
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe development and validation of an HPLC-MS/MS method for the determination of eriocitrin in rat plasma and its application to a pharmacokinetic study
Luya Li†
 a,
Rui Feng†b,
Xue Fenga,
Yuting Chena,
Xin Liua,
Wenjing Suna and
Lantong Zhang*a
a,
Rui Feng†b,
Xue Fenga,
Yuting Chena,
Xin Liua,
Wenjing Suna and
Lantong Zhang*a
aDepartment of Pharmaceutical Analysis, School of Pharmacy, Hebei Medical University, Shijiazhuang 050017, P.R. China. E-mail: zhanglantong@263.net; Fax: +86-311-86266419; Tel: +86-311-86266419
bDepartment of Pharmacy, The Fourth Hospital of Hebei Medical University, Shijiazhuang 050011, P.R. China
First published on 12th March 2020
Abstract
Eriocitrin is one of the major active constituents of lemon fruit, and it possesses strong antioxidant, lipid-lowering, anticancer and anti-inflammatory activities and has long been used in food, beverages and wine. In this study, for the first time, a rapid, selective, and sensitive liquid chromatography-tandem mass spectrometry method (LC/MS/MS) with protein precipitation was developed and validated for the analysis of eriocitrin in rat plasma. Chromatographic separation was achieved using a mobile phase, comprising 0.1% formic acid aqueous solution and acetonitrile eluted at a flow rate of 0.8 mL min−1. In multiple reaction monitoring (MRM) modes, eriocitrin and internal standard (IS) were quantified using precursor-to-product ion transitions of m/z 595.4 → 287.1 and m/z 252.0 → 155.9, respectively. The intra- and inter-day precision (RSD) were below 6.79% in plasma, while accuracy (RE) was within ±7.67%. The matrix effect, recovery and stability were also demonstrated to be within acceptable limits. This method was successfully employed in the pharmacokinetic study on rats after the oral administration of eriocitrin. The pharmacokinetic parameters show that the maximum plasma concentration (Cmax) of eriocitrin was 299.833 ± 16.743 μg L−1, while the corresponding time to reach Cmax(Tmax) was 0.094 ± 0.019 h, and the half-time (T1/2) was 1.752 ± 0.323 h. The present results would be valuable for further research and development of eriocitrin.
1. Introduction
Epidemiological and clinical studies show that diets with a high antioxidant capacity are closely related to significant decreases in the plasma C-reactive protein, ischemic stroke, or the overall risk of cardiovascular disease or colorectal cancer.1 Eriocitrin, (eriodictyol 7-O-beta-rutinoside), as the main ingredient in citrus fruits (lemon, citrus, and grapefruit),2–5 vegetables, and processed products (drinks, wine),6 has aroused extensive interests of the scientific community due to its strong antioxidant activity,7–9 which is stronger than that of hesperidin and naringin. Modern pharmacological studies show that eriocitrin has strong lipid-lowering, anticancer and anti-inflammatory activities. It also plays an important role in effectively preventing and reducing oxidative stress,10,11 hyperlipidaemia,12,13 cardiovascular and cerebrovascular diseases, cancer14 as well as inflammation.15,16 A recent report also confirmed that it could be a potential hit candidate for antiobesity treatment and prevention due to its inhibition profile against the hCAVA isoform.17 As eriocitrin has potential applications and value in clinics, research on it is very valuable and meaningful.Pharmacokinetics is the study of the absorption, distribution, metabolism and excretion of drugs in vivo. It has vital implications for pharmacology and clinical medicine, elucidating the mechanism of drug action. Pharmacokinetic studies have played an important role in the evaluation of the concentration-effect relationship of drugs, the design of new drug delivery systems, and the optimization of dosing regimens.18 However, up to now, no pharmacokinetic studies on eriocitrin have been reported; therefore, the purpose of this study was to determine the pharmacokinetic properties of eriocitrin in rats.
As we know, LC-MS/MS is widely applied in the pharmaceutical analysis due to its high sensitivity and structural specificity.19 In previous studies, a variety of analytical methods, including high performance liquid chromatography (HPLC),6 ultra-performance liquid chromatography-triple quadrupole mass spectrometry (UPLC-QqQ/MS),5 capillary electrochromatography (CEC)20 and electrochemical methods,21,22 have been reported to quantify eriocitrin in raw materials and eriocitrin-based products. However, there are still no published simple and rapid analytical methods for the determination of eriocitrin in rat plasma. This is the first report on the development and validation of an LC-MS/MS method for the determination of eriocitrin in rat plasma, and it was successfully applied to a pharmacokinetic study of eriocitrin. The experimental results obtained in this study will provide a better understanding of the pharmacokinetic properties of eriocitrin in rats, and further help to understand the pharmacokinetic effects of eriocitrin in animals as a whole, which will provide greater possibilities for the development of new drugs.
2. Materials and methods
2.1. Chemicals and reagents
Eriocitrin (13463-28-0, purity > 98.5%) was obtained from Chengdu Desite Co., Ltd. (Chengdu, China). Sulfamethoxazole was provided by National Institutes for Food and Drug Control (Beijing, China). Acetonitrile, methanol and formic acid were all of HPLC grade and were purchased from Fisher Scientific (Waltham, MA, USA). Ether anesthesia was purchased from Shijiazhuang Reagent Factory (China). Purified water was purchased from Wahaha (Hangzhou Wahaha Group Co., Ltd.). Sodium carboxymethyl cellulose (CMC-Na) was obtained from Tianjin Guangfu Technology Development Co., Ltd. (Tianjin, China).2.2. Instruments and conditions
The analysis was performed on an Agilent Technology Series 1200 system (Agilent, USA), which was combined with a 3200 QTRAPTM system from Applied Biosystems/MDS SCIEX (Applied Biosystems, Foster City, CA, USA). The chromatographic separation was achieved on a Wonda Cract ODS-2 C18 Column (150 mm × 4.6 mm, 5 μm). The mobile phase was composed of 0.1% aqueous formic acid (eluent A) and acetonitrile (eluent B) at a flow rate of 0.8 mL min−1. The injection volume was set at 10 μL. The gradient elution programme was as follows: 10–80% B from 0 to 2 min and 80–100% B from 2 to 10 min.The mass spectrometric detection was performed using a QTRAP-3200 system with Turbo V sources and turbo ionspray interface in the negative electrospray ionization (ESI) mode. Also, the mass spectrometry was carried out in a multiple reaction monitoring (MRM) mode. The following mass spectrometry parameter settings were applied: ion spray voltage (IS), −4.5 kV; turbo spray temperature, 650 °C; nebulizing gas (GAS1), 60 psi; heating gas (GAS2), 65 psi; and curtain gas, 35 psi. The selected precursor/product ion pairs were 595.4 → 287.1 for eriocitrin and 252.0 → 155.9 for sulfamethoxazole.
2.3. Preparation of calibration standards and QC standards
An appropriate amount of eriocitrin was precisely weighed and diluted with methanol to 0.16 mg mL−1 as a stock solution. Working stock solutions were prepared via a serial dilution method in methanol including calibration standards (3.125, 6.25, 12.5, 25, 50, 100, 200, 400 and 800 ng mL−1), and QC samples: lower quality control (LQC, 6.25 ng mL−1), medium quality control (MQC, 50 ng mL−1) and higher quality control (HQC, 600 ng mL−1). An IS working solution was prepared at a concentration of 400 ng mL−1 in methanol. All the above solutions were stored at 4 °C.2.4. Sample preparation
Protein precipitation was applied to extract eriocitrin and IS from plasma samples via the addition of methanol. The 100 μL of plasma samples were spiked with 100 μL of IS working solution and 300 μL of methanol, the mixture was vortexed for 3 min and centrifuged at 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 380 × g for 10 min at 4 °C. The supernatant was removed and slowly dried under nitrogen at room temperature. The residue was redissolved with 100 μL methanol and centrifuged twice at 21380 × g for 10 min. 10 μL of the supernatant was injected into the LC/MS/MS system for analysis.
380 × g for 10 min at 4 °C. The supernatant was removed and slowly dried under nitrogen at room temperature. The residue was redissolved with 100 μL methanol and centrifuged twice at 21380 × g for 10 min. 10 μL of the supernatant was injected into the LC/MS/MS system for analysis.
2.5. Method validation for pharmacokinetics
In order to demonstrate the suitability of the developed method, the method validation was performed, including the specificity and selectivity, linearity and sensitivity, precision and accuracy, recovery and matrix effect, and the stability according to the guidelines of the US Food and Drug Administration (FDA) for a bioanalytical method validation.232.6. Pharmacokinetic study in rats
Eighteen male Sprague-Dawley rats (certificate no. 1904020, weighing 200–250 g, 12–14 weeks old) were purchased from the Experimental Animal Research Center of Hebei Medical University. All the protocols and procedures for animal handling were performed as per the guidelines of the Hebei Committee for Care and Use of Laboratory Animals and were approved by the Animal Experimentation Ethics Committee of the Hebei Medical University (Hebei, China). The conditions of temperature (22–25 °C), humidity (55–60%) and light (12 h light/dark cycle) were standardised for a minimum of 1 week prior to the use. All rats were fasted but allowed water for 12 h before the experiments. These rats were randomly divided into three groups with six rats per group. All rats were given eriocitrin by gavage, which was dissolved in a 0.5% CMC-Na solution at a dose of 50 mg kg−1.19,31 All rat experiments were conducted in accordance with the committee's guidelines on the Care and Use of Laboratory Animals.The plasma sample collection was performed as follows: approximately 0.3 mL of blood sample was collected from the eye canthus of rats under the condition of mild ether anesthesia into 1.5 mL heparinized tubes at 0, 0.033, 0.083, 0.117, 0.167, 0.25, 0.5, 1, 2, 3, 4, 6, 9 and 12 h after gavage.32,33 Blood samples were immediately centrifuged at 1920 × g for 10 min to obtain the plasma. Then, all collected plasma samples were mixed and frozen at −80 °C until analysis. The pharmacokinetic parameters of eriocitrin were calculated by the DAS 3.0 software (Beijing, China) in the non-compartmental mode.
3. Results and discussion
3.1. Optimization of chromatographic and mass conditions
In this study, the chromatographic and mass parameters were carefully optimized for the determination of the analyte and IS. To the best of our knowledge, chromatographic conditions, particularly the composition of the mobile phase, played a pivotal role in achieving good chromatographic behavior (including peak symmetry, sensitivity and short run time) and appropriate ionization.31,34 Finally, the best peak shape and ionization were achieved using the mobile phase composed of a formic acid aqueous solution (0.1%, v/v) and acetonitrile with gradient elution at a flow rate of 0.8 mL min−1. As for mass conditions, the positive and negative modes were employed to obtain the precursor and product ions, and it was discovered that the response of the analytes and IS observed in the negative ionization mode was higher than that in positive ionization mode. The parent ion m/z of eriocitrin was 595.4, and the characteristic product ion was 287.1, while the m/z of the parent ion and product ion of IS were 252.0 and 155.9, respectively. The MS/MS spectra of eriocitrin and IS are shown in Fig. 1.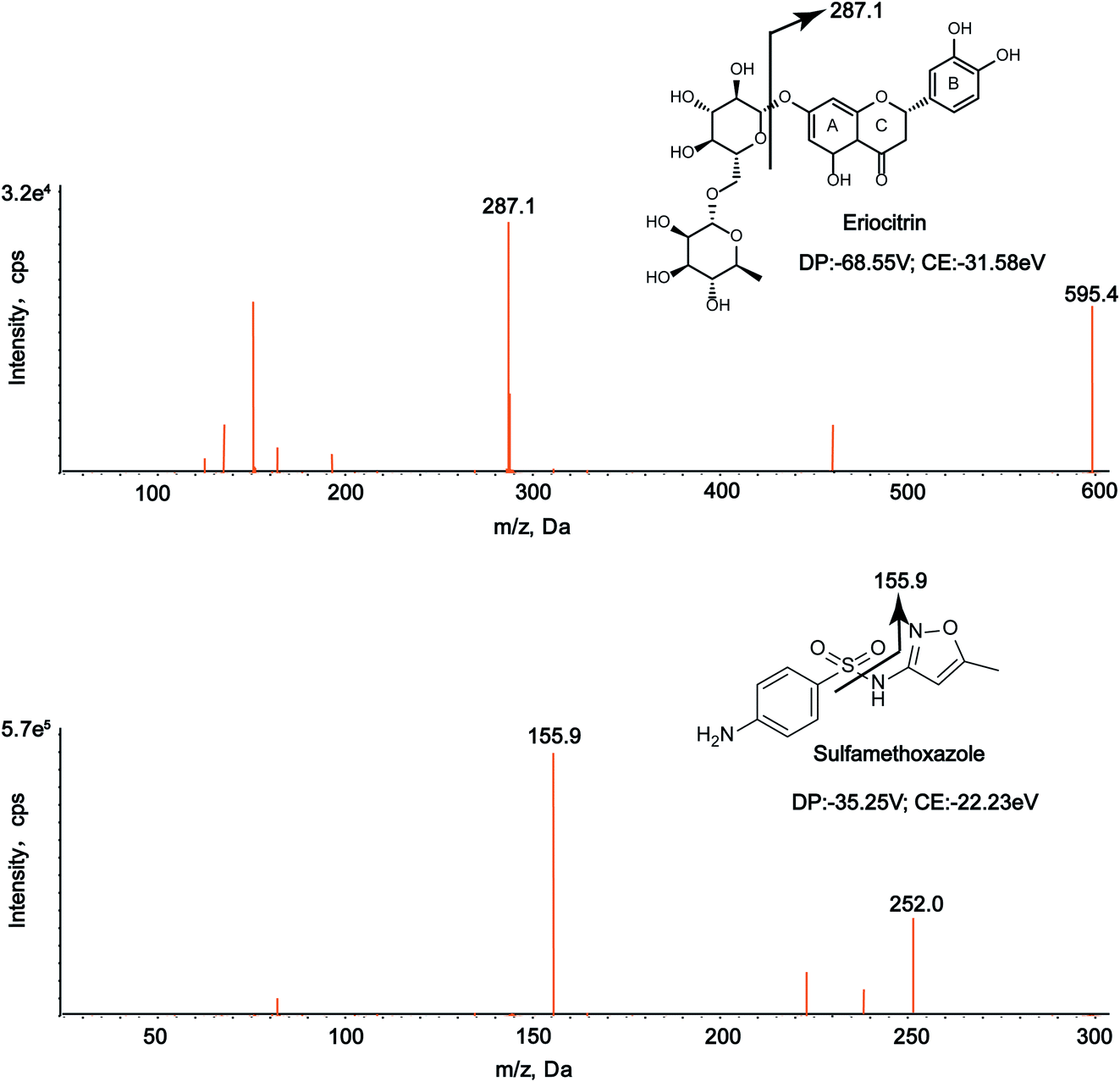 | ||
| Fig. 1 MS/MS spectra of eriocitrin and IS (sulfamethoxazole) in negative mode. | ||
3.2. Method validation
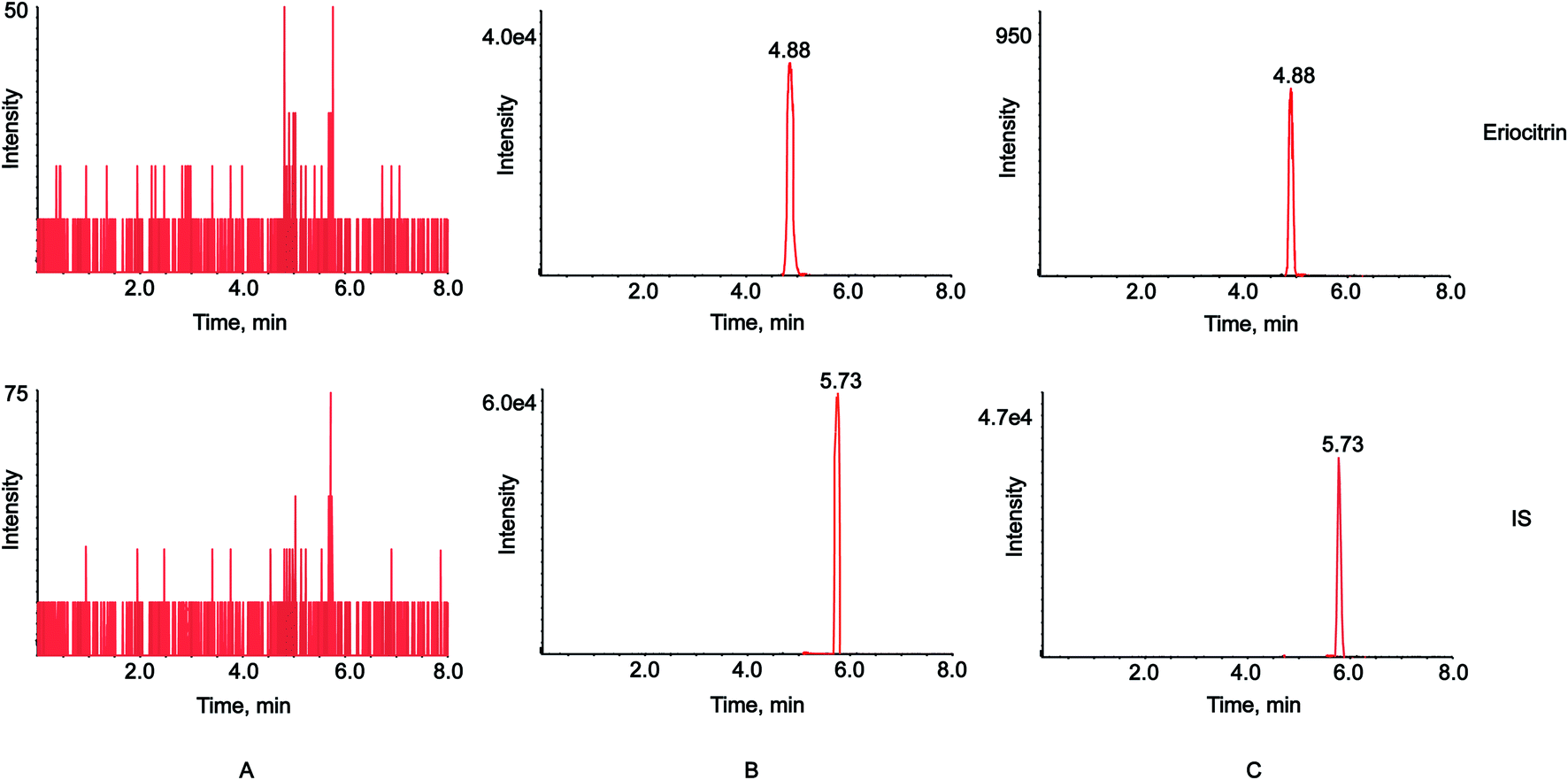 | ||
| Fig. 2 MRM chromatograms of the blank plasma (A), plasma spiked with eriocitrin and IS (B), and plasma samples after oral administration of eriocitrin (C). | ||
| Concentration (ng mL−1) | Intra-day (n = 6) | Inter-day (n = 6) | ||||
|---|---|---|---|---|---|---|
| Measured conc., (ng mL−1) | Precision, RSD (%) | Accuracy, RE (%) | Measured conc., (ng mL−1) | Precision, RSD (%) | Accuracy, RE (%) | |
| 6.25 | 6.41 ± 0.36 | 5.64 | 2.56 | 6.50 ± 0.31 | 4.79 | 4.06 |
| 50 | 53.04 ± 3.20 | 6.02 | 6.09 | 53.83 ± 3.65 | 6.79 | 7.67 |
| 600 | 593.01 ± 12.30 | 2.07 | −1.17 | 596.42 ± 11.15 | 1.87 | −0.60 |
| Analyte | Spiked concentration (ng mL−1) | Extraction recovery | Matrix effect | ||
|---|---|---|---|---|---|
| Mean (%) | RSD (%) | Mean (%) | RSD (%) | ||
| Eriocitrin | 6.25 | 82.13 | 4.17 | 82.23 | 5.61 |
| 50 | 93.14 | 7.63 | 88.65 | 8.41 | |
| 600 | 87.16 | 8.09 | 90.59 | 1.96 | |
| Storage condition | Eriocitrin | ||
|---|---|---|---|
| Nominal conc. (ng mL−1) | Found conc. (ng mL−1) | RE (%) | |
| Short-term stability (room temperature for 24 h) | 6.25 | 6.29 ± 0.315 | 0.69 |
| 50 | 52.25 ± 3.208 | 4.51 | |
| 600 | 594.38 ± 12.715 | −0.94 | |
| Long-term stability (−20 °C for 30 days) | 6.25 | 6.12 ± 0.241 | −2.06 |
| 50 | 49.43 ± 0.291 | −1.13 | |
| 600 | 605.61 ± 4.378 | 0.93 | |
| Three freeze–thaw cycles stability | 6.25 | 6.32 ± 0.265 | 1.17 |
| 50 | 51.60 ± 2.927 | 3.20 | |
| 600 | 589.09 ± 9.952 | −1.82 | |
| Stock solution stability (4 °C for 30 days) | 6.25 | 6.33 ± 0.340 | 1.28 |
| 50 | 53.35 ± 3.134 | 6.71 | |
| 600 | 584.61 ± 5.330 | −2.56 | |
3.3. Pharmacokinetic study
The validated LC-MS/MS method was successfully applied to the pharmacokinetic study of eriocitrin in rat plasma after oral administration at a single dose of 50 mg kg−1. The average plasma concentration–time curve of eriocitrin in rats is illustrated in Fig. 3. The pharmacokinetic parameters including the maximum plasma concentration (Cmax), the time to reach the maximum concentrations (Tmax), the half-time (T1/2), area after gavage in rats under the concentration–time curve (AUC0–t, AUC0–∞), the plasma clearance (CLz/F) and volume of distribution (Vz/F) are listed in Table 4. They were analyzed by the non-compartment model and shown as mean ± SD.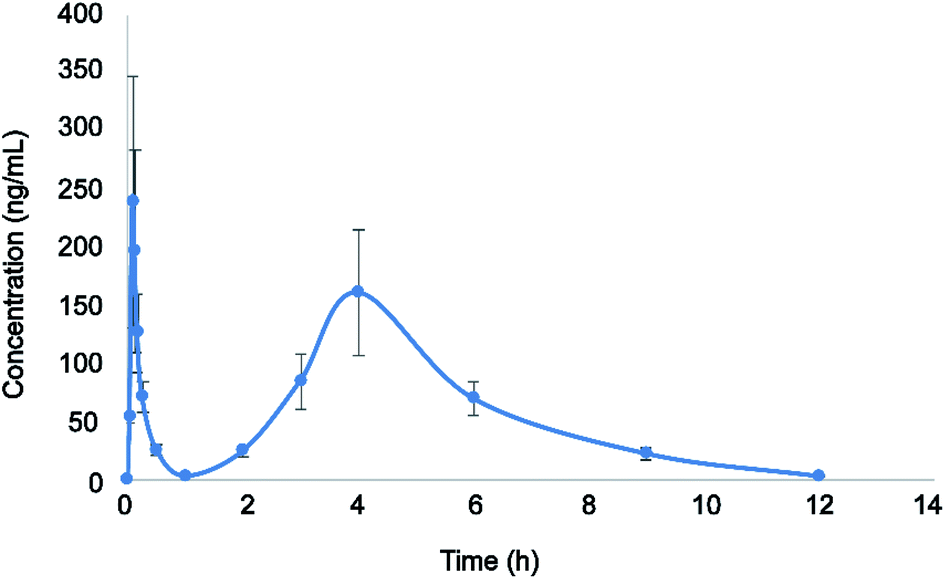 | ||
| Fig. 3 Plasma concentration–time profile after a single oral administration of eriocitrin (50 mg kg−1) to rats. Data are expressed as the mean ± SD (n = 6). | ||
| Pharmacokinetic parameters | Eriocitrin |
|---|---|
| Cmax (μg L−1) | 299.833 ± 16.743 |
| Tmax (h) | 0.094 ± 0.019 |
| T1/2 (h) | 1.752 ± 0.323 |
| AUC0–t (μg h L−1) | 653.852 ± 143.341 |
| AUC0–∞ (μg h L−1) | 670.847 ± 141.634 |
| CLz/F (L kg−1 h−1) | 77.139 ± 18.52 |
| Vz/F (L h−1) | 198.656 ± 75.89 |
As shown above, the maximum plasma concentration (Cmax) of eriocitrin was 299.833 ± 16.743 μg L−1, while the corresponding time to reach the Cmax(Tmax) was 0.094 ± 0.019 h, demonstrating rapid absorption from the gastrointestinal tract. The half-time (T1/2) was 1.752 ± 0.323 h, indicating that it would remain in the body for a short time and can be given many times to enhance the clinical efficacy. In addition, double peaks were detected in mean plasma concentration profiles after the oral administration of eriocitrin; the first peak appeared at about 0.094 h, and the second peak appeared at about 4 h, which was lower than the first peak. This phenomenon was almost the same as that reported in a previous study,28,35,36 and it may have been caused by a variety of factors, such as fractionated gastric emptying, enterohepatic circulation and separated “absorption windows”.37–39 However, the mechanism of this phenomenon in the pharmacokinetic study needs further detailed investigation. All in all, these results should drive further studies of eriocitrin and promote research into developing efficacy in clinical therapeutic studies.
We know some flavanones, such as eriocitrin, naringin, and hesperidin, possess an additional d-configured mono or disaccharide sugar in the C7 position on ring A on the basis of the chemical structure of the C6–C3–C6 configuration.40 Based on a study in the pharmacokinetics of eriocitrin, it is of great significance to understand and compare the pharmacokinetics of drugs with similar structures to eriocitrin. In previous studies, naringin was quickly absorbed into a serum by reaching the first concentration peak at 15 min and another at 3 h after orally administering a naringin monomer.41 Similarly, the plasma levels of free hesperidin quickly reached Cmax within 5 min but dropped rapidly afterwards after a single oral dose of hesperidin. Then, there was a second peak.42 By comparing the results of their pharmacokinetic studies, we found that eriocitrin, naringin and hesperidin were rapidly absorbed after oral administration, and all of them showed double peaks, which may be related to their structures; however, further investigation is needed.
3.4. The impact of bleeding and anaesthesia on the eriocitrin distribution
Long term anaesthesia has an effect on the absorption, distribution, metabolism and excretion of drugs.43 However, in this study, we collected each blood sample from the eye canthus of rats under the condition of mild ether anesthesia.32,33 The dosage of ether was relatively small, the concentration was relatively low, and it was short-term anaesthesia, so it was speculated that anaesthesia had little effect on the distribution of eriocitrin. For the extraction of blood from the eye canthus of rats, it is required that the operator should be well trained.44 First of all, our blood collection skill was relatively professional, the blood collection was accurate and fast, and the blood collection volume was not large. In addition, the blood collection points were not dense, which may not have a great impact on the distribution of drugs.4. Conclusions
In the study, for the first time, a rapid and sensitive LC/MS/MS analytical method was developed and validated for eriocitrin quantification in rat plasma, and the present method was successfully employed to establish the oral pharmacokinetic profile of eriocitrin. The pharmacokinetic parameters showed that eriocitrin reached its Cmax very fast and showed secondary absorption. The obtained results would provide reference and valuable evidence for the further development of new pharmaceuticals and pharmacological mechanisms, and lay a foundation for preclinical and clinical studies of eriocitrin.Conflicts of interest
All the authors have declared no conflict of interest.Acknowledgements
The project was financially supported by the National Natural Science Foundation of China (No. 81473180). Thanks to the Department of Pharmaceutical Analysis, School of Pharmacy, Hebei Medical University for the instrument support.References
- J. Perez-Jimenez, M. E. Diaz-Rubio and F. Saura-Calixto, Plant Foods Hum. Nutr., 2015, 70, 365–370 CrossRef CAS PubMed.
- M. Makni, R. Jemai, W. Kriaa, Y. Chtourou and H. Fetoui, BioMed Res. Int., 2018, 2018, 6251546 Search PubMed.
- X. Dong, Y. Hu, Y. Li and Z. Zhou, Sci. Hortic., 2019, 243, 281–289 CrossRef CAS.
- J. J. Peterson, G. R. Beecher, S. A. Bhagwat, J. T. Dwyer, S. E. Gebhardt, D. B. Haytowitz and J. M. Holden, J. Food Compos. Anal., 2006, 19, S74–S80 CrossRef CAS.
- Y. Xue, L. S. Qing, L. Yong, X. S. Xu, B. Hu, M. Q. Tang and J. Xie, Molecules, 2019, 24, 3016 CrossRef CAS PubMed.
- E. Mesquita and M. Monteiro, Food Res. Int., 2018, 106, 54–63 CrossRef CAS PubMed.
- M. Hajimahmoodi, G. Moghaddam, S. M. Mousavi, N. Sadeghi, M. R. Oveisi and B. Jannat, Trop. J. Pharm. Res., 2014, 13, 951–956 CrossRef.
- X. Cao, Y. He, Y. Kong, X. Mei, Y. Huo, Y. He and J. Liu, Food Hydrocolloids, 2019, 94, 63–70 CrossRef CAS.
- X. Cao, Z. Yang, Y. He, Y. Xia, Y. He and J. Liu, J. Mol. Recognit., 2019, 32, e2779 CrossRef PubMed.
- P. S. Ferreira, L. C. Spolidorio, J. A. Manthey and T. B. Cesar, Food Funct., 2016, 7, 2675–2681 RSC.
- K.-i. Minato, Y. Miyake, S. Fukumoto, K. Yamamoto, Y. Kato, Y. Shimomura and T. Osawa, Life Sci., 2003, 72, 1609–1616 CrossRef CAS.
- Y. Miyake, E. Suzuki, S. Ohya, S. Fukumoto, M. Hiramitsu, K. Sakaida, T. Osawa and Y. Furuichi, J. Food Sci., 2006, 71, S633–S637 CrossRef CAS.
- M. Hiramitsu, Y. Shimada, J. Kuroyanagi, T. Inoue, T. Katagiri, L. Zang, Y. Nishimura, N. Nishimura and T. Tanaka, Sci. Rep., 2014, 4, 3708 CrossRef PubMed.
- Z. Wang, H. Zhang, J. Zhou, X. Zhang, L. Chen, K. Chen and Z. Huang, Cancer Chemother. Pharmacol., 2016, 78, 1143–1150 CrossRef CAS PubMed.
- G. Guo, W. Shi, F. Shi, W. Gong, F. Li, G. Zhou and J. She, J. Biochem. Mol. Toxicol., 2019, 33, e22400 CAS.
- J. Liu, H. Huang, Z. Huang, Y. Ma, L. Zhang, Y. He, D. Li, W. Liu, S. Goodin, K. Zhang and X. Zheng, J. Funct. Foods, 2019, 56, 321–332 CrossRef CAS.
- M. C. Gidaro, F. Alcaro, S. Carradori, G. Costa, D. Vullo, C. T. Supuran and S. Alcaro, Planta Med., 2015, 81, 533–540 CrossRef CAS PubMed.
- I. H. Baek, Xenobiotica, 2019, 49, 734–739 CrossRef CAS PubMed.
- C. Liang, J. Yin, Y. Ma, X. Zhang and L. Zhang, J. Pharm. Biomed. Anal., 2020, 177, 112835 CrossRef CAS PubMed.
- C. Desiderio, A. D. Rossi and M. Sinibaldi, J. Chromatogr. A, 2005, 1081, 99–104 CrossRef CAS PubMed.
- L. Wang, Q. Wang, K. Sheng, G. Li and B. Ye, J. Electroanal. Chem., 2017, 785, 96–102 CrossRef CAS.
- S. Yao, W. Cai, L. Liu, X. Liao, K. Tao, F. Feng and G. Yang, Anal. Methods, 2016, 8, 3722–3729 RSC.
- U.S. Department of Health and Human Services, Food and Drug Administration and Center for Drug Evaluation and Research (CDER), Guidance for Industry, Bioanalytical Method Validation, 2018 Search PubMed.
- F. Zhang, L. Sun, J. Zhai, T. Xia, W. Jiang, M. Li, S. Gao, X. Tao, W. Chen and Y. Chai, BioMed Res. Int., 2019, 2019, 1854323 Search PubMed.
- Z. Guo, B. Li, J. Gu, P. Zhu, F. Su, R. Bai, X. Liang and Y. Xie, Molecules, 2019, 24, 3057 CrossRef PubMed.
- L. Zhao, W. Li, Y. Li, H. Xu, L. Lv, X. Wang, Y. Chai and G. Zhang, J. Chromatogr. Sci., 2015, 53, 1185–1192 CAS.
- B. Yang, L. Xie, S. Peng, K. Sun, J. Jin, Y. Zhen, K. Qin and B. Cai, J. Sep. Sci., 2019, 42, 2341–2350 CrossRef CAS PubMed.
- X. Zhang, J. Guan, H. Zhu and T. Niu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 971, 126–132 CrossRef CAS.
- E. Ezzeldin, M. Iqbal, R. Al-Salahi and T. El-Nahhas, J. Pharm. Biomed. Anal., 2019, 166, 244–251 CrossRef CAS PubMed.
- C. Zhao, S. Han, S. Yang and W. Xin, Biomed. Chromatogr., 2019, 33, e4667 CAS.
- Z. Wang, Q. Wu, Y. Meng, Y. Sun, Q. Wang, C. Yang, Q. Wang, B. Yang and H. Kuang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 985, 164–171 CrossRef CAS.
- S. T. Dong, Y. Li, H. T. Yang, Y. Wu, Y. J. Li, C. Y. Ding, L. Meng, Z. J. Dong and Y. Zhang, Molecules, 2018, 23, 2894 CrossRef.
- S. Lata and S. K. Mittal, Int. J. Pharmacol., 2017, 13, 563–572 CAS.
- M. Ding, W. Ma, X. Wang, S. Chen, S. Zou, J. Wei, Y. Yang, J. Li, X. Yang, H. Wang, Y. Li, Q. Wang, H. Mao, X. M. Gao and Y. X. Chang, J. Ethnopharmacol., 2019, 242, 112055 CrossRef PubMed.
- R. Shi, S. Qiao, D. Yu, X. Shi, M. Liu, X. Jiang, Q. Wang and L. Zhang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1625–1632 CrossRef CAS PubMed.
- Z. Zhu, L. Zhao, X. Liu, J. Chen, H. Zhang, G. Zhang and Y. Chai, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2184–2190 CrossRef CAS PubMed.
- S. K. Thapa, M. Upadhyay, T. H. Kim, S. Shin, S. J. Park and B. S. Shin, Molecules, 2019, 24, 2037 CrossRef CAS PubMed.
- B. Ji, Y. Zhao, P. Yu, B. Yang, C. Zhou and Z. Yu, Talanta, 2018, 190, 450–459 CrossRef CAS PubMed.
- H. Ding, Y. Liu, Z. Zeng, H. Si, K. Liu, Y. Liu, F. Yang, Y. Li and D. Zeng, Res. Vet. Sci., 2012, 93, 374–377 CrossRef CAS PubMed.
- J. A. Yanez, C. M. Remsberg, N. D. Miranda, K. R. Vega-Villa, P. K. Andrews and N. M. Davies, Biopharm. Drug Dispos., 2008, 29, 63–82 CrossRef CAS PubMed.
- S. Q. Li, S. Dong, Z. H. Su, H. W. Zhang, J. B. Peng, C. Y. Yu and Z. M. Zou, Metabolites, 2013, 3, 867–880 CrossRef PubMed.
- J. T. Lee, L. H. Pao, C. D. Hsieh, P. W. Huang and Y. P. Hu, Anal. Methods, 2017, 9, 3329–3337 RSC.
- Y. C. Wong, S. Qian and Z. Zuo, Pharm. Res., 2014, 31, 2107–2123 CrossRef CAS PubMed.
- H. Du, S. Li, Y. Zhang, H. Guo, L. Wu, H. Liu, A. Manyande, F. Xu and J. Wang, Molecules, 2019, 24, 2542 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |