polyisobutylenes (PIBs), the most important industrial products of cationic polymerization, are characterized by their thermal stability, flexibility at ambient temperature, and impermeability to gases.1–5 Therefore, PIBs have been widely used in automobile tires, medical bottle plugs, additives of fuels, and lubricants.6–9 Highly reactive polyisobutylenes (HRPIBs),10,11 which are PIBs with high contents of exo-olefin end groups (≥60 mol%) and a specific molecular weight distribution (Mn = 500–5000), are highly reactive intermediates in the preparation of additives for lubricants and fuels. Thus, HRPIBs attract considerable attention from both industry and academia. The commercial synthesis of HRPIBs is dominated by the cationic polymerization of isobutylene (IB) using BF3 as a coinitiator and traces of alcohol or water as an initiator at temperatures slightly below 0 °C in n-hexane.10,12 However, BF3 is costly and strongly corrosive, resulting in serious economic and safety concerns.13In a series of publications, novel and economic catalyst systems were reported to produce HRPIBs with high exo-olefin content,14 such as FeCl3/iPrOH,8,15 FeCl3/iPr2O,7,16–18 and AlCl3/ether. Among the various coinitiators, solid AlCl3 has the advantages of low cost and high activity; however, it is always accompanied by the use of chlorinated solvents like CH2Cl2,9,19–26 at least in the preparation of initiator solution. Considering the environmental and health impacts of chlorinated solvents, it is highly desirable to completely replace them with green solvents such as n-hexane. However, the low stability of the carbenium ions in nonpolar solvents generally results in low conversion and poor controllability,27 making it difficult to effectively obtain HRPIBs in pure n-hexane.
The proper stabilization of carbenium ions and effective β-proton elimination, which are known as the keys to preparing HRPIBs,21,23,25,28,29 depend on the careful regulation of active centers and chain reactions. Our previous investigations have shown that introducing nucleophilic reagents into the initiator solution is an effective and direct method to adjust the active centers and chain reactions when using AlCl3 as a coinitiator.29–31 In detail, strongly basic Et2O can decrease the acidity of AlCl3 via complexation and inhibit its catalysis of H2O dissociation, which can stabilize carbenium ions and decrease the polymerization rate in CH2Cl2/n-hexane solvent mixtures. Meanwhile, the free Et2O significantly promotes proton elimination and isomerization, resulting in a high conversion rate and a low content of exo-olefin. Similarly, iPr2O can stabilize carbenium ions and promote proton elimination in CH2Cl2. However, iPr2O has little influence on isomerization due to steric hindrance, leading to a high content of exo-olefin.32 From these results, it can be concluded that Et2O may have a stronger influence on the rate of β-H abstraction in pure n-hexane, while iPr2O may better stabilize the carbocations.
Herein, we present a novel and simple method to prepare effective initiation solutions in n-hexane with a special focus on the synergistic effects of nucleophilic reagents (iPr2O and Et2O) on AlCl3-catalyzed IB polymerization. HRPIBs with high conversion rates and exo-olefin contents were successfully synthesized by comprehensively regulating carbenium ion stability, reactivity, and proton elimination. Moreover, attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy, and 27Al nuclear magnetic resonance (NMR) spectroscopy were combined with density functional theory (DFT) simulation to reveal how the controlled polymerization process was achieved in pure n-hexane.
Experimental methods
Materials
n-Hexane (C6H6, 97.5+%, anhydrous), isopropyl ether (iPr2O, 99.0+%), and aluminum chloride (AlCl3, 99+%, anhydrous) were purchased from J&K Scientific (China). Diethyl ether (Et2O, 99.5+%) and ethanol (analytical reagent) were obtained from Sinopharm Chemical Reagent Co. Ltd (China). IB (99.9+%, anhydrous) was obtained from Dalian Special Gases Co., LTD (China) and used directly as received. n-Hexane was dried over Solvent Purification Assembly (VAC, USA), and the content of water was determined using a coulometric Karl Fischer moisture meter (Mettler Toledo, Switzerland). iPr2O and Et2O were distilled to remove stabilizer and then dried over Molecular Sieves 5A overnight. iPr2O, Et2O, AlCl3, and n-hexane were preserved in a glovebox (Mikrouna, China). The content of AlCl3 in the initiator solution was measured by ultraviolet-visible spectrophotometry (UV-2450, Shimadzu).Polymerization of IB
The polymerization of IB was performed in a microflow system composed of three T-shaped micromixers (M1 for the mixing of IB and diluent in n-hexane; M2 for the mixing of IB solution and initiator solution; and M3 for the injection of terminator agent in ethanol), two precooling (or preheating) coiled stainless tubes (C1 and C2, inner diameter = 900 μm), and a microtube reactor (R, inner diameter = 900 μm), as shown in Fig. 1. The polymerization of IB proceeded in R1, and the reaction time could be adjusted by the flow rate and the length of R1. Four syringe pumps were used to deliver IB, n-hexane, initiator solution, and terminator at flow rates of 2, 6, 8, and 2 mL min−1, respectively. IB was transferred as a liquid from the bottom of the IB cylinder into the syringe and then mixed with n-hexane in the tube as a liquid under a pressure of 3 bar.29,31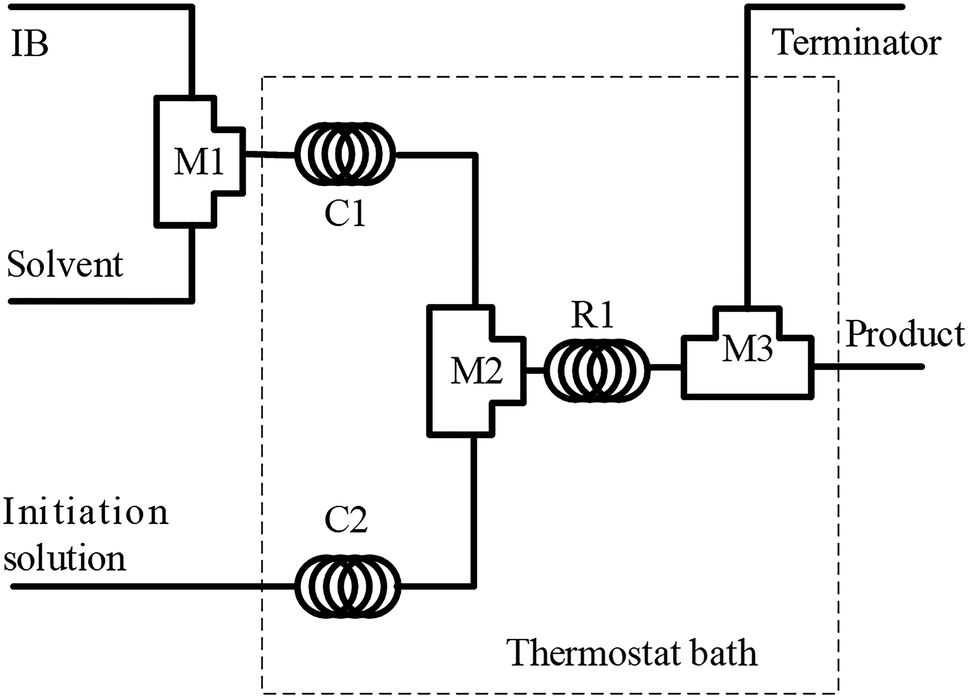 | ||
| Fig. 1 Schematic diagram of the flow synthesis setup. M1, M2, and M3 are T-shaped micromixers; C1 and C2 are curved tubes for achieving the pre-set temperature; and R1 is a microtube reactor. | ||
Preparation of initiation solution
The initiation solution was prepared just before polymerization in a glove box under an argon atmosphere. Seven preparation methods with single or double ethers were studied in this work. (a) Method 1: dry n-hexane was added to AlCl3 powder, and then slight excess Et2O was added to form the initial solution. (b) Method 2: dry n-hexane was added to AlCl3 powder, and then slight excess iPr2O was added. Since AlCl3 powder could not be completely dissolved in this case, the upper layer was withdrawn as an initiation solution, denoted as solution A. (c) Method 3: dry n-hexane was added to AlCl3 powder, and an appropriate amount of Et2O was then added. After several minutes, dry iPr2O was added. The solution was used as an initiation solution. (d) Method 4: dry n-hexane was added to AlCl3 powder, and then an appropriate amount of iPr2O was added. After stirring for 2 h, dry Et2O was added. The solution was used as an initiation solution. (e) Method 5: dry Et2O was added into solution A to form the initial solution. (f) Methods 6 and 7. Dry Et2O (method 6), n-hexane/Et2O mixture [5/2 (v/v), method 7], or separate n-hexane and Et2O (method 7–1) was added to AlCl3 powder and then vacuumed to form a colorless crystal B. Solution A was added to crystal B under stirring to form the initiation solution (Fig. 2).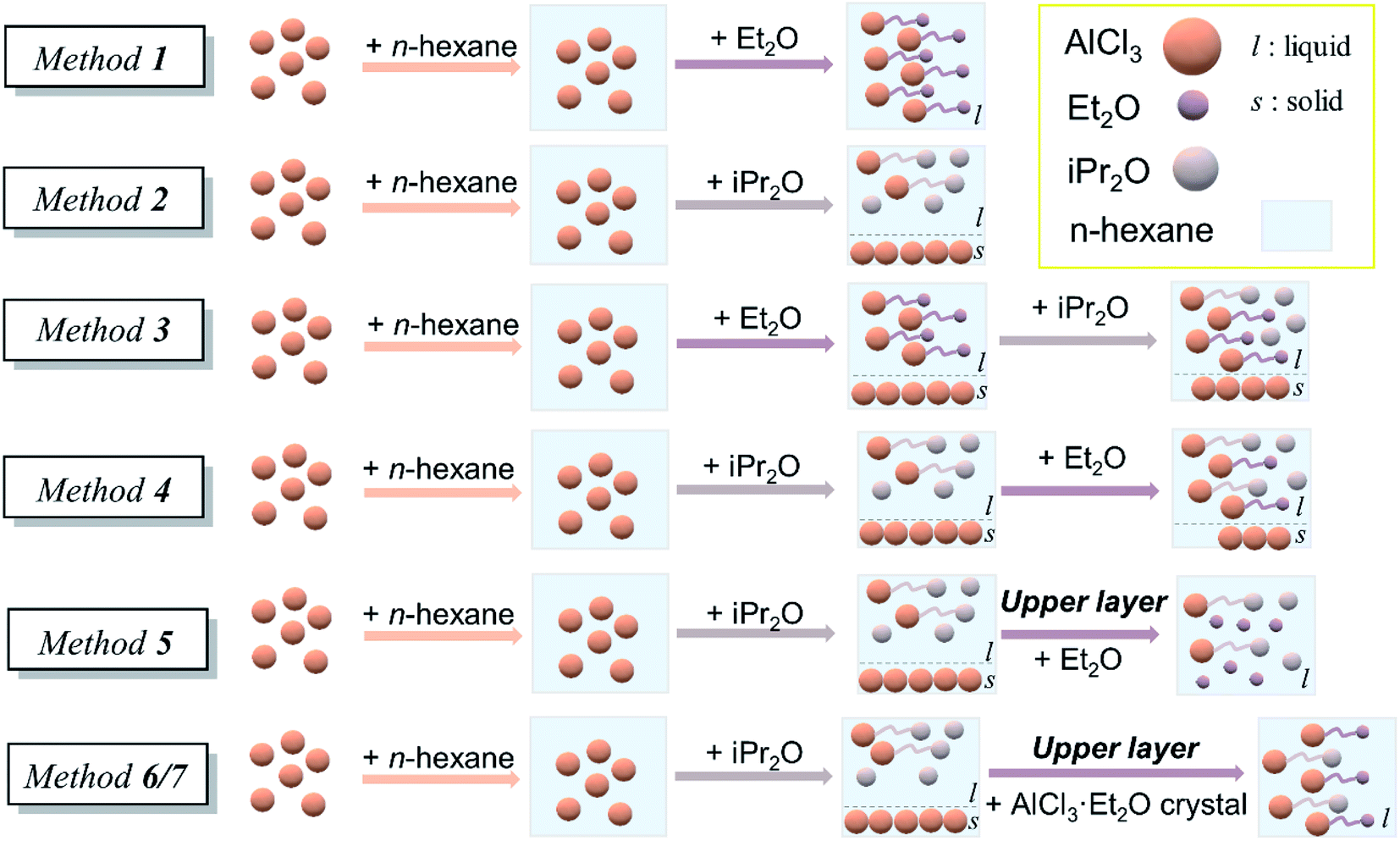 | ||
| Fig. 2 Schematic of various preparation methods of initiation solution. | ||
Characterization
1H NMR spectra were recorded on a JNM-ECA 600 MHz spectrometer with CDCl3 as the solvent. The PIB end-group content was calculated from the 1H NMR spectra. Fig. 3 shows a typical 1H NMR spectrum; the main resonance signals are located at δ = 1.1 ppm (z), 1.41 ppm (y), 0.99 ppm (x), 4.85 ppm (a1), 4.64 ppm (a2), 5.17 ppm (c1), 5.37 ppm (c2), 5.15 ppm (d), and 2.83 ppm (e). The two characteristic protons of the exo-olefin end group (structure A, protons a1 and a2) appear as two well-resolved peaks at 4.85 and 4.64 ppm, respectively. Small amounts of the E and Z configurations of the tri-substituted olefin end group (structure C, protons c1 and c2) appear at 5.37 and 5.17 ppm, respectively. The one characteristic proton of the endo-olefin end group (structure D, proton d) appears at 5.15 ppm. The signal corresponding to the tetra-substituted olefin end group (structure E, proton e) appears as a broad multiplet at 2.85 ppm. The methylene, methyl, and end methyl protons of the PIB chains (structure A, protons y, z, and x, respectively) typically appear at 1.41, 1.11, and 0.99 ppm, respectively.
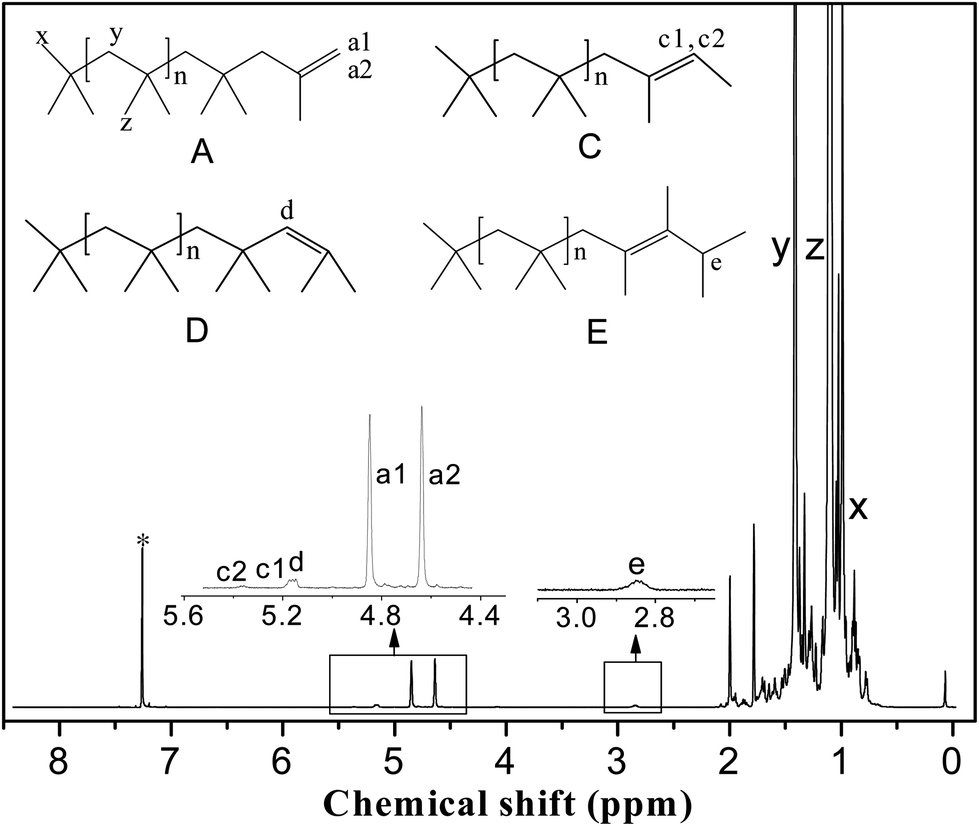 | ||
| Fig. 3 Typical 1H NMR spectrum of PIB. The asterisk denotes the CHCl3 resonance. | ||
Results and discussion
Synergistic effects of Et2O and iPr2O on AlCl3-initiated IB polymerization in pure n-hexane
We first determined the polymerization characteristics using only Et2O or iPr2O. The polymerization results are shown in Table 1; method 1 and method 2 correspond to the introduction of only Et2O and only iPr2O, respectively. The ether/AlCl3 molar ratio was controlled over unity as a prerequisite to ensuring a high content of exo-olefin according to previous reports on HRPIB synthesis.8,29,38 As shown in Table 1, the exo-olefinic end-group content (∼24%) and yield (∼18%) obtained using method 1 are relatively high. In contrast, with method 2, the IB conversion and exo-olefinic end-group content were only 3–5% and 15%, respectively. However, in contrast to the initiation solution prepared in CH2Cl2 (ε = 9.08),29 the initiation solution prepared in pure n-hexane (ε = 1.89) afforded PIBs mainly containing endo-, tri-, and tetra-substituted double bonds and having polydispersity indices (PDIs) around 2.0 or higher. Meanwhile, the IB conversions were low (<20%) and seemed to be independent of residence time in the range of 60–600 s. We supposed that the AlCl3·Et2O or AlCl3·iPr2O complex could only generate ionic species with low reactivity or poor stability in pure n-hexane, which may be attributed to the weak ionization in the nonpolar solvent.39–41| Entry | Method | t (s) | [AlCl3]I (mM) | Conv. (%) | Mn | Đ | [PIB]/mM | Exo (%) | Tri + endo (%) | Tetra (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Ftotal = 16 mL min−1; [IB] = 1.3 M; T = 0 °C; [H2O] = 0 (control) mM, for method 1: [Et2O] = 15 mM, for method 2: [iPr2O] = 15 mM. Conv.: gravimetric conversion.b Not determined. | ||||||||||
| 1 | 1 | 60 | 10.51 | 12 | 5189 | 2.76 | 0.82 | NDb | ND | ND |
| 2 | 600 | 10.51 | 18 | 4664 | 4.15 | 1.41 | 24 | 48 | 28 | |
| 3 | 2 | 60 | 2.20 | 3 | 3828 | 2.05 | 0.25 | ND | ND | ND |
| 4 | 600 | 2.20 | 5 | 4074 | 2.75 | 0.46 | 15 | 85 | 0 | |
Considering that Et2O or iPr2O alone did not work for HRPIB preparation in pure n-hexane, we attempted to use double ethers to simultaneously regulate the reactivity and stability of ionic species in initiation solution. iPr2O was expected to stabilize carbenium ions, while Et2O was expected to promote proton elimination as the key for end group control.
The results of polymerizations utilizing various preparation strategies for initiation solution are summarized in Table 2. For method 3, the IB conversion was similar to that of method 1 (only Et2O), while the exo-olefin content was reduced to 13%. The subsequent addition of iPr2O had little influence on the stability and reactivity of growing species mainly coinitiated by AlCl3·Et2O and weakened the β-H elimination effect of Et2O to some extent. In contrast, when reversing the addition sequence of the two ethers (method 4), the obtained PIBs had a much higher exo-olefin content (up to 35%). This may be attributed to the effect of Et2O on the proton elimination of growing species mainly coinitiated by AlCl3·iPr2O. In addition, since approximately half of the added AlCl3 remained undissolved in method 4, all existing forms of Et2O in the upper layer were AlCl3·Et2O complexes, which might also play a role in inducing chain initiation and β-H elimination. To confirm this assumption, we used the initiation solution prepared by method 5, in which undissolved AlCl3 at the bottom was removed followed by the addition of Et2O. As seen in entry 10, the content of exo-olefin end groups decreased slightly to 29%, and the conversion decreased seriously. This indicates that free Et2O inhibits the reactivity of the growing species coinitiated by AlCl3·iPr2O, and AlCl3·Et2O may be critical to facilitate conversion and end group control. Thus, for the growing species coinitiated by AlCl3·iPr2O, we speculated that weak ionization in conjunction with slow chain transfer could be overcome by introducing AlCl3·Et2O only. To this end, we removed the free Et2O during AlCl3·Et2O preparation via vacuum to obtain AlCl3·Et2O crystals. Meanwhile, n-hexane was added to the AlCl3 powders before adding Et2O to regulate the interaction between AlCl3 and Et2O and facilitate the removal of free Et2O (methods 6 and 7).
| Entry | Method | t (s) | [AlCl3]I (mM) | Conv. (%) | Mn | Đ | [PIB] (mM) | Exo (%) | Tri + endo (%) | Tetra (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Ftotal = 16 mL min−1; [IB] = 1.3 M; T = 0 °C; [H2O] = 0 (control) mM. [Et2O]0 = 7.86 mM, [iPr2O]0 = 7.14 mM. Conv.: gravimetric conversion.b Not determined. | ||||||||||
| 5 | 3 | 60 | 11.09 | 3 | ND | ND | NDb | ND | ND | ND |
| 6 | 600 | 11.09 | 17 | 4183 | 2.94 | 1.44 | 13 | 87 | 0 | |
| 7 | 4 | 60 | 8.92 | 7 | 3382 | 3.27 | 0.77 | 32 | 52 | 16 |
| 8 | 600 | 8.92 | 15 | 3576 | 2.20 | 1.54 | 35 | 57 | 8 | |
| 9 | 5 | 60 | 3.30 | 3 | 4126 | 2.01 | 0.25 | ND | ND | ND |
| 10 | 600 | 3.30 | 4 | ND | ND | ND | 29 | 71 | 0 | |
As seen in Table 3, the cationic polymerization of IB with methods 6 and 7 in pure n-hexane proceeded smoothly; relatively high monomer conversions (21–89%) were achieved within 10 min, affording HRPIBs with comparable contents of exo-olefin terminal groups in the range of 60–75%. The number-average molecular weight (Mn) increased slightly with polymerization time, and the polydispersity index was less than 2.0 in most cases. These results indicate that isomerization via carbenium ion rearrangement could be suppressed to some extent. From entries 15 and 16, it is apparent that adding n-hexane/Et2O mixture is important to achieve higher conversion. The addition of n-hexane in method 7 may weaken the interaction between AlCl3 and Et2O, resulting in higher conversion (∼89%). This confirmed that the appropriate activity and stability of active species could be obtained by combining double ethers and precise control of the ether/AlCl3 ratio. As a result, HRPIBs could be obtained in under a wide range of IB concentrations (Table 4). To the best of our knowledge, this is the first example of the effective preparation of HRPIBs with AlCl3 as a coinitiator in pure n-hexane.
| Entry | Method | t (s) | [AlCl3]I (mM) | Conv. (%) | Mn | Đ | [PIB] (mM) | Exo (%) | Tri + endo (%) | Tetra (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Ftotal = 16 mL min−1; [IB] = 1.3 M; T = 0 °C; [H2O] = 0 (control) mM. [Et2O]0 = 7.86 mM, [iPr2O]0 = 7.14 mM. [AlCl3]A = 1.62 mM, Conv.: gravimetric conversion.b Not determined. | ||||||||||
| 11 | 6 | 60 | 12.98 | 5 | 1271 | 1.61 | 1.39 | NDb | ND | ND |
| 12 | 600 | 12.98 | 21 | 1464 | 2.01 | 5.15 | 75 | 19 | 6 | |
| 13 | 7 | 60 | 13.21 | 36 | 1629 | 1.91 | 8.03 | 60 | 30 | 10 |
| 14 | 600 | 13.21 | 89 | 1647 | 2.23 | 19.66 | 52 | 29 | 20 | |
| 15 | 7–1 | 60 | 13.31 | 6 | 4117 | 1.23 | 0.554 | ND | ND | ND |
| 16 | 600 | 13.31 | 33 | 4302 | 1.23 | 2.755 | 68 | 20 | 12 | |
| Entry | [IB] (M) | t (s) | [AlCl3]I (mM) | [AlCl3]A (mM) | Conv. (%) | Mn | Đ | Exo (%) | Tri (%) | Tetra (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Ftotal = 16 mL min−1; T = 0 °C; [H2O]0 = 0.45 mM; [iPr2O]0 = 7.14 mM. Conv.: gravimetric conversion. | ||||||||||
| 17 | 0.33 | 60 | 10.98 | 3.80 | 29 | 1880 | 2.01 | 73 | 13 | 14 |
| 18 | 0.33 | 600 | 10.98 | 3.80 | 65 | 1460 | 2.08 | 72 | 15 | 13 |
| 19 | 0.65 | 60 | 11.28 | 3.75 | 23 | 4440 | 1.91 | 63 | 22 | 15 |
| 20 | 0.65 | 600 | 11.28 | 3.75 | 56 | 3730 | 2.06 | 58 | 26 | 16 |
| 21 | 1.30 | 60 | 11.06 | 3.79 | 15 | 6020 | 2.20 | 63 | 20 | 17 |
| 22 | 1.30 | 600 | 11.06 | 3.79 | 30 | 6500 | 2.25 | 61 | 21 | 18 |
Proposed mechanism of IB polymerization under dual ethers in pure n-hexane
In this section, ATR-FTIR and 27Al NMR spectroscopies were used to gain further insight into the role of the dual ethers (Et2O and iPr2O). The ATR-FTIR spectra are presented in Fig. 4. The characteristic peak of the C–O bond in free Et2O is located at 1126 cm−1, while the characteristic peaks of free iPr2O are situated at 1113, 1126, and 1171 cm−1. For AlCl3·Et2O solution using method 1, the peak at 1126 cm−1 was not detected, and two new and broad peaks appeared at 884 and 1005 cm−1. For AlCl3·iPr2O solution using method 2, free iPr2O molecules still existed. This may be because iPr2O is present in excess with respect to AlCl3 due to the poor solubility of AlCl3 in iPr2O with relatively large steric hindrance.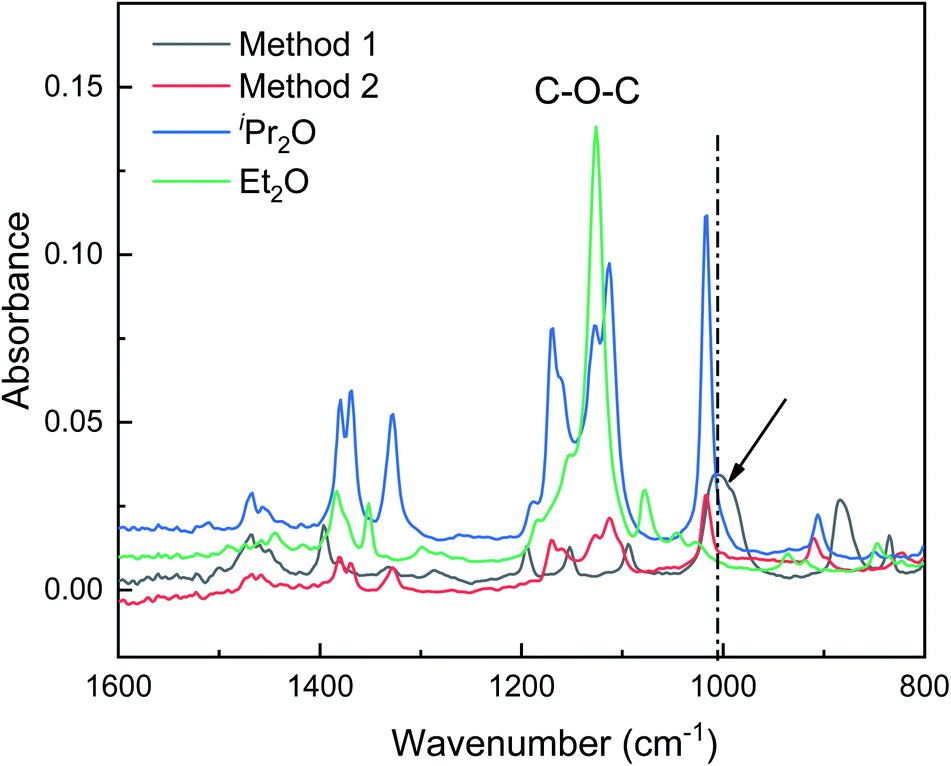 | ||
| Fig. 4 ATR-FTIR spectra showing the interactions between AlCl3 and ether in initiation solutions prepared with methods 1 and 2. The arrow and dash line indicate the peak corresponding to Al–O bond. | ||
As shown in Fig. 5, when using dual ethers, the characteristic peaks for ether C–O bond stretching were redshifted, and a peak emerged at 999 cm−1 (methods 3, 4, 6, and 7), suggesting that the interaction between AlCl3 and C–O was enhanced. More interestingly, the intensities of the peaks around 999 cm−1, which correspond to the Al–O bond, were significantly enhanced in the initiation solutions prepared with methods 6 and 7. This enhancement was positively correlated with the IB conversion. These results suggest that the coinitiators formed in methods 6 and 7 were beneficial for ionizing H2O and stabilizing the carbocations. However, compared to the different polymerization results obtained using methods 3, 4, 6, and 7, their ATR-FTIR spectra were difficult to distinguish; thus, it is necessary to consider other methods to characterize the interaction between AlCl3 and ether.
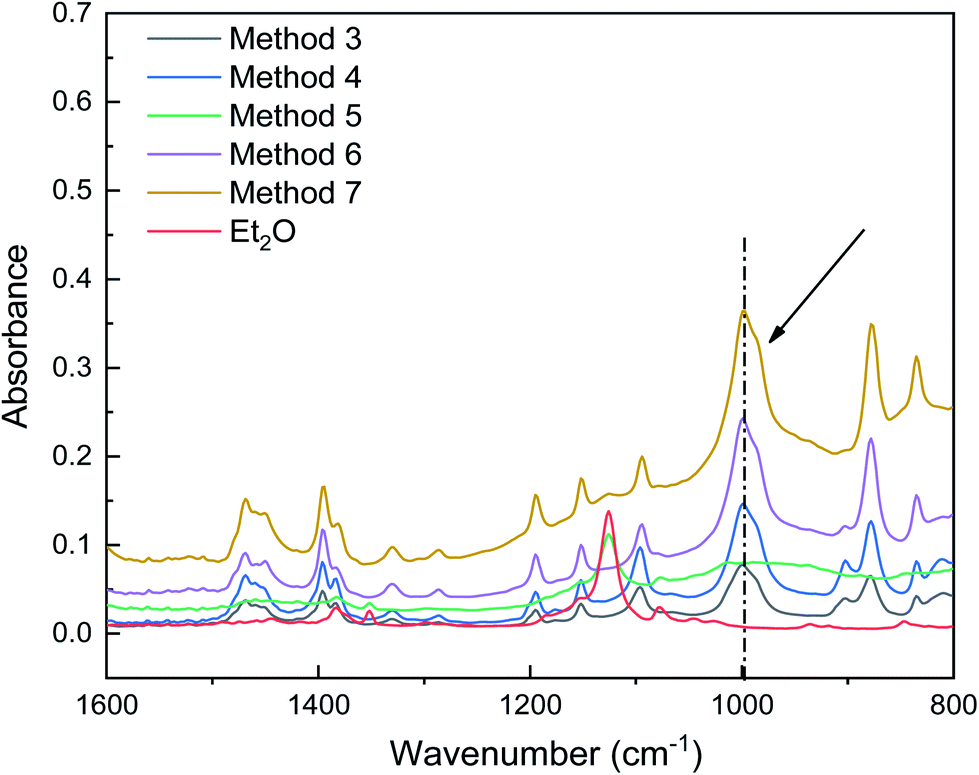 | ||
| Fig. 5 ATR-FTIR spectra showing the interaction between AlCl3 and ether in initiation solutions prepared using methods 3–7. | ||
To clarify the nature of different AlCl3·ether complexes, ab initio calculations were conducted to determine the AlCl3·ether binding energies. As shown in Fig. 6, the binding energies between AlCl3 and iPr2O were 33.07 kcal mol−1 in CH2Cl2 and 30.49 kcal mol−1 in n-hexane; the binding energies between AlCl3 and Et2O were 30.95 kcal mol−1 in CH2Cl2 and 28.14 kcal mol−1 in n-hexane. On the other hand, for the same complexes, the binding energies in nonpolar solvent (n-hexane) were relatively low. These results indicate that the stability of AlCl3·Et2O was less than that of AlCl3·iPr2O, and the stability further decreased with decreasing solvent polarity.
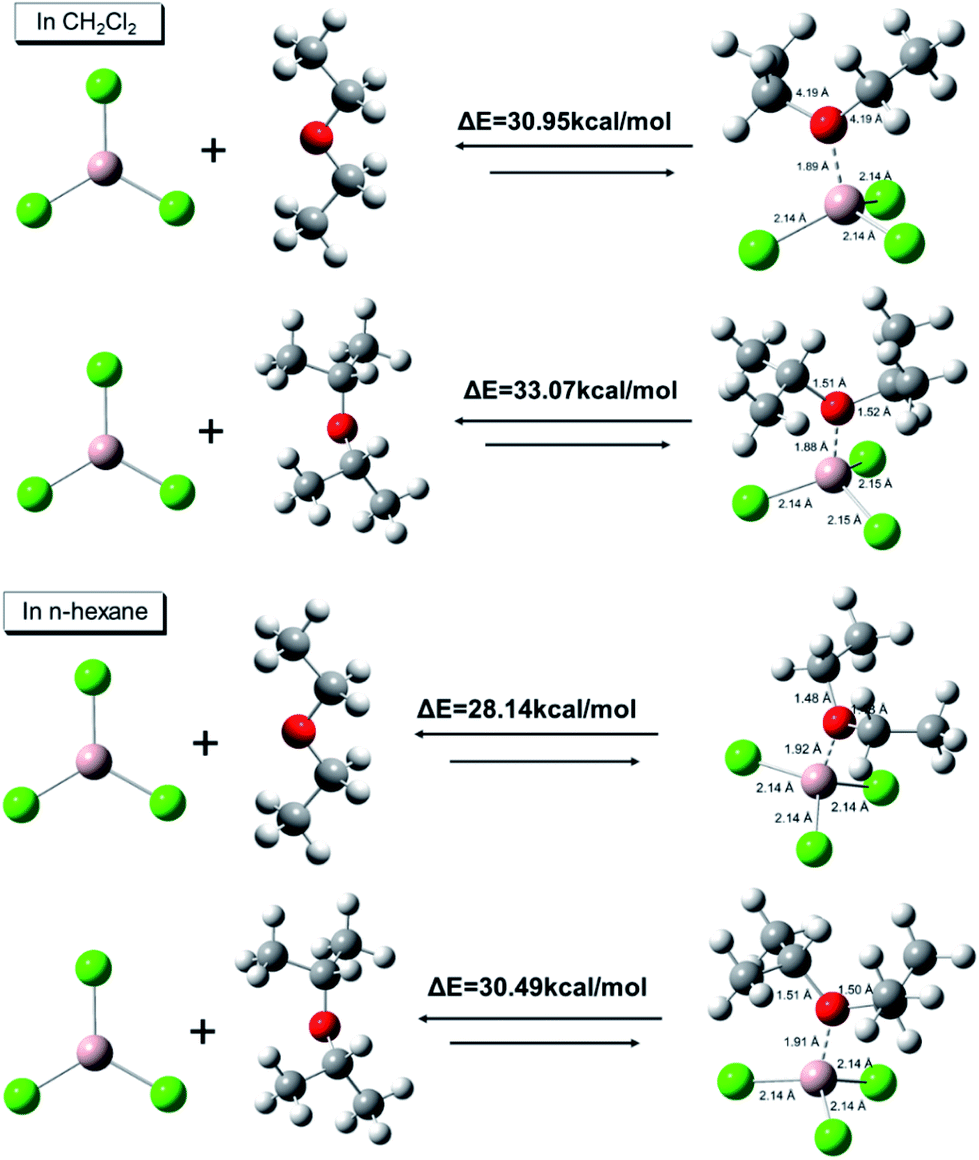 | ||
| Fig. 6 DFT binding energies and optimized structures of AlCl3·ether calculated using Gaussian 09W [B3LYP/6-311G (++, d, p), solvation model: SMD]. Distances are in Å. | ||
Herein, we further studied the interaction between AlCl3 and ether in n-hexane by 27Al NMR spectroscopy (Fig. 7). A single resonance was detected in all cases, indicating that the complexes of AlCl3 and ether or the microenvironment around AlCl3 were uniform. In addition, according to the position of this characteristic peak, the stabilities of the different AlCl3·ether complexes decreased in the order of method 2, method 4, method 3, method 6, method 7, method 5, and method 1, consistent with the DFT simulations. Methods 6 and 7, which permitted faster polymerization and effective β-H abstraction, resulted in intermediate complex stability.
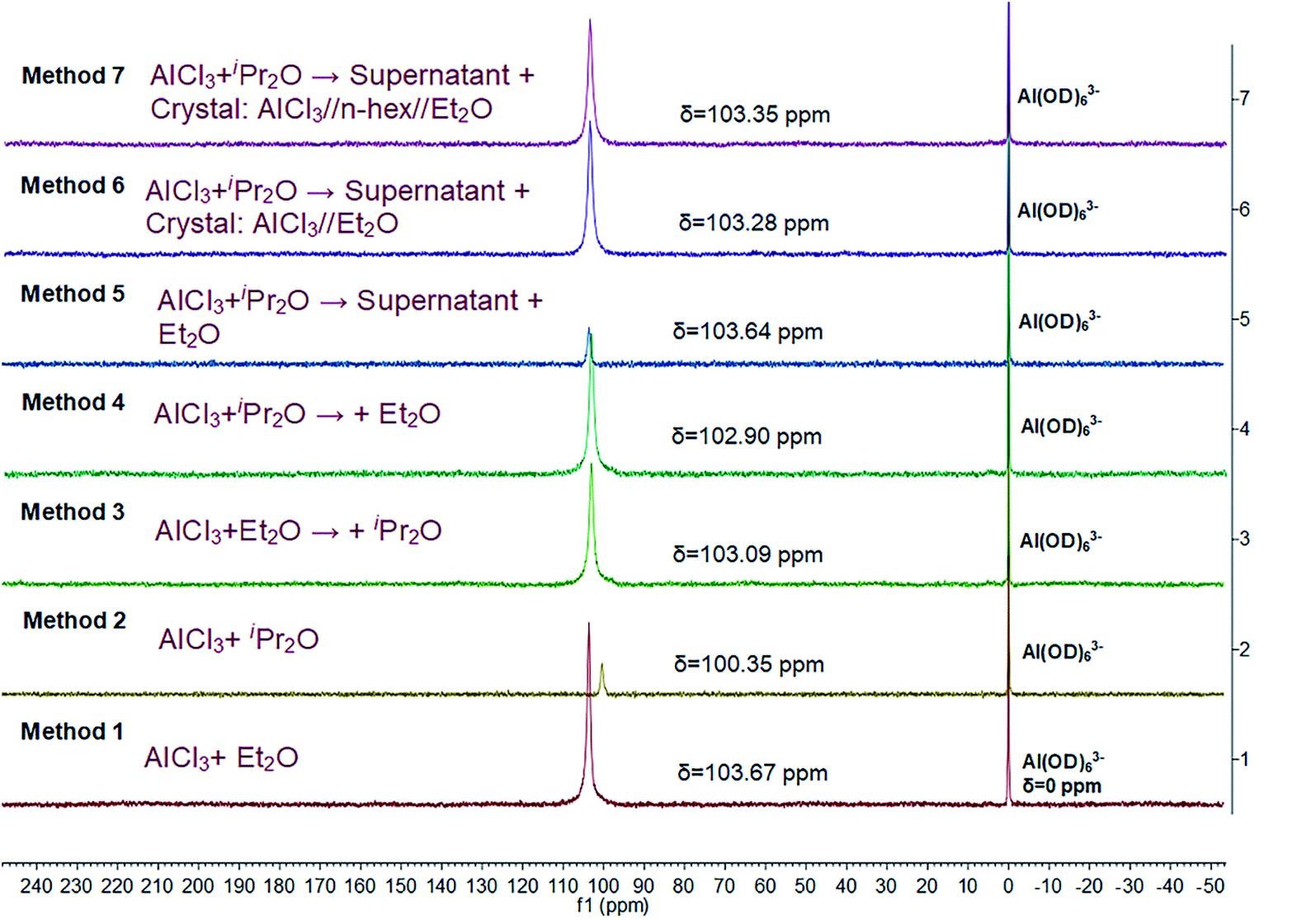 | ||
| Fig. 7 27Al NMR spectra of initiation solutions prepared using different methods. | ||
In detail, the 27Al NMR signal of AlCl3·iPr2O (method 2) shifted even further toward lower frequency (higher field) compared to that of AlCl3·Et2O (method 1). This means that iPr2O has a stronger electron-donating ability and a greater effect on the microenvironment around the Al atom. In other words, the interaction between AlCl3 and iPr2O is relatively strong in n-hexane, resulting in low reactivity for the ionization of H2O and inducing transfer side reactions (isomerizations). The interaction between AlCl3 and Et2O is comparatively weak; thus, the AlCl3·Et2O in H2O/AlCl3·Et2O initiation system (method 1) has high reactivity for H2O ionization. However, free Et2O may promote isomerization and increase the amount of endo-double bond terminal groups in the products. For method 3, weaker AlCl3·Et2O was formed at the first stage, and the subsequently added iPr2O may distribute around AlCl3·Et2O; however, its effect on Al atom may be weak due to steric hindrance. Thus, the microenvironment of the Al atoms was dominated by Et2O. In contrast, in method 4, stronger AlCl3·iPr2O was formed first, and some added Et2O bound with AlCl3 powder; thus, the microenvironment of Al atom in solution reflected the effect of iPr2O to some extent. For method 5, all the added free Et2O was distributed around AlCl3·iPr2O, and the interaction between them caused the 27Al shift to be closer to that of AlCl3·Et2O. Under the precise control of the AlCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O ratio (methods 6 and 7), higher activity for AlCl3·iPr2O was endowed by introducing suitable AlCl3·Et2O complex to regulate the micro surroundings of AlCl3·iPr2O properly. In general, the interaction between AlCl3 and Et2O inhibited the isomerization effect of free Et2O and regulated the effect of Et2O on AlCl3·iPr2O toward compromised stability and reactivity.
:Et2O ratio (methods 6 and 7), higher activity for AlCl3·iPr2O was endowed by introducing suitable AlCl3·Et2O complex to regulate the micro surroundings of AlCl3·iPr2O properly. In general, the interaction between AlCl3 and Et2O inhibited the isomerization effect of free Et2O and regulated the effect of Et2O on AlCl3·iPr2O toward compromised stability and reactivity.
Based on the above observations, we proposed that AlCl3·Et2O might play two roles: participate in the initiation step, which is attributed to the weak interactions between AlCl3 and Et2O; and accelerate effective β-H elimination since Et2O was modified by AlCl3. The complexation between AlCl3 and Et2O may also inhibit the effect of Et2O on internal proton transfer and abstraction. Meanwhile, AlCl3·iPr2O at a relatively low concentration rarely produced active centers. However, it could modify the stability of cation centers via solvation.
Scheme 1 shows the proposed mechanism of the cationic polymerization of IB using the AlCl3/Et2O/iPr2O system in pure n-hexane. In detail, modified Et2O with the assistance of iPr2O provided the proper microenvironment around AlCl3. AlCl3 then catalyzed the ionization of H2O and generated considerable stable ionic species. Subsequently, the chain reactions were controlled by the selective β-H abstraction of modified Et2O, resulting in higher conversion and a greater content of exo-olefin. In addition, the use of n-hexane weakened the interaction between AlCl3 and Et2O, facilitating the removal of free Et2O and inhibiting undesired proton transfer.
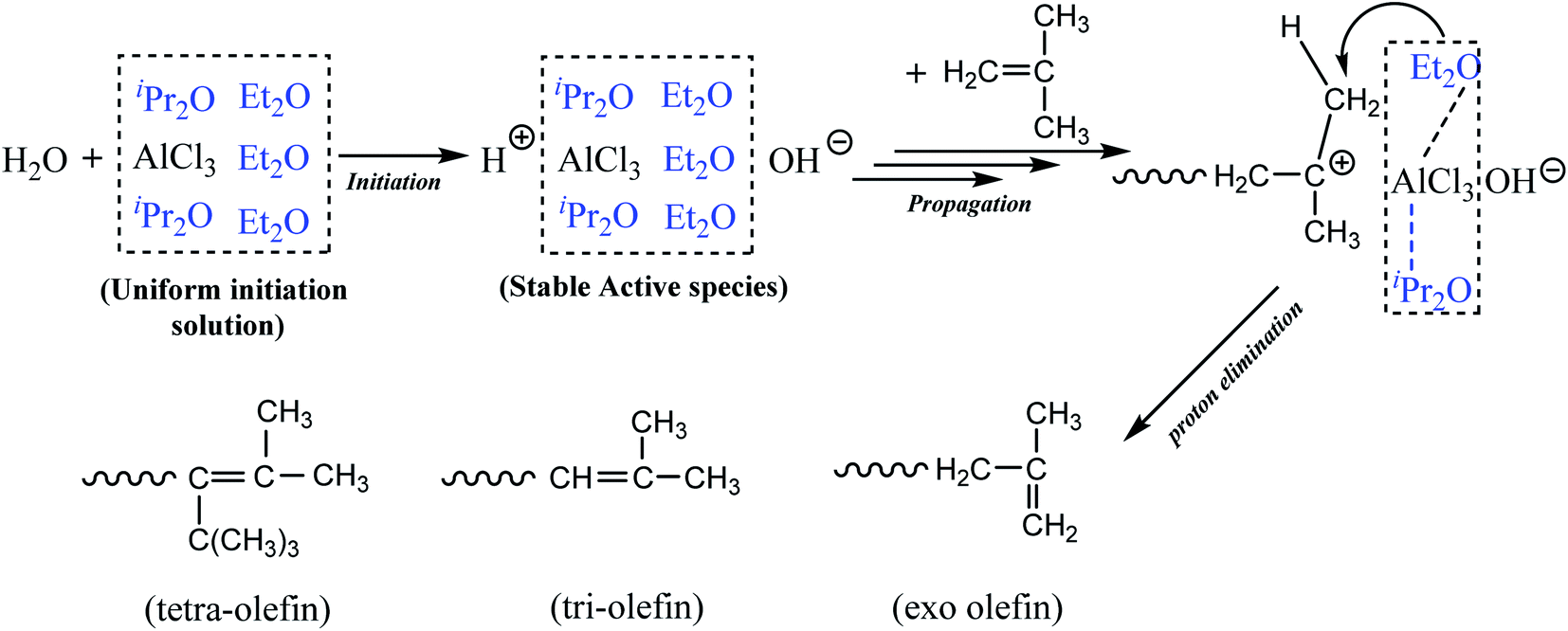 | ||
| Scheme 1 Proposed mechanism of the cationic polymerization of IB using dual ethers. | ||
Conclusion
In summary, an efficient strategy combining the precise control of the ether/AlCl3 ratio and the use of two nucleophilic reagents (iPr2O and Et2O) was developed to synthesize HRPIBs in pure n-hexane, resulting in 89% conversion with 60–75% exo-double bond content within 10 min. Among the AlCl3·ether complexes prepared with different preparation methods, good performance in terms of both IB conversion and exo-olefin content was achieved by preparing AlCl3·iPr2O solution and AlCl3·Et2O crystals separately. The various functions of iPr2O and Et2O in the initiator solution were comprehensively revealed based on the polymerization results, ATR-FTIR and 27Al NMR spectra, and DFT simulations. The results confirmed that: (1) AlCl3·iPr2O complexes were the key component that stabilized carbenium ions; (2) AlCl3·Et2O complexes were the key component that promoted proton elimination; and (3) free Et2O had a negative effect on isomerization and should be removed to the extent possible. This new strategy may provide an alternative to commercial BF3-based initiating systems and lead to commercial interest in HRPIB synthesis in pure green solvent. In addition, the new method can potentially be extended to other initiation systems containing solid Lewis acids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the support of the National Natural Science Foundation of China (Grant No. 21176136, 21422603, and 21978152).Notes and references
- K. Kunal, M. Paluch, C. M. Roland, J. E. Puskas, Y. Chen and A. P. Sokolov, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1390–1399 CrossRef CAS.
- G. K. J. E. Puskas, in Encyclopedia of Polymer Science and Technology, Wiley-InterScience, New York, 2003, vol. 5, pp. 382–418 Search PubMed.
- J. P. Kennedy and B. Ivan, Theory and Practice, Hanser, Munich, Germany, 1991, pp. 173–177 Search PubMed.
- B. Giese, Bunsen-Ges. Phys. Chem., Ber., 1983, 87, 289 CrossRef.
- S. Rach and F. Kühn, Sustainability, 2009, 1, 35–42 CrossRef CAS.
- I. V. Vasilenko, D. I. Shiman and S. V. Kostjuk, Polym. Chem., 2014, 5, 3855–3866 RSC.
- R. Kumar, P. Dimitrov, K. J. Bartelson, J. Emert and R. Faust, Macromolecules, 2012, 45, 8598–8603 CrossRef CAS.
- Q. Liu, Y. Wu, P. Yan, Y. Zhang and R. Xu, Macromolecules, 2011, 44, 1866–1875 CrossRef CAS.
- P. Dimitrov, J. Emert and R. Faust, Macromolecules, 2012, 45, 3318–3325 CrossRef CAS.
- H. Mach and P. Rath, Lubr. Sci., 1999, 11, 175–185 CrossRef CAS.
- J. J. Harrison, C. M. Mijares, M. T. Cheng and J. Hudson, Macromolecules, 2002, 35, 2494–2500 CrossRef CAS.
- A. G. Evans and G. W. Meadows, J. Polym. Sci., 1949, 4, 359–376 CrossRef CAS.
- L.-b. Zhang, Y.-x. Wu, P. Zhou, G.-y. Wu, W.-t. Yang and D.-s. Yu, Chin. J. Polym. Sci., 2011, 29, 360–367 CrossRef CAS.
- S. V. Kostjuk, RSC Adv., 2015, 5, 13125–13144 RSC.
- A.-R. Guo, X.-J. Yang, P.-F. Yan and Y.-X. Wu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4200–4212 CrossRef CAS.
- K. J. Bartelson, P. De, R. Kumar, J. Emert and R. Faust, Polymer, 2013, 54, 4858–4863 CrossRef CAS.
- R. Kumar, J. Emert and R. Faust, Polym. Bull., 2014, 72, 49–60 CrossRef.
- R. Kumar, P. De, B. Zheng, K.-W. Huang, J. Emert and R. Faust, Polym. Chem., 2015, 6, 322–329 RSC.
- Y. Li, Y. Wu, X. Xu, L. Liang and G. Wu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3053–3061 CrossRef CAS.
- Y. Li, Y.-x. Wu, L.-h. Liang, Y. Li and G.-y. Wu, Chin. J. Polym. Sci., 2009, 28, 55–62 CrossRef CAS.
- Q. Liu, Y.-X. Wu, Y. Zhang, P.-F. Yan and R.-W. Xu, Polymer, 2010, 51, 5960–5969 CrossRef CAS.
- I. V. Vasilenko, A. N. Frolov and S. V. Kostjuk, Macromolecules, 2010, 43, 5503–5507 CrossRef CAS.
- I. V. Vasilenko, D. I. Shiman and S. V. Kostjuk, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 750–758 CrossRef CAS.
- L. B. Zhang, Y. X. Wu, P. Zhou and R. W. Xu, Polym. Adv. Technol., 2012, 23, 522–528 CrossRef CAS.
- D. I. Shiman, I. V. Vasilenko and S. V. Kostjuk, Polymer, 2013, 54, 2235–2242 CrossRef CAS.
- S. V. Kostjuk, I. V. Vasilenko, D. I. Shiman, A. N. Frolov and L. V. Gaponik, Macromol. Symp., 2015, 349, 94–103 CrossRef CAS.
- M. Vierle, Y. Zhang, E. Herdtweck, M. Bohnenpoll, O. Nuyken and F. E. Kuhn, Angew. Chem., Int. Ed. Engl., 2003, 42, 1307–1310 CrossRef CAS PubMed.
- D. I. Shiman, I. V. Vasilenko and S. V. Kostjuk, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2386–2393 CrossRef CAS.
- S. Zhu, Y. C. Lu, K. Wang and G. S. Luo, RSC Adv., 2016, 6, 9827–9834 RSC.
- S. Zhu, K. Wang and Y. Lu, ACS Omega, 2018, 3, 2033–2039 CrossRef CAS PubMed.
- S. Zhu, Y. Lu, K. Wang and G. Luo, RSC Adv., 2016, 6, 97983–97989 RSC.
- D. I. Shiman, I. V. Vasilenko and S. V. Kostjuk, Polymer, 2016, 99, 633–641 CrossRef CAS.
- J. W. Akitt, Prog. Nucl. Magn. Reson. Spectrosc., 1989, 21, 1–149 CrossRef.
- Y. A. Buslaev and S. P. Petrosyants, Polyhedron, 1984, 3, 265–270 CrossRef CAS.
- D. E. O'Reilly, J. Chem. Phys., 1960, 32, 1007–1012 CrossRef.
- H. Haraguch and S. Fujiwara, J. Phys. Chem., 1969, 73, 3467–3473 CrossRef.
- D. Chen, K. P. Taylor, Q. Hall and J. M. Kaplan, Genetics, 2016, 204, 1151–1159 CrossRef CAS PubMed.
- Y. Wu and G. Wu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2209–2214 CrossRef CAS.
- M. Givehchi, M. Tardi, A. Polton and P. Sigwalt, Macromolecules, 2000, 33, 710–716 CrossRef CAS.
- K. Matyjaszewski, Macromol. Symp., 1996, 107, 53–63 CrossRef CAS.
- I. Dimitrov and R. Faust, Macromolecules, 2004, 37, 9753–9760 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |