DOI:
10.1039/C9RA10917J
(Review Article)
RSC Adv., 2020,
10, 6678-6724
Phosphorus-containing amino acids with a P–C bond in the side chain or a P–O, P–S or P–N bond: from synthesis to applications
Received
2nd December 2019
, Accepted 22nd January 2020
First published on 13th February 2020
Abstract
Since the discovery of (L)-phosphinothricin in the year 1970, the development of α-amino acids bearing a phosphorus group has been of renewed interest due to their diverse applications, including their use in [18F]-fluorolabeling, as fluorescent probes, as protecting groups and in the reversible immobilization of amino acids or peptide derivatives on carbon nanomaterials. Considerable progress has also been achieved in the field of antiviral agents, through the development of phosphoramidate prodrugs, which increase significantly the intracellular delivery of nucleoside monophosphate and monophosphonate analogues. This review aims to summarize the strategies reported in the literature for the synthesis of P(III), P(IV) and P(V) phosphorus-containing amino acids with P–C, P–O, P–S or P–N bonds in the side chains and their related applications, including their use in natural products, ligands for asymmetric catalysis, peptidomimetics, therapeutic agents, chemical reagents, markers and nanomaterials. The discussion is organized according to the position of the phosphorus atom linkage to the amino acid side chain, either in an α-, β-, γ- or δ-position or to a hydroxyl, thiol or amino group.
1. Introduction
Heterosubstituted amino acids containing P, B, Si and F elements are important classes of unnatural compounds that have gained considerable attention in peptide engineering, because they improve stability toward enzymatic degradation, they can modulate bioactivity and they can also be used for labeling purposes. Among the available heterosubstituted amino acids, those containing a phosphorus atom in a tri-, tetra-, penta- or even hexavalent form are a source of structural diversity, as illustrated by compounds 1–7 in Fig. 1. α-Amino acids bearing a phosphate or a phosphinate group on the side chain give rise to biologically active scaffolds, because this type of functionality is isosteric with the carboxylate group in the transition state formed during hydrolysis. (L)-2-Amino-3-phosphonopropionic acid, (L)-AP3, 1 was identified as an antagonist of the metabotropic glutamate receptor (NMDAR) and was evaluated for the treatment of central nervous system disorders. A wide range of acyclic and cyclic derivatives, such as Selfotel 2 (CGS-19755), have also been identified as competitive antagonists to glutamate for binding to NMDAR. Besides these synthetic phosphonic α-amino acids, (L)-phosphinothricin 3 (glufosinate) is a natural phosphinic acid analogue of (L)-glutamate that exhibits herbicidal properties due to its ability to inhibit glutamine synthetase. In this latter case, (L)-phosphinothricin has engendered the development of numerous drugs for neurodegenerative disease treatment. The α-phosphonoglycinate 4 developed in the early 1990s has emerged as a powerful reagent for the synthesis of α,β-dehydroamino acids via the Horner–Wadsworth–Emmons (HWE) reaction. Those compounds led to enantiomerically enriched α-amino acids after asymmetric hydrogenation, which are key intermediates in the total synthesis of bioactive compounds. More recently, phosphine borane fullerene amino esters such as 5 were obtained via the hydrophosphination of [60]fullerene using sec-phosphine boranes under phase transfer catalysis. The electrochemical behavior of the C60 phosphine borane amino esters demonstrates that retro-hydrophosphination into free [60]fullerene is possible, thus offering a new strategy for the reversible immobilization of amino acid or peptide derivatives on carbon nanomaterials.
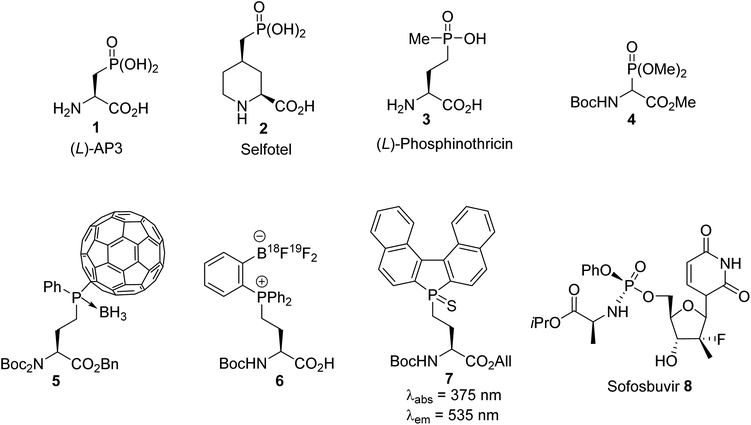 |
| | Fig. 1 Representative examples of recent phosphorus-containing amino acids. | |
The synthesis of o-trifluoroborato phosphonium salts of (L)-amino acids 6 was achieved via quaternization, starting from o-(pinacolato)boronato phenylphosphine with γ-iodoamino ester followed by fluorination. The usefulness of this compound was demonstrated in fast nucleophilic [18F]-radiolabeling, leading to [18F]-6 for peptide labeling via positron emission tomography (PET). Finally, the grafting of a fluorescent phosphole to an amino acid side chain led to the phospholyl sulfur amino ester 7, which exhibits remarkable fluorescence emission at 535 nm (green) with a large Stokes shift of 160 nm. Such phospholyl amino acids constitute a new class of probe for the fluorescent labeling of peptides. In addition to the α-amino acids 1–7 mentioned above, which are useful in the fields of medicinal chemistry, organic synthesis and labeling, phosphoramidate prodrugs, such as the anti-hepatitis C agent sofosbuvir 8 (Sovaldi), have been intensively developed for the intracellular delivery of nucleoside monophosphate and monophosphonate analogues.
This review aims to summarize the strategies reported in the literature for the synthesis of phosphorus-containing α- and β-amino acids with P–C, P–O, P–S or P–N bonds and it is organized according to the position of the phosphorus atom linkage to the amino acid side chain at an α-, β-, γ- or δ-position.
The preparation of other stable phosphino- and phosphono-amino acids is also reported in this review, along with their introduction into peptides; this provides ligands for asymmetric catalysis or tools for understanding the phosphorylation and dephosphorylation processes that regulate diverse cellular signalling pathways.
2. Phosphorus linked to carbon in the α-position
2.1. α-Phosphonium α-amino acids
The N-acyl-α-triphenylphosphonium α-amino esters 11a–b were reported for the first time by Kober and Steglich in 1983 and they were prepared via the quaternization of triphenylphosphine with N-acetyl-α-bromoglycinate 10, which was previously generated via in situ photochemical bromination in the presence of bromine or N-bromosuccinimide (Scheme 1).1
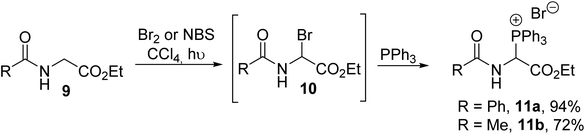 |
| | Scheme 1 The synthesis of α-triphenylphosphonium α-amino esters 11 from the α-bromoglycinate 9. | |
Other strategies to access the α-triphenylphosphonium α-amino esters 14a–b were later investigated by Mazurkiewicz et al., involving the transformation of the phosphoranylidene oxazolone 13, which resulted from the reaction of the oxazolone 12 with triphenylphosphine dibromide in the presence of triethylamine (Scheme 2).2 Quaternary triphenylphosphonium amino esters 15 were prepared via the alkylation of the phosphoranylidene oxazolone 13 with alkyl halides, followed by ring-opening with methanol. These N-acyl-α-triphenylphosphonium α-amino esters 15 are stable and crystalline compounds and they can further lead to α,α′-disubstituted glycine 16 via the addition of a nucleophile.3
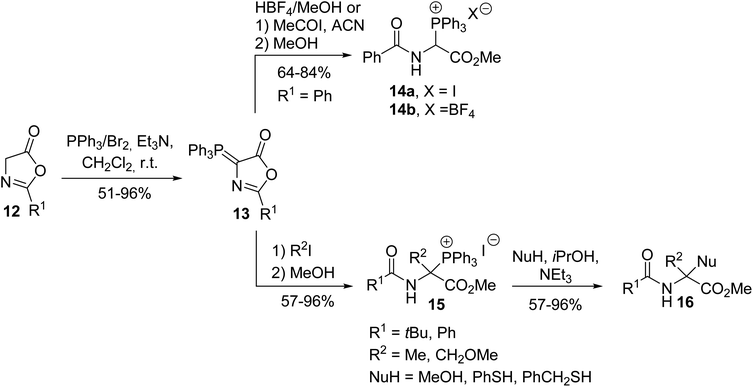 |
| | Scheme 2 The synthesis of the α-triphenylphosphonium α-amino esters 14 and 15 from the oxazolone 12. | |
Such compounds are of great interest in organic synthesis due to their diverse reactivity, either as synthetic equivalents to glycine α-cations or as precursors to α,β-dehydroamino acid derivatives, involving their corresponding phosphonium ylides in Wittig olefination.4,5 Thus, the treatment of α-triphenylphosphonium glycinate 14b with triethylamine resulted in the formation of its corresponding ylide 18 and N-acylimino acetate 17, which are highly reactive compounds, both in equilibrium in solution. When the ylide 18 reacted with an aliphatic or aromatic aldehyde or ketone, the Wittig reaction proceeded under mild conditions, leading to α,β-dehydroamino esters 19 with yields of up to 53% and when the ylide 18 reacted in situ with N-acylimino acetate 17, the dimer 20 was obtained with 80% yield (Scheme 3).4 Reactions involving di- or tri-peptides have also been performed, illustrating the interest in this methodology for designing peptidic and non-peptidic structures as original tools to stabilize the three-dimensional folding of backbone chains in proteins.5
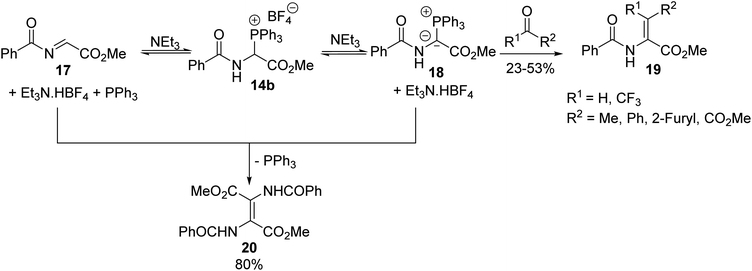 |
| | Scheme 3 The Wittig reaction using the α-triphenylphosphonium glycinate 14b as a reagent. | |
Compared to the ylide 18 reported above that could not be isolated because it was generated along with N-acylimino acetate 17, the other phosphonium ylide 22 with a nitrogen atom inserted into a β-lactam ring was more stable and was used for the synthesis of the bicyclic β-lactam antibiotic 24 (Scheme 4).6 The bicyclic β-lactam 24 was obtained via an intramolecular Wittig reaction with the thioester moiety, followed by the removal of the carboxylic acid protecting group via hydrogenation.
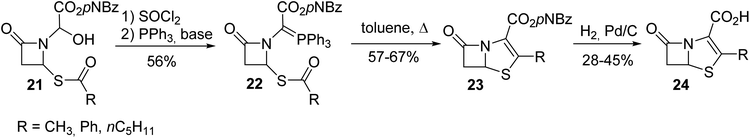 |
| | Scheme 4 The synthesis of the β-lactam 24 using the Wittig reagent 22. | |
2.2. α-Phosphino α-amino acids
α-Phosphino α-amino acids and peptides are particularly useful ligands for a variety of transition metal catalyzed or organocatalyzed chemical transformations and for providing water-soluble conjugates for biomedical applications. The first N-aryl α-diphenylphosphinoglycines were reported by Heinicke and coworkers via the one-pot three-component condensation of diphenylphosphine 25 with glyoxylic acid hydrate (GAH) in the presence of the aniline derivatives 26a–e.7,8 The reactions were performed at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 reagent ratio at room temperature via the addition of a mixture of diphenylphosphine and amine to an ethereal solution of GAH. The resulting ammonium salts 27a–e led to the α-diphenylphosphinoglycines 28a–e after dehydration, with yields of up to 97% (Scheme 5).
:1:1 reagent ratio at room temperature via the addition of a mixture of diphenylphosphine and amine to an ethereal solution of GAH. The resulting ammonium salts 27a–e led to the α-diphenylphosphinoglycines 28a–e after dehydration, with yields of up to 97% (Scheme 5).
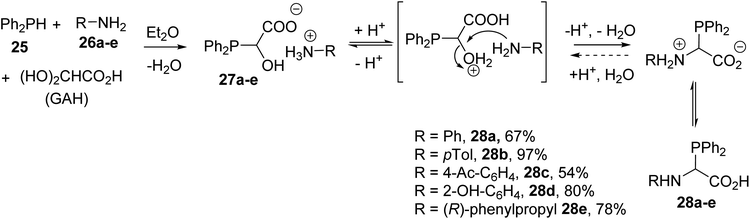 |
| | Scheme 5 The synthesis of N-aryl α-phosphinoglycines 28a–e from glyoxylic acid via a one-pot three-component condensation. | |
α-Phosphinoglycines possess two Lewis base P- and N-substituents at the α-carbon position; the presence of a carboxylic acid group destabilizes these compounds, leading to equilibrium in solution (Scheme 5). Another point to be mentioned is that α-phosphinoglycines are much more sensitive to decarboxylation and hydrolysis compared with conventional α-amino acids. N-Aryl derivatives are less sensitive to hydrolysis than those bearing N-alkylated substituents and can easily be stabilized either via sulfuration or oxidation with aqueous hydrogen peroxide, providing the corresponding P(V) compounds 29 and 30 with 84% and 93% yields, respectively (Scheme 6). Complexation with tungsten, chromium or molybdenum pentacarbonyl was also demonstrated to afford the transition-metal complexes 31–33.8 The complexation of such ligands with Ni(cod)2 was successfully achieved, leading to highly active catalysts for the oligomerization of ethylene.
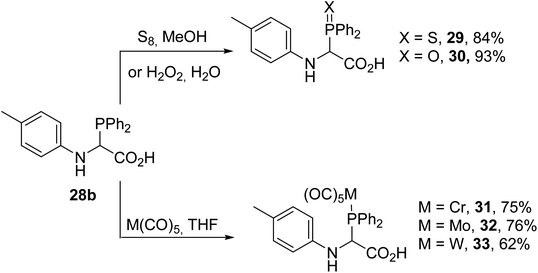 |
| | Scheme 6 Chemical transformations at the P-center of N-aryl α-phosphinoglycine 28b. | |
The influence of more basic alkylphosphines (PR1H2) on reactions with p-toluidine and glyoxylic acid was also studied; this did not afford phosphino-bis(amino acids) as expected, but the lactams 34–35 were formed (Scheme 7).9 This could be explained based on the higher P-basicity of the alkylphosphines, which are easily protonated, leading to intramolecular cyclization into a lactam ring at room temperature.
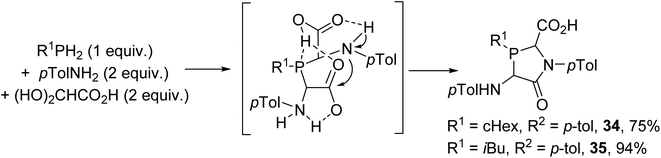 |
| | Scheme 7 One-pot three-component condensation from glyoxylic acid using primary alkylphosphines. | |
This reaction was also extended to ortho-phosphanylanilines 36. Metallation and subsequent P-alkylation in liquid ammonia followed by cyclocondensation with GAH led to the 1,3-benzazaphospholine carboxylic acids 38a–b (Scheme 8).10 In spite of the presence of a bulky neopentyl substituent on the P-atom, low diastereoselectivity was observed in the ring closure step. Only the condensation of N-neopentyl-o-phosphanylaniline with glyoxylic acid in the presence of p-toluidine afforded the defined trans diastereoisomer phosphanyl-bis(amino acid) 40, having the P-lone pair and carboxylic acid functionality on the same side (Scheme 8). This N-substituted benzazaphospholine 2-carboxylate, when complexed with nickel, allowed ethylene polymerization at a high conversion rate.
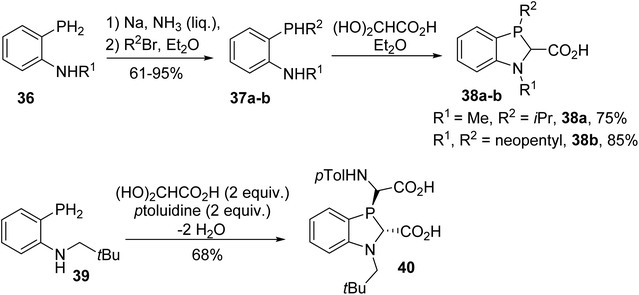 |
| | Scheme 8 The cyclocondensation of glyoxylic acid with the ortho-phosphanylanilines 36 and 39. | |
Among the α-amino acids bearing a phosphine group at the α-carbon position, the phenylphosphaprolines 42a–b and 43a–b, containing a trivalent phosphorus atom within the five-membered proline ring, were also reported by Heinicke et al. These compounds were obtained with up to 95% yields via the condensation of the 2-phenylphosphanyl ethylamines 41a–b with an equimolar amount of glyoxylic acid or pyruvic acid in diethyl ether or 1,4-dioxane, respectively (Scheme 9).11 The structures and diastereoisomeric ratios of these compounds were determined via 1H, 31P and 13C NMR and the structures of trans 42a and trans 43a were assigned via X-ray diffraction.
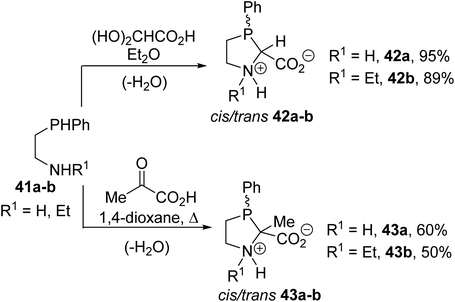 |
| | Scheme 9 The synthesis of the phenylphosphaprolines 42 and 43. | |
2.3. α-Phosphono α-amino acids
2.3.1. Via the Michaelis–Arbuzov reaction with α-chloroglycinates. One of the first synthesis procedures for the α-phosphonoglycinates 47a–e was reported by Schmidt et al. in 1982 and consists of a Michaelis–Arbuzov reaction involving trimethyl phosphite and chloroglycinate 45, which is more reactive than the methoxyamino ester precursor 44 (Scheme 10).12,13 After the removal of the Cbz protecting group, the free amine was then transformed into a series of N-Boc, N-acetyl or N-formyl derivatives, with the dipeptides 47a–e showing good yields from 78 to 91%.13
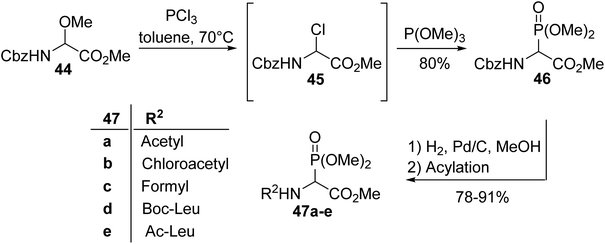 |
| | Scheme 10 The synthesis of the α-phosphono α-amino esters 47a–e from α-chloroglycinate 45. | |
As an alternative to the chlorination step, α-methoxyglycinate 48 was activated with boron trifluoride etherate Lewis acid and dialkyl phosphites bearing O-trimethylsilyl as a leaving group were also used (Scheme 11).14 Under these conditions, the α-phosphono α-amino esters 49a–c were obtained with good to excellent yields, from 42 to 95%, depending on the reactivity of the phosphite.
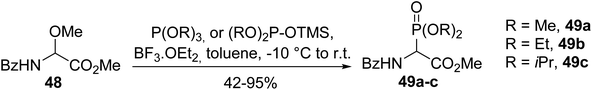 |
| | Scheme 11 The synthesis of the α-phosphono α-amino esters 49a–c from α-methoxyglycinate 48. | |
2.3.2. Via the rhodium catalyzed N–H insertion of diaza-phosphonoacetate. In 1995, Moody et al. described an efficient strategy to prepare N-acyl phosphonoglycinate, starting with diethyl diaza-phosphonoacetate as a carbenoid precursor.15 This method is based on an N–H insertion reaction involving rhodium carbenoids derived from the readily available diaza-phosphonate 50 and the amide, carbamate or urea compounds 51a–e. This method is as similarly effective as the Michaelis–Arbuzov reaction described above and provides the N-acyl phosphonoglycinates 52a–e with yields of up to 79% (Scheme 12).
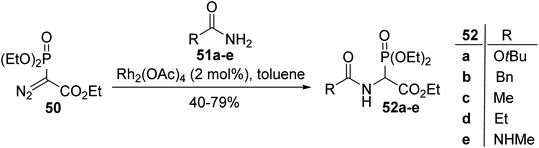 |
| | Scheme 12 The synthesis of the α-phosphonoglycinates 52a–e from diethyl diaza-phosphonoacetate 50. | |
Later, this methodology was extended to a range of N-protected α-amino amides 53, which were prepared from the corresponding acids. The N–H insertion reaction was performed in the presence of a Rh(II) catalyst and resulted in the formation of the phosphonate dipeptide derivatives 54 with 81% to 88% yields. Those phosphonates were subjected to the Horner–Wadsworth–Emmons (HWE) reaction with aldehydes to furnish a variety of dehydrodipeptides 55 (Scheme 13).16,17
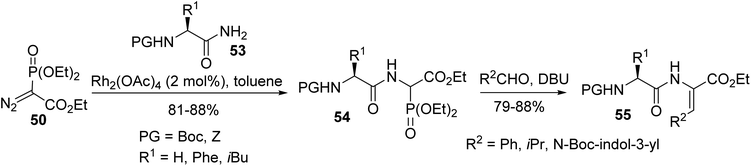 |
| | Scheme 13 The formation of dehydrodipeptides 55 via HWE olefination. | |
2.3.3. Via the nucleophilic addition of dimethyl phosphonate to a Schiff base. The preparation of quaternary α-phosphonoglycinate was also possible via the addition of dimethyl phosphite 57 to the electrophilic trifluoromethyl N-(pyridin-2-yl) Schiff base 56. This strategy allows the corresponding α-phosphonoglycinate 58 to be obtained with a good yield of 76% and it provides perspectives for the design of 2-aminopyridine derivatives, which are a structural fragment of many bioactive substances (Scheme 14).18
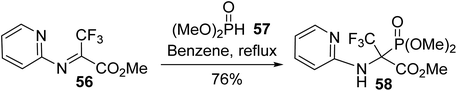 |
| | Scheme 14 The synthesis of α-phosphonoglycinate 58 via the hydrophosphonylation of the Schiff base 56. | |
2.3.4. Via the asymmetric cyanation of α-ketiminophosphonates. While efficient preparations of racemic α-phosphonoglycinates were achieved according to the above strategies (the Michaelis–Arbuzov reaction, the N–H insertion of rhodium carbenoids derived from diaza-phosphonates and the hydrophosphonylation of Schiff base), only one asymmetric synthesis of such compounds has been reported to date. In this context, Palacios et al. reported the asymmetric cyanation of α-ketiminophosphonates 59 with aryl cyanoformate, catalyzed by the Cinchona alkaloid 61.19 Tetrasubstituted (S)-α-amino nitriles 62 were obtained with good yields and enantioselectivities of up to 92%. Finally, treatment with hydrochloric acid afforded the corresponding (S)-phosphonated tetrasubstituted amino acids 63 bearing various aromatic substituents (Scheme 15).
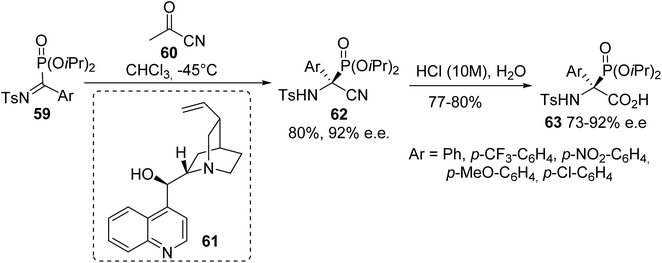 |
| | Scheme 15 The synthesis of α-phosphonoamino acids 63 via asymmetric cyanation. | |
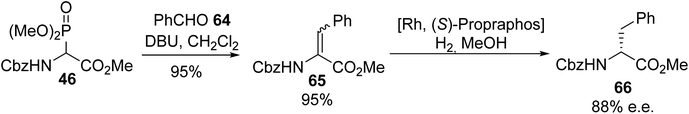
2.3.5. The utilization of α-phosphonoamino acids. The use of α-phosphonoglycinates to afford α,β-dehydroamino acids via the Horner–Wadsworth–Emmons (HWE) reaction was first reported in the early 1990s and it is still of great interest nowadays. This olefination, which proceeds between an aldehyde or a ketone and a phosphonate carbanion, has been widely explored, as illustrated by the following examples. The initial conditions developed by Schmidt et al. to generate α-phosphonoglycinate ylides in the presence of DBU as a base were also used by other authors for reactions with aldehydes or ketones to access a variety of α,β-dehydroamino acids and peptide derivatives.20 Under these conditions, the HWE reaction involving the ylide obtained from α-phosphonoglycinate 46 was performed with benzaldehyde to furnish the corresponding α,β-dehydroamino ester 65 almost quantitatively and asymmetric hydrogenation catalyzed by rhodium led to the (D)-enantiomer of phenylalanine 66 with 88% e.e. (Scheme 16).21
 |
| | Scheme 16 The formation of the dehydroamino acid 65 using the α-phosphonoglycinate 46 as the HWE reagent. | |
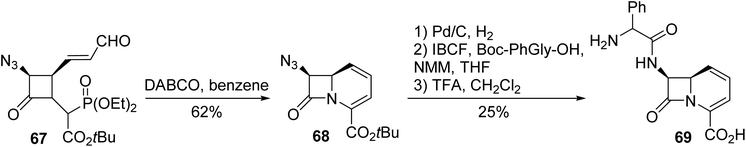
Cephalosporine is a β-lactamine antibiotic isolated from Cephalosporium acremonium and carbacephem 69 is an analogue in which the sulfur atom is substituted for a simple methylene isosteric group. It was obtained via an intramolecular HWE reaction starting from the phosphonate lactamine derivative 67, followed by the reduction of the azido group and coupling with N-Boc phenylglycine, with 30% overall yield (Scheme 17).22,23 Studies performed with this analogue demonstrate that the presence of a sulfur atom in the cephem nucleus is not essential for antimicrobial activity.
 |
| | Scheme 17 The stereoselective synthesis of carbacephem 69 via an intramolecular HWE reaction. | |
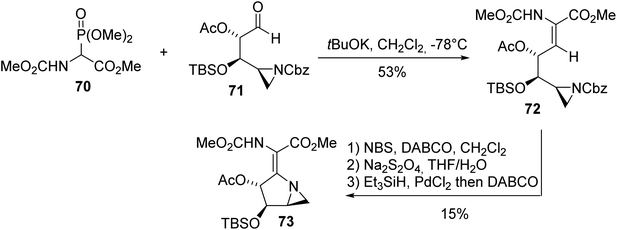
The azinomycins A and B, which exhibit antitumor properties, were isolated from a culture of Streptomyces griseofuscus S42227 in 1986. These densely functionalized molecules contain an aziridine motif and pyrrolidine ring connected to each other, making their synthesis challenging. In this context, Carpenter et al. found that HWE olefination involving α-phosphonoglycinate 70 and the aldehyde aziridine derivative 71 is a convenient method for the construction of the intermediate α,β-dehydroamino ester 72 (Scheme 18).24 Then, radical bromination of the olefin, deprotection of the amine and intramolecular cyclization led successfully to the key building block 73 with 15% yield.
 |
| | Scheme 18 The stereoselective synthesis of the dehydroamino acid 73 using the HWE reagent 70. | |
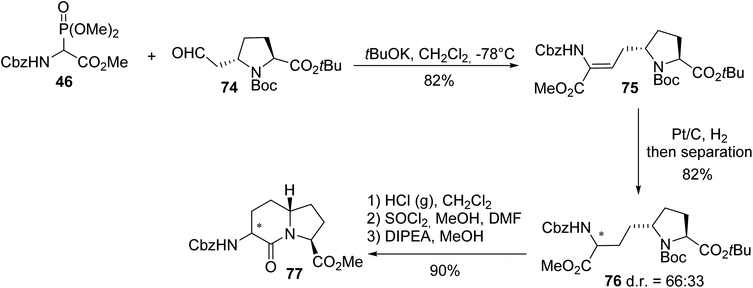
Gonadotropin-releasing hormone (GnRH) is a neurohormone that controls gonadotropin synthesis and is released with a very short half-life. It is therefore necessary to develop analogues that are stable enough in vivo to be administered to patients who suffer from endocrine disorders. In line with this objective, the constrained dipeptide mimic AZABIC type I 77 was synthesized and its affinity towards the GnRH receptor was evaluated. First, the ylide of phosphonoamino ester 46 was reacted with the aldehyde derived from (L)-proline 74 to afford the unsaturated dipeptide 75 with 82% yield. Then, hydrogenation gave a mixture of the diastereoisomers of 76 in a 66:33 ratio, which were separated via column chromatography, deprotected and subjected to intramolecular cyclization to provide AZABIC type I 77 with 90% yield. The absolute configurations of each stereocenter were determined via X-ray diffraction analysis of the crystals (Scheme 19).25
 |
| | Scheme 19 The synthesis of the constrained dipeptide mimic 77 via the HWE reaction using 46. | |
Two enantiomers of AZABIC were also prepared via HWE olefination using the ylide of phosphonoglycinate 46 and the dialdehyde 78, followed by asymmetric hydrogenation of the unsaturated intermediate 79 catalyzed using a Rh(I)/Et-DUPHOS system.26 Then, hydrogenolysis of the N-protected amine, removal of the acetal and intramolecular cyclization afforded both enantiomers (S,S)-81a and (R,R)-81b with 71% and 74% yields, respectively (Scheme 20).
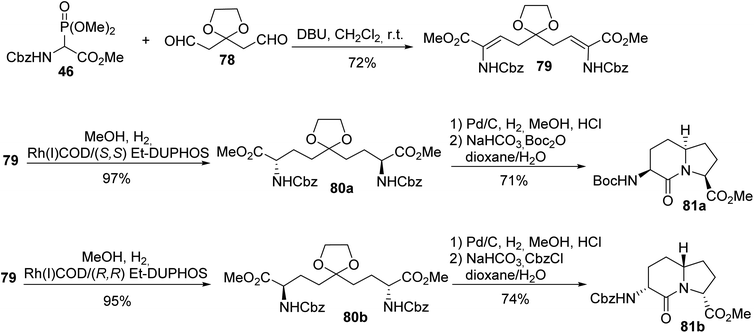 |
| | Scheme 20 The synthesis of constrained dipeptides 81 via the asymmetric hydrogenation of the dehydroamino acid 79 prepared using the HWE reagent 46. | |
The introduction of rigid non-natural amino acids into bioactive peptides induces conformational constraints into peptide backbones, tuning their pharmacological properties. Among those amino acids, an original synthesis of the conformationally restricted cubane alanine 84 was accomplished via HWE olefination under the conditions reported by Schmidt et al. The grafting of a cubane moiety onto the glycinate 4, followed by the hydrogenation of the resulting α,β-dehydroamino ester 83, the deprotection of the amine with TFA and the saponification of the methyl ester led to the cubane alanine 84 (Scheme 21).27,28
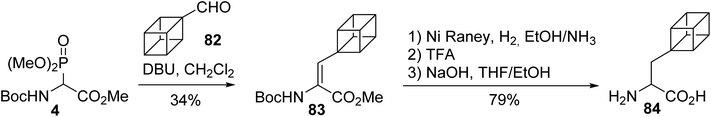 |
| | Scheme 21 The synthesis of the cubane alanine 84 via the hydrogenation of a dehydroamino ester prepared using the HWE reagent 4. | |
An approach for the treatment of chronic hepatitis C virus (HCV) infection consists of inhibiting the protein HCV NS5A with a small and highly potent molecule named BMS-986097. This compound has been selected as a lead candidate for preclinical toxicology studies, requiring its synthesis on a large scale. In 2017, the key intermediates 87a and 87b were developed to access the BMS-986097 inhibitor, based on HWE olefination involving the ylide of the readily accessible α-phosphonoglycinate 46 with the cyclopropyl tetrahydropyran ketone 85. Asymmetric hydrogenation of a Z/E mixture of 86 followed by separation provided the diastereoisomers 87a and 87b with 32% and 47% yields, respectively (Scheme 22).29
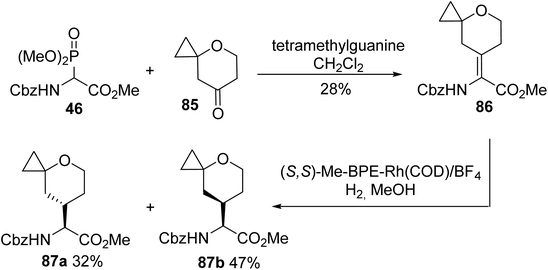 |
| | Scheme 22 The synthesis of the tetrahydropyranoamino esters 87 via the asymmetric hydrogenation of the dehydro-precursor 86 prepared using the HWE reagent 46. | |
Coelenterazine is a luciferin found in many aquatic organisms and its analogues are of considerable interest for bioluminescence assays. While known methods employ drastic reaction conditions, limiting the access to such compounds, the recent work of Kirkland et al. has reported an efficient strategy based on a HWE reaction between α-phosphonoglycinate 88 and pyridyl aldehyde 89, followed by the reduction of the olefin to afford the coelenterazine analogues 91 with a yield of up to 75% (Scheme 23).30 Following this strategy, derivatives containing various heterocyclic motifs and substituted aromatic groups with diverse electronic R2 substituents were characterized and used for the development of improved bioluminescence systems.
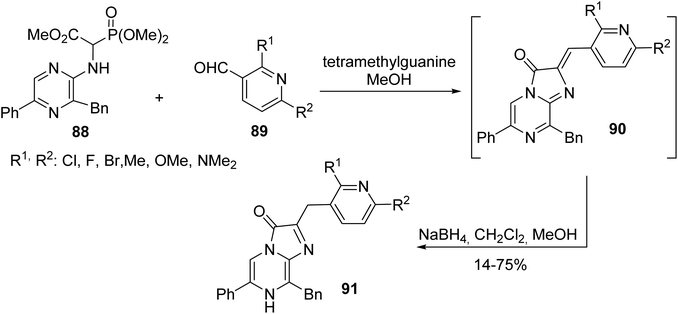 |
| | Scheme 23 The synthesis of the coelenterazine analogues 91 via olefination using the HWE reagent 88. | |
Among the known ionotropic glutamate receptors (iGluRs), the N-methyl-(D)-aspartate receptor (NMDAR) subtype is known to regulate signal transduction in the central nervous system. It has also been demonstrated that Alzheimer's and Huntington's neurodegenerative diseases are related to NMDAR dysfunction, due to their prolonged activation periods. Kaitocephalin (KCP), isolated form Eupenicillium shearii PF1191, was found to selectively antagonize NMDAR, displaying neuroprotective effects. Considering these results, the synthesis of the potent KCP analogues (S)-94 and (R)-94 was achieved to perform other structure–activity relationship studies.31,32 The amino acid moiety was introduced via an olefination reaction between the α-phosphonoglycinate 92 and aldehyde 93. Finally, reduction with a rhodium catalyst and deprotection using AlCl3 and Me2S led to the resulting mixture of diastereoisomers, which was further separated via chiral HPLC (Scheme 24).
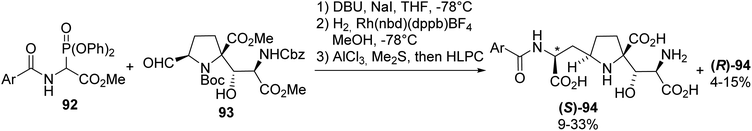 |
| | Scheme 24 The synthesis of the kaitocephalin analogue 94 via the asymmetric hydrogenation of a dehydro-precursor prepared using the HWE regent 92. | |
In the field of ligands designed to target receptors of the central nervous system that are related to neurodegenerative diseases, N-hydroxy-1,2-pyridone (Hop) amino acid, which exhibits similar metal-chelating properties to DOPA (3,4-dihydroxy phenylalanine) but without its unwanted redox activity, was readily obtained via a key HWE reaction between the ylide of α-phosphonoglycinate 46 and pyridyl aldehyde 95. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the intermediate 96 was reduced and the nitrogen atom of pyridine was oxidized, to afford the hydroxy-1,2-pyridone derivative 97 (Scheme 25). This amino ester was further transformed into the desired enantiopure protected Fmoc-(L)-Hop(tBu)-OH molecule 98 via enantioselective enzymatic hydrolysis and the product was used in peptide synthesis for the assembly of metallopeptides.33
C bond of the intermediate 96 was reduced and the nitrogen atom of pyridine was oxidized, to afford the hydroxy-1,2-pyridone derivative 97 (Scheme 25). This amino ester was further transformed into the desired enantiopure protected Fmoc-(L)-Hop(tBu)-OH molecule 98 via enantioselective enzymatic hydrolysis and the product was used in peptide synthesis for the assembly of metallopeptides.33
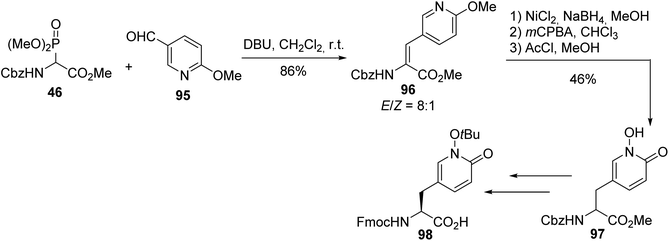 |
| | Scheme 25 The synthesis of the N-hydroxy-1,2-pyridone amino acid 98 via HWE olefination with the α-phosphonoglycinate 46. | |
Dysiherbaine, a natural product isolated from the Marine sponge Dysidea herbacea, is a selective agonist of CNS kainate subtype receptors. The enantioselective synthesis of (−)-dysiherbaine reported in 2015 by Gilbertson et al.34 is based on HWE olefination involving the α-phosphonoglycinate 46 and aldehyde 99, leading to the corresponding α,β-dehydroamino ester 100. Then, asymmetric hydrogenation using rhodium DuPhos in ethanol provided the protected version of dysiherbaine 101. Saponification with NaOH followed by ion exchange and reverse phase chromatography afforded (−)-dysiherbaine 102 with a quantitative yield (Scheme 26).35
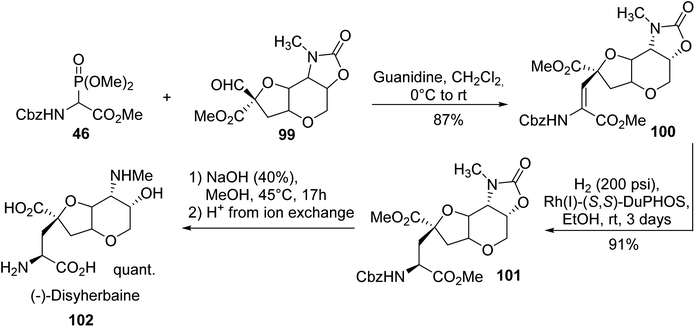 |
| | Scheme 26 The synthesis of (−)-dysiherbaine 102 via the asymmetric hydrogenation of the dehydro-precursor 100 prepared using the HWE reagent 46. | |
As exemplified above, the conditions developed by Schmidt et al. for HWE olefination provide a wide variety of α,β-dehydroamino ester derivatives efficiently, but usually, excess of organic base and low temperature are required for the deprotonation step. Alternatively, Lamaty et al. explored this reaction in the absence of solvent in a ball-mill for the synthesis of unsaturated amino esters. Under these conditions, α-phosphonoglycinate 4 was mixed with a series of aromatic and aliphatic aldehydes 103 in the presence of Cs2CO3 or K2CO3 as a base (Scheme 27). The corresponding α,β-dehydroamino esters 104 were obtained with good to excellent yields; however, two ketones, acetone and butanone, tested in this reaction were unreactive.36
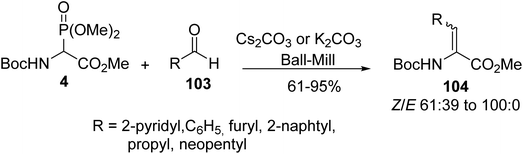 |
| | Scheme 27 The preparation of the dehydroamino acid 104 using the HWE reagent 4 via a ball-milling process. | |
3. Phosphorus linked to carbon in the β-position
3.1. β-Phosphino and β-phospholyl α-amino acids
3.1.1. Via nucleophilic phosphination with secondary phosphines. The first synthesis of β-phosphino α-amino acids was reported by Gilbertson et al. and consists of the hydrophosphination of acrylic acid with diphenylphosphine 25 in the presence of tetramethylammonium hydroxide as a base, leading to the thiophosphine acid 106 with an almost quantitative yield. Then, it was coupled with the Evans oxazolidinone lithium salt 107 and the diastereoselective transfer of an azido group to the enolate of oxazolidinone 108 gave the corresponding azide oxazolidinone 109 with 85% yield. Cleavage of the chiral auxiliary and the reduction of the azide with tin(II) chloride afforded the amino acid, which was protected to form the N-Fmoc (L)-diphenylphosphinoserine sulfide 110, which was ready for peptide synthesis (Scheme 28).37
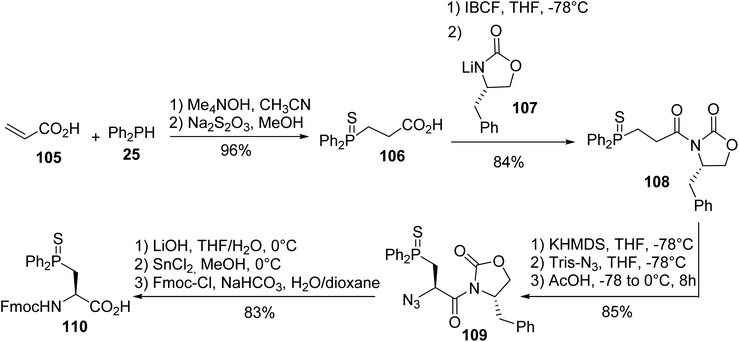 |
| | Scheme 28 The diastereoselective synthesis of (L)-diphenylphosphinoserine sulfide 110 using the Evans oxazolidinone 107. | |
The same strategy was applied to the preparation of the dicyclohexylphosphinoserine sulfide 116.38 First, the 1,4-addition of the dicyclohexylphosphide sulfide 111 anion to acrylic acid 105 provided the thiophosphine acid 112 for coupling to the Evans oxazolidinone lithium salt 107, according to the above-mentioned conditions. The treatment of compound 113 with Tris-N3, the reduction of the azide oxazolidinone 114 and the protection of the resulting amine with tert-butyl anhydride afforded the corresponding α-amino ester 115 with 93% yield. Finally, hydrolysis with lithium hydroxide gave the N-protected (L)-amino acid 116, a key building block in the syntheses of peptidomimetics with phosphine-containing side chains (Scheme 29).
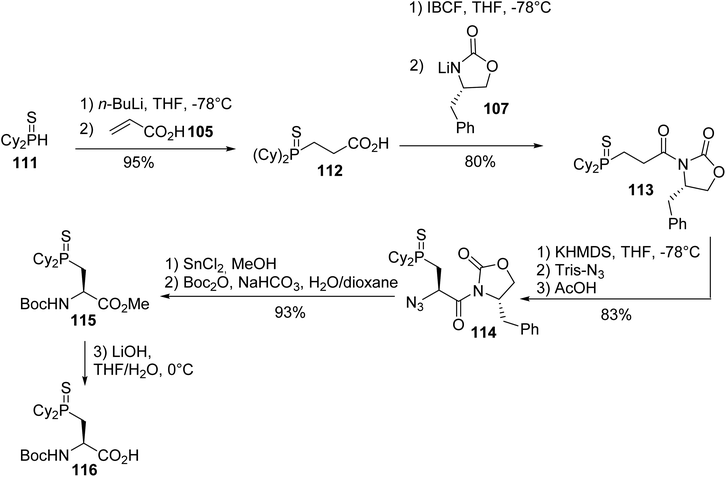 |
| | Scheme 29 The diastereoselective synthesis of (L)-dicyclohexylphosphinoserine sulfide 116 using the Evans oxazolidinone 107. | |
Burgess reported another strategy based on the nucleophilic substitution of the tosyl oxazolidine 117 with the diphenylphosphide anion 118, followed by the sulfuration of the phosphorus atom to furnish the corresponding thiophosphino oxazolidine 119 with 87% yield. Then, acidolysis cleaved the oxazolidine ring, but it also removed the N-Boc protection; this was reinstalled to give the alcohol 120. Finally, the optimized conditions for the oxidation of the primary alcohol were found, using pyridinium dichromate to afford the enantiopure (D)-diphenylphosphinoserine 121 (Scheme 30).39
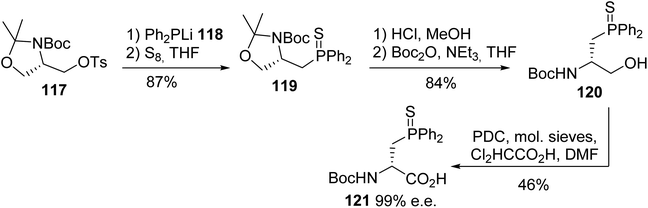 |
| | Scheme 30 The diastereoselective synthesis of the (D)-diphenylphosphinoserine 121 from the oxazolidine 117. | |
With the aim of preparing β-phosphinoamino acids on a larger scale compared to the multistage synthesis involving a chiral auxiliary reported by Gilbertson and Burgess, the nucleophilic substitution of the readily accessible (L)-iodoalanine 122, using a phosphide anion generated in situ under solid–liquid phase transfer conditions, was performed. Unfortunately, although the Ph2PH/K2CO3 heterogeneous system provided phosphide anions only at a low concentration, extensive racemization at the α-carbon atom of the starting material occurred and the phosphinoamino ester (±)-123 was observed (Scheme 31).40
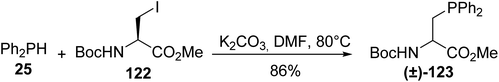 |
| | Scheme 31 The synthesis of the β-phosphinoamino ester 123 from sec-phosphine 25 under solid–liquid phase transfer conditions. | |
Based on these results, Le Floch et al. examined this approach for the nucleophilic substitution of iodoalanine using phospholide species. While no reaction was observed with the lithium 2,5-diphenylphospholide anion, probably due to the stabilization of negative charge within the ring, satisfactory results were obtained using the more nucleophilic 3,4-dimethylphospholide anion 124′. This anion was generated via the cleavage of the P–phenyl bond of the phenylphosphole 124 in the presence of lithium wires. After the removal of the metal excess, iodoalanine 122 was added to the anion and the tertiary phosphole was trapped with sulfur, leading to the corresponding 3,4-dimethylphospholyl sulfide amino ester 125 with 92% yield (Scheme 32).41 However, under these experimental conditions, where iodoalanine was in the presence of phospholide anion excess, racemization at the α-carbon position couldn't be excluded.
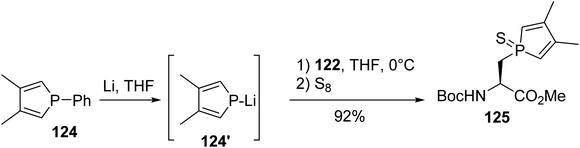 |
| | Scheme 32 The stereoselective synthesis of the phospholyl amino acid 125 via P–C bond formation. | |
3.1.2. Via coupling a nucleophilic zinc/copper amino ester with chlorophosphine. In addition to methodologies based on using chiral oxazolidinones to prepare enantiopure β-phosphino α-amino acids, a more direct route consists in coupling a nucleophilic zinc/copper reagent derived from iodoalanine 122 with electrophilic chlorophosphine. This reaction proceeds with a range of aromatic and aliphatic chlorophosphines, except for when using the less reactive chloro-di-tert-butylphosphine. The resulting tertiary phosphines were derivatized with sulfur in order to avoid their oxidation. The corresponding enantiopure β-phosphinoamino esters were isolated with 52 to 75% yields, depending on the electrophilicity of the chlorophosphine (Scheme 33).42,43
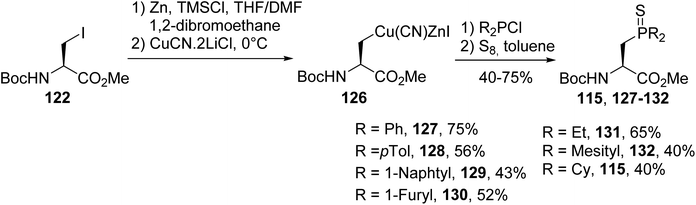 |
| | Scheme 33 The stereoselective synthesis of β-phosphinoamino esters via P–C coupling using the zinc/copper reagent 126. | |
As phosphines are widely used either as ligands or as catalysts to promote organic reactions, the development of β-phosphino α-amino acids paves the way to a new class of highly selective transition-metal catalysts and organocatalysts. In this context, Miller et al. examined the [3 + 2]-cycloaddition of the allene 133 and enone 134 catalyzed using (R)-diphenylphosphinylalanine 123. Indeed, this reaction is of particular interest for generating regioselectively in the conjugate-addition product or the cycloadduct 135a–b via switching between amine and phosphine catalysts. Under optimized conditions, the cycloaddition of the allenic ester 133 and enone 134 was achieved with high regio- and enantio-selectivity using the catalyst (R)-123 (Scheme 34).44 The formation of the major regioisomer 135a was rationalized using a transition state in which the enone approaches the zwitterion, which results from the addition of phosphine to the allene, via the π-face opposite to one of the phenyl rings of the catalyst.
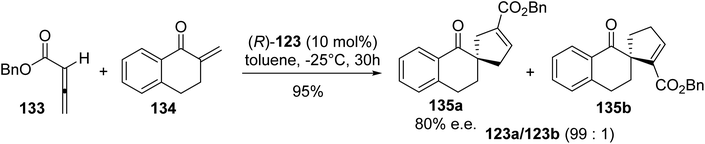 |
| | Scheme 34 The stereoselective [3 + 2]-cycloaddition reaction promoted by the diphenylphosphinylalanine (R)-123. | |
A library of peptide-based bisphosphine ligands, which are expected to form β-turn secondary structures, was designed and evaluated for palladium-catalyzed reactions between cyclic allyl acetates and dimethyl malonate (Scheme 35). Ligands bearing modified amino acids at each end of the turn were first evaluated, then phosphines with various substituents on the aromatic moiety were introduced into the most promising sequences. The reactivity and selectivity of the ligands were determined either in solution or when immobilized on a solid support. As an example, the addition of 3-acetoxycyclopentene 136 to dimethyl malonate 137 catalyzed by ligand 139 was achieved with 90% yield and 85% enantiomeric excess, which is a comparable result to when the catalyst was immobilized on a resin support (91% yield and 86% e.e.). When this reaction was performed at 0 °C in THF, the alkylated malonate 138 was obtained with excellent enantioselectivity of 95%.45–48
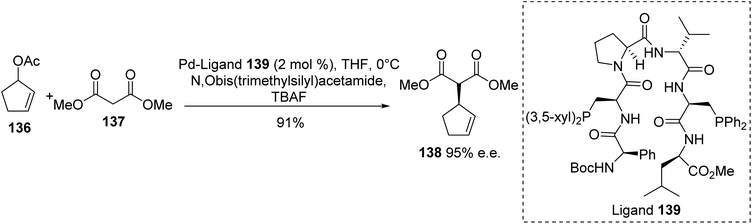 |
| | Scheme 35 Asymmetric Pd-catalyzed allylic alkylation using the bisphosphine peptide 139 as a chiral ligand. | |
3.1.3. Via the quaternization of triphenylphosphine with β-halogenoalanines. Phosphonium salts derived from amino acids are of particular interest as Wittig reagents for preparing β,γ-unsaturated amino acids with a variety of substituents on the side chain. The pioneering work of Itaya et al. consists of the quaternization of triphenylphosphine with the iodoalanines 140 and 141 in toluene at 50 °C for several days to afford the corresponding triphenylphosphonium iodide amino esters 142 and 143 with good yields of 76% and 97%, respectively (Scheme 36).49 Unfortunately, attempts to release the free carboxylic acid via the treatment of 142 with an aqueous solution of sodium hydroxide in methanol resulted in β-elimination. In addition, compound 143 can be converted into a chloride using an ion exchange resin to perform hydrogenolysis over Pd/C. In this case, the corresponding phosphonium amino acid 144 was obtained almost quantitatively.
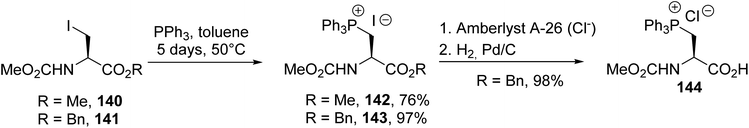 |
| | Scheme 36 The synthesis of the phosphonium amino acid derivative 144 from β-iodoalanine. | |
Its utility as an equivalent of the nucleophilic alanine synthon was exemplified by Wittig olefination reactions that led to β,γ-unsaturated α-amino acid derivatives. This method was applied to the synthesis of (S)-wybutine, a fluorescent base isolated from yeast phenylalanine transfer ribonucleic acids. It was necessary to use a phosphonium salt bearing the free carboxylic acid because, under basic conditions, the resulting carboxylate ylide protects against racemization. Furthermore, when the phosphonium amino ester 142 was used, β-elimination was observed in preference to the Wittig reaction. Under optimized conditions, the Wittig reaction between the phosphonium chloride amino acid 144 and the aldehyde derived from the modified nucleic base 145, followed by methylation, afforded the (E)-isomer of the β,γ-unsaturated amino ester 146 with 16% yield (Scheme 37). Finally, the CC bond and imidazole ring were hydrogenated to provide the corresponding wyosine α-amino ester 147.50–53
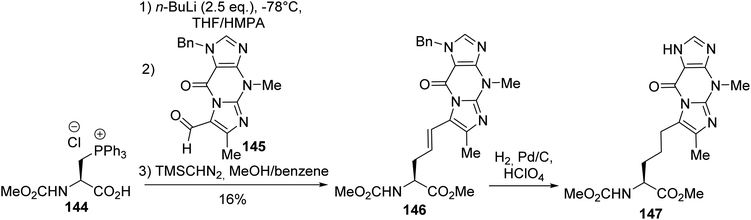 |
| | Scheme 37 A Wittig reaction using the β-triphenylphosphonium amino acid 144. | |
Subsequently, Jugé et al. reported a shorter synthesis protocol for the N-protected phosphonium salt amino acids 152 and 153. N-Benzoyl-β-bromoalanine 150 or N-benzoyl-β-iodoalanine 151, obtained from the ring-opening of oxazoline 149, was refluxed with triphenylphosphine in chloroform (Scheme 38). The appeal of this strategy arises from the mild conditions used for the quaternization of triphenylphosphine with the β-halogeno amino acid. The desired phosphonium salts were obtained without side products or racemization, as demonstrated from 31P NMR analysis with TRISPHAT as the chiral counter-anion and by comparison with a racemic sample.54 Unfortunately, their use in Wittig reactions with aldehydes resulted in poor reactivity and the unsaturated amino acid was obtained as a racemic mixture.
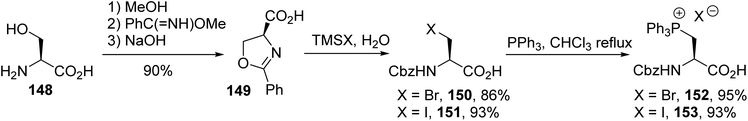 |
| | Scheme 38 The synthesis of triphenylphosphonium amino acids via the ring-opening of the oxazoline 149 and quaternization using PPh3. | |
3.2. β-Phosphono α-amino acids and peptides
3.2.1. Via the reductive amination of phosphonopyruvate. The pioneering work of Savignac et al. in 1980 involved the synthesis of β-phosphono α-amino esters via the reductive amination of phosphonopyruvates 154 in the presence of NaBH3CN (Scheme 39). The substituents (OR) carried by the phosphorus atom have no influence on this reaction; however, the replacement of oxygen (X) with sulfur leads to lower yields, probably due to steric hindrance. Finally, the corresponding phosphonic α-amino acids 156a–e were obtained after hydrolysis with hydrochloric acid (6 N) in satisfactory yields.55
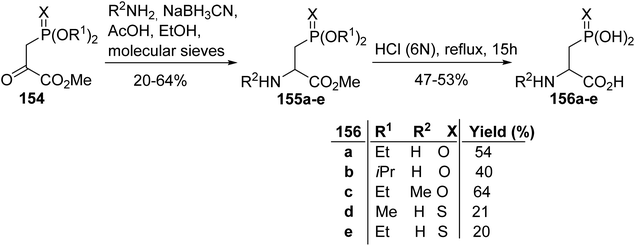 |
| | Scheme 39 The synthesis of the β-phosphono α-amino esters 156a–e from the phosphonopyruvates 154. | |
3.2.2. Via the alkylation of a nickel(II) complex of a glycine Schiff base. Among the chiral auxiliaries commonly involved in the asymmetric synthesis of unnatural α-amino acids, a nickel(II) complex of a glycine Schiff base was used for the preparation of enantiopure β-phosphono α-amino acids.56 When the complex 157 was deprotonated with potassium hydroxide, the resulting enolate reacted successfully with iodomethyl diisopropylphosphite 158 to afford the corresponding alkylated Schiff base as a single diastereoisomer. After hydrolysis, the β-phosphono α-amino acid (L)-159 was obtained with 30% yield and excellent enantiomeric purity of 99% (Scheme 40).
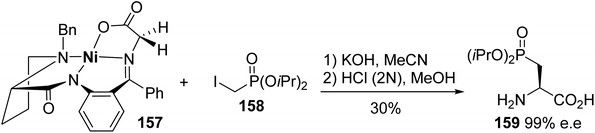 |
| | Scheme 40 The synthesis of the β-phosphono α-amino acid (L)-159 via the asymmetric alkylation of the Schiff base complex 157. | |
4. Phosphorus linked to carbon in the γ-position
4.1. γ-Phosphino and γ-phospholyl α-amino acids
4.1.1. Via the quaternization of triphenylphosphine with γ-iodoamino esters. The synthesis procedures for β-phosphonium salt α-amino acids reported above involve demanding strategies. In addition, their use in Wittig reactions results in racemic mixtures of unsaturated amino acids with low yields. Considering these limitations, another class of α-amino acid Wittig reagent 172, having both free carboxylic functionality and a phosphonium moiety in the γ-position on the lateral chain, was reported with the objective of avoiding racemization or eliminating the triphenylphosphine during Wittig olefination. The stereoselective synthesis of the γ-phosphonium salt amino acid 172 was achieved with 58% overall yield via the quaternization of triphenylphosphine with the γ-iodoamino ester 170 at 80 °C without solvent, followed by deallylation with Pd2dba3. The enantiomeric excess was checked via 31P NMR studies with BINPHAT as the chiral counter-anion, proving that no racemization occurred during the reaction.61 Wittig reactions with aromatic or aliphatic aldehydes were performed under solid–liquid phase-transfer conditions in chlorobenzene and in the presence of K3PO4 as a weak base, affording the unsaturated amino acids 173a–d and 174–178 as Z/E mixtures from 30:70 to 10:90, without racemization (Scheme 44). Under these conditions, the inorganic base deprotonated the carboxylic acid functionality and methylene substituent to form the ylide. In chlorobenzene, as a non-dissociative solvent, the phosphonium salt 172 favored the formation of an ion pair with the carboxylate moiety, enabling better solubility and reactivity of the ylide with the aldehyde. The amino acids 176 and 177, bearing aryl boronate and silylated moieties on the side chains, were also obtained under these conditions and they are potentially useful for further functionalization or for 18F-labeling (Scheme 44).62,63
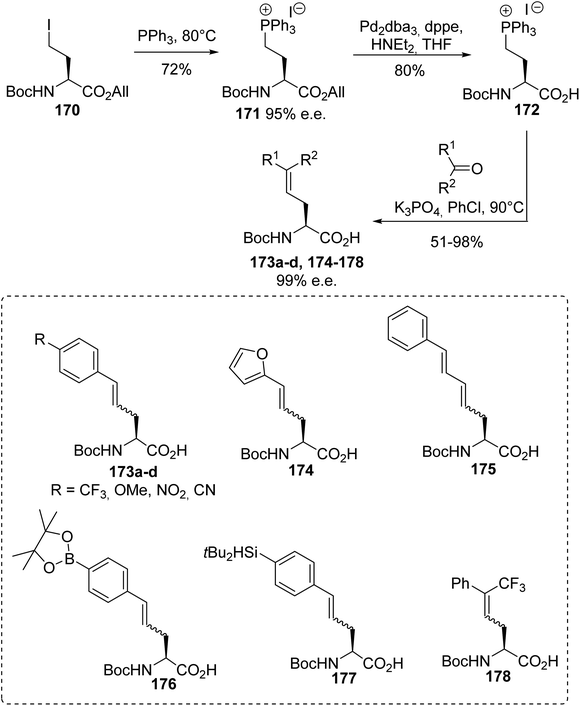 |
| | Scheme 44 The γ-phosphonium α-amino acid Wittig reagent 172: its synthesis and application to the preparation of γ,δ-dehydroamino acids. | |
Ortho-boronato-phenylphosphonium amino acids were also prepared according to this strategy via the quaternization of ortho-boronato-diphenylphosphine 180 with the γ-iodoamino ester 179. Then, saponification led to compound 182 with a free carboxylic acid and the deprotection of the amine was performed via acidolysis into the corresponding ammonium salt 183 (Scheme 45). The feasibility of peptide coupling was demonstrated, as well as transformation into trifluoroborates. Finally, carrying out radiolabeling to obtain the [18F]-trifluoroborato-phenylphosphonium α-amino acid 6 demonstrated the utility of such compounds for peptide labeling.64
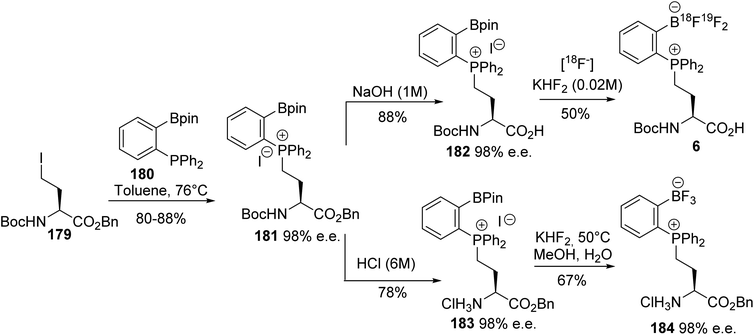 |
| | Scheme 45 The ortho-borato-phenylphosphonium α-amino acid 181: its synthesis and application to 18F-radiolabeling. | |
4.1.2. Via the substitution of γ-iodoamino esters with a phosphide anion. A glycopeptide CSF114 (Glc) modified with an α-amino acid bearing a ferrocenyl thiophosphine substituent on its side chain was developed for the detection of autoantibodies in the serum of patients suffering from multiple sclerosis disease (MSD).65 The ferrocenyl group of this marker allowed for the electrochemical detection of autoantibodies at potentials compatible with the biological medium and the thiophosphine substituent was attached to a gold surface for the development of a sensor. The diastereoselective synthesis of the amino acid marker 189 relies on the condensation of the enantio-enriched secondary ferrocenylphosphine borane 185 (86% e.e.), which was generated according to (−)-ephedrine methodology, with the N,N-diprotected γ-iodoamino ester 186. The double protection of the amine with tert-butyloxycarbonyl (Boc) groups was necessary to avoid secondary reactions under basic conditions. The resulting phosphine borane amino ester was subjected to deprotection with DABCO and sulfurization to give the corresponding thiophosphine derivative 188 with 80% yield. Finally, the removal of one Boc-protecting group and the saponification of the benzyl ester led to the ferrocenyl thiophosphinoamino acid 189, which was ready for peptide coupling with 76% yield and 86% diastereomeric purity (Scheme 46). After electrochemical grafting of the labeled antigen (FcPS-CSF114 (Glc)) 190 and incubation with autoantibodies, antigen/antibody recognition was possible via electrochemistry at very low concentrations, comparable to ELISA testing.
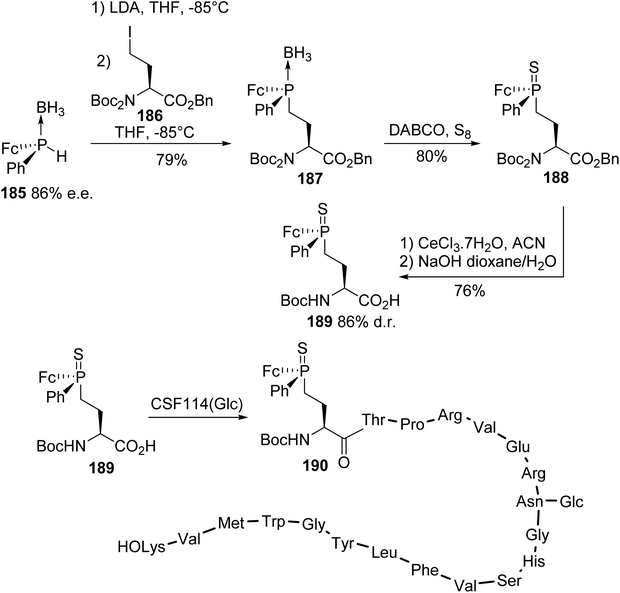 |
| | Scheme 46 The ferrocenyl thiophosphinoamino acid 189: its diastereoselective synthesis via P–C bond formation and application to the electrochemical detection of MSD autoantibodies. | |
The examples reported above demonstrate the interest in phosphorus and boron chemistry for creating customized or labeled peptides. In the following examples, phosphine boranes are used as the precursors for grafting [60]fullerene to an amino acid side chain.66 Fullerene derivatives have been studied for their biological activities and for medical diagnosis, as they are without known toxicity. Phenylphosphine borane 191 was linked to the side chain of the γ-iodoamino ester 186 under solid–liquid phase transfer conditions, leading to the corresponding phosphine borane amino ester 192 with 98% yield. The resulting sec-phosphine borane was subsequently grafted to [60]fullerene under the same conditions to give the [60]fullerene γ-phosphine borane α-amino ester 5 as an epimeric mixture (1:1), due to the existence of a P-chirogenic center without racemization (Scheme 47). The reversibility of the hydrophosphination was demonstrated via cyclic voltammetry, demonstrating the ability to immobilize and release amino acids or peptides on carbon nanomaterials.
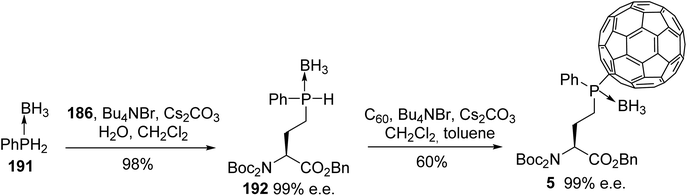 |
| | Scheme 47 Grafting α-amino acids to [60]fullerene using phosphine borane as a linker. | |
For our part, we recently reported the first synthesis of phospholyl(borane) α-amino acids, as a new class of fluorescent amino acids, via the nucleophilic substitution of β- or γ-iodoamino esters with phospholide anions.67 The phospholide anion 193′ was generated via the deprotonation of dibenzophosphole or via the cleavage of the P–phenyl bond starting from the phenylphosphole in the presence of potassium. After the removal of the excess metal, the anion was added to a solution of γ-iodoamino ester 170 and the resulting tertiary phospholyl amino ester was complexed in situ with BH3.DMS (Scheme 48).67 Under these conditions, the phospholyl(borane) (L)-α-amino ester derivatives 194–197 were isolated as stable compounds, which were easy to store and handle for further use.
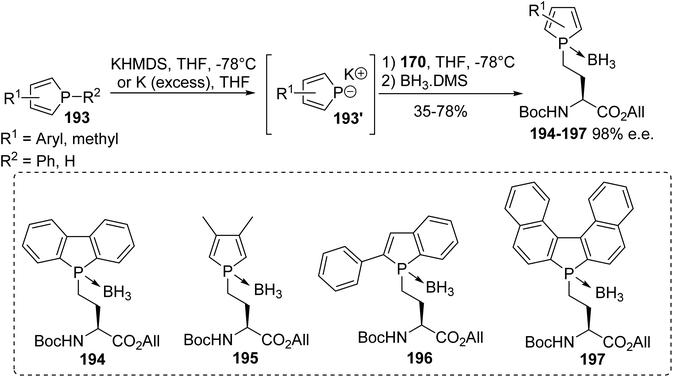 |
| | Scheme 48 The stereoselective synthesis of the phospholyl(borane) (L)-α-amino esters 194–197 via P–C bond formation. | |
The phospholyl(borane) α-amino ester 194 was successfully transformed via decomplexation, oxidation, sulfuration, complexation with gold and quaternization, respectively, to provide the derivatives 198–202 with tunable fluorescence emission.67 Via a tandem reaction involving decomplexation and sulfuration, the binaphtyl sulfur amino ester 7 was obtained with 64% yield. It exhibits remarkable fluorescence emission in the green range at 535 nm, with a large Stokes shift of 160 nm. Such phospholyl amino acids were also selectively deprotected for carrying out peptide C- and N-coupling, either in solution or on a solid support. These molecules constitute a promising new class of probe for the fluorescence labeling of peptides (Scheme 49).67
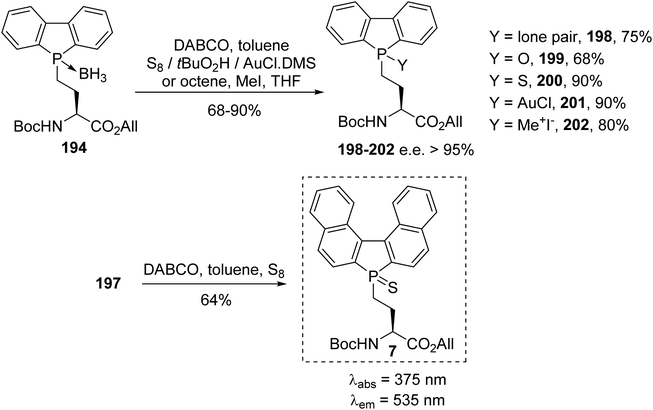 |
| | Scheme 49 Chemical transformations at the P-center of the phospholyl(borane) amino acid 194. | |
The incorporation of phosphino α-amino acids that can coordinate transition metals, such as rhodium, ruthenium, palladium and platinum, at specific positions into peptides provides a method to stabilize their secondary structures to control their reactivity. In this context, phosphine-containing proline derivatives were developed to coordinate with rhodium and ruthenium into β-turn structural motifs. This synthesis is based on the substitution of γ-mesylate prolinol 203 using the diphenylphosphide anion 204, followed by the sulfuration of the phosphorus center. Then, the amine of the resulting phosphine sulfide prolinol was protected and the alcohol was oxidized, leading to the N-Boc- and N-Fmoc-γ-thiophosphinoprolines 206 and 207, with 21% and 27% overall yields, respectively (Scheme 50). These phosphinoprolines and a diphenylphosphine alanine derivative were introduced at the i and i + 2 positions, forming a tetramer with a type II β-turn conformation in which both phosphines are on the same face to bind with transition metals.68
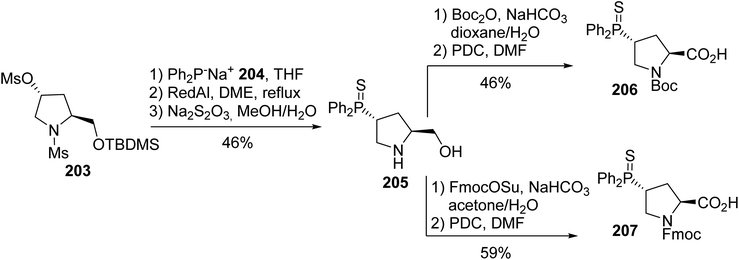 |
| | Scheme 50 The stereoselective synthesis of the diphenylphosphinoprolines 206 and 207. | |
4.2. γ-Phosphinate and γ-phosphono α-amino acids
4.2.1. Via a Michaelis–Arbuzov reaction with phosphite and γ-bromoamino esters. (L)-Phosphinothricin is a natural phosphinic acid analogue of (L)-glutamate, which is isolated from two different Streptomyces species; it is shown to have antibiotic and herbicidal properties and to be an inhibitor of glutamine synthetase, blocking the incorporation of ammonia into amino acids and pyrimidines. Various γ-phosphonic α-amino acid analogues, (L)-AP4, were also developed for structure–activity relationship studies. Racemic (D,L)-phosphinotricin and one analogue were prepared via a Michaelis–Arbuzov reaction between diethyl methylphosphonite or triethyl phosphite and the γ-bromoamino ester 208.69 After heating in toluene, the resulting phosphonate was treated with HCl (6 N) to give the corresponding (D,L)-phosphinotricin (±)-3 or its free phosphonic α-amino acid 211 with 85% yield (Scheme 51).
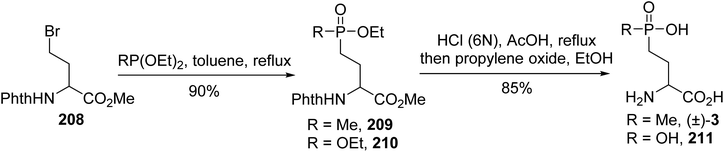 |
| | Scheme 51 The synthesis of phosphinotricin (±)-3 via a Michaelis–Arbuzov reaction. | |
4.2.2. Via the hydrophosphination of vinyl oxazolidinone. Later, the enantiopure (L)-phosphinotricin was synthesized based on the regioselective hydrophosphination of the vinyl oxazolidinone 212 with methyl phosphonate 213, in the presence of a perester catalyst.70 The intermediate phosphonate oxazolidinone 214 was isolated with 77% yield. Acidic treatment led to the ammonium chloride, which was neutralized via the addition of propylene oxide to obtain the enantiopure (L)-phosphinothricin 3 (Scheme 52).
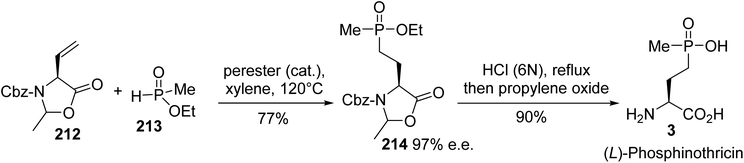 |
| | Scheme 52 The stereoselective synthesis of (L)-phosphinotricin 3 via the hydrophosphination of the vinyl oxazolidinone 212. | |
4.2.3. Via the palladium catalyzed C(sp3)–H alkylation of aminoquinoline derivatives. In 2017, an efficient procedure for the construction of chiral γ-phosphono-α-amino acids via the Pd catalyzed C(sp3)–H alkylation of alanine aminoquinoline derivatives was developed.71 Coupling between the methylene C(sp3)–H bond of the substrate 215 and phosphonated alkyl iodides led to the alkylated products 218 and 219 (Scheme 53). Then, the auxiliary group was removed via treatment with Boc2O and DMAP and LiOH and H2O2 in two steps. Finally, after the removal of the phthaloyl and ester groups in the presence of HCl (6 N), (L)-phosphinothricin 3 and (L)-AP4 were obtained with 61 and 72% yields, respectively (Scheme 53).
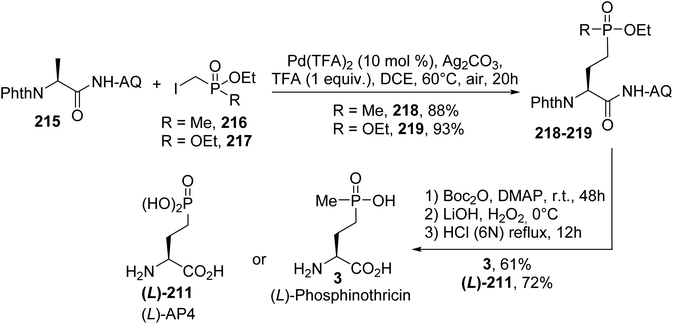 |
| | Scheme 53 The stereoselective synthesis of (L)-phosphinothricin 3 and (L)-AP4 211 via alkylation at the β-position of the alanine derivative 215. | |
4.2.4. Diastereoselective synthesis via the Schöllkopf method. Another strategy for the asymmetric synthesis of γ-phosphono α-amino esters relies on the use of Schöllkopf bis-lactim ether as a chiral auxiliary.72 After deprotonation by n-BuLi, the conjugate addition of the corresponding lithium salt to the vinyl phosphonate 222 leads exclusively to the diastereoisomer trans-223 with 81% yield. In solution, the alkylation adduct was protonated by diethyl 2-bromoethylphosphonate 221, which regenerated the vinyl phosphonate 222 in situ after dehydrobromination.73 Acidolysis, followed by the protection of the amine with Boc2O, led to the γ-phosphonate 224 with 74% yield (Scheme 54). This amino ester was used to prepare analogues of sphingosine-1-phosphate and sphinganine-1-phosphate, which are present in cell membranes as intermediates during the degradation of ceramide and the biosynthesis of sphingolipids, respectively.
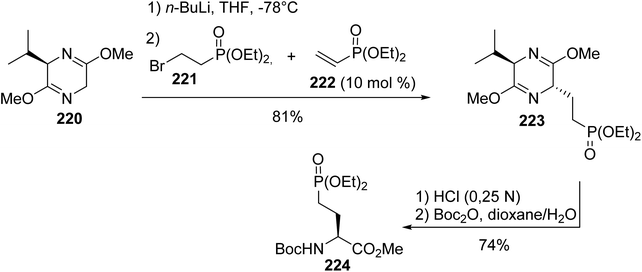 |
| | Scheme 54 The diastereoselective synthesis of the γ-phosphonoamino ester 224 from the Schöllkopf bis-lactim ether 220. | |
Later, this methodology was applied to the synthesis of a variety of enantiomerically pure 4-substituted AP4 derivatives, which are useful for elucidating the role of metabotropic glutamate receptors in the central nervous system and for the development of potent and selective pharmacological agents.74 When the electrophilic substitution of the phosphonate bis-lactim ether 225 was performed via trapping the anion at the α-position of the phosphonate with alkyl halides or other electrophile sources, compounds syn-226 and anti-226, bearing a functional group, were obtained with up to 85% yield. Then, after separation, the bis-lactim ether of the intermediate syn-226 was cleaved and the resulting phosphonate amino ester was treated with HCl (12 N) to provide the corresponding free phosphonic amino acid 228 (Scheme 55).
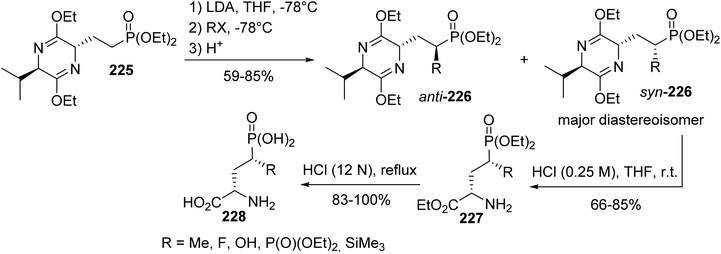 |
| | Scheme 55 Functionalizing the α-position of the γ-phosphonoamino acid 228 obtained from the bis-lactim ether precursor 225. | |
To explore the scope of these electrophilic substitutions, olefination reactions were examined involving α-stabilized phosphonate carbanions. As an example, tin-Peterson olefinations with the α-triphenylstannyl derivative 229 and aryl or alkyl aldehydes were performed to give the (Z)-vinyl phosphonates 230 in moderate yields. Bis-lactim ether cleavage and a final deprotection step under strongly acidic conditions afforded the corresponding enantiopure unsaturated phosphonic α-amino acids 232 (Scheme 56).
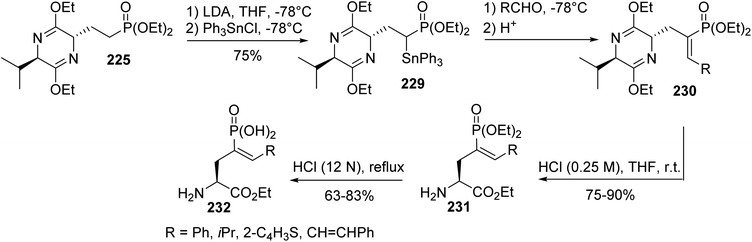 |
| | Scheme 56 The diastereoselective synthesis of the phosphonic α-amino acid 232 via the functionalization of the phosphono bis-lactim ether 225. | |
4.2.5. Via the condensation of methylene bisphosphonate using Garner's aldehyde. An alternative synthetic route to AP4 derivatives was based on the Horner–Wadsworth–Emmons reaction via the condensation of a methylene bisphosphonate anion, using Garner's aldehyde 234 as the chiral pool. The resulting olefins 235a–b, bearing α-methylene- or α-fluoro-phosphonates, were then reduced via hydrogenation over Pd/C to give 236a–b with 99% and 88% yields, respectively. Finally, deprotection and oxidation steps using Jones reagent were simultaneously performed to afford the corresponding γ-phosphonoamino acids (R)-237a–b (Scheme 57).75 Moreover, such amino acids are involved as key intermediates in the design of N-arylamide phosphonates, which are selective agonists or antagonists of sphingosine 1-phosphate (S1P) receptors.
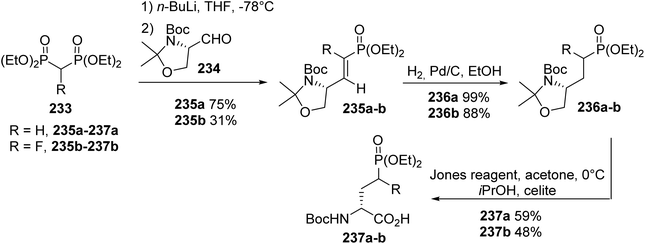 |
| | Scheme 57 The stereoselective synthesis of γ-phosphonoamino acids 237a–b from Garner's aldehyde 234. | |
4.2.6. Via the Abramov reaction of phosphite with β-formylamino esters. Besides the asymmetric synthesis routes reported above for the preparation of (D)- and (L)-AP4 derivatives involving oxazolidinone, Schöllkopf bis-lactim ether and Garner's aldehyde as chiral auxiliaries, a hemisynthetic approach for accessing such compounds relies on the Abramov reaction of the aldehyde 238, prepared from (L)-aspartic acid, with dimethyl trimethylsilylphosphite.76,77 The resulting 4-hydroxy-4-dimethylphosphono derivative 239 was obtained with 98% yield after the in situ hydrolysis of the trimethylsilyl group. Hydride substitution of the hydroxyl group was achieved via homolytic deoxygenation through the treatment of the thiocarbonylimidazolide derivative with tributyltin hydride. Finally, the Boc and tert-butyl protecting groups were removed with TFA; Boc was then reintroduced under standard conditions, leading to the enantiopure γ-phosphono α-amino acids 242 and 243 with 63% and 68% yields, respectively (Scheme 58). The optical purity was determined via chiral HPLC in comparison with the dipeptides derived from (L)-leucine and the racemate using (D,L)-leucine.
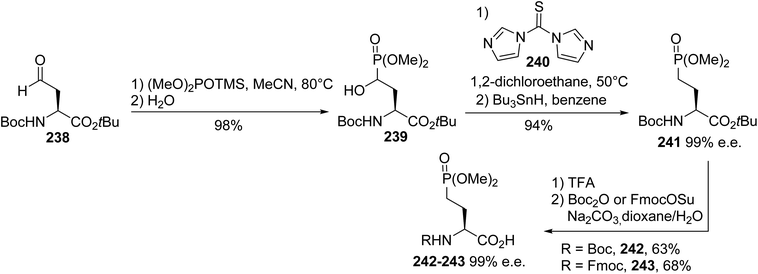 |
| | Scheme 58 The stereoselective synthesis of the γ-phosphonoamino acids 242 and 243 via the Abramov reaction involving the β-formylamino ester 238. | |
4.2.7. Via the enantioselective acylation of prochiral phosphonodiols. The enantiocontrolled syntheses of phosphonate α-amino acids in (L)- and (D)-forms have been considerably developed, but desymmetrization to provide chiral analogues of naturally occurring biological phosphates is less explored. For this purpose, Shibuya et al. reported the first desymmetrization of prochiral phosphono-l,3-propanediols, although this strategy was previously applied to other 2-substituted 1,3-propanediols.78 The nucleophilic substitution of triflate 244 using the methyl phosphonate anion 245 afforded the phosphono ketal 246 with 70% yield. Then, this was subjected to transesterification with lipase forming the intermediate 247 with high enantioselectivity of 93%. The amino phosphonate 248 was obtained after Jones oxidation and amination with diphenylphosphoryl azide (DPPA), followed by protection with benzyl carbamate. Finally, the deprotection of the acetate and a second Jones oxidation step led to the γ-phosphonate (L)-amino acid 249 with 47% yield and 93% e.e. (Scheme 59).
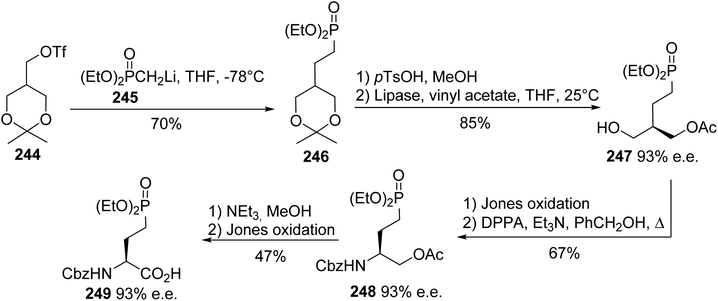 |
| | Scheme 59 The asymmetric synthesis of the γ-phosphonoamino acid 249 via the enzymatic desymmetrization of the substrate 246. | |
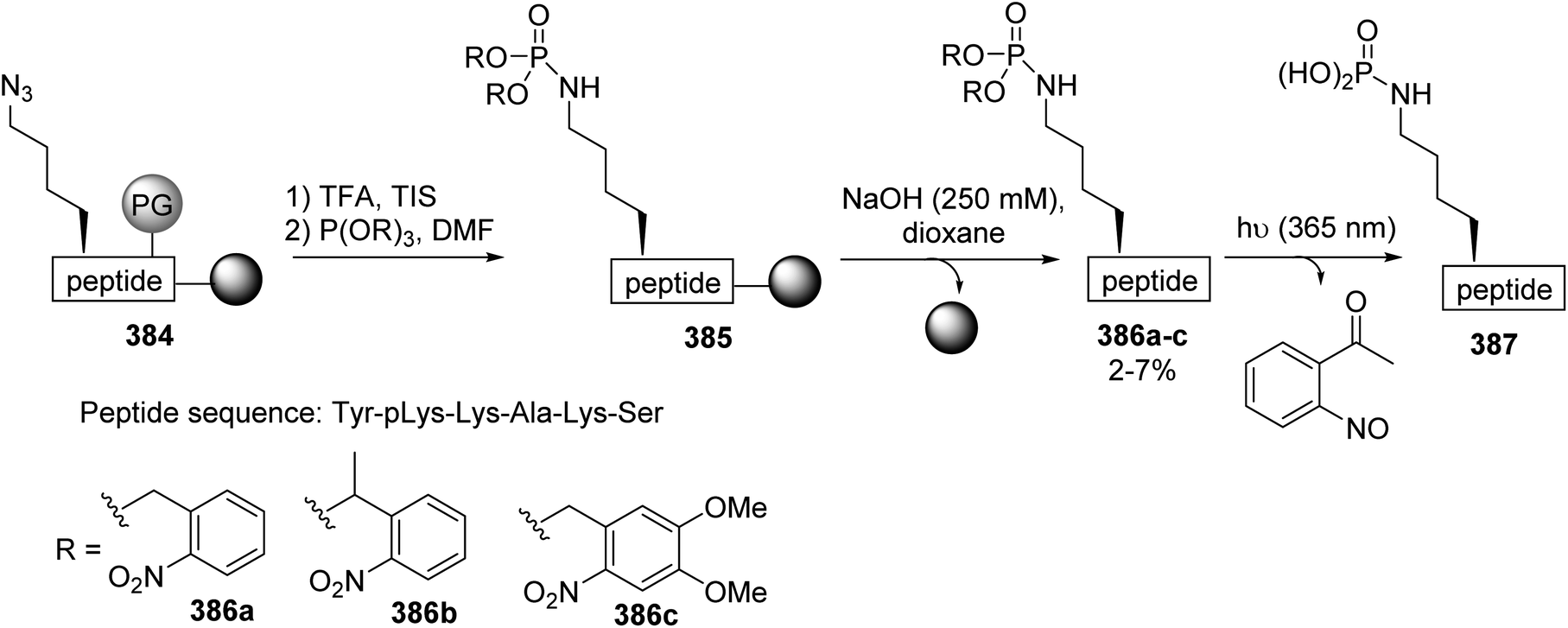
Phosphopeptides are tools for understanding the biological functions of the phosphorylation regulating phosphoserine (pSer) and phosphothreonine (pThr) residues that are involved in protein–protein interactions. As phosphate moieties are easily hydrolyzed by phosphatases in vivo, several mimics containing non-hydrolysable methylene phosphonate or difluoromethyl phosphonate units with similar physicochemical properties to the phosphate moiety were developed. The synthesis of CF2-substituted pSer and pThr mimics reported in 2018 by Chen et al. illustrates recent progress in this area.79 The key step of this synthesis is the substitution of the iodine and bromine α,β-dehydroamino esters 250 and 255, using the difluorophosphonate copper/zinc reagent 251, to afford the unsaturated intermediates 252 and 256 with 85% and 70% yields, respectively. These were subjected to asymmetric hydrogenation catalyzed by a rhodium/DuPhos complex and deprotected using TFA or HCl (9 N). The resulting free amines were finally protected with FmocOSu, affording the corresponding N-Fmoc difluoro-substituted pSer 254 and pThr 258 mimics with high enantioselectivity (Scheme 60a and b). Furthermore, those amino acids were readily used in SPPS to provide phosphatase-resistant peptides that exhibit remarkable binding efficacy compared to the parent phosphopeptides.
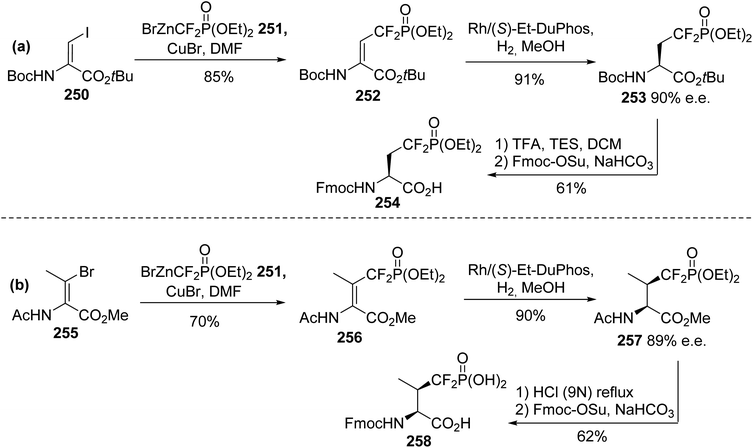 |
| | Scheme 60 The synthesis of the difluorophosphonoamino acids 254 and 258 via cross-coupling reactions using the difluorophosphonate copper/zinc reagent 251 (a) or 255 (b). | |
4.2.8. Via the Michael addition of azalactone to vinylidene bisphosphonate. Quaternary amino acids are relevant structures for improving stability when incorporated into peptides. Geminal bisphosphonates constitute another important class of pharmacologically active compounds and they are involved in the treatment of bone disorders such as Paget's disease, myeloma, bone metastases and osteoporosis.80 In this context, Albrecht et al. developed a new class of quaternary α-amino acids bearing a bisphosphonate moiety on the side chain in order to improve the properties of both these compounds. The strategy consists of an enantioselective Michael reaction between the α-substituted azalactone 259 and vinylidene bisphosphonate 260 in the presence of Cinchona alkaloids as chiral Brønsted bases, followed by ring opening under acidic conditions (Scheme 61). Among the different Cinchona alkaloids evaluated, the highest enantioselectivity (37% e.e.) was obtained using catalyst 261, which suggests that further optimization of this process is possible.
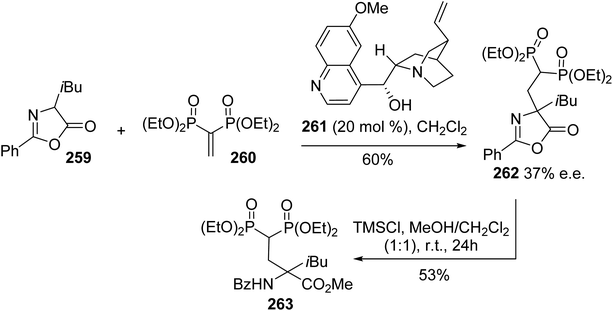 |
| | Scheme 61 The asymmetric synthesis of the bisphosphonate amino ester 263 via an organocatalyzed Michael-type reaction. | |
5. Phosphorus linked to carbon in the δ-position
5.1. δ-Phosphono α-amino acids
5.1.1. Via four-component Ugi condensation. One of the first synthetic route for δ-phosphonic α-amino acids was reported in 1988 by Natchev et al. The key step for introducing the phosphorus moiety is based on a Michaelis–Arbuzov reaction between the bromopropene acetal 265, readily obtained from the aldehyde 264 and triethyl phosphite.81 The corresponding unsaturated phosphonate 266 was obtained and the acetal was deprotected into the aldehyde 267. Finally, compound 267 was condensed with ammonium formate, cyclohexyl isonitrile and sodium hydroxide to provide the unsaturated δ-phosphonic α-amino acid 269 in a single step with 79% yield. Enzymatic approaches were used to separate the (L)- and (D)-isomers (Scheme 62).
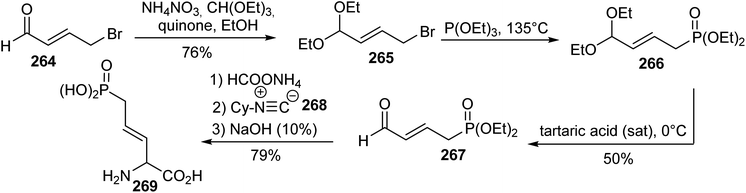 |
| | Scheme 62 The synthesis of the δ-phosphonic α-amino acid 269 via the four-component Ugi condensation of the phosphonoaldehyde 267. | |
5.1.2. Via the diastereoselective alkylation of Seebach's imidazolidinone. (S)-2-Amino-5-phosphonopentanoic acid, (L)-AP5, is a potent antagonist for the N-methyl-(D)-aspartic acid receptor (NMDAR) and can be synthesized starting from Seebach's imidazolidinone.82 The resulting enolate, obtained via the treatment of the imidazolidinone 270 with LDA, was added to 3-bromopropyl phosphonate 271 to obtain the trans-substitution product 272 with 65% yield and high diastereoselectivity, as assigned via 1H NMR. Hydrolysis under acidic conditions led to the enantiopure aminophosphonic acid 273 (Scheme 63). This strategy was also applied to the preparation of the enantiomer (D)-AP5, taking care with the choice of chiral inductor; this is because when starting from N-methyl imidazolidinone, racemization due to the assistance of the neighbouring phosphonate group was observed during hydrolysis.
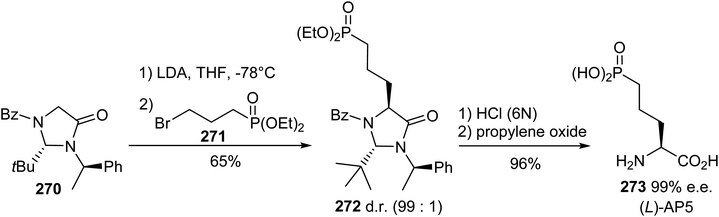 |
| | Scheme 63 The diastereoselective synthesis of the δ-phosphonic α-amino acid 273 via the alkylation of the Seebach imidazolidinone 270. | |
5.2. β-Keto-δ-phosphonate α-amino acids
5.2.1. Via the addition of cuprate phosphonate to acid chloride oxazolidinone. The (L)-AP5 antagonist exhibits suitable affinity (IC50 = 5.5 μM) towards NMDAR, but its weak blood–brain barrier crossing ability results in weak systemic activity. Considering this limitation and on the basis of molecular modeling studies, the analogue (R)-4-oxo-5-phosphononorvaline 278 was identified as a potent ligand and it was prepared via a convergent synthesis procedure reported by Whitten et al.83 The N-protected oxazolidinone acid chloride 275, obtained from (D)-aspartic acid 274, reacted with the nucleophilic diethyl methylphosphonate cuprate 276 to provide the alkylated imidazolidinone 277 with 71% yield. Finally, ring opening with TMSI and ion exchange gave the (R)-γ-keto-δ-phosphonic α-amino acid 278 with 68% yield and 95% e.e. (Scheme 64).
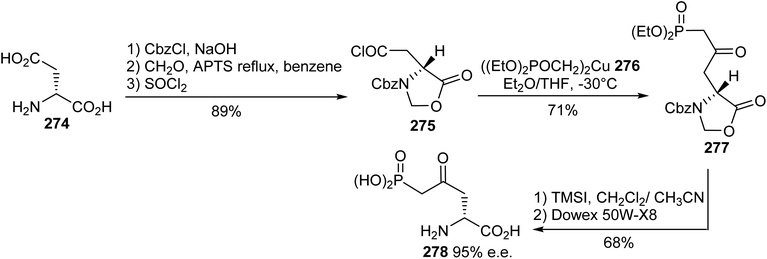 |
| | Scheme 64 The stereoselective synthesis of the γ-keto-δ-phosphonic α-amino acid 278 via P–C bond coupling with the acid chloride 275. | |
A more straightforward strategy consists of opening the β-lactam with the diethyl methylphosphonate lithium salt 245.84 Under these conditions, the enantiopure δ-phosphono-γ-ketoamino esters 280a–b, bearing Boc and Cbz protecting groups, respectively, were obtained with yields of up to 94% (Scheme 65). The optical purity was determined via 19F and 1H NMR studies on the Mosher salts after deprotection of the amine.
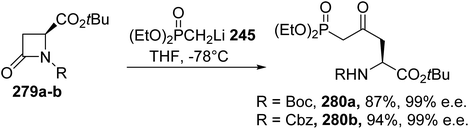 |
| | Scheme 65 The stereoselective synthesis of the δ-phosphono-γ-ketoamino esters 280 from the β-lactam 279. | |
Sutherland et al. exploited another approach based on the nucleophilic addition of the methyl diethylphosphonate lithium salt 245 to the methyl ester on the side chain of the aspartate derivative 281. It must be noted that a sterically hindered trityl protecting group is necessary for regioselective addition to the ester of the side chain (Scheme 66).85–87 These authors also reported the synthesis of optically active β-pyridyl α-amino acids displaying fluorescence properties.88 First, (E)-enones 284 were obtained via Horner–Wadsworth–Emmons reactions between the δ-phosphono-γ-keto α-amino ester 282 and various substituted benzaldehydes 283. Then, the trityl group was removed and replaced by an N-protecting group (Cbz) under standard conditions. A regioselective hetero-Diels–Alder reaction between the enone derived from the α-amino ester 285 and ethyl vinyl ether 286 was performed using an ytterbium catalyst. Finally, a modified Knoevenagel–Stobbe reaction with hydroxylamine hydrochloride at 70 °C led to pyridines 288 bearing various aromatic substituents with good yields from 58% to 71% (Scheme 66). The removal of both the amine and carboxylic acid protecting groups was performed to give the free amino acids. These amino acids exhibit fluorescence emission from 380 to 460 nm, with large Stokes shifts and intensities that depend on the nature of the electron-deficient or -rich substituents.
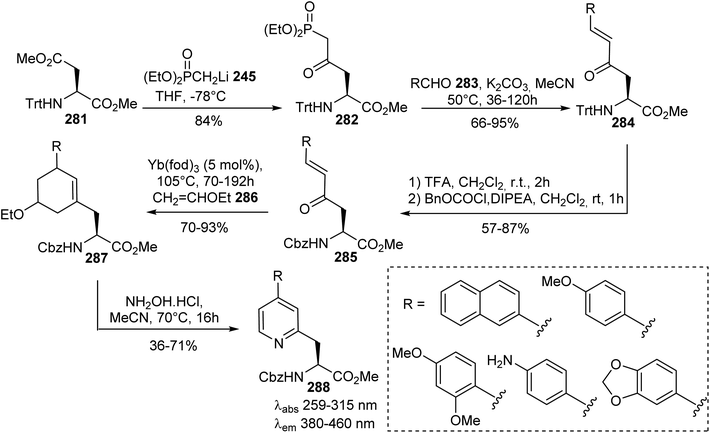 |
| | Scheme 66 The stereoselective synthesis of fluorescent β-pyridyl α-amino acids 288 via a Knoevenagel–Stobbe type reaction from the aspartate precursor 281. | |
The same method was employed by Lubell et al. to prepare the δ-phosphono-γ-ketoamino ester 291 via the addition of the dimethyl methylphosphonate anion 290 to aspartates 289.89 Then, Horner–Wadsworth–Emmons olefination reactions with the aldehyde 292 led to the corresponding α,ω-diamino dicarboxylates 293 with carbon chain lengths of nine to eleven and 78% to 87% yields (Scheme 67). These dimers were involved in the preparation of the bicyclic indolizidin-9-one derivative 294, which could introduce rigidity into peptide structures.
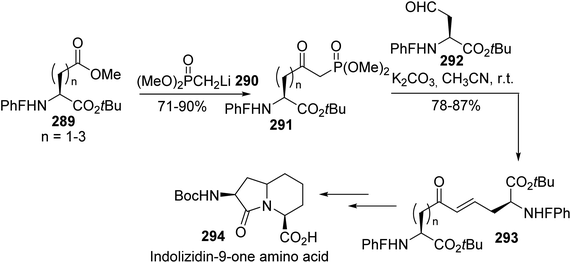 |
| | Scheme 67 The stereoselective synthesis of the indolizidinone amino acid 294 via a HWE reaction. | |
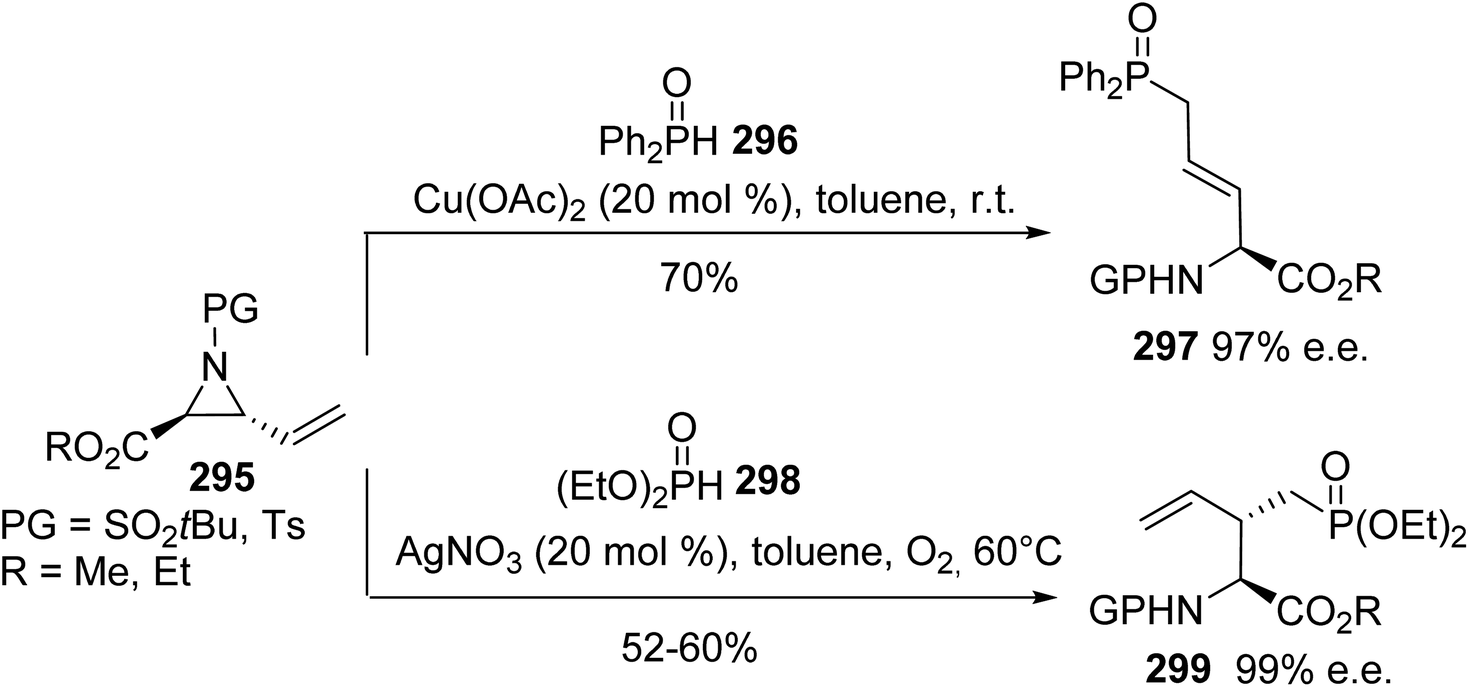
5.2.2. Via the phosphatation of vinyl aziridines. As exemplified above, efficient phosphonation and phosphatation reactions involving α-amino acids have been reported in the literature, however divergent and stereoselective methodologies have not been explored. In 2018, a regiodivergent approach for the ring-opening of vinyl aziridines using phosphorus nucleophiles, depending on the catalyst and the reaction atmosphere, was reported.90 The regioselective addition of the P-radical obtained from diphenylphosphine oxide 296 enabled the 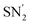 ring-opening of vinyl aziridines to afford the δ-phosphinoamino esters 297 with 70% yield and 97% e.e. at room temperature (Scheme 68).
ring-opening of vinyl aziridines to afford the δ-phosphinoamino esters 297 with 70% yield and 97% e.e. at room temperature (Scheme 68).
 |
| | Scheme 68 The regiodivergent hydrophosphonation of vinyl aziridines 295. | |
On the other hand, when phosphate anions generated in situ from a dialkylphosphite reagent and a Ag catalyst were used under an oxygen atmosphere, the SN2 phosphonation of vinyl aziridines led to the enantiopure branched α-amino esters 299 (Scheme 68). Via this divergent strategy, it is possible to access various optically active phosphorus-containing α-amino acids, which can act as useful building blocks for the design of more relevant compounds in the field of medicinal chemistry.
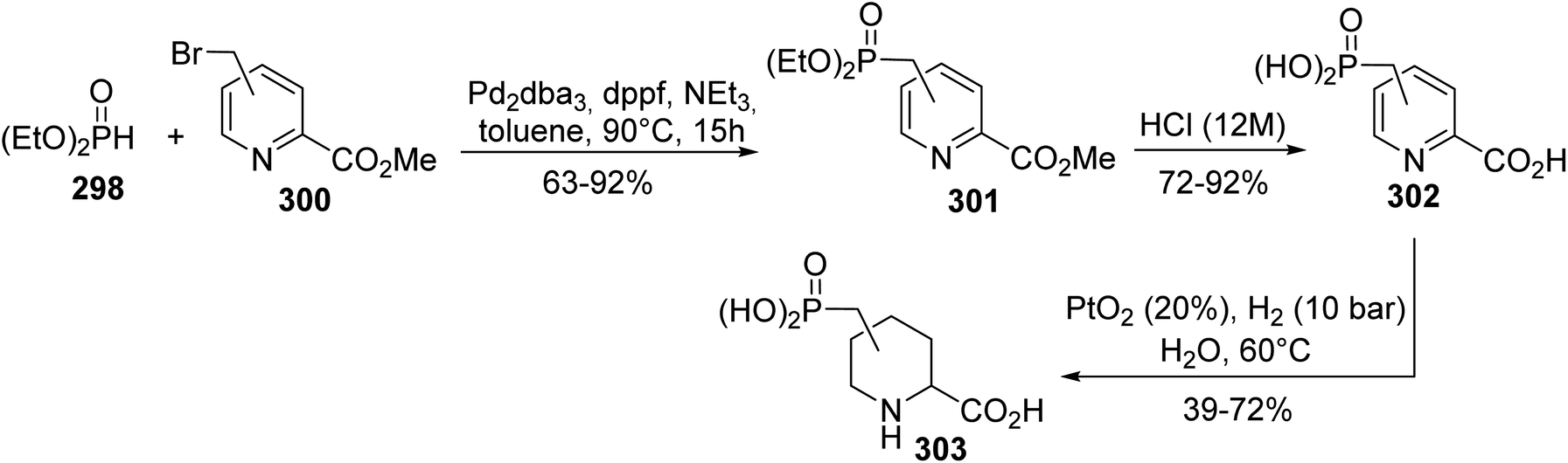
5.2.3. Via Hirao cross-coupling starting from bromomethylpicolinate. Selfotel (CGS 19755) is one of the most potent NMDAR antagonists, but it was finally rejected during clinical trials due to its toxic side effects. In this context, the synthesis of Selfotel analogues with limited side effects has been explored according to a procedure developed by Kafarski et al.91 The first step consists of palladium-catalyzed Hirao cross-coupling between the bromomethylpicolinate regioisomers 300 and diethyl phosphite 298 to give a series of phosphorylpyridyl carboxylates with isolated yields of up to 92% (Scheme 69). Acidic hydrolysis with HCl (12 N) led to the desired acids 302, which were then subjected to hydrogenation over PtO2. The reaction proceeded smoothly and the aliphatic acids 303 were obtained as a mixture of stereoisomers, identified via NMR, with the cis addition product being the major one. This methodology was also extended to piperidyl diphosphinic acid derivatives.
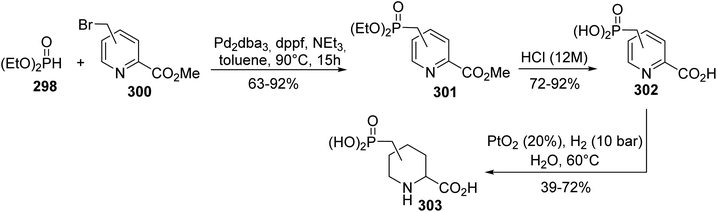 |
| | Scheme 69 The synthesis of the Selfotel analogue 303 via P–C Hirao cross-coupling. | |
6. Phosphorus-containing phenylalanine derivatives
6.1. Via palladium-catalyzed cross-coupling reactions
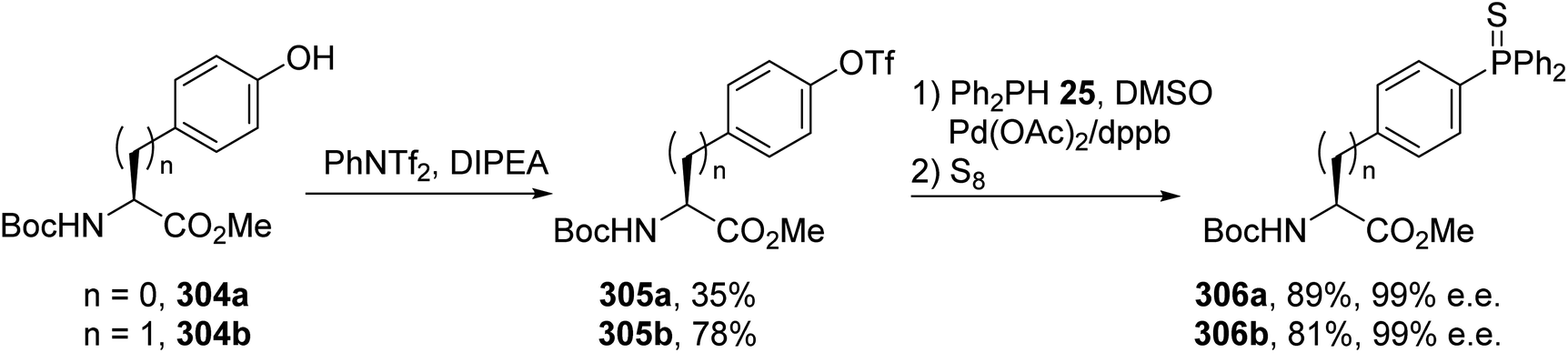
Amino acids containing functionalized aromatic substituents are interesting building blocks for the design of modified peptides with unusual properties. In connection with their research in the field of peptide-based phosphine ligands, Gilbertson et al. developed a catalytic method to convert the phenol moiety of tyrosine into the corresponding arylphosphine α-amino acids.92 N-Protected hydroxyphenylglycine or tyrosine was transformed into a triflate to act as a suitable reagent for palladium-catalyzed Stille cross-coupling via a reaction with N-phenyltrifluoromethane sulfonamide and diisopropylethylamine. While the first attempts using triflate and (trimethylstannyl)diphenylphosphine reagents in the presence of a palladium catalyst were unsuccessful, the use of diphenylphosphine and Pd(OAc)2 led to the corresponding phosphine α-amino esters. Besides, the choice of protecting group is important for the stereoselectivity because starting from N-Boc hydroxyphenylglycine or tyrosine 304a–b, this reaction proceeds without racemization, unlike in the case of N-acetyl precursors (Scheme 70). Under optimized conditions, this reaction was also extended to the ortho and meta isomers of tyrosine and to hydroxyphenylglycine to provide new phosphine ligands. Finally, the air-sensitive phosphine groups were protected in the form of the phosphine sulfur derivatives 306a–b, which are convenient for purification via chromatography and removable with RANEY® nickel. The method was also performed on a pentapeptide containing a tyrosine residue, leading to the corresponding phosphine peptide with 78% yield (Scheme 70).
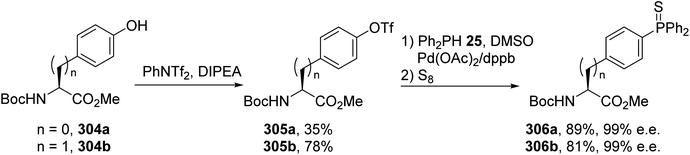 |
| | Scheme 70 The synthesis of the arylphosphine ligands 306a–b via a palladium-catalyzed P–C cross-coupling reaction. | |
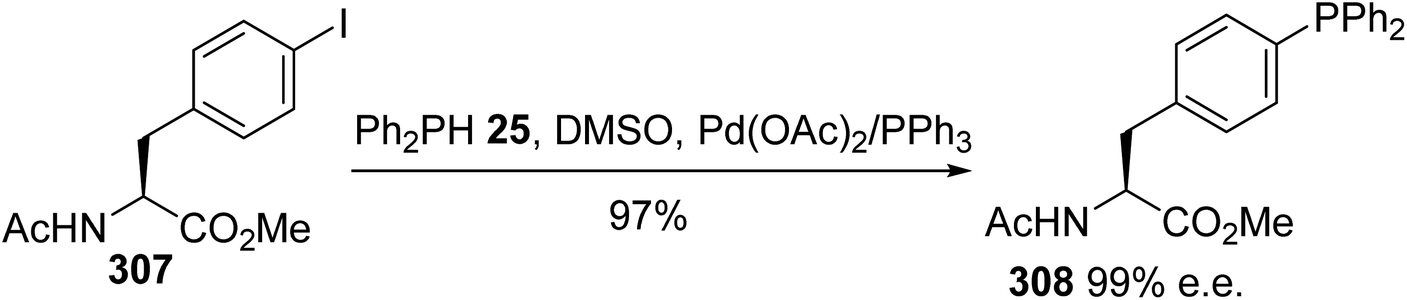
Alternatively, Kraatz et al. reported similar conditions for the cross-coupling of the N-acetyl iodophenylalanine methyl ester 307 with diphenylphosphine in the presence of triethylamine as a base.93 In this case, the free phosphine derived from phenylalanine 308 was isolated with an almost quantitative yield after purification via column chromatography in a glove box, without racemization (Scheme 71).
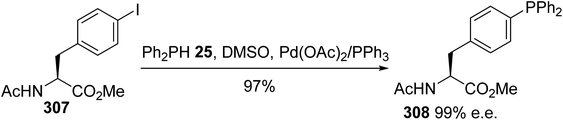 |
| | Scheme 71 The synthesis of the diphenylphosphine (L)-phenylalanine 308 via a palladium-catalyzed P–C cross-coupling reaction. | |
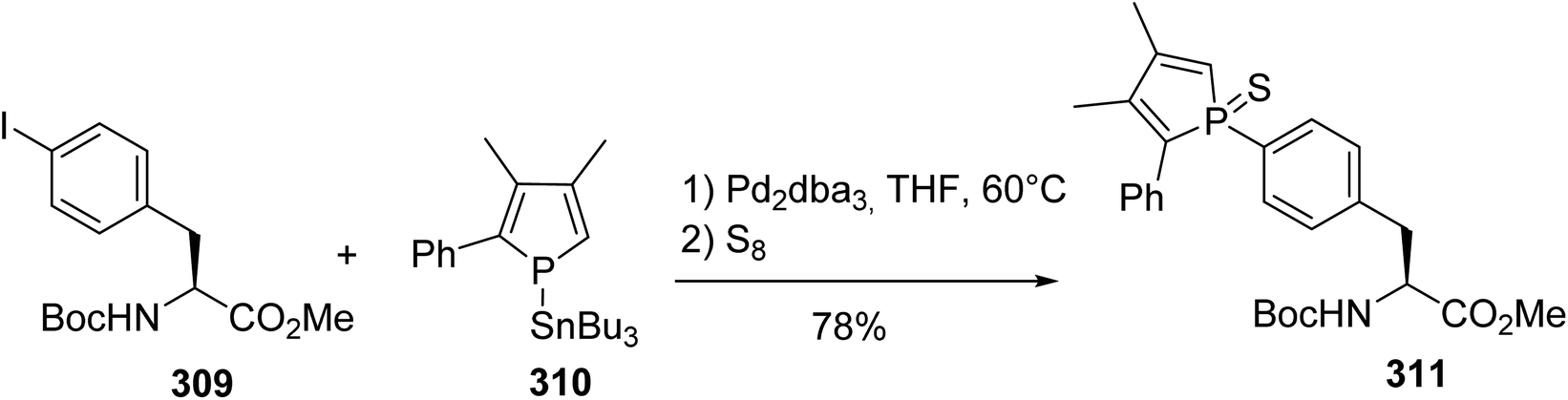
Considering the extensive use of phospholes as ligands that can bind a large variety of transition metals for applications in catalysis and optoelectronics, the incorporation of such compounds into the side chains of phenylalanine and tyrosine was investigated.94 While the Stille cross-coupling of tyrosine triflate failed with stannylphosphole 310, the reaction with N-protected 4-iodophenylalanine 309 was successfully achieved in the presence of Pd2dba3 (Scheme 72). Then, the phosphole was protected with elemental sulfur and purified via column chromatography to generate the phosphole sulfur phenylalanine 311 with 78% yield as a mixture of two diastereoisomers due to the chirality of the P-center.
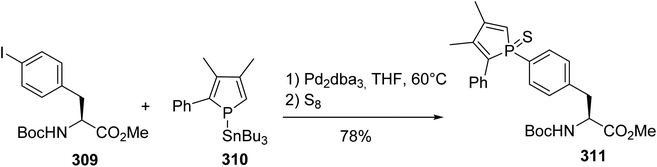 |
| | Scheme 72 The synthesis of the phosphole (L)-phenylalanine 311 via Stille cross-coupling. | |
6.2. Via aromatic nucleophilic substitution starting from fluorophenylglycine
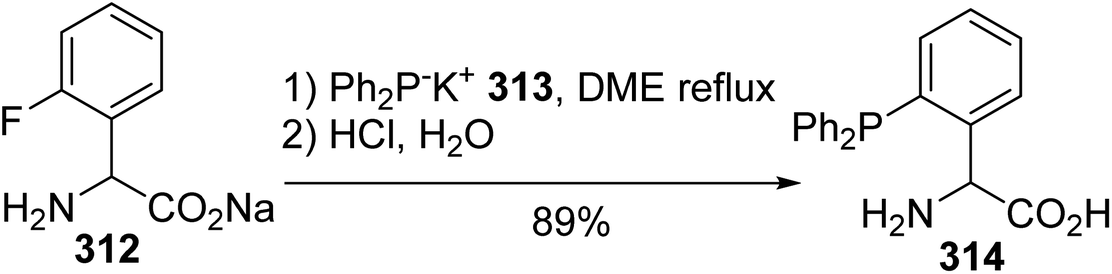
Compared to methods wherein a palladium catalyst or stannyl reagent was required to promote P–C bond formation between the phosphorus moiety and aryl triflate or halide amino esters, the reaction involving sodium 2-fluorophenylglycinate 312 proceeds directly with phosphide anions.95 Indeed, the presence of fluorine as a good leaving group enhances the reactivity of the aromatic ring towards the nucleophilic diphenylphosphide potassium salt 313, without the need for a transition metal catalyst, to provide the 2-diphenylphosphine phenylglycine 314 with 89% yield (Scheme 73).
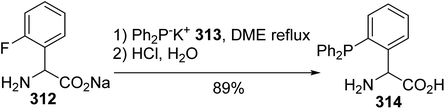 |
| | Scheme 73 The synthesis of the phosphinophenylglycine 314 via P–C bond formation using SNAr. | |
6.3. Via the diastereoselective alkylation of iminolactone
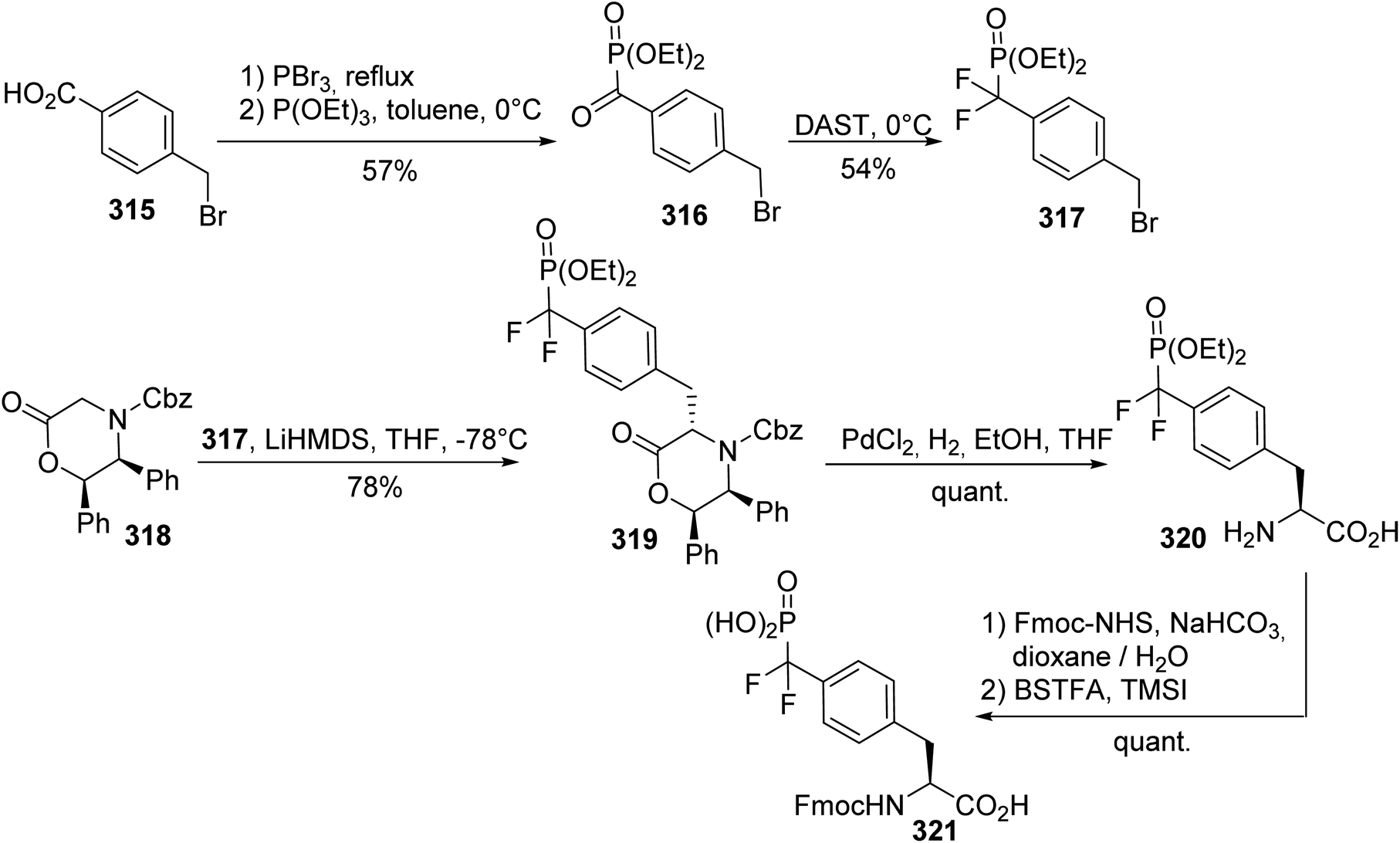
The development of non-hydrolyzable phosphotyrosine analogues that could be introduced into peptides is of considerable interest for studying the cellular signaling events involved in diseases such as diabetes and cancer. For this purpose, the efficient asymmetric synthesis of phosphonodifluoromethyl (L)-phenylalanine 321 was reported using the commercially available diphenyloxazinone 318 as a chiral auxiliary. The alkylating reagent 317 was obtained via two steps: the reaction of triethyl phosphite with a benzoic acid bromide reagent 316, generated in situ via the treatment of toluic acid with PBr3, followed by the addition of (diethylamido)sulfur trifluoride (DAST) to the resulting ketophosphonate 316. The enolate of the chiral auxiliary 318 was then alkylated in the benzylic position, leading to compound 319 with 78% yield. The hydrogenation and protection of the amine with an Fmoc-NHS ester, followed by the deprotection of the phosphonate, provided the N-Fmoc phosphonic (L)-phenylalanine derivative 321 with a quantitative yield (Scheme 74).96,97 Analysis of compound 320 via HPLC after derivatization with a chiral amine proves that no racemization occurs during the alkylation step.
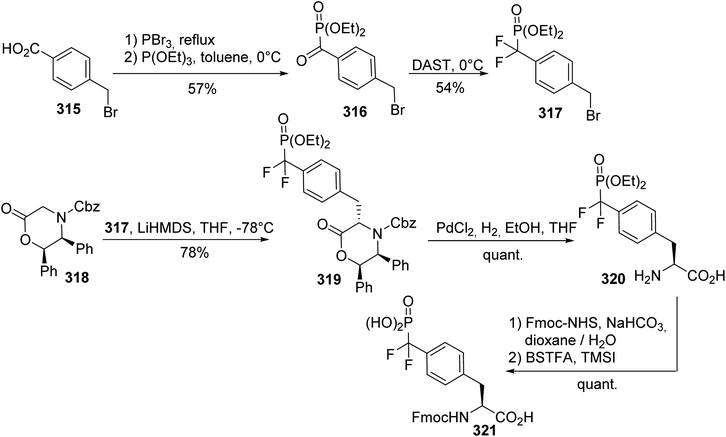 |
| | Scheme 74 The synthesis of the phosphonodifluoromethyl phenylalanine 321 via the diastereoselective alkylation of the diphenyloxazinone 318. | |
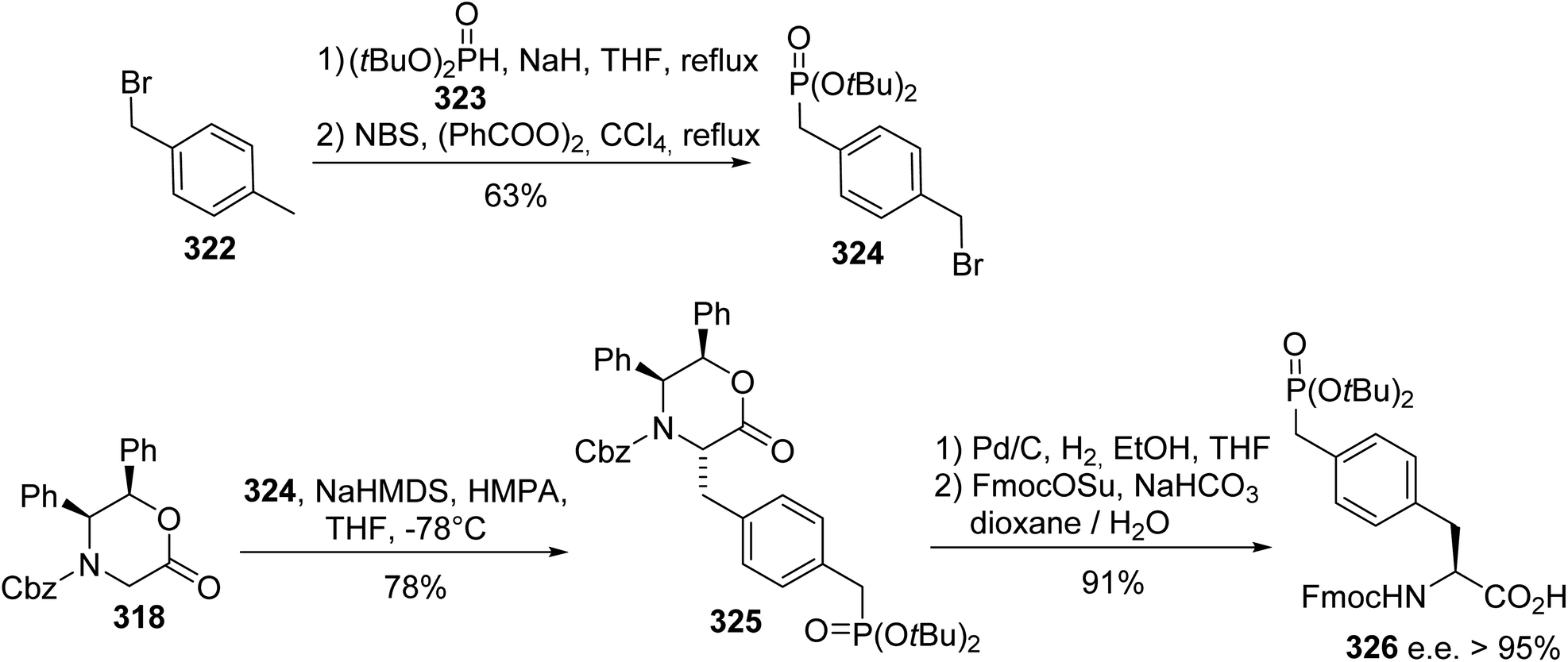
A similar approach was also employed for the synthesis of the (L)-phosphonomethyl phenylalanine 326, a phosphatase-resistant phosphotyrosine analogue that exhibits similar biological properties to phosphotyrosine.98 The phosphonate motif was obtained via the reaction of 4-methylbenzyl bromide with nucleophilic sodium di-(tert-butyl)phosphite 323, followed by bromination with N-bromosuccinimide. The enolate of the chiral auxiliary 318 was generated under the conditions reported above and, then, trans-alkylation was performed in the presence of HMPA as a solvating agent, leading to the compound 325 with 78% yield. Finally, the (L)-phosphonomethyl phenylalanine 326 was obtained with high enantioselectivity (e.e. > 95%) after hydrogenolysis and Fmoc-protection of the free amine (Scheme 75).
 |
| | Scheme 75 The synthesis of the phosphonomethyl phenylalanine 326 via the diastereoselective alkylation of the diphenyloxazinone 318. | |
7. Phosphohistidine analogues
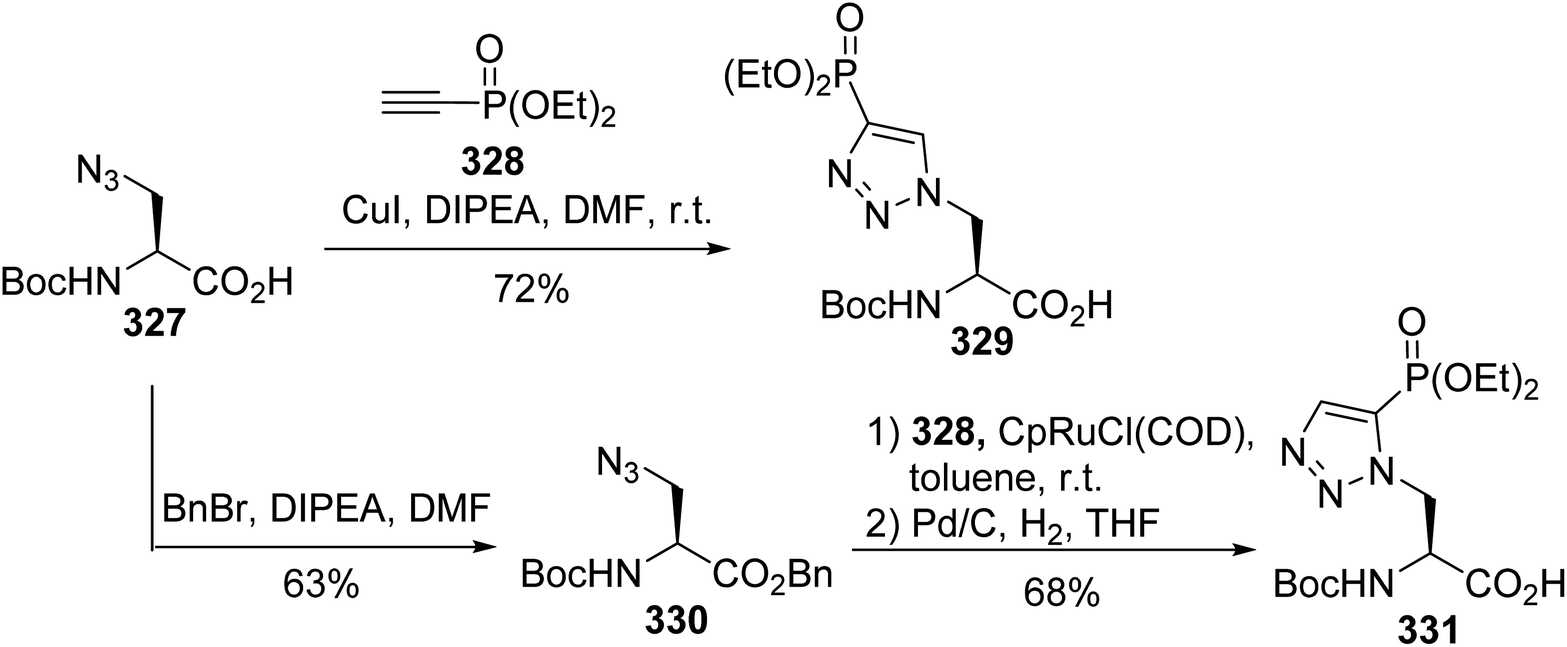
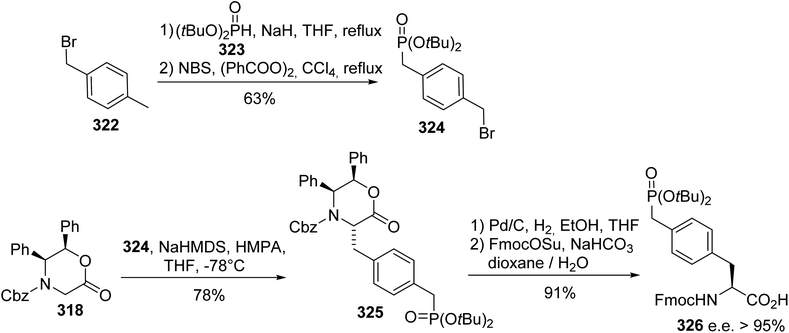
Protein phosphorylation is one of the most studied post-translational modifications. Compared to serine, threonine and tyrosine O-phosphorylation, obtaining an understanding of histidine phosphorylation is more difficult due to the instability and isomerism of phosphohistidine (pHis), making its detection and isolation from biological sources challenging. On the basis of molecular modeling, isomers 329 and 331, having non-hydrolysable C–P bonds as replacements for the labile P–N bond, are expected to mimic the geometry and electronic properties of phosphohistidine. The principle of the synthesis is based on the cycloaddition of the azide alanine derivative 327 with ethynyl phosphonate 328. When CuI was used as a catalyst, the regioisomer 329 was directly obtained with 72% yield, while for the Ru-catalyzed cycloaddition, it was necessary to protect carboxylic acid with benzyl bromide. After the reaction and debenzylation, the isomer 331 was isolated with 68% yield (Scheme 76).99 These stable analogues were then incorporated into peptides via solid-phase peptide synthesis using a Boc-strategy to study histidine phosphorylation in histones.
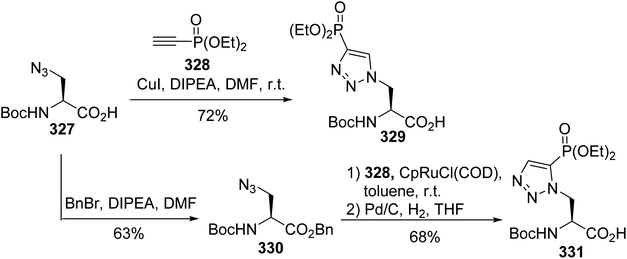 |
| | Scheme 76 The stereoselective synthesis of the phosphohistidine analogues 329 and 331 via click chemistry. | |
8. The phosphorylation and phosphination of hydroxyamino acids with a P–O bond
Serine phospholipid analogues are of crucial interest for understanding the physiological activity of phosphatidylserine (PS), a naturally occurring amphiphilic phospholipid that is involved in the activation of several membrane-associated enzymes. In addition, recent studies have reported the interactions of such negatively charged phospholipids with polynuclear platinum anticancer agents to facilitate cellular uptake.100
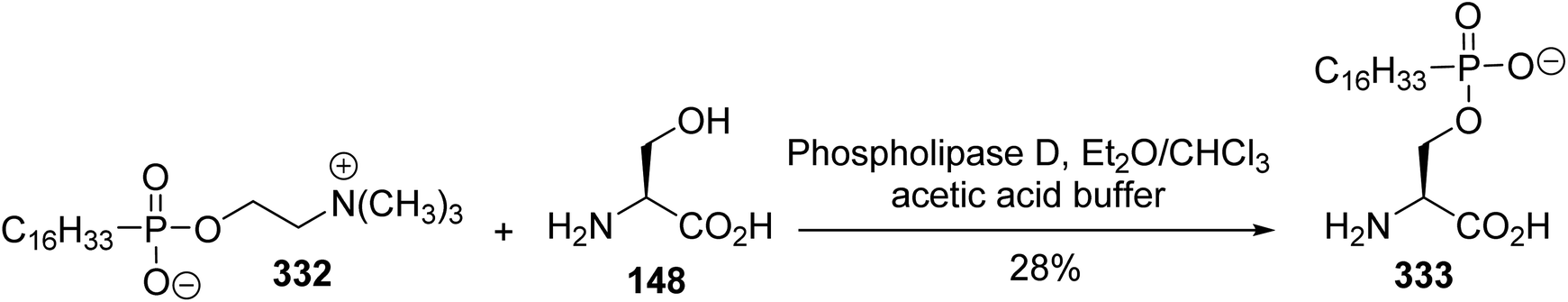
The synthesis of hexadecylphosphono-(L)-serine 333 was reported by Brachwitz et al. The transesterification reaction was carried out by stirring a mixture of hexadecyl phosphonocholine 332 and excess (L)-serine, at a ratio of 1:100, with phospholipase D, which is known to convert phosphatidylcholine into phosphatidylserine (Scheme 77).101 After treatment, the crude substance was purified via chromatography on cellulose to furnish the phosphonoserine 333 with 28% yield.
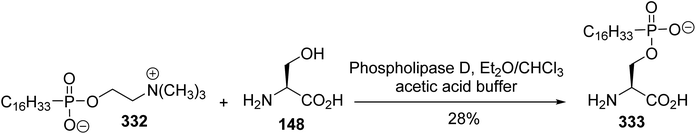 |
| | Scheme 77 The synthesis of the lipophilic phosphonoserine 333 via enzymatic transesterification. | |
The one-pot synthesis of another important building block, Fmoc-O-benzyl-(L)-phosphoserine 337, bearing convenient protecting groups for solid-phase peptide synthesis, was reported more recently by Petrillo et al.; its incorporation into the phosphopeptide Forigerimod was also studied for the treatment of systemic lupus erythematosus (SLE).102 It must also be noted that the use of a monobenzyl protecting group was necessary to avoid the elimination of the phosphate into dehydroalanine under the basic conditions required for Fmoc deprotection. The reaction between phosphorus trichloride and benzyl alcohol afforded benzyl dichlorophosphite 334, which reacted efficiently with Fmoc-(L)-serine 335 in the presence of 2,6-lutidine as a base without the deprotection of the amine. When the cyclic intermediate 336 was observed via 31P NMR studies, hydrolysis was performed in the presence of NaBr, leading to the acyclic phosphite 337 with 71% yield (Scheme 78). As an alternative to phosphoramidite chemistry, the phosphorylation of other Fmoc-hydroxyamino acids was also performed based on this one-pot strategy.
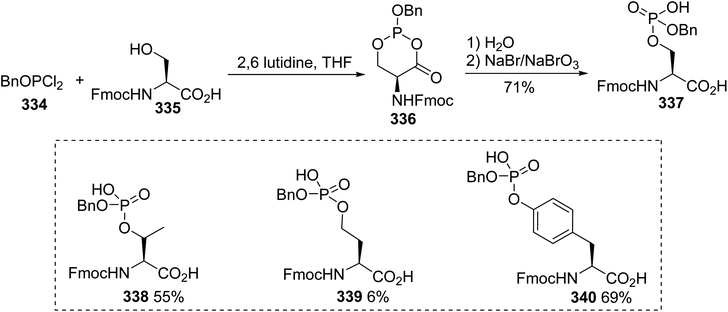 |
| | Scheme 78 The synthesis of the O-phosphorylated amino acids 337–340. | |
Phosphinites constitute a class of phosphorus-based ligands, leading to robust complexes for transition metal catalysis. They exhibit stronger metal-phosphorus bonds compared with the related phosphines, due to the presence of the electron-withdrawing alkoxy group. Phosphinite ligands derived from naturally occurring (L)-hydroxyamino acids were conveniently prepared by reacting an alcoholic amino acid with one equivalent of chlorodiphenylphosphine in the presence of NEt3 and a catalytic amount of DMAP.103,104 After purification on basic alumina, phosphinites based on serine 343–344, on tyrosine 345 and on threonine 347, were obtained with up to 60% yield (Scheme 79). Then, a platinum complex of a phosphinite ligand 348 was also prepared with a quantitative yield and X-ray structure analysis indicates a square planar coordination environment around the metal center, with phosphinite amino acids positioned cis to each other.
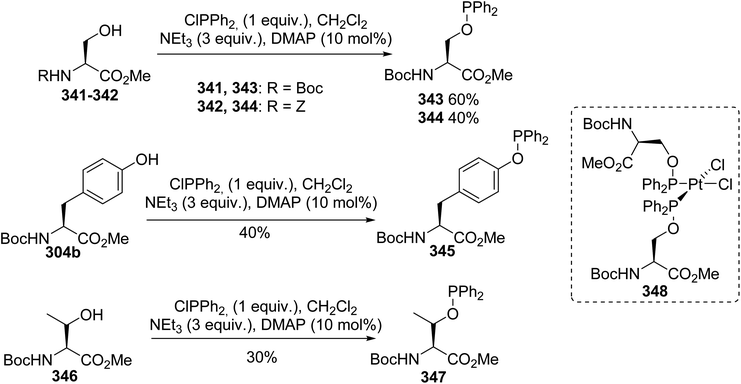 |
| | Scheme 79 The stereoselective synthesis of phosphinite ligands from (L)-hydroxyamino acids. | |
Cidofovir (HPMPC) 349 is an antiviral agent for the treatment of human cytomegalovirus (HCMV) retinitis, aimed at immune-suppressed individuals, which contains an ionisable P(O)(OH)2 moiety at physiological pH, resulting in low internalization and biodisponibility. To address these limitations, prodrugs based on cyclic Cidofovir (cHPMPC) conjugated to serine and dipeptides have been developed.105 The interest in this approach arises from the possibility of coupling a variety of amino acids with serine to fine-tune the prodrug, minimize its toxicity and improve its lipophilicity, which is necessary for its transportation in the gastrointestinal tract. The cHPMPC conjugates 350, 352 and 354 were obtained using PyBOP to promote the intramolecular cyclization of Cidofovir 349 into cHPMPC, followed by the grafting of serine or dipeptides and Boc-deprotection with TFA (Scheme 80). The target compounds 352 and 354 were isolated after purification as a pair of diastereoisomers, owing to the chiral phosphorus atom. This strategy was also applied to the synthesis of an adenine conjugate (cHPMPA) but, in this case, the amine functionality of serine was anchored to a solid support. Studies with a peptide-specific intestinal transporter (hPEPT1) suggest that the presence of a dipeptide in the conjugate is necessary to increase its affinity towards the transporter and that (L)-amino terminal prodrugs are stereospecifically recognized by hPEPT1 but not transported due to steric hindrance or the polar properties.
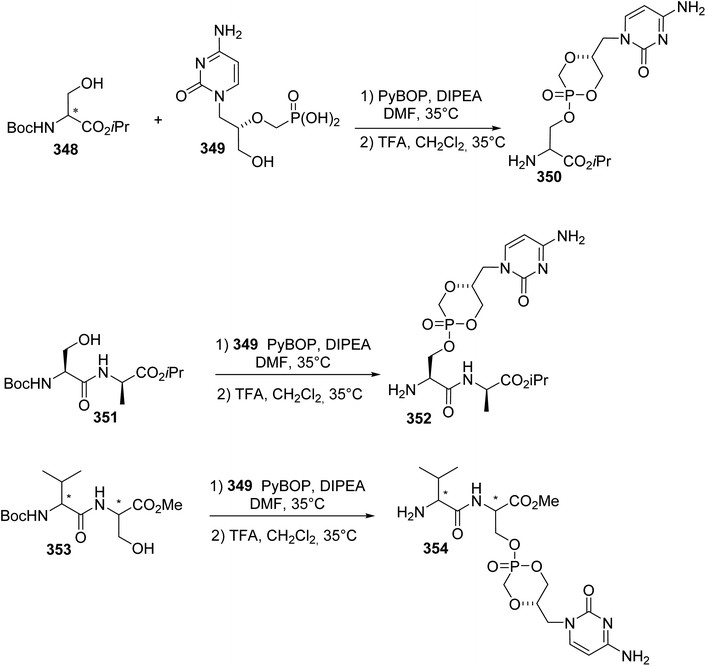 |
| | Scheme 80 The synthesis of cyclo-Cidofovir conjugates with serine to obtain the prodrugs 352 and 354. | |
9. Phosphocysteine and its derivatives with a P–S bond
S-Phosphocysteine is one of the lesser-known phosphoamino acids, discovered in both prokaryotic and eukaryotic systems, which is involved in mechanisms related to protein tyrosine phosphatases (PTPases) that are responsible for the dephosphorylation of tyrosine side chains. It is of major importance to understand the role of S-phosphocysteine in the regulation of tyrosine phosphorylation, which is associated with cell growth and division processes and is therefore of interest to cancer researchers.106–108 Only a few methodologies have been developed for the synthesis of S-phosphocysteine derivatives. The first strategy is based on a Michaelis–Arbuzov reaction involving a readily available disulfanyl derivative and trimethyl phosphite, leading to the corresponding phosphorothioate 356 with an almost quantitative yield after purification (Scheme 81).109,110
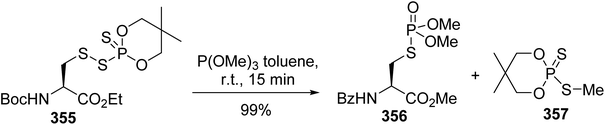 |
| | Scheme 81 The synthesis of the S-phosphocysteine derivative 356 via a Michaelis–Arbuzov type reaction. | |
A more recent strategy that consists of reactions between S-nitrosothiols and phosphite esters was reported last year.111 Interestingly, this reaction led to two different products, thiophosphoramidates and phosphorothioates, depending on the structure of the S-nitrosothiol reagent. When S-nitrosocysteine 358 was used, the phosphite directly attacks the sulfur atom of the S–N bond, leading to the thiophosphonium salt intermediates 359–360, which produced the corresponding phosphocysteine derivatives 361 and 362 with 82% and 83% yields, respectively, after hydrolysis (Scheme 82). The derivative 362 was also obtained with a higher yield of 90% through the reaction of nucleophilic P(OEt)3 with a cystine precursor.
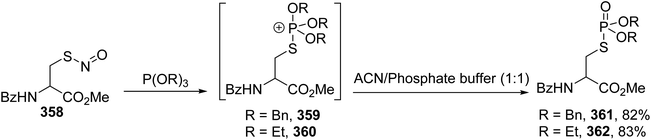 |
| | Scheme 82 The synthesis of the phosphocysteines 361 and 362 using S-nitrosocysteine 358. | |
Chiral thiophosphinite ligands derived from cysteine and their stable complexes with platinum(II) have been prepared and characterized. The reaction of (L)-cysteine methyl ester hydrochloride with chlorodiphenylphosphine in the presence of NEt3 proceeds quantitatively, leading to the corresponding thiophosphinite derivative 364, as shown by 31P NMR analysis (Scheme 83).112,113 The platinum complex 365 was prepared via the direct addition of [PtCl2(1,5-COD)] at a ratio of 2:1 to the non-isolated ligand and equilibrium with the bis-chelate 366 was observed in solution.
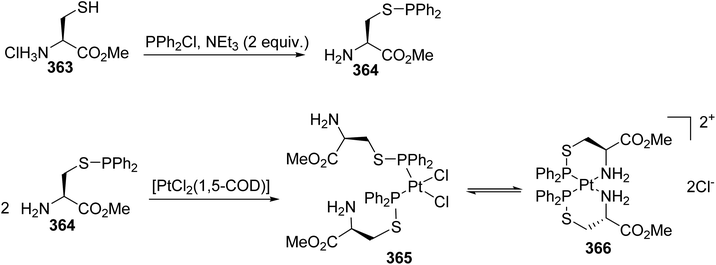 |
| | Scheme 83 The synthesis of the β-thiophosphinito amino acid ligands used in Pt-coordination chemistry. | |
10. Phosphorus-containing α-amino acids with a P–N bond
10.1. N-Phosphinothioyl α-amino acid derivatives
Diphenylphosphinothioyl (Ppt) and dimethylphosphinothioyl (Mpt) groups were used to protect the amine groups of α-amino acids, taking advantage of the P–N bond lability under acidic conditions. While the removal of the Ppt group via hydrogen chloride is inappropriate for the synthesis of peptides on a large scale, more efforts have been devoted to the preparation of Mpt-amino acids and their deprotection under mild conditions.114 They were obtained either via the alkaline hydrolysis of Mpt-amino esters (Method A) or via the direct phosphinothioylation of the free amino acids (Method B) (Scheme 84). The chloride 367 reacted more efficiently with the amino esters 368, which were subsequently hydrolyzed using aqueous sodium hydroxide into Mpt-amino acids and isolated as dicyclohexylamine (DCHA) salts 370, compared to direct synthesis using the amino acids 371 due to the partial hydrolysis of 367 under basic conditions.
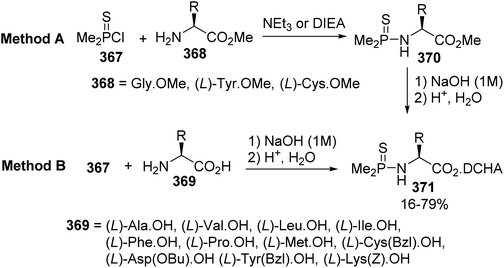 |
| | Scheme 84 The preparation of N-phosphinothioyl protected α-amino acids. | |
The removal of the Mpt group was successfully performed using PPh3/HCl in dichloromethane. Interestingly, Mpt removal was increased by a factor of 60 compared to the tert-butoxycarbonyl (Boc) group.114 Considering the stability of the P–O bond under acidic conditions, a good orthogonal relationship between the N- and O-phosphinothioyl groups was observed. The N-Mpt group was also selectively removed to give H-(L)-Cys(Mpt)-OH with 89% yield. Mild conditions for the deprotection of the S-Mpt group, suitable for use with a Boc-strategy, were found when using tetrabutylammonium fluoride hydrate (TBAF·H2O).115,116 These conditions present an interesting alternative to the use of heavy metal catalysts such as AgNO3 and Hg(OAc)2.
10.2. N-Phosphoramidate α-amino acid derivatives
Nucleoside analogues have been developed for a number of decades to treat various cancers and viral infections.117,118 The in vivo activation of such nucleosides is based on their phosphorylation into active di- or tri-phosphate counterparts. However, several limitations are associated with their administration as drugs, due to poor cellular permeability and because the first phosphorylation step into 5′-O-monophosphate derivatives is the rate-limiting step. To overcome these limitations, one of the most applied phosphate prodrug approaches is ProTide technology, which consists in masking the hydroxyl moieties of the monophosphate or monophosphonate groups using an aryl motif and an amino ester.119,120 Then, the ProTides are metabolized inside cells by the action of an esterase and a phosphoramidase enzyme to release the free nucleoside monophosphate and monophosphonate. ProTide prodrug technology has led to the discovery of two FDA-approved antiviral drugs, sofosbuvir (Sovaldi) 8 and tenofovir alafenamide (TAF) 372 for clinical use against hepatitis C (HCV) and for human immunodeficiency virus (HIV) therapy, respectively (Fig. 2). Other ProTide prodrugs bearing various esters of (L)-alanine and aryloxy substituents in a phosphoramidate system have also been approved by the FDA and are being subjected to clinical trials for the treatment of cancers and the Ebola virus.
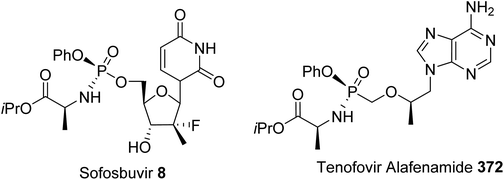 |
| | Fig. 2 Representative FDA-approved antiviral ProTides. | |
As the biological activity of phosphoramidate prodrugs depends on the (S)P or (R)P configuration at the phosphorus center, the development of stereoselective synthesis procedures is of major importance. The conventional preparation of phosphoramidates involves a chlorophosphoramidate electrophilic reagent 373, which is subjected to nucleophilic substitution with a nucleoside to give a diastereomeric mixture of isomers 375 that are further separated via column chromatography (Scheme 85a).121 Another approach consists of the reaction of the stereochemically pure phosphordiamidate (S)P or (R)P 376, derived from the chiral auxiliary (S)-4-isopropylthiazolidine-2-thione, with the nucleoside analogue 377. Via this strategy, the almost diastereomerically pure (S)P or (R)P phosphoramidate prodrug 378 (95% d.e.) was obtained (Scheme 85b).122
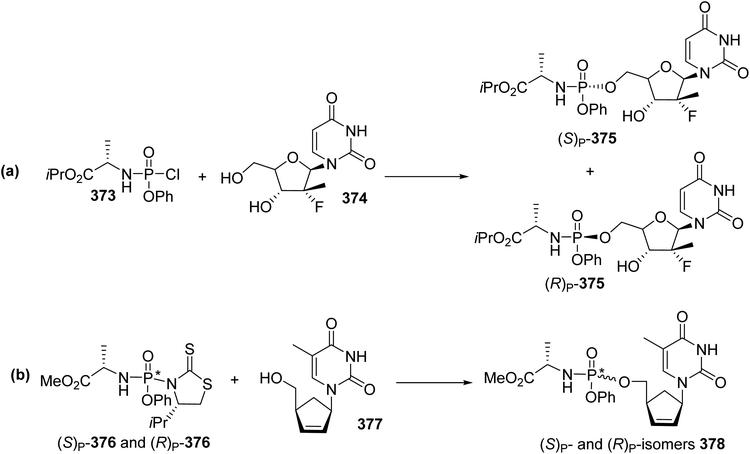 |
| | Scheme 85 The stereoselective synthesis of aryloxy phosphoramidate prodrugs 375 (a) and 378 (b). | |
Highly stereoselective and regioselective nucleoside 5′-phosphorylation was recently reported without the use of a protecting group.123 The strategy consists of the activation of stable chiral pentafluorophenyl reagents 379 by a dimethylaluminium chloride Lewis acid to afford the ProTides without employing 3′ protection. Optimization of 5′-selective phosphorylation was first performed to regioselectively target sofosbuvir 8 without the undesired 3′,5′-bis-ProTide. In the presence of Me2AlCl (0.5 equiv.) and pyridine as a solvent, sofosbuvir was obtained with 84% yield and excellent diastereo- and regio-selectivity (>500:1; 110:1) (Scheme 86). This method was extended to other relevant phosphoramidate prodrugs that have been investigated for the treatment of viral infections or cancer. Methyl and benzyl phosphoryl-(L)-alaninates were used to access AZT ProTide 381 and Acelarin 382 with 94% and 80% yields, respectively. Finally, INX-08189 383, which is a liver targeting prodrug in phase II of clinical studies, was obtained with 81% yield and with excellent diastereo- and regio-selectivities.
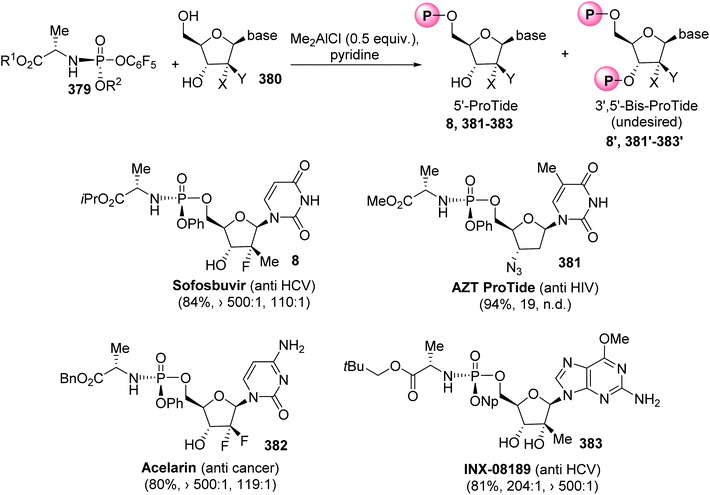 |
| | Scheme 86 The regioselective synthesis of ProTides using the pentafluorophenyl phosphoramidate reagents 379. | |
10.3. Phosphoramidate peptide derivatives via Staudinger reactions
Phospholysine (pLys) is one of the less studied phosphorylated amino acids due to the lability of the P(O)–N bond under acidic conditions, which is an obstacle both for its identification and its synthesis. To overcome this issue, a strategy based on the chemoselective Staudinger reaction of an azidonorleucine-containing peptide was reported by Hackenberger et al.124 Considering the stability of the phosphoramidate bond under alkaline conditions, it was envisaged that the Staudinger reaction could be performed starting from the azidonorleucine peptide 384, fixed on base-labile 4-hydroxymethyl benzoic acid (HMBA) TG resin and ortho-nitrobenzylphosphite (Scheme 87). The amine side chain was first deprotected by treatment with TFA in the presence of triisopropylsilane (TIS), then the unprotected peptide was subjected to the Staudinger reaction via incubation for 48 h with phosphite. The excess reagent was removed and the peptide was cleaved from the resin using NaOH (250 mM). After neutralization with HCl, the corresponding phosphoramidate esters 386a–c were identified via LCMS analysis and the ortho-nitrobenzyl moieties were released upon UV light irradiation to provide the expected phospholysine peptide 387 (Scheme 87).
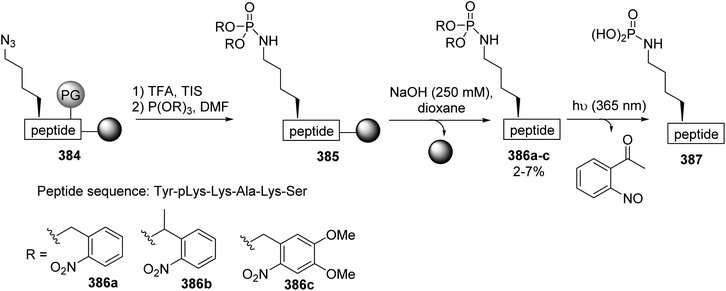 |
| | Scheme 87 The synthesis of phospholysine-containing peptides via the Staudinger reaction. | |
11. Phosphorus-containing β-amino acids
A few examples of β-amino acids containing a phosphorus moiety have been reported in the literature to date. To access such amino acids, asymmetric organocatalysis has been considered, owing to the efficacy of this approach in the development of optically active organophosphorus compounds. Herein, the reaction of the substituted cyanoacetates 388 with electrophilic diarylphosphine chloride led to the corresponding optically active α-phosphinocyano acetates 389, which were further transformed into the quaternary α-phosphino β-amino acids 390 via a one-pot procedure (Scheme 88).125 A Cinchona alkaloid (DHQD)2PYR catalyst was used in the presence of 1,8-bis(dimethylamino)naphthalene as a scavenger to consume the hydrochloric acid formed during the reaction; otherwise the catalyst would become protonated and remain inactive. Under the optimized conditions, various substituents on the cyanoacetate and different diaryl phosphines were tolerated in this reaction, providing the β-amino esters 390 with yields from 52% to 55% and enantioselectivities ranging from 88% to 92% (Scheme 88).
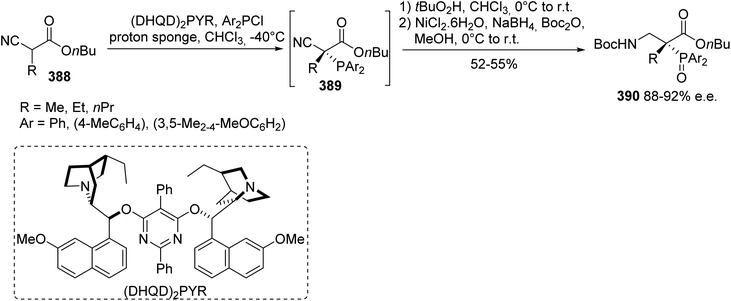 |
| | Scheme 88 The asymmetric synthesis of the α-phosphino β-amino acids 390 via the phosphination of the α-cyanoacetates 389. | |
As the formation of a P–C bond could be achieved via the nucleophilic addition of phosphites to hybridized sp2 carbon atoms in polarized π-bond electrophiles, this strategy was applied to the coupling of α-ketoesters with imines in the presence of phosphite under basic conditions.126 The addition of the diethyl phosphite anion to the α-ketoester 391 followed by the [1,2]-phospha-Brook rearrangement of the intermediate 392 generated the α-phosphonyloxy enolate 393, which was subsequently trapped by imines 394 through Mannich reactions. The use of aryl and heteroaryl N-sulfonyl imines as coupling partners permits the syn α-hydroxy-β-amino esters 395 to be selectively obtained with excellent diastereoselectivities (d.r. > 20:1) (Scheme 89). This reaction was also successfully performed using aliphatic imines and heteroaromatic ketoesters and constitutes a tunable approach, since trapping the intermediate from the phosphate addition/[1,2]-phospha-Brook rearrangement with N-diphenylphosphinyl imine results in an aziridine derivative.
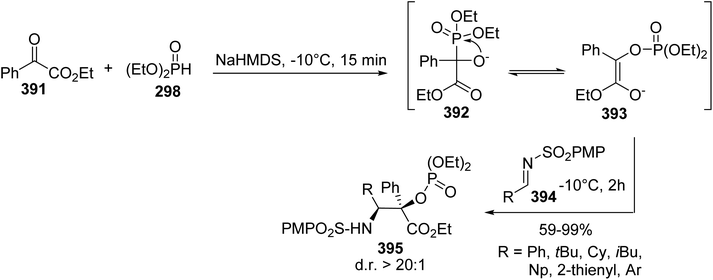 |
| | Scheme 89 The synthesis of the α-phosphonyloxy-β-amino esters 395 from the phenylglyoxylate 391 using a three-component reaction. | |
[4 + 2] Diels–Alder cycloaddition is a method of choice for the construction of regio- and stereo-selective six-membered rings. In this context, this strategy was exploited for the reaction of aminodienes with phosphonate dienophiles to afford β-aminophosphonic acids.127 The cycloaddition of the succinimide diene 396 to the activated vinyl phosphonate dienophile 397 bearing an electron-withdrawing ester led to the cycloadducts 398a–b with 93% yield as a mixture of endo/exo stereoisomers at a ratio of 65:35. Then, hydrolysis of the succinimide and the phosphonate gave the corresponding ammonium salts 399a–b with 84% yield (Scheme 90). Functionalization of the CC bond in the cyclohexene ring was also reported via oxidation with m-chloroperbenzoic acid, resulting in the trans-oxirane 400. This was finally transformed into the trans-dihydroxycyclohexane 401 via the nucleophilic opening of the oxirane with lithium hydroxide, followed by acidic hydrolysis of the protected functional groups.
 |
| | Scheme 90 The synthesis of cyclo-β-aminophosphonic acid derivatives using Diels–Alder cycloaddition. | |
12. Conclusions
The chemistry of amino acids containing a phosphorus atom emerged as a field of study fifty years ago and it is still subject to developments that provide reagents for hemisynthesis, ligands for coordination chemistry and asymmetric catalysis, precursors for peptide labeling and bioactive compounds and bioconjugates.
Such amino acids are obtained via P–C, P–O, P–S or P–N bond formation. In the former case, phosphorus amino acids were reported based on phosphorus atoms in the α-, β-, γ- and δ-positions of the side chain. α-Amino acids with a Cα–P bond have been extensively used as HWE reagents to obtain α,β-dehydro-α-amino acids, which are key intermediates in the total synthesis of biomolecules, dehydropeptides or β-lactam antibiotics. Considerable efforts have been made relating to the synthesis of enantiopure alanine and proline derivatives containing a phosphine moiety in the β-position for use as chiral ligands in asymmetric catalysis with transition metal complexes. Since the discovery of the natural product (L)-phosphinothricin, numerous phosphonic analogues of aspartic and glutamic acid have been developed for their application as antagonists of the ionotropic glutamate receptors associated with neurodegenerative diseases. More recent developments concern the stereoselective synthesis of amino acids bearing a phosphorus group in the γ-position, leading to stable phosphine borane, trifluoroboronato phosphonium salt and phospholyl derivatives. Such compounds have been used as electrochemical markers or for the radio- and fluorescence-labeling of peptides. The synthesis of phosphine phenylalanine derivatives has also been investigated either through palladium-catalyzed cross coupling or via the direct nucleophilic substitution of halogenophenyl moieties.
In the α-amino acid series containing P–O bonds, those obtained via the phosphorylation of hydroxyamino acids have been extensively studied, as this is one of the most important post-translational modifications. Conjugates with serine and dipeptide derivatives based on the cyclic Cidofovir antiviral agent have been developed for minimizing its toxicity and improving its lipophilicity.
Recent methodologies to access stable analogues of phosphohistidine (pHis) and S-phosphocysteine, associated with signaling processes in living cells, have also recently been achieved.
Among the α-amino acids containing a P–N bond, the synthetic utility of N-phosphinothioyl derivatives has been demonstrated in solid-phase peptide synthesis. Also, the linkage of a phosphoryl group to the nitrogen atom of an amino acid in the form of phosphoramidate ProTides, highly potent antiviral and anticancer agents, constitutes one of the major forms of progress in the prodrug field over the last twenty years.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank Montpellier University for the grant to Mathieu Arribat. They also thank Dr Jean-Pierre Vors.
References
- R. Kober and W. Steglich, Untersuchungen zur Reaktion von Acylaminobrommalonestern und Acylaminobromessigestern mit Trialkylphosphiten – eine einfache Synthese von 2-Amino-2-(diethoxyphosphoryl)essigsäure-ethylester, Liebigs Ann. Chem., 1983, 1983, 599–609 CrossRef.
- R. Mazurkiewicz and M. Grymel, N-Acyl-a-triphenylphosphonioglycinates: A Novel Cationic Glycine Equivalent and its Reactions with Heteroatom Nucleophiles, Monatsh. Chem., 1999, 130, 597–604 CAS.
- M. Grymel, A. Kuźnik and R. Mazurkiewicz, N-Acyl-α-Triphenylphosphonio-α-Amino Acid Esters as Synthetic Equivalents of α-Amino Acid α-Cations, Phosphorus, Sulfur Silicon Relat. Elem, 2015, 190, 429–439 CrossRef CAS.
- R. Mazurkiewicz, A. Kuźnik, M. Grymel and N. Kuźnik, N-Acyl-α-triphenylphosphonioglycinates in the Synthesis of α,β-Dehydro-α-amino Acid Derivatives, Monatsh. Chem., 2004, 135, 807–815 CrossRef CAS.
- S. Schumann, K. Zeitler, M. Jäger, K. Polborn and W. Steglich, Stereocontrolled Backbone Connection of Peptides by CC-Double Bonds, Tetrahedron, 2000, 56, 4187–4195 CrossRef CAS.
- M. Lang, K. Prasad, W. Holick, J. Gosteli, I. Ernest and R. B. Woodward, The penems, a new class of .beta.-lactam antibiotics. 2. Total synthesis of racemic 6-unsubstituted representatives, J. Am. Chem. Soc., 1979, 101, 6296–6301 CrossRef CAS.
- J. Heinicke, N. Peulecke and P. G. Jones, Novel [small alpha]-functionally substituted amino acids: diphenylphosphinoglycines, Chem. Commun., 2005, 262–264 RSC.
- J. Lach, C.-Y. Guo, M. K. Kindermann, P. G. Jones and J. Heinicke, α-Phosphanyl Amino Acids: Synthesis, Structure and Reactivity of N-Aryl-α-phosphanylglycines, Eur. J. Org. Chem., 2010, 1176–1186 CrossRef CAS.
- J. Lach, G. J. Palm, P. G. Jones and J. W. Heinicke, One-Pot Synthesis of Phosphanylbis(N-arylglycines) and Spontaneous Diastereoselective Lactamization of P-Alkyl Derivatives To Form Five-Membered P,N-Heterocyclic Amino Acids, Eur. J. Inorg. Chem., 2016, 3417–3422 CrossRef CAS.
- N. Peulecke, D. G. Yakhvarov and J. W. Heinicke, Chemistry of α-Phosphanyl α-Amino Acids, Eur. J. Inorg. Chem., 2019, 1507–1518 CrossRef CAS.
- K. R. Basvani, M. K. Kindermann, H. Frauendorf, C. Schulzke, P. G. Jones and J. W. Heinicke, 3-Phenylphosphaprolines – Synthesis, structure and properties of heterocyclic α-phosphanyl amino acids, Polyhedron, 2017, 130, 195–204 CrossRef CAS.
- U. Schmidt, A. Lieberknecht, U. Schanbacher, T. Beuttler and J. Wild, Facile Preparation of N-Acyl-2-(diethoxyphosphoryl)glycine Esters and Their Use in the Synthesis of Dehydroamino Acid Esters, Angew. Chem., Int. Ed., 1982, 21, 776–777 CrossRef.
- U. Schmidt, A. Lieberknecht and J. Wild, Amino Acids and Peptides; XLIII1. Dehydroamino Acids; XVIII2. Synthesis of Dehydroamino Acids and Amino Acids from N-Acyl-2-(dialkyloxyphosphinyl)-glycin Esters; II, Synthesis, 1984, 53–60 CrossRef CAS.
- B. Ku and D. Y. Oh, Facile synthesis of α-phosphorylated α-amino acids, Tetrahedron Lett., 1988, 29, 4465–4466 CrossRef CAS.
- L. Ferris, D. Haigh and C. J. Moody, A Simple Route to N-Acylaminophosphonoacetates (N-protected phosphonylglycine esters), Synlett, 1995, 921–922 CrossRef CAS.
- C. J. Moody, E. Swann, C. J. Moody, L. Ferris and D. Haigh, A new approach to peptide synthesis, Chem. Commun., 1997, 2391–2392 RSC.
- R. T. Buck, P. A. Clarke, D. M. Coe, M. J. Drysdale, L. Ferris, D. Haigh, C. J. Moody, N. D. Pearson and E. Swann, The Carbenoid Approach to Peptide Synthesis, Chem.–Eur. J., 2000, 6, 2160–2167 CrossRef CAS.
- V. B. Sokolov and A. Y. Aksinenko, Reactions of methyl 3,3,3-trifluoro-2-(pyridin-2-ylimino)-propanoates with mono- and difunctional nucleophiles, Russ. J. Gen. Chem., 2010, 80, 112–116 CrossRef CAS.
- J. Vicario, J. M. Ezpeleta and F. Palacios, Asymmetric Cyanation of α-Ketiminophosphonates Catalyzed by Cinchona Alkaloids: Enantioselective Synthesis of Tetrasubstituted α-Aminophosphonic Acid Derivatives from Trisubstituted α-Aminophosphonates, Adv. Synth. Catal., 2012, 354, 2641–2647 CrossRef CAS.
- U. Schmidt, H. Griesser, V. Leitenberger, A. Lieberknecht, R. Mangold, R. Meyer and B. Riedl, Diastereoselective Formation of (Z)-Didehydroamino Acid Esters, Synthesis, 1992, 1992, 487–490 CrossRef.
- H.-J. Kreuzfeld, C. Döbler, H. W. Krause and C. Facklam, Unusual amino acids V. Asymmetric hydrogenation of (Z)-N-acylaminocinnamic acid derivatives bearing different protective groups, Tetrahedron: Asymmetry, 1993, 4, 2047–2051 CrossRef CAS.
- R. W. Ratcliffe and B. G. Christensen, Total synthesis of β-lactam antibiotics I, Tetrahedron Lett., 1973, 14, 4645–4648 CrossRef.
- T. Ogasa, H. Saito, Y. Hashimoto, K. Sato and T. Hirata, Synthesis and Biological Evaluation of Optically Active 3-H-1-Carbacephem Compounds, Chem. Pharm. Bull., 1989, 37, 315–321 CrossRef CAS.
- R. Coleman and A. Carpenter, The development of strategies for construction of the aziridine core of the antitumor agents azinomycins A and B, Tetrahedron, 1997, 53, 16313–16326 CrossRef CAS.
- J. Mulzer, F. Schülzchen and J.-W. Bats, Rigid Dipeptide Mimetics. Stereocontrolled Synthesis of All Eight Stereoisomers of 2-Oxo-3-(N-Cbz-amino)-1-azabicyclo[4.3.0]nonane-9-carboxylic Acid Ester, Tetrahedron, 2000, 56, 4289–4298 CrossRef CAS.
- W. Wang, C. Xiong and V. J. Hruby, An efficient approach to asymmetric synthesis of dipeptide β-turn mimetics: indolizidinone amino acids, Tetrahedron Lett., 2001, 42, 3159–3161 CrossRef CAS.
- J. Wlochal, R. D. M. Davies and J. Burton, Synthesis of Novel Amino Acids Containing Cubane, Synlett, 2016, 27, 919–923 CrossRef CAS.
- Q. I. Churches, R. J. Mulder, J. M. White, J. Tsanaktsidis and P. J. Duggan, The Synthesis of a Cubane-Substituted Dipeptide, Aust. J. Chem., 2012, 65, 690–693 CrossRef CAS.
- A. Mathur, B. Wang, D. Smith, J. Li, J. Pawluczyk, J.-H. Sun, M. K. Wong, S. Krishnananthan, D.-R. Wu, D. Sun, P. Li, S. Yip, B.-C. Chen, P. S. Baran, Q. Chen, O. D. Lopez, Z. Yong, J. A. Bender, V. N. Nguyen, J. L. Romine, D. R. S. Laurent, G. Wang, J. F. Kadow, N. A. Meanwell, M. Belema and R. Zhao, Development of the Large-Scale Synthesis of Tetrahydropyran Glycine, a Precursor to the HCV NS5A Inhibitor BMS-986097, J. Org. Chem., 2017, 82, 10376–10387 CrossRef CAS.
- A. Shakhmin, M. P. Hall, J. R. Walker, T. Machleidt, B. F. Binkowski, K. V. Wood and T. A. Kirkland, Three Efficient Methods for Preparation of Coelenterazine Analogues, Chem.–Eur. J., 2016, 22, 10369–10375 CrossRef CAS.
- Y. Yasuno, M. Hamada, M. Kawasaki, K. Shimamoto, Y. Shigeri, T. Akizawa, M. Konishi, Y. Ohfune and T. Shinada, (7S)-Kaitocephalin as a potent NMDA receptor selective ligand, Org. Biomol. Chem., 2016, 14, 1206–1210 RSC.
- Y. Yasuno, M. Hamada, Y. Yoshida, K. Shimamoto, Y. Shigeri, T. Akizawa, M. Konishi, Y. Ohfune and T. Shinada, Structure–activity relationship study at C9 position of kaitocephalin, Bioorg. Med. Chem. Lett., 2016, 26, 3543–3546 CrossRef CAS PubMed.
- J. Reutzel, T. M. Diogo and A. Geyer, Reversible Folding of a β-Hairpin Peptide by a Metal-Chelating Amino Acid, Chem.–Eur. J., 2017, 23, 8450–8456 CrossRef CAS PubMed.
- H. Do, C. W. Kang, J. H. Cho and S. R. Gilbertson, Enantioselective Synthesis of (−)-Dysiherbaine, Org. Lett., 2015, 17, 3972–3974 CrossRef CAS PubMed.
- K. Fukushima, Y. Ishikawa, R. Sakai and M. Oikawa, A monocyclic neodysiherbaine analog: Synthesis and evaluation, Bioorg. Med. Chem. Lett., 2016, 26, 5164–5167 CrossRef CAS PubMed.
- A. Baron, J. Martinez and F. Lamaty, Solvent-free synthesis of unsaturated amino esters in a ball-mill, Tetrahedron Lett., 2010, 51, 6246–6249 CrossRef CAS.
- S. R. Gilbertson, G. Chen and M. McLoughlin, Versatile Building Block for the Synthesis of Phosphine-Containing Peptides: The Sulfide of Diphenylphosphinoserine, J. Am. Chem. Soc., 1994, 116, 4481–4482 CrossRef CAS.
- S. R. Gilbertson and X. Wang, Synthesis of (Dicyclohexylphosphino)serine, Its Incorporation into a Dodecapeptide, and the Coordination of Rhodium, J. Org. Chem., 1996, 61, 434–435 CrossRef CAS.
- A. M. Porte, W. A. van der Donk and K. Burgess, New and Efficient Synthesis of an Amino Acid for Preparing Phosphine-Functionalized Peptidomimetics, J. Org. Chem., 1998, 63, 5262–5264 CrossRef CAS.
- D. J. Brauer, K. W. Kottsieper, S. Schenk and O. Stelzer, Chiral Phosphine Ligands from Amino Acids. II. A Facile Synthesis of Phosphinoserines by Nucleophilic Phosphination Reactions, Z. Anorg. Allg. Chem., 2001, 627, 1151–1156 CrossRef CAS.
- S. van Zutphen, V. J. Margarit, G. Mora and P. Le Floch, Readily available amino acid building blocks for the synthesis of phosphole-containing peptides, Tetrahedron Lett., 2007, 48, 2857–2859 CrossRef CAS.
- S. J. Greenfield and S. R. Gilbertson, Preparation of Diphenylphosphinoserine and Synthesis of Other Phosphine Containing Amino Acids Using Zinc/Copper Reagents, Synthesis, 2001, 2001, 2337–2340 CrossRef.
- A. Agarkov, S. Greenfield, D. Xie, R. Pawlick, G. Starkey and S. R. Gilbertson, Synthesis of phosphine containing amino acids: Utilization of peptide synthesis in ligand design, Pept. Sci., 2006, 84, 48–73 CrossRef CAS.
- B. J. Cowen and S. J. Miller, Enantioselective [3 + 2]-Cycloadditions Catalyzed by a Protected, Multifunctional Phosphine-Containing α-Amino Acid, J. Am. Chem. Soc., 2007, 129, 10988–10989 CrossRef CAS.
- S. R. Gilbertson, S. E. Collibee and A. Agarkov, Asymmetric Catalysis with Libraries of Palladium β-Turn Phosphine Complexes, J. Am. Chem. Soc., 2000, 122, 6522–6523 CrossRef CAS.
- A. Agarkov, S. J. Greenfield, T. Ohishi, S. E. Collibee and S. R. Gilbertson, Catalysis with Phosphine-Containing Amino Acids in Various “Turn” Motifs, J. Org. Chem., 2004, 69, 8077–8085 CrossRef CAS PubMed.
- S. J. Greenfield, A. Agarkov and S. R. Gilbertson, High Asymmetric Induction with β-Turn-Derived Palladium Phosphine Complexes, Org. Lett., 2003, 5, 3069–3072 CrossRef CAS PubMed.
- S. R. Gilbertson and S. Yamada, A study of catalyst selectivity with polymer bound palladium phosphine complexes on various solid phase synthesis supports, Tetrahedron Lett., 2004, 45, 3917–3920 CrossRef CAS.
- T. Itaya, A. Mizutani and T. Iida, Synthesis and Absolute Configuration of Wybutine, the Fluorescent Minor Base from Phenylalanine Transfer Ribonucleic Acids, Chem. Pharm. Bull., 1991, 39, 1407–1414 CrossRef CAS.
- T. Itaya and A. Mizutani, Synthesis of (S-(−)-wybutine, the fluorescent minor base from yeast phenylalanine transfer ribonucleic acids, Tetrahedron Lett., 1985, 26, 347–350 CrossRef CAS.
- T. Itaya, M. Shimomichi and M. Ozasa, Access to the synthesis of wybutosine, the first tricyclic fluorescent nucleoside isolated from phenyllalanine transfer ribonucleic acids, Tetrahedron Lett., 1988, 29, 4129–4132 CrossRef CAS.
- T. Itaya, T. Iida, S. Shimizu, A. Mizutani, M. Morisue, Y. Sugimoto and M. Tachinaka, Wittig Reaction with N-Protected 3-(Triphenylphosphonio)alaninates : Synthesis of Optically Active (E)-(2-Arylvinyl)glycine Derivatives, Chem. Pharm. Bull., 1993, 41, 252–261 CrossRef CAS.
- T. Itaya, T. Kanai and T. Iida, Synthesis of [R-(R*,S*)]- and [S-(R*,R*)]β-hydroxy-3-(β-d-ribofuranosyl)-wybutines, the most probable alternatives for the hypermodified nucleoside of rat liver phenylalanine transfer ribonucleic acid, Tetrahedron Lett., 1997, 38, 1979–1982 CrossRef CAS.
- F. Meyer, J. Uziel, A. M. Papini and S. Jugé, Triphenylphosphonium salts bearing an l-alanyl substituent: short synthesis and enantiomeric analysis by NMR, Tetrahedron Lett., 2001, 42, 3981–3984 CrossRef CAS.
- J.-M. Varlet, N. Collignon and P. Savignac, Synthèse et amination réductrice de phosphonopyruvates: préparation d'acides amino-2 carboxy-2 alkylphosphoniques (β-phosphonoalanine), Can. J. Chem., 1979, 57, 3216–3220 CrossRef CAS.
- V. A. Soloshonok, Y. N. Belokon, N. A. Kuzmina, V. I. Maleev, N. Y. Svistunova, V. A. Solodenko and V. P. Kukhar, Asymmetric synthesis of phosphorus analogues of dicarboxylic [small alpha]-amino acids, J. Chem. Soc., Perkin Trans. 1, 1992, 1525–1529 RSC.
- E. Smith, L. McQuaid, J. Paschal and J. DeHoniesto, An enantioselective synthesis of D-(-)- and L-(+)-2-amino-3-phosphonopropanoic acid, J. Org. Chem., 1990, 55, 4472–4474 CrossRef CAS.
- M. Cortes-Clerget, O. Gager, M. Monteil, E. Migianu-Griffoni, J. Deschamp and M. Lecouvey, Peptides holding a phosphonic acid: Easily recyclable organocatalysts for enantioselective C–C bond creation, Phosphorus, Sulfur, Silicon Relat. Elem., 2016, 191, 1593–1594 CrossRef CAS.
- M. Cortes-Clerget, O. Gager, M. Monteil, J.-L. Pirat, E. Migianu-Griffoni, J. Deschamp and M. Lecouvey, Novel Easily Recyclable Bifunctional Phosphonic Acid Carrying Tripeptides for the Stereoselective Michael Addition of Aldehydes with Nitroalkenes, Adv. Synth. Catal., 2016, 358, 34–40 CrossRef CAS.
- M. Cortes-Clerget, J. Jover, J. Dussart, E. Kolodziej, M. Monteil, E. Migianu-Griffoni, O. Gager, J. Deschamp and M. Lecouvey, Bifunctional Tripeptide with a Phosphonic Acid as a Brønsted Acid for Michael Addition: Mechanistic Insights, Chem.–Eur. J., 2017, 23, 6654–6662 CrossRef CAS.
- E. Rémond, J. Bayardon, M.-J. Ondel-Eymin and S. Jugé, Stereoselective Synthesis of Unsaturated and Functionalized l-NHBoc Amino Acids, Using Wittig Reaction under Mild Phase-Transfer Conditions, J. Org. Chem., 2012, 77, 7579–7587 CrossRef.
- H. Audi, E. Rémond, M.-J. Eymin, A. Tessier, R. Malacea-Kabbara and S. Jugé, Modular Hemisyntheses of Boronato- and Trifluoroborato-Substituted L-NHBoc Amino Acid and Peptide Derivatives, Eur. J. Org. Chem., 2013, 2013, 7960–7972 CrossRef CAS.
- B. Rugeri, H. Audi, P. Jewula, R. Koudih, R. Malacea-Kabbara, D. Vimont, J. Schulz, P. Fernandez and S. Jugé, Designing Silylated l-Amino Acids using a Wittig Strategy: Synthesis of Peptide Derivatives and 18F-Labelling, Eur. J. Org. Chem., 2017, 2017, 5399–5409 CrossRef CAS.
- J. Bernard, R. Malacea-Kabbara, G. S. Clemente, B. P. Burke, M.-J. Eymin, S. J. Archibald and S. Jugé, o-Boronato- and o-Trifluoroborato–Phosphonium Salts Supported by l-α-Amino Acid Side Chain, J. Org. Chem., 2015, 80, 4289–4298 CrossRef CAS PubMed.
- F. Real-Fernández, A. Colson, J. Bayardon, F. Nuti, E. Peroni, R. Meunier-Prest, F. Lolli, M. Chelli, C. Darcel, S. Jugé and A. M. Papini, Ferrocenyl glycopeptides as electrochemical probes to detect autoantibodies in multiple sclerosis patients' sera, Pept. Sci., 2008, 90, 488–495 CrossRef PubMed.
- P. Minois, J. Bayardon, R. Meunier-Prest and S. Jugé, [60]Fullerene l-Amino Acids and Peptides: Synthesis under Phase-Transfer Catalysis Using a Phosphine–Borane Linker. Electrochemical Behavior, J. Org. Chem., 2017, 82, 11358–11369 CrossRef CAS PubMed.
- M. Arribat, E. Rémond, S. Clément, A. Van der Lee and F. Cavelier, Phospholyl(borane) Amino Acids and Peptides: Stereoselective Synthesis and Fluorescent Properties with Large Stokes Shift, J. Am. Chem. Soc., 2018, 140, 1028–1034 CrossRef CAS PubMed.
- S. Gilbertson and R. Pawlick, Synthesis of Thiophosphoryl Derivatives of Proline: Building Blocks for Phosphanyl-Substituted Peptides with β-Turns, Angew. Chem., Int. Ed., 1996, 35, 902–904 CrossRef CAS.
- E. W. Logusch, Facile synthesis of D,L-phosphinothricin from methyl 4-bromo-2-phthalimidobutyrate, Tetrahedron Lett., 1986, 27, 5935–5938 CrossRef CAS.
- H.-J. Zeiss, Enantioselective synthesis of L-phosphinothricin from L-methionine and L-glutamic acid via L-vinylglycine, Tetrahedron, 1992, 48, 8263–8270 CrossRef CAS.
- Q. Yang and S.-D. Yang, Highly Efficient and Divergent Construction of Chiral γ-Phosphono-α-Amino Acids via Palladium-Catalyzed Alkylation of Unactivated C(sp3)–H Bonds, ACS Catal., 2017, 7, 5220–5224 CrossRef CAS.
- A. Schick, T. Kolter, A. Giannis and K. Sandhoff, Synthesis of phosphonate analogues of sphinganine-1-phosphate and sphingosine-1-phosphate, Tetrahedron, 1995, 51, 11207–11218 CrossRef CAS.
- G. Shapiro, D. Buechler, V. Ojea, E. Pombo-Villar, M. Ruiz and H.-P. Weber, Synthesis of both D- and L-Fmoc-Abu[PO(OCH2CH=CH2)2]-OH for solid phase phosphonopeptide synthesis, Tetrahedron Lett., 1993, 34, 6255–6258 CrossRef CAS.
- M. C. Fernández, A. Díaz, J. J. Guillín, O. Blanco, M. Ruiz and V. Ojea, Diastereoselective Synthesis of 2-Amino-4-phosphonobutanoic Acids by Electrophilic Substitution and Tin–Peterson Olefination of Bis-lactim Ethers Derived from cyclo-[l-AP4-d-Val], J. Org. Chem., 2006, 71, 6958–6974 CrossRef PubMed.
- F. W. Foss, A. H. Snyder, M. D. Davis, M. Rouse, M. D. Okusa, K. R. Lynch and T. L. Macdonald, Synthesis and biological evaluation of γ-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists, Bioorg. Med. Chem., 2007, 15, 663–677 CrossRef CAS PubMed.
- G. Tong, J. Perich and R. Johns, The Improved Synthesis of Boc-Abu(PO3Me2)-OH and Its Use for the Facile Synthesis of Glu-Abu(P)-Leu, Aust. J. Chem., 1992, 45, 1225–1240 CrossRef CAS.
- J. W. Perich, The Facile Synthesis of 2-(Fluorenylmethoxycarbonylamino)-4-(O′,O′′-dimethylphosphono)-L-butanoic Acid {Fmoc-Abu(PO3Me2)-OH}, Synlett, 1992, 595–596 CrossRef CAS.
- T. Yokomatsu, M. Sato and S. Shibuya, Lipase-catalyzed enantioselective acylation of prochiral 2-(ω-phosphono)alkyl-1,3-propanediols: Application to the enantioselective synthesis of ω-phosphono-α-amino acids, Tetrahedron: Asymmetry, 1996, 7, 2743–2754 CrossRef CAS.
- H.-X. Chen, J. Kang, R. Chang, Y.-L. Zhang, H.-Z. Duan, Y.-M. Li and Y.-X. Chen, Synthesis of α,α-Difluorinated Phosphonate pSer/pThr Mimetics via Rhodium-Catalyzed Asymmetric Hydrogenation of β-Difluorophosphonomethyl α-(Acylamino)acrylates, Org. Lett., 2018, 20, 3278–3281 CrossRef CAS PubMed.
- M. Dzięgielewski, J. Hejmanowska and Ł. Albrecht, A Convenient Approach to a Novel Group of Quaternary Amino Acids Containing- a Geminal Bisphosphonate Moiety, Synthesis, 2014, 46, 3233–3238 CrossRef.
- I. A. Natchev, Total synthesis, enzyme-substrate interactions and herbicidal activity of plumbemicin A and B (N-1409), Tetrahedron, 1988, 44, 1511–1522 CrossRef CAS.
- O. García-Barradas and E. Juaristi, Highly enantioselective synthesis of (R)- and (S)-2-amino-5-phosphonopentanoic acids [(R)- and (S)-AP5] via modified Seebach imidazolidinones, Tetrahedron, 1995, 51, 3423–3434 CrossRef.
- J. P. Whitten, B. M. Baron, D. Muench, F. Miller, H. S. White and I. A. McDonald, (R)-4-Oxo-5-phosphononorvaline: a new competitive glutamate antagonist at the NMDA receptor complex, J. Med. Chem., 1990, 33, 2961–2963 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, A. T. Russell and M. L. Smith, Carbon based nucleophilic ring opening of activated monocyclic β-lactams; synthesis and stereochemical assignment of the ACE inhibitor WF-10129, Tetrahedron, 1995, 51, 4733–4762 CrossRef CAS.
- D. E. Rudisill and J. P. Whitten, Synthesis of (R)-4-Oxo-5-phosphononorvaline, an N-Methyl-D-aspartic Acid Receptor Selective β-Keto Phosphonate, Synthesis, 1994, 851–854 CrossRef CAS.
- L. S. Fowler, D. Ellis and A. Sutherland, Synthesis of fluorescent enone derived [small alpha]-amino acids, Org. Biomol. Chem., 2009, 7, 4309–4316 RSC.
- L. S. Fowler, L. H. Thomas, D. Ellis and A. Sutherland, A one-pot, reductive amination/6-endo-trigcyclisation for the stereoselective synthesis of 6-substituted-4-oxopipecolic acids, Chem. Commun., 2011, 47, 6569–6571 RSC.
- A. H. Harkiss, J. D. Bell, A. Knuhtsen, A. G. Jamieson and A. Sutherland, Synthesis and Fluorescent Properties of β-Pyridyl α-Amino Acids, J. Org. Chem., 2019, 84, 2879–2890 CrossRef CAS PubMed.
- F. Gosselin and W. D. Lubell, An Olefination Entry for the Synthesis of Enantiopure α,ω-Diaminodicarboxylates and Azabicyclo[X.Y.0]alkane Amino Acids, J. Org. Chem., 1998, 63, 7463–7471 CrossRef CAS PubMed.
- D. Kang, T. Kim, H. Lee and S. Hong, Regiodivergent Ring-Opening Cross-Coupling of Vinyl Aziridines with Phosphorus Nucleophiles: Access to Phosphorus-Containing Amino Acid Derivatives, Org. Lett., 2018, 20, 7571–7575 CrossRef CAS PubMed.
- Z. A. Dziuganowska, K. Ślepokura, J.-N. Volle, D. Virieux, J.-L. Pirat and P. Kafarski, Structural Analogues of Selfotel, J. Org. Chem., 2016, 81, 4947–4954 CrossRef CAS PubMed.
- S. Gilbertson and G. Starkey, Palladium-Catalyzed Synthesis of Phosphine-Containing Amino Acids, J. Org. Chem., 1996, 61, 2922–2923 CrossRef CAS PubMed.
- H.-B. Kraatz and A. Pletsch, P–C bond formation: synthesis of phosphino amino acids by palladium-catalysed cross-coupling, Tetrahedron: Asymmetry, 2000, 11, 1617–1621 CrossRef CAS.
- F. Bisaro and P. Le Floch, Incorporation of Phosphole Moieties into the Side Chain of Tyrosine and Phenylalanine, Synlett, 2010, 2010, 3081–3085 CrossRef.
- M. Tepper, O. Stelzer, T. Häusler and W. S. Sheldrick, A systematic synthetic approach to phosphinophenyl-glycine and -alanine chiral phosphine ligands with amino acid moieties, Tetrahedron Lett., 1997, 38, 2257–2258 CrossRef CAS.
- D. Solas, R. L. Hale and D. V. Patel, An Efficient Synthesis of N-α-Fmoc-4-(Phosphonodifluoromethyl)-l-phenylalanine, J. Org. Chem., 1996, 61, 1537–1539 CrossRef CAS.
- C. Meyer and M. Köhn, Efficient Scaled-Up Synthesis of N-α-Fmoc-4-Phosphono(difluoromethyl)-l-phenylalanine and Its Incorporation into Peptides, Synthesis, 2011, 2011, 3255–3260 CrossRef.
- P. Li, M. Zhang, M. L. Peach, H. Liu, D. Yang and P. P. Roller, Concise and Enantioselective Synthesis of Fmoc-Pmp(But)2-OH and Design of Potent Pmp-Containing Grb2-SH2 Domain Antagonists, Org. Lett., 2003, 5, 3095–3098 CrossRef CAS PubMed.
- J.-M. Kee, B. Villani, L. R. Carpenter and T. W. Muir, Development of Stable Phosphohistidine Analogues, J. Am. Chem. Soc., 2010, 132, 14327–14329 CrossRef CAS PubMed.
- A. K. Gorle, J. Zhang, Q. Liu, S. J. Berners-Price and N. P. Farrell, Structural Factors Affecting Binding of Platinum Anticancer Agents with Phospholipids: Influence of Charge and Phosphate Clamp Formation, Chem.–Eur. J., 2018, 24, 4643–4652 CrossRef CAS PubMed.
- H. Brachwitz, M. Ölke, J. Bergmann and P. Langen, Alkylphospho-L-serine analogues: Synthesis of cytostatically active alkylphosphono derivatives, Bioorg. Med. Chem. Lett., 1997, 7, 1739–1742 CrossRef CAS.
- D. E. Petrillo, D. R. Mowrey, S. P. Allwein and R. P. Bakale, A General Preparation of Protected Phosphoamino Acids, Org. Lett., 2012, 14, 1206–1209 CrossRef CAS PubMed.
- P. W. Galka and H.-B. Kraatz, Synthesis and study of amino acid based phosphinite ligands, J. Organomet. Chem., 2003, 674, 24–31 CrossRef CAS.
- J. I. Murray, R. Woscholski and A. C. Spivey, Highly efficient and selective phosphorylation of amino acid derivatives and polyols catalysed by
2-aryl-4-(dimethylamino)pyridine-N-oxides – towards kinase-like reactivity, Chem. Commun., 2014, 50, 13608–13611 RSC.
- L. W. Peterson, M. Sala-Rabanal, I. S. Krylov, M. Serpi, B. A. Kashemirov and C. E. McKenna, Serine Side Chain-Linked Peptidomimetic Conjugates of Cyclic HPMPC and HPMPA: Synthesis and Interaction with hPEPT1, Mol. Pharm., 2010, 7, 2349–2361 CrossRef CAS PubMed.
- M. J. Piggott and P. V. Attwood, Focus on O-phosphohydroxylysine, O-phosphohydroxyproline, N1-phosphotryptophan and S-phosphocysteine, Amino Acids, 2017, 49, 1309–1323 CrossRef CAS PubMed.
- A. K. Buchowiecka, Puzzling over protein cysteine phosphorylation – assessment of proteomic tools for S-phosphorylation profiling, Analyst, 2014, 139, 4118–4123 RSC.
- D. Asthagiri, T. Liu, L. Noodleman, R. L. Van Etten and D. Bashford, On the Role of the Conserved Aspartate in the Hydrolysis of the Phosphocysteine Intermediate of the Low Molecular Weight Tyrosine Phosphatase, J. Am. Chem. Soc., 2004, 126, 12677–12684 CrossRef CAS PubMed.
- A. Kertmen, S. Lach, J. Rachon and D. Witt, Novel and Efficient Methods for the Synthesis of Symmetrical Trisulfides, Synthesis, 2009, 2009, 1459–1462 CrossRef.
- S. Lach and D. Witt, A New and Convenient Method for the Preparation of Functionalized Phosphorothioates, Synthesis, 2011, 2011, 3975–3978 CrossRef.
- C. Liu, C.-M. Park, D. Wang and M. Xian, Phosphite Esters: Reagents for Exploring S-Nitrosothiol Chemistry, Org. Lett., 2018, 20, 7860–7863 CrossRef CAS PubMed.
- A. M. Z. Slawin, J. D. Woollins and Q. Zhang, Novel chiral phosphine ligands and complexes from amino acid esters, J. Chem. Soc., Dalton Trans., 2001, 621–632 RSC.
- P. Bergamini, V. Bertolasi and R. Giordani, Platinum-assisted preparation of PS ligands containing chiral centres; X-ray crystal structure of a dithiophosphinite platinum complex, Inorg. Chim. Acta, 2005, 358, 2031–2039 CrossRef CAS.
- U. Masaaki, I. Toshiyuki and I. Shigeru, Phosphinyl- and Phosphinothioylamino Acids and Peptides. V. Preparation of Dimethylphosphinothioylamino Acids and Solid Phase Peptide Synthesis, Bull. Chem. Soc. Jpn., 1979, 52, 2424–2427 CrossRef.
- U. Masaaki and S. Kozo, Phosphinyl and Phosphinothioyl Amino Acids and Peptides. VII. The Use of the Dimethylphosphinothioyl Group as a Thiol-protecting Group of Cysteine, Bull. Chem. Soc. Jpn., 1983, 56, 1187–1191 CrossRef.
- M. Ueki, T. Ikeo, K. Hokari, K. Nakamura, A. Saeki and H. Komatsu, A New Efficient Method for S–CH2–S Bond Formation and Its Application to a Djenkolic Acid-Containing Cyclic Enkephalin Analog, Bull. Chem. Soc. Jpn., 1999, 72, 829–838 CrossRef CAS.
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases, Nat. Rev. Drug Discovery, 2013, 12, 447 CrossRef CAS PubMed.
- M. Slusarczyk, M. H. Lopez, J. Balzarini, M. Mason, W. G. Jiang, S. Blagden, E. Thompson, E. Ghazaly and C. McGuigan, Application of ProTide Technology to Gemcitabine: A Successful Approach to Overcome the Key Cancer Resistance Mechanisms Leads to a New Agent (NUC-1031) in Clinical Development, J. Med. Chem., 2014, 57, 1531–1542 CrossRef CAS PubMed.
- Y. Mehellou, The ProTides Boom, ChemMedChem, 2016, 11, 1114–1116 CrossRef CAS PubMed.
- Y. Mehellou, H. S. Rattan and J. Balzarini, The ProTide Prodrug Technology: From the Concept to the Clinic, J. Med. Chem., 2018, 61, 2211–2226 CrossRef CAS PubMed.
- B. S. Ross, P. Ganapati Reddy, H.-R. Zhang, S. Rachakonda and M. J. Sofia, Synthesis of Diastereomerically Pure Nucleotide Phosphoramidates, J. Org. Chem., 2011, 76, 8311–8319 CrossRef CAS PubMed.
- C. A. Roman, J. Balzarini and C. Meier, Diastereoselective Synthesis of Aryloxy Phosphoramidate Prodrugs of 3′-Deoxy-2′,3′-didehydrothymidine Monophosphate, J. Med. Chem., 2010, 53, 7675–7681 CrossRef CAS.
- B. Simmons, Z. Liu, A. Klapars, A. Bellomo and S. M. Silverman, Mechanism-Based Solution to the ProTide Synthesis Problem: Selective Access to Sofosbuvir, Acelarin, and INX-08189, Org. Lett., 2017, 19, 2218–2221 CrossRef CAS.
- J. Bertran-Vicente, M. Schümann, P. Schmieder, E. Krause and C. P. R. Hackenberger, Direct access to site-specifically phosphorylated-lysine peptides from a solid-support, Org. Biomol. Chem., 2015, 13, 6839–6843 RSC.
- M. Nielsen, C. B. Jacobsen and K. A. Jørgensen, Asymmetric Organocatalytic Electrophilic Phosphination, Angew. Chem., Int. Ed., 2011, 50, 3211–3214 CrossRef CAS PubMed.
- J. Jiang, H. Liu, C.-D. Lu and Y.-J. Xu, Diethyl Phosphite Initiated Coupling of α-Ketoesters with Imines for Synthesis of α-Phosphonyloxy-β-amino Acid Derivatives and Aziridine-2-carboxylates, Org. Lett., 2016, 18, 880–883 CrossRef CAS PubMed.
- N. Defacqz, R. Touillaux, A. Cordi and J. Marchand-Brynaert, [small beta]-Aminophosphonic compounds derived from methyl 1-dimethoxyphosphoryl-2-succinimidocyclohex-3-ene-1-carboxylates, J. Chem. Soc., Perkin Trans. 1, 2001, 2632–2637 RSC.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Emmanuelle Rémond
*b and
Emmanuelle Rémond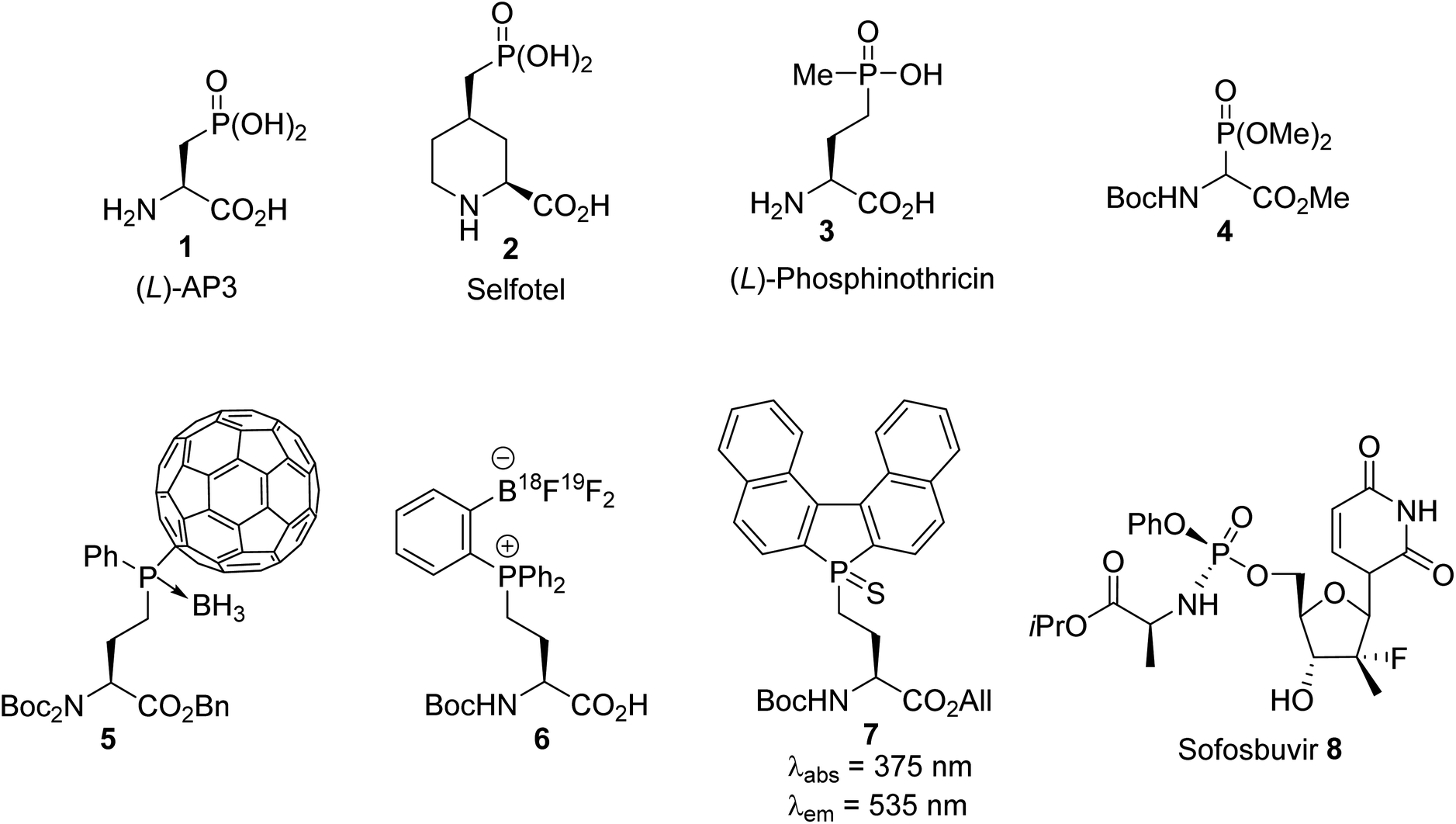
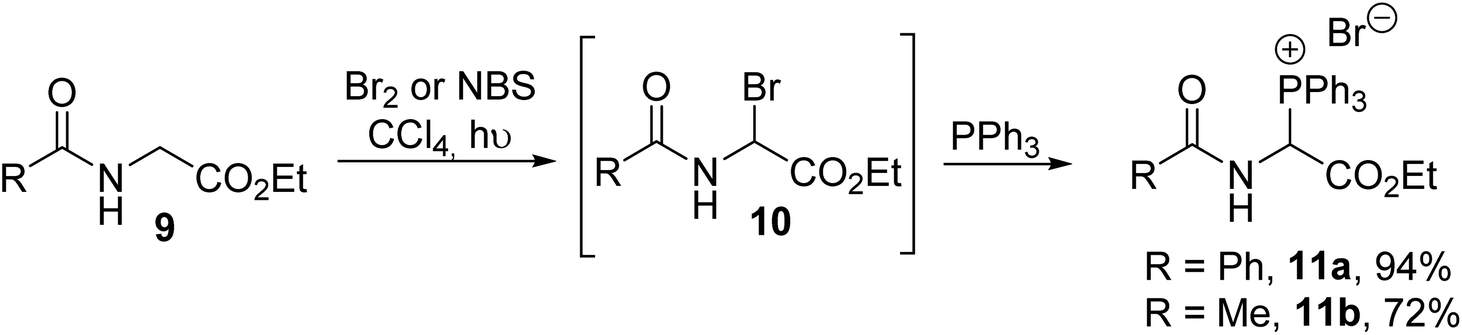
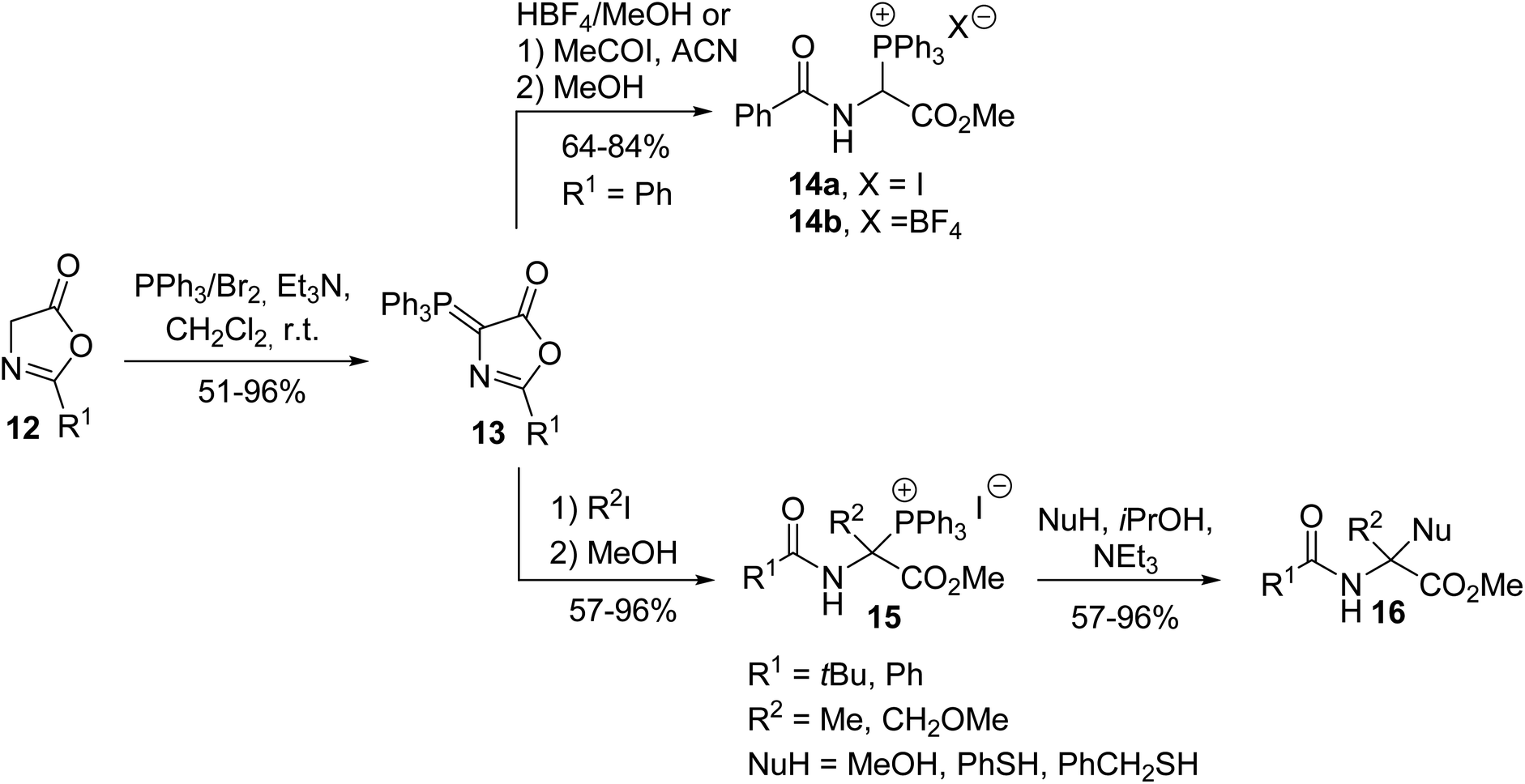
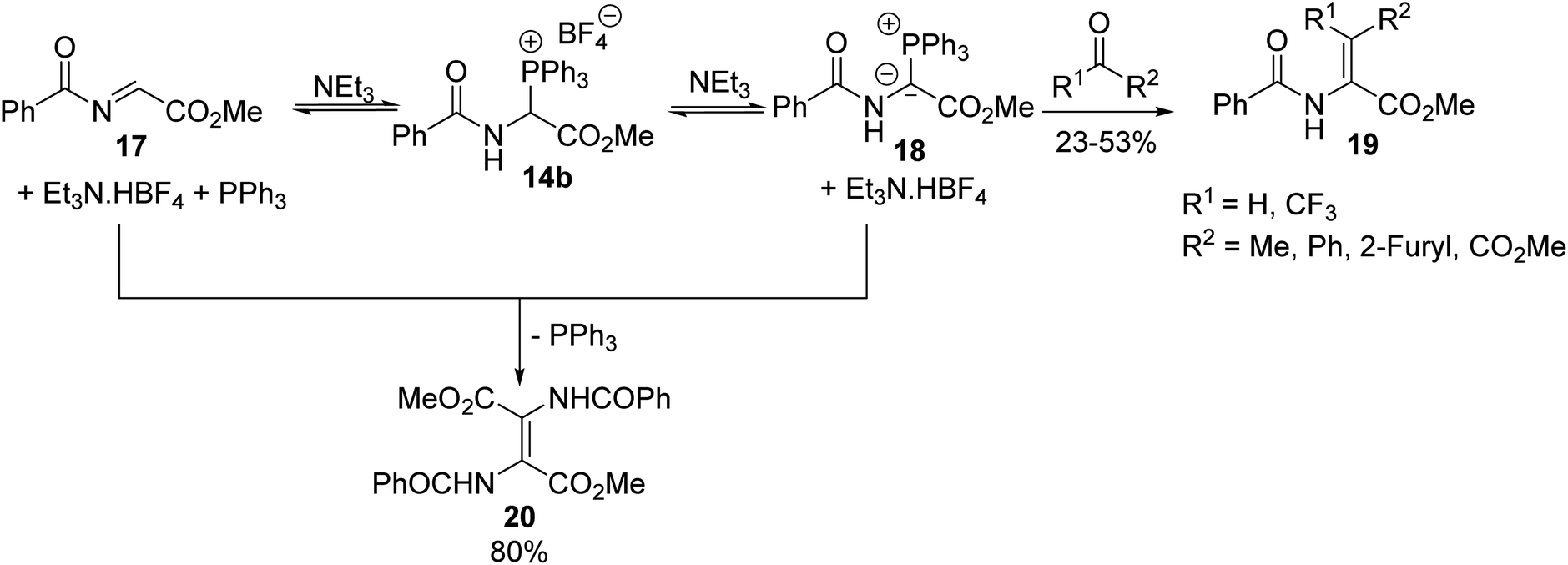
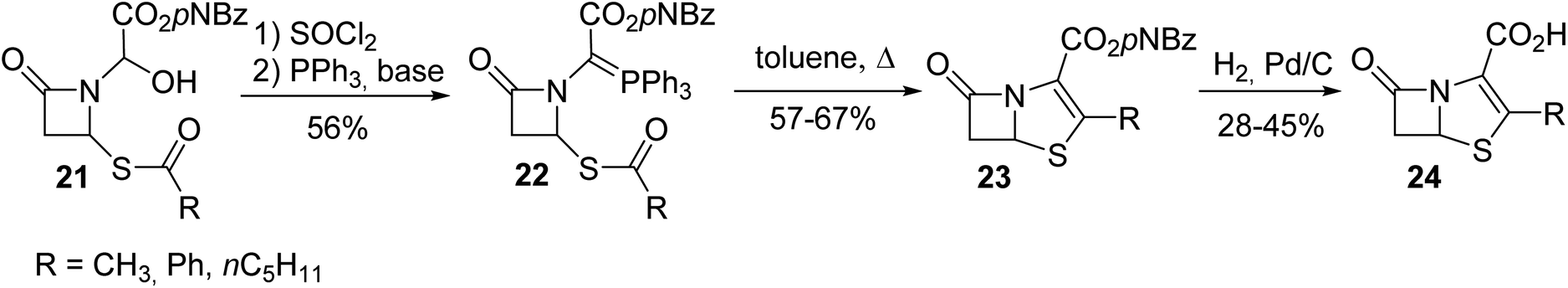
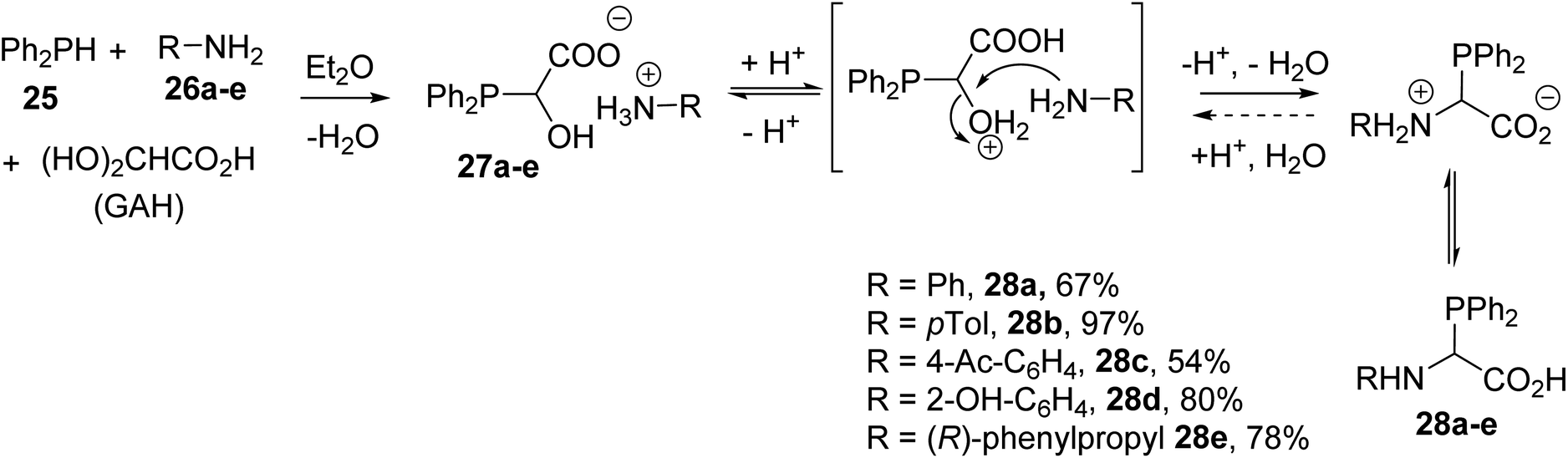

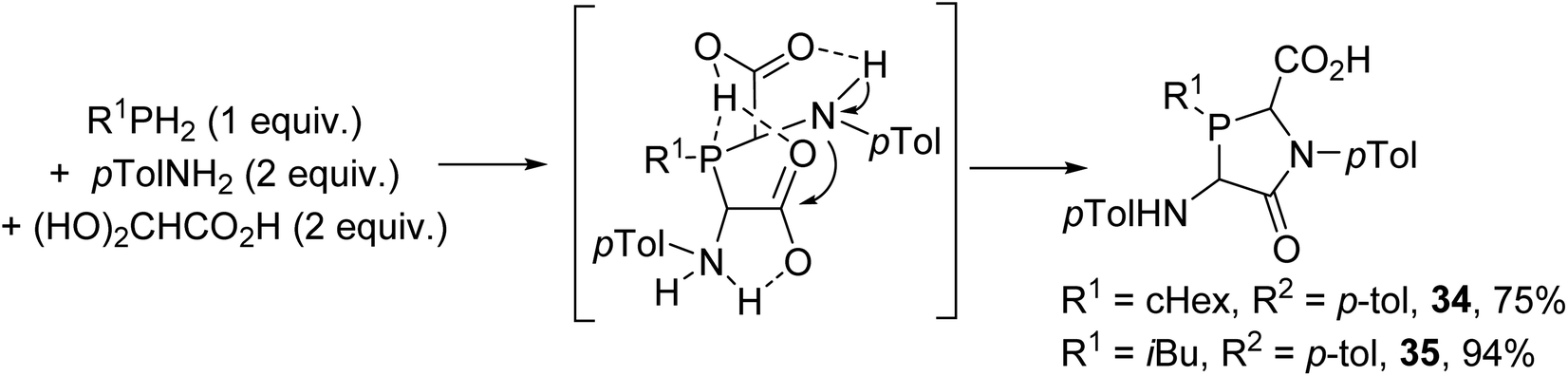
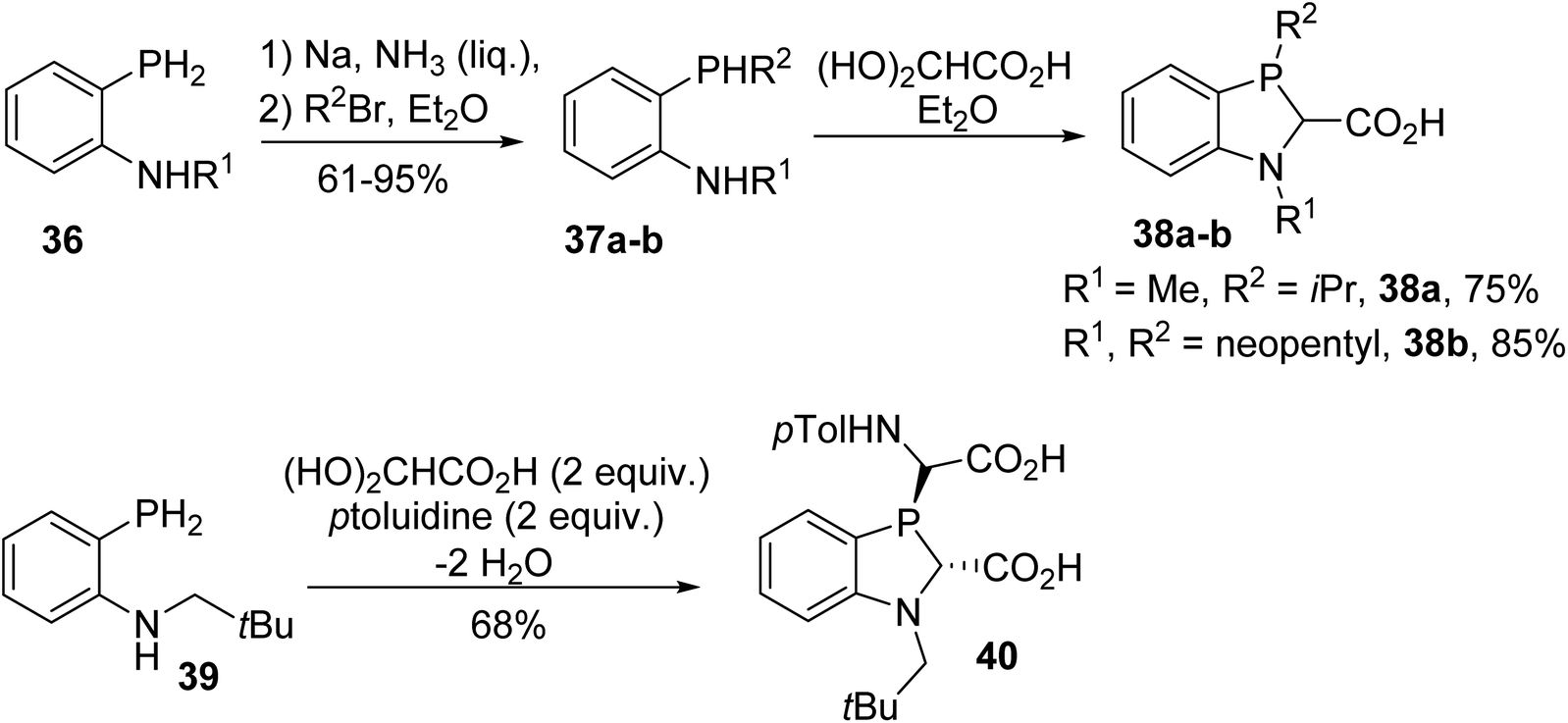
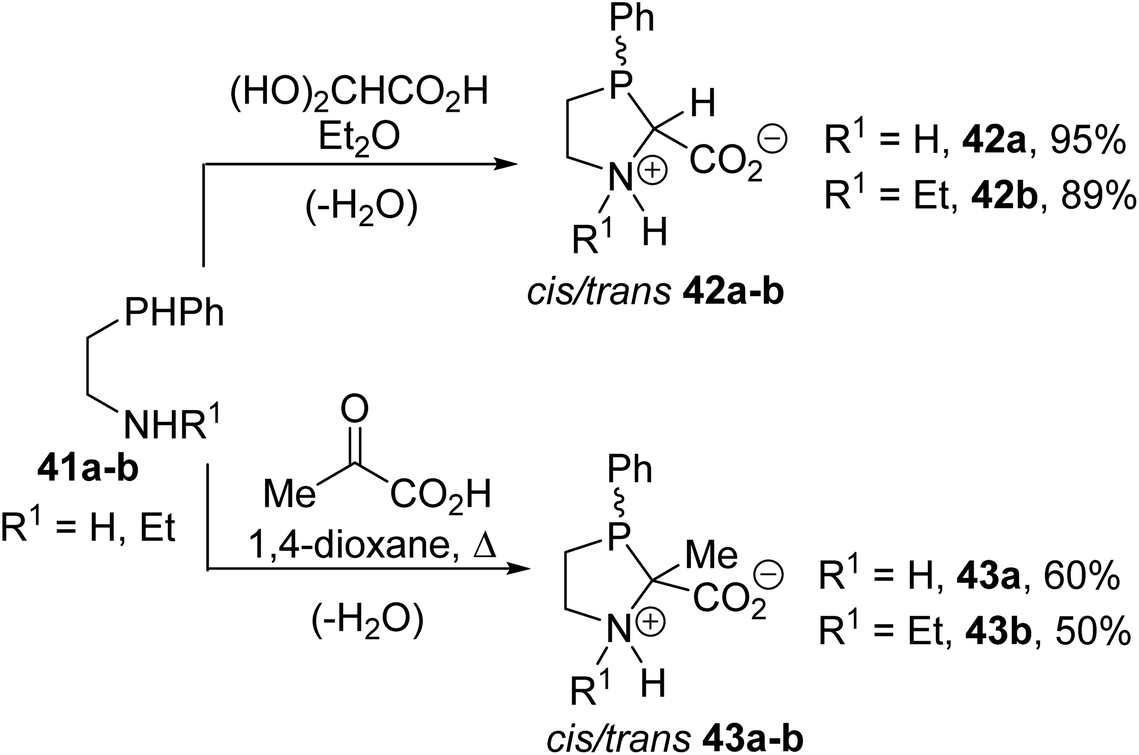
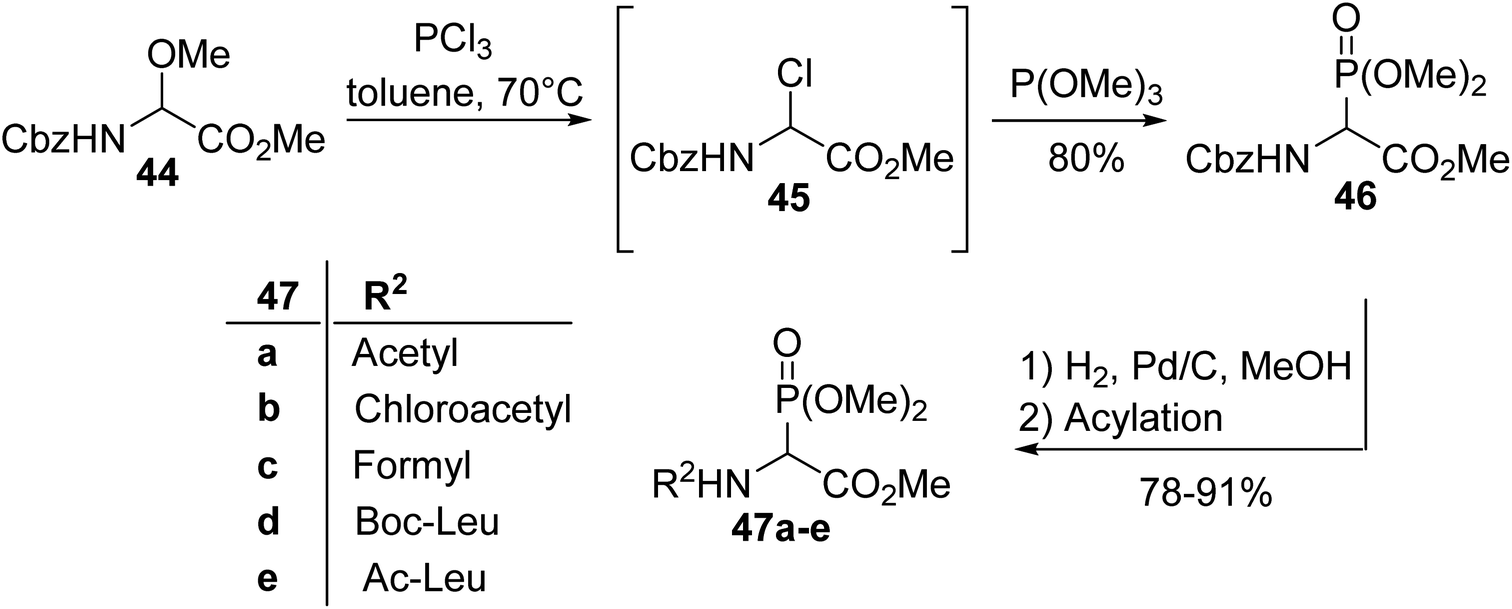
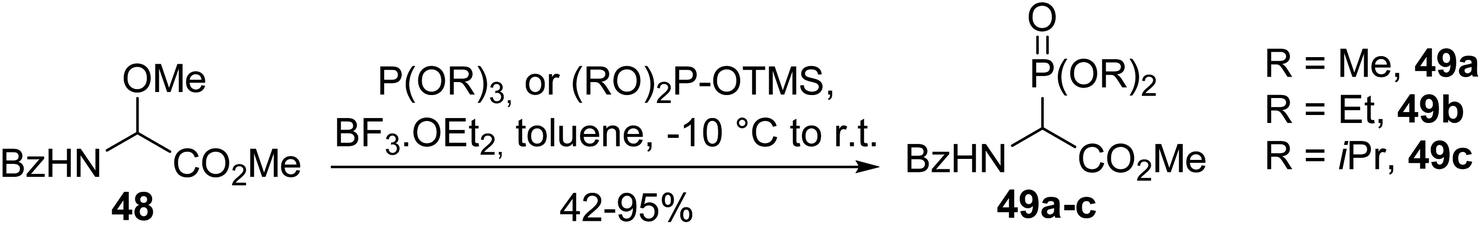
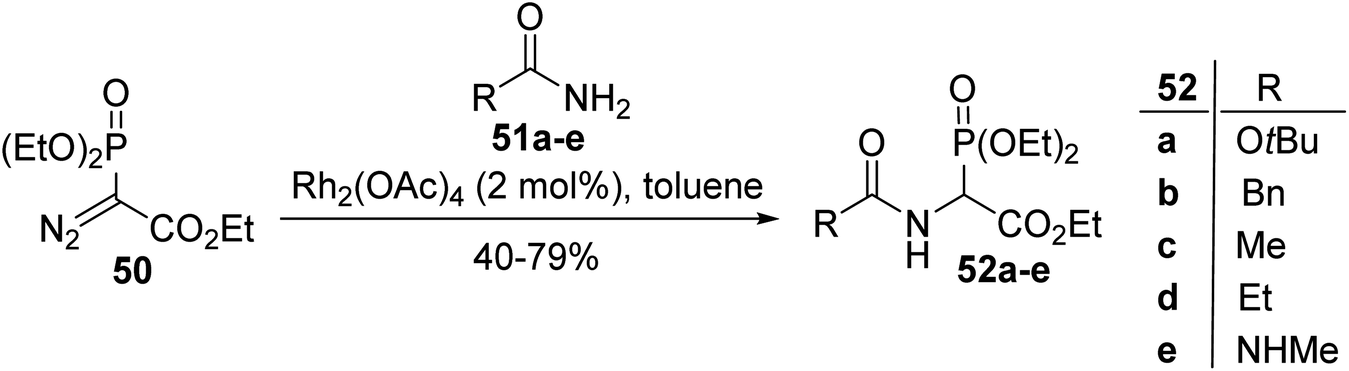
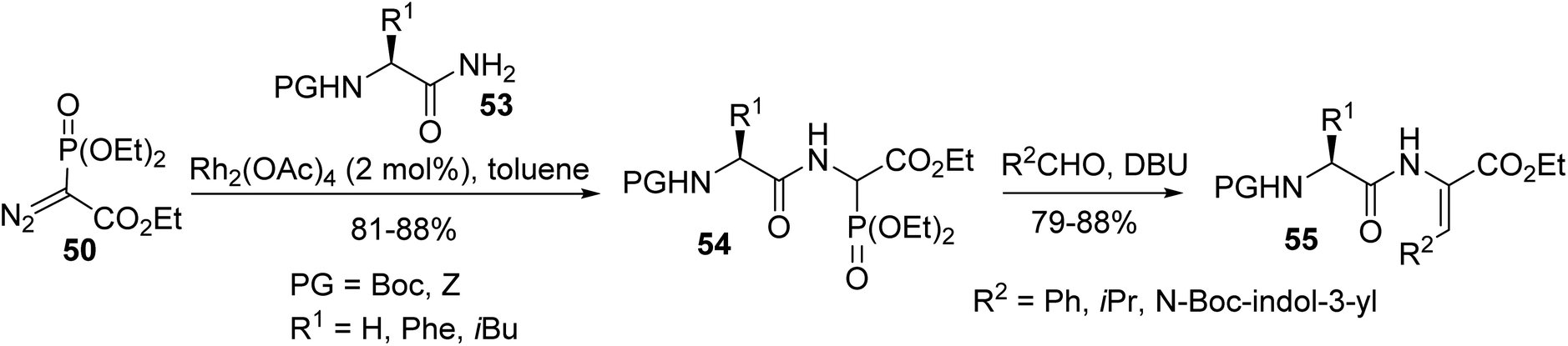

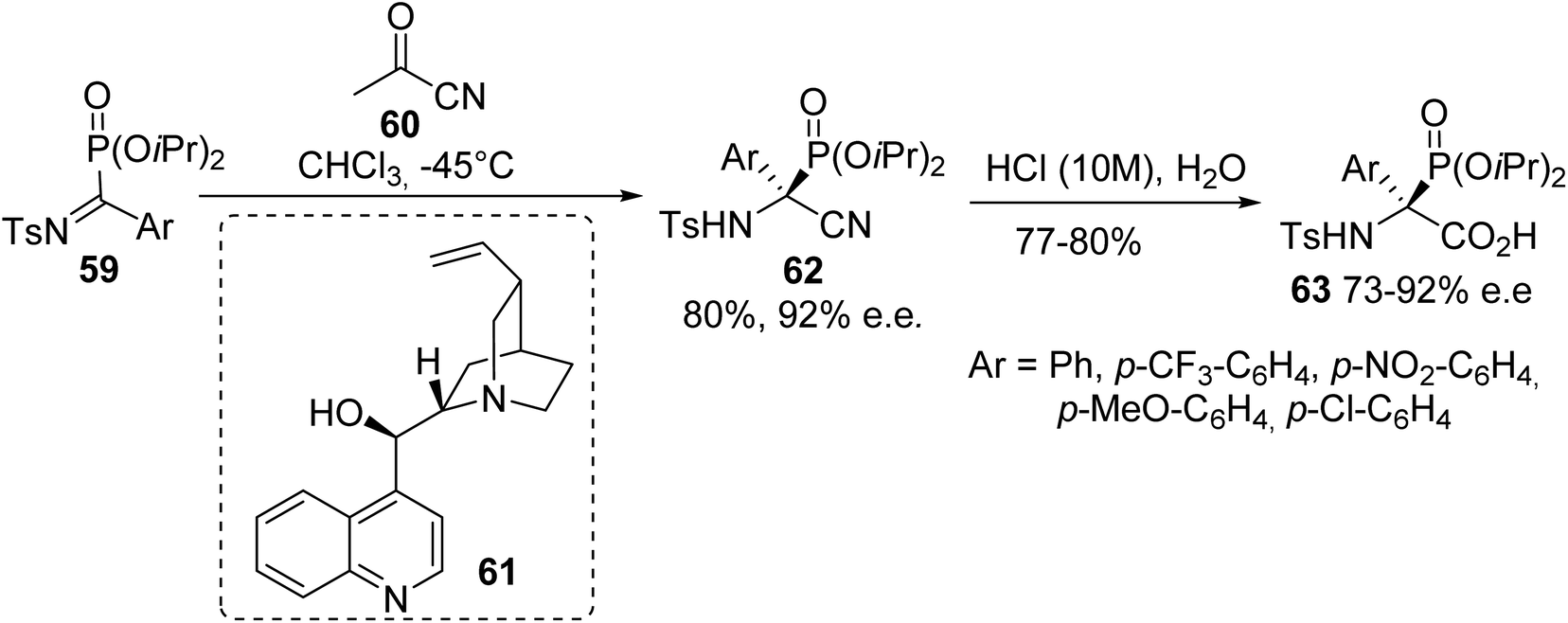
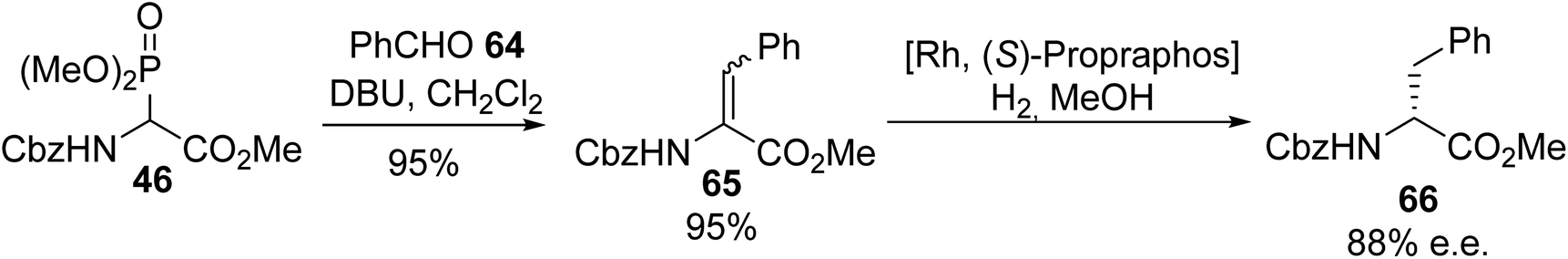

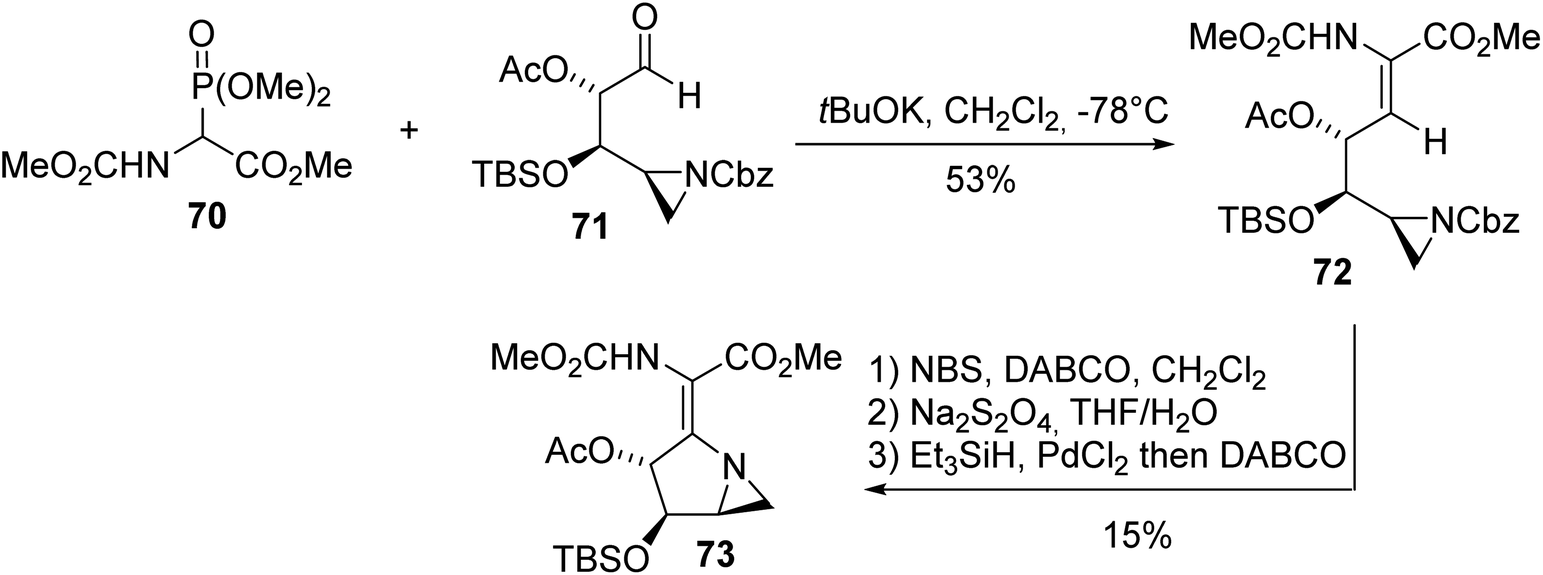
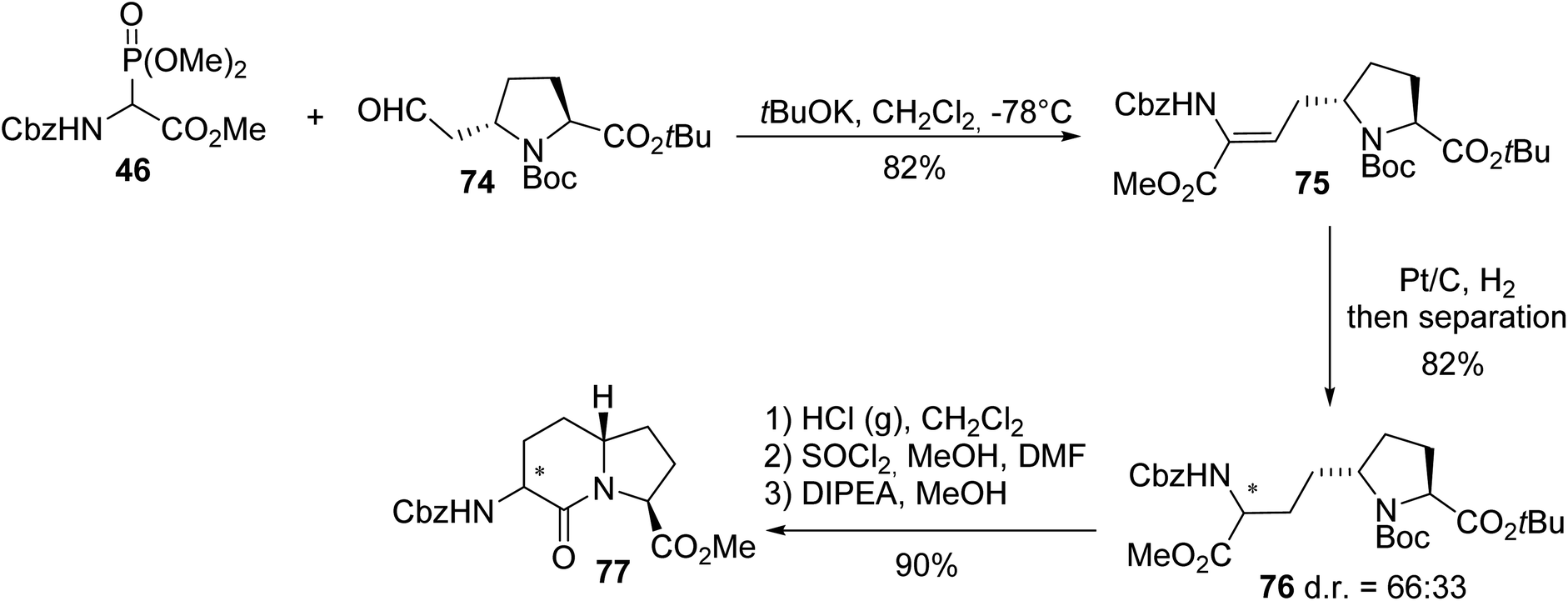
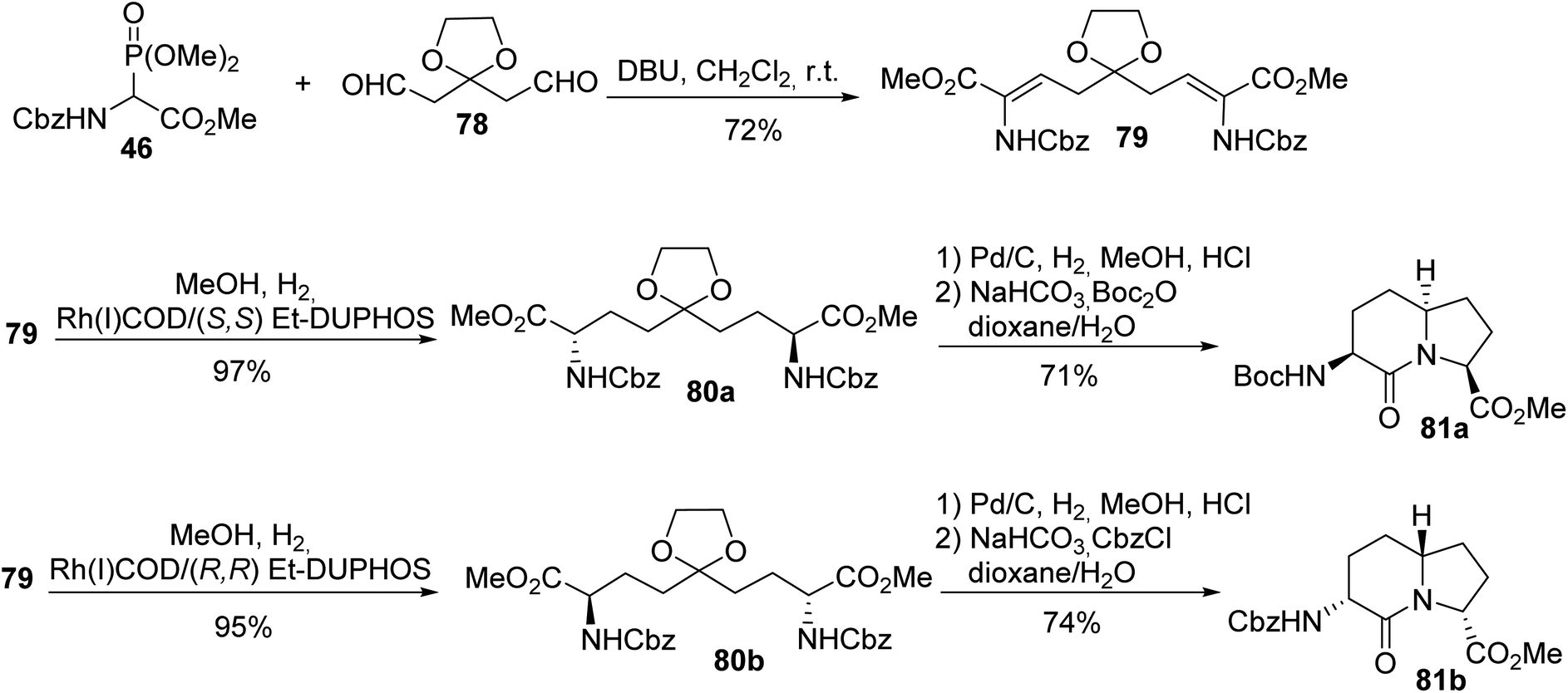
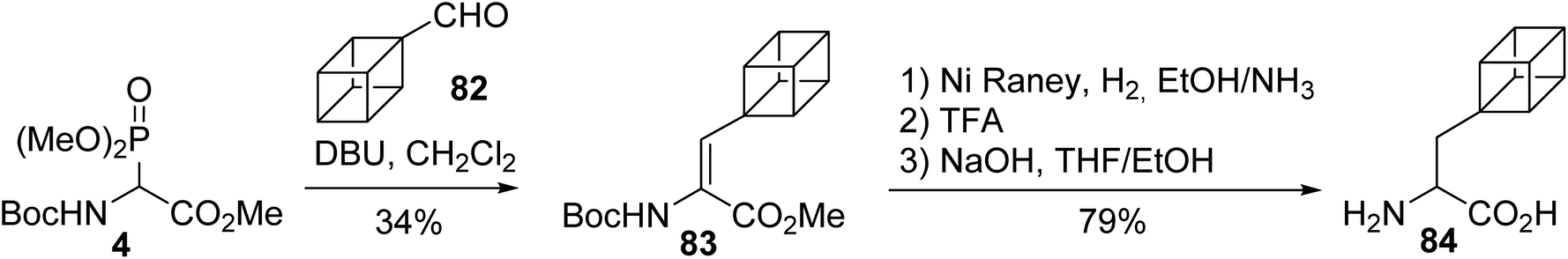
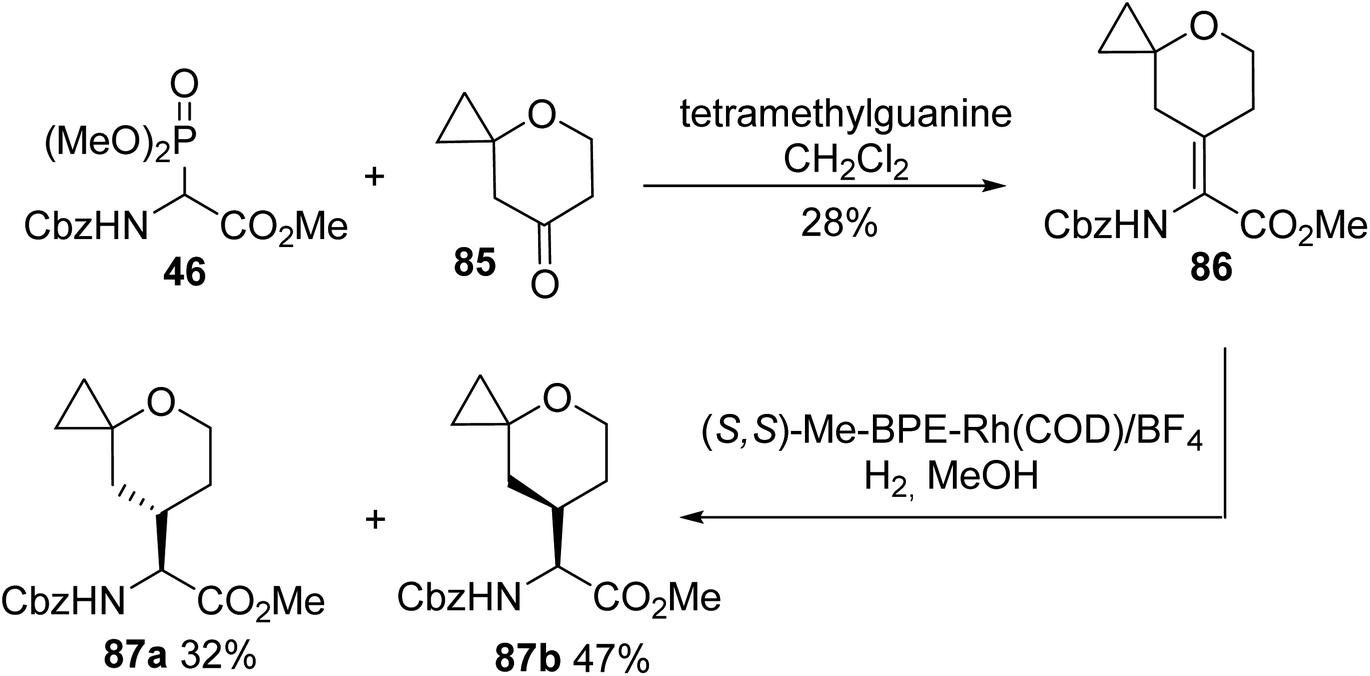
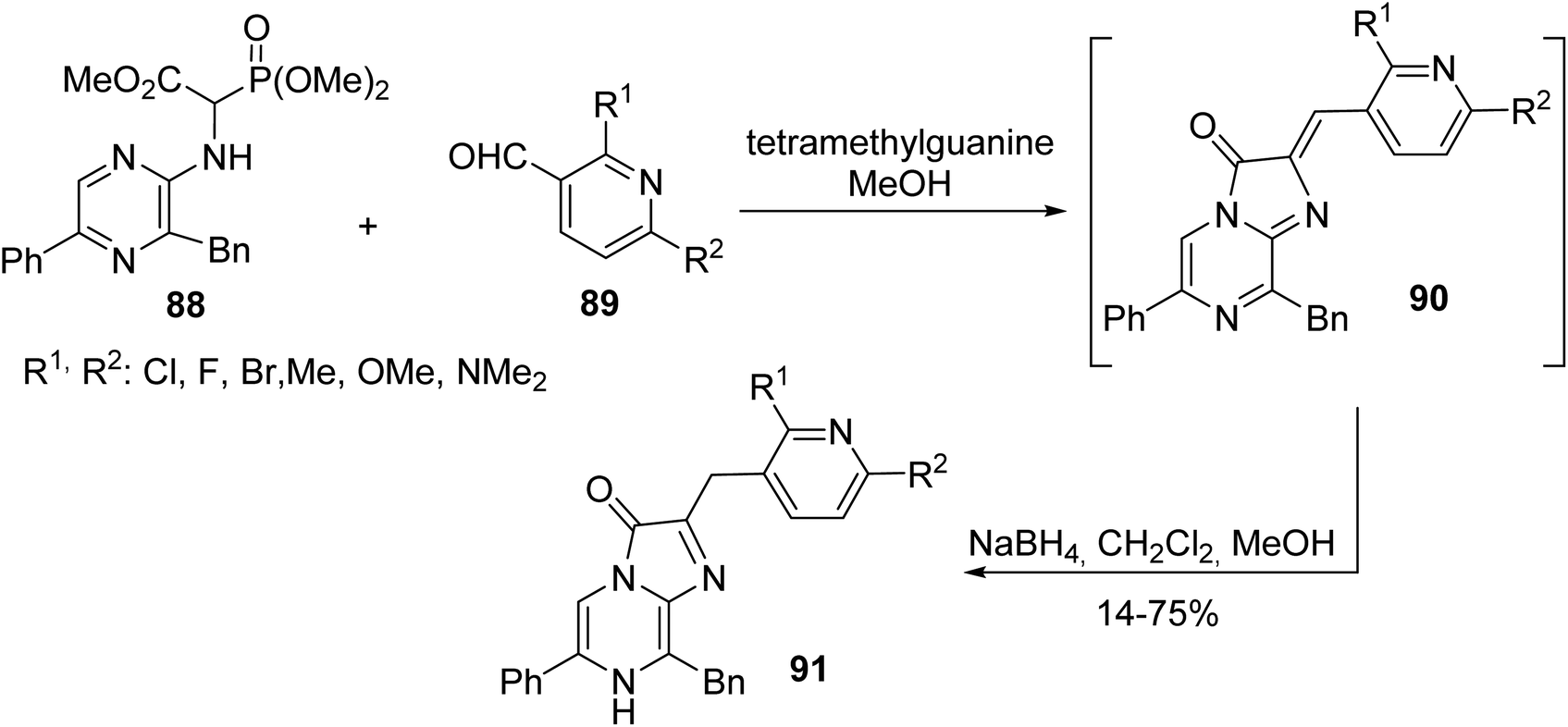
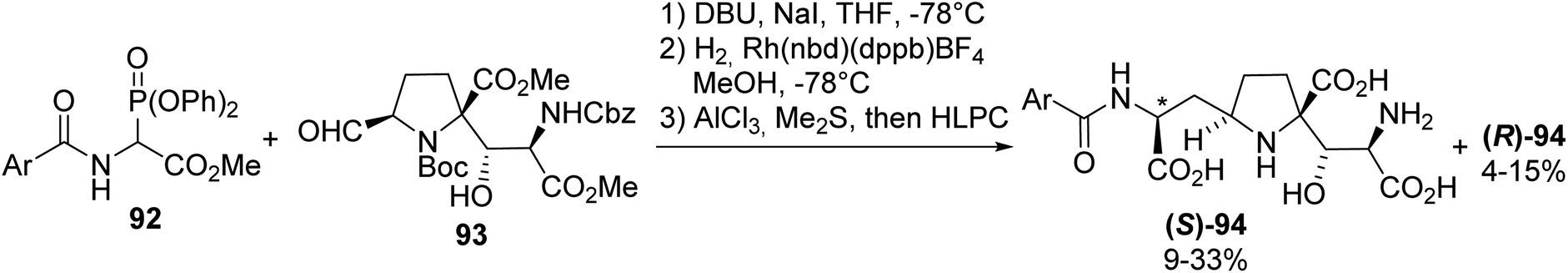
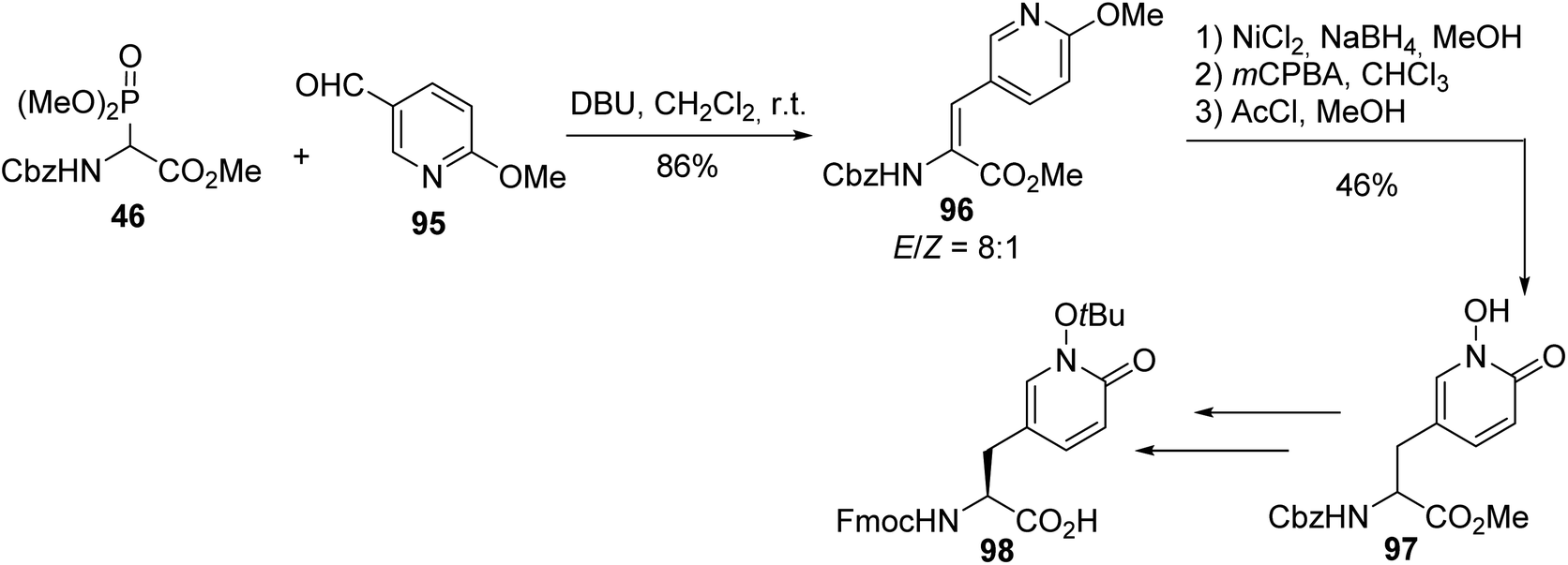
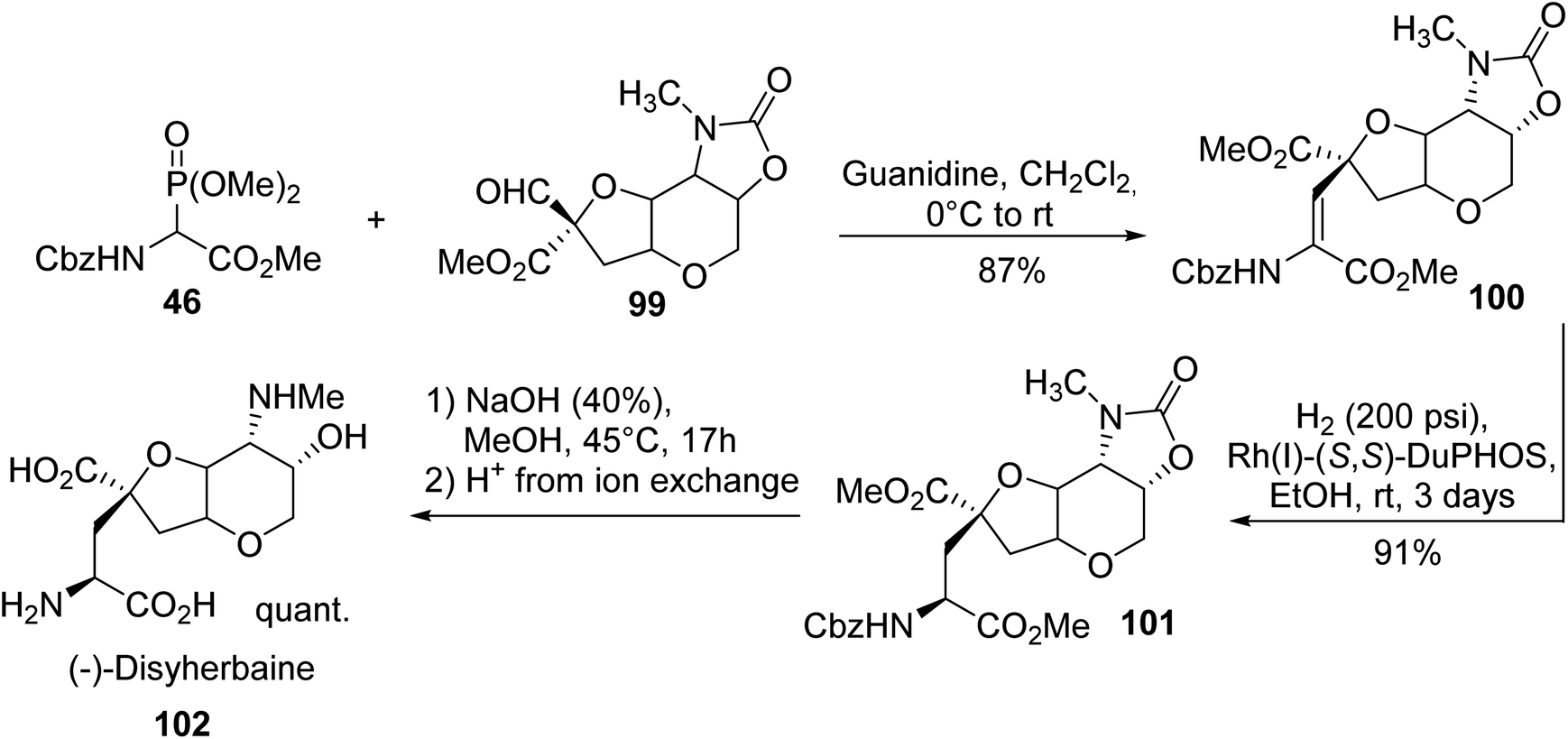
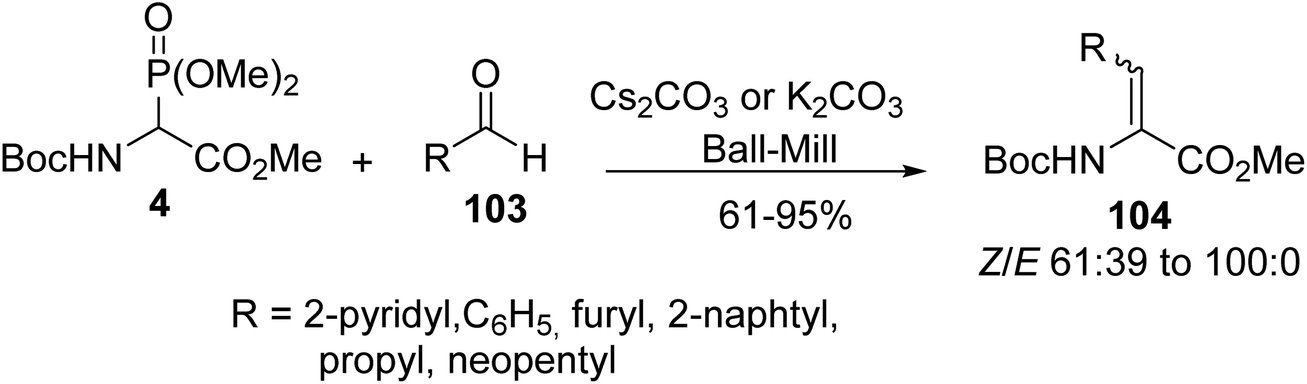
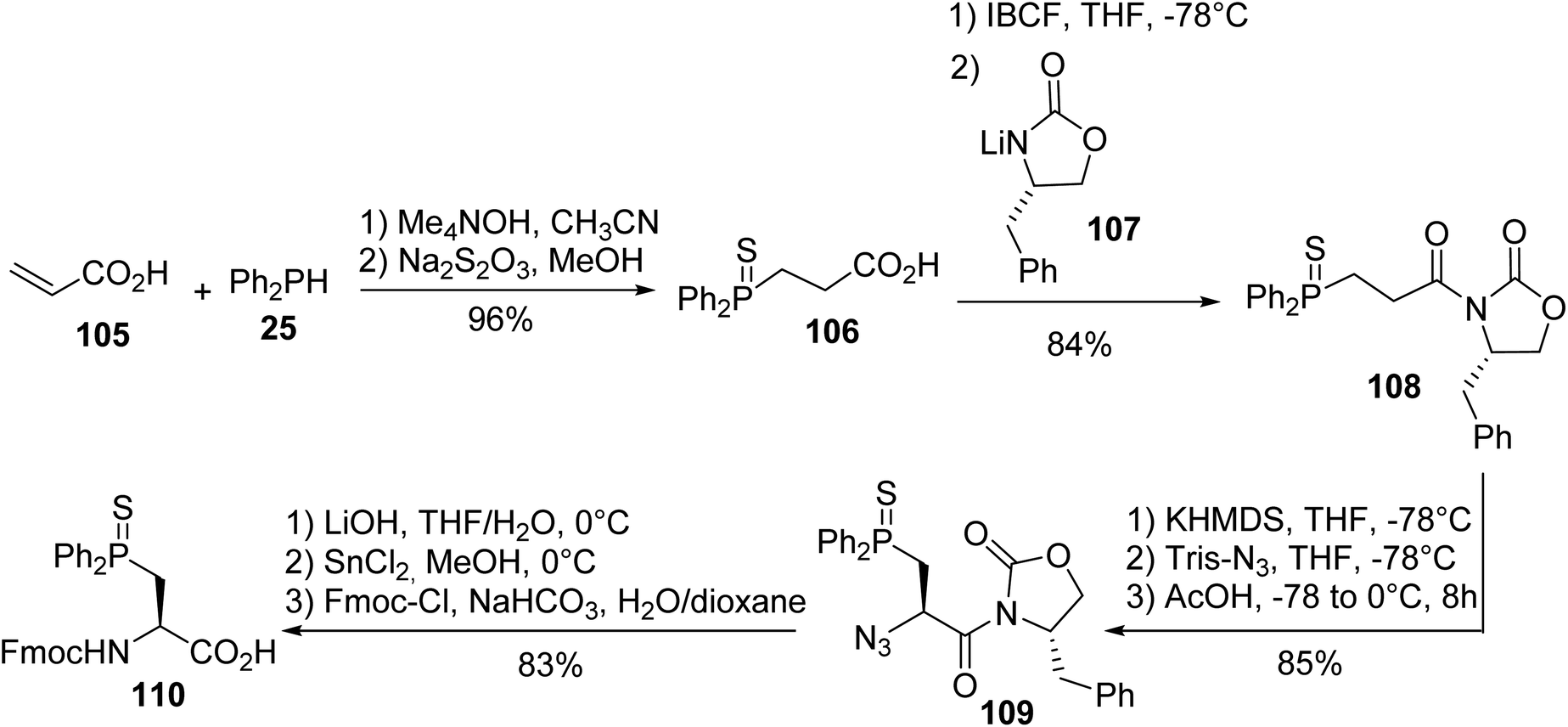
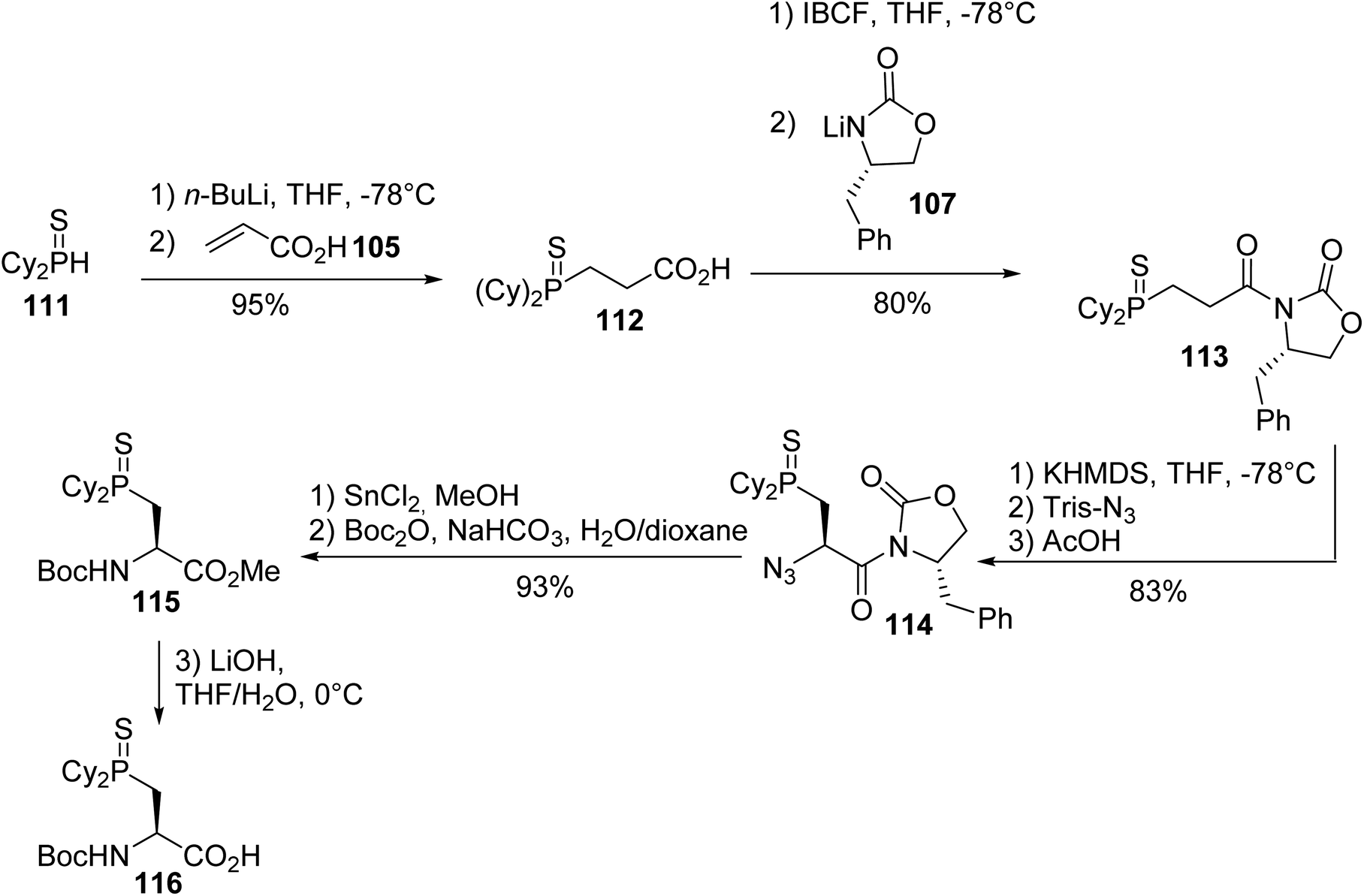
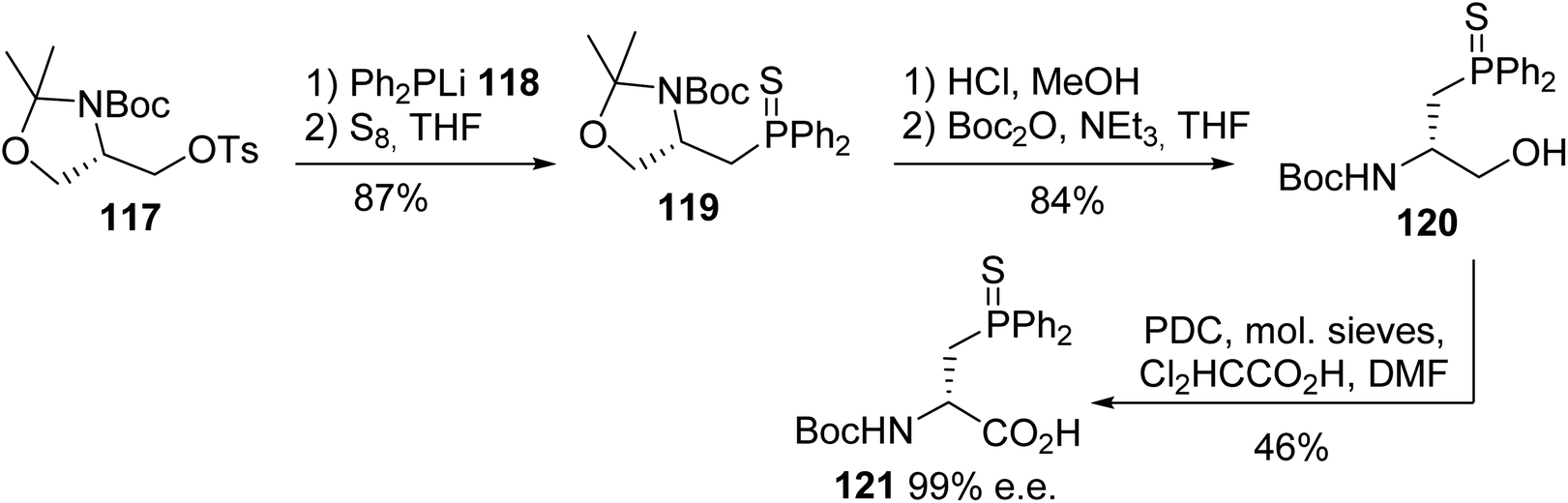
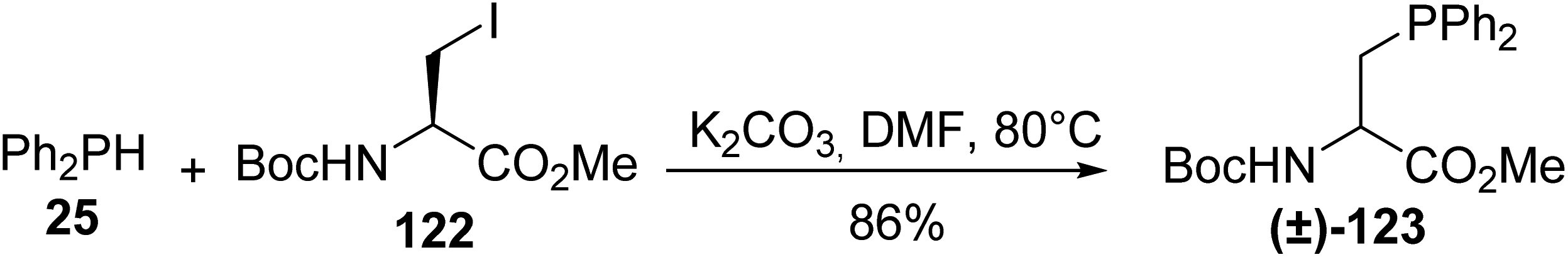
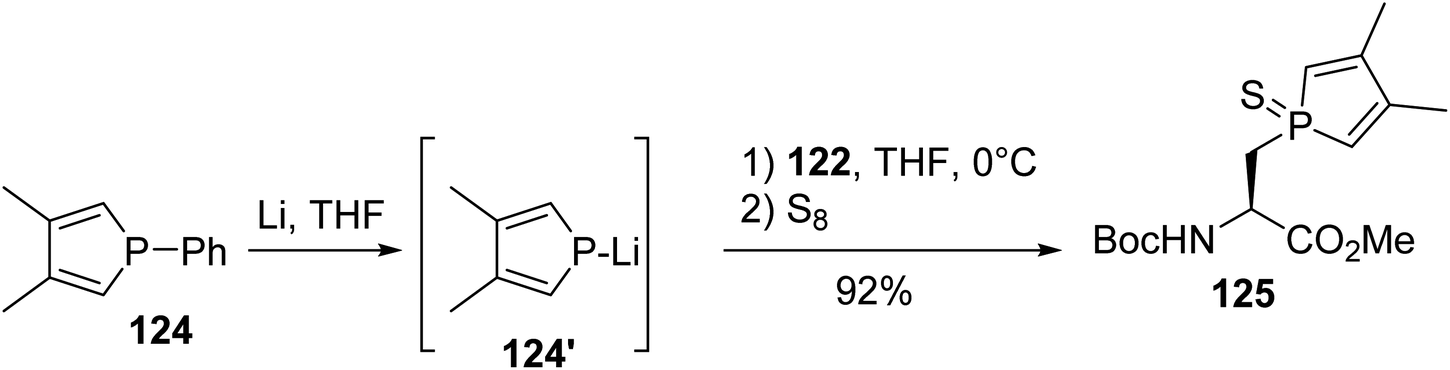
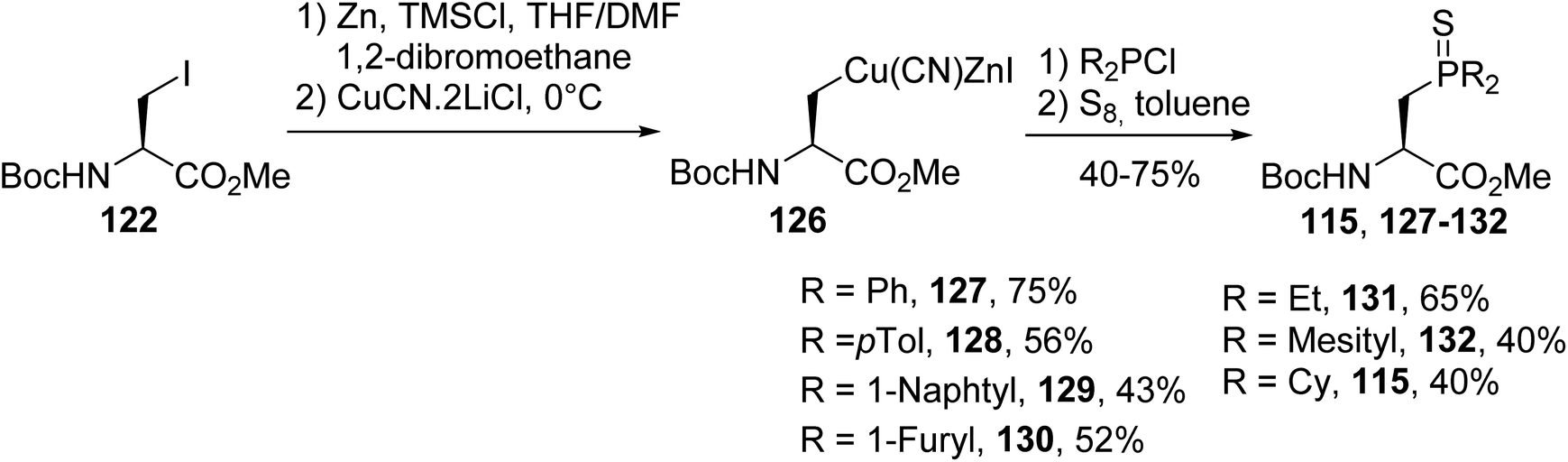
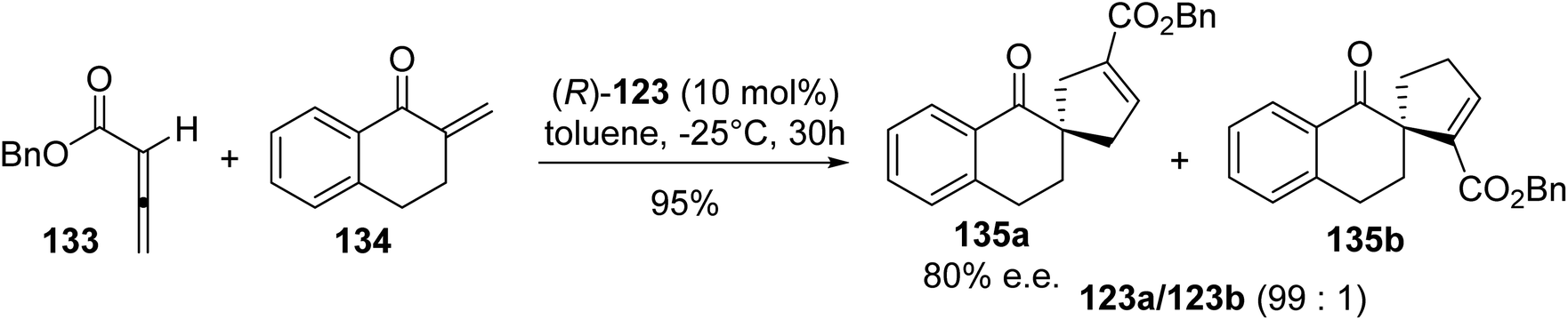

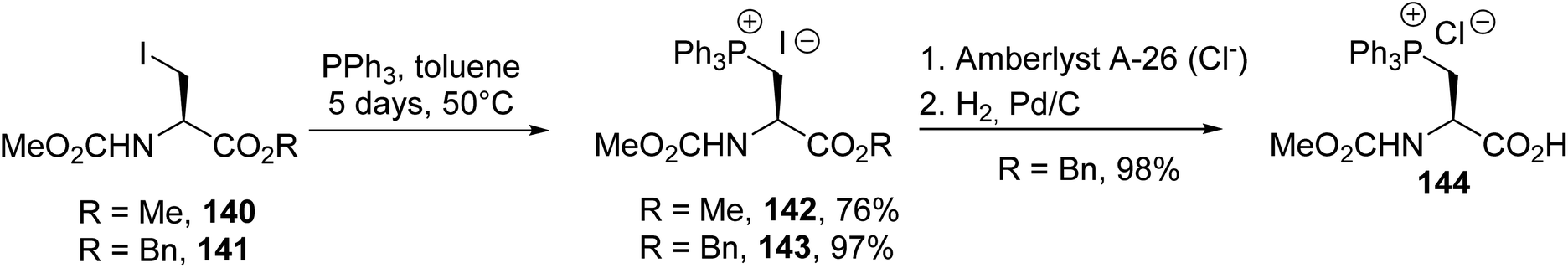
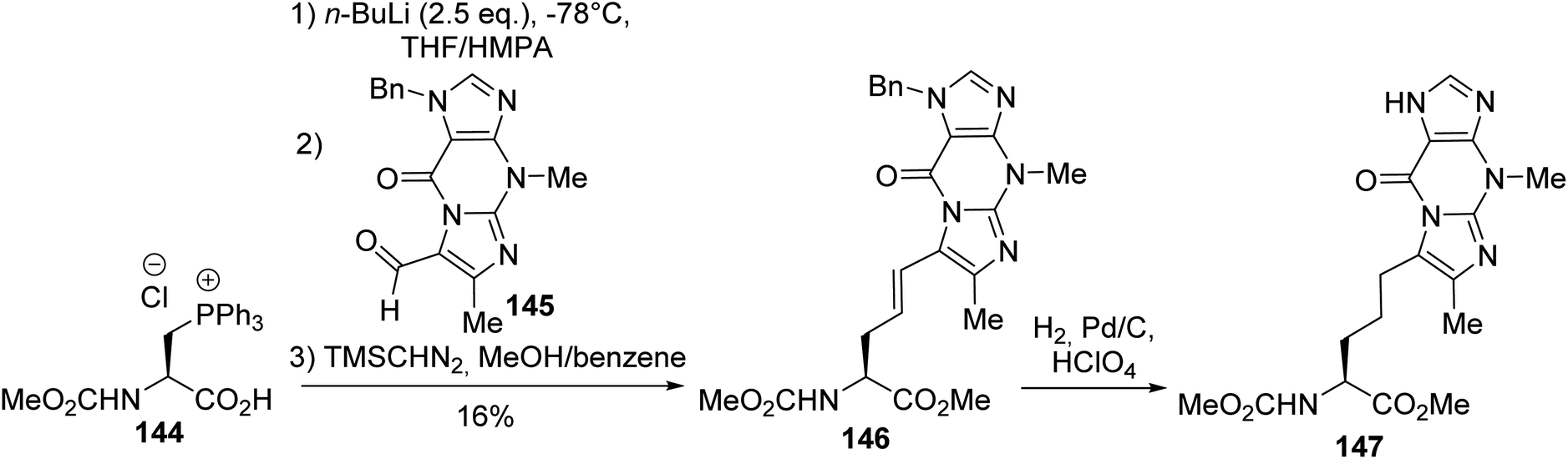
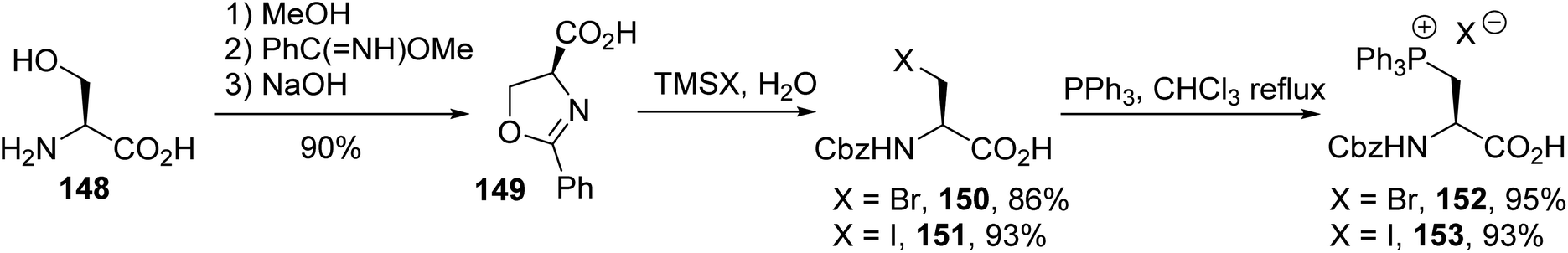
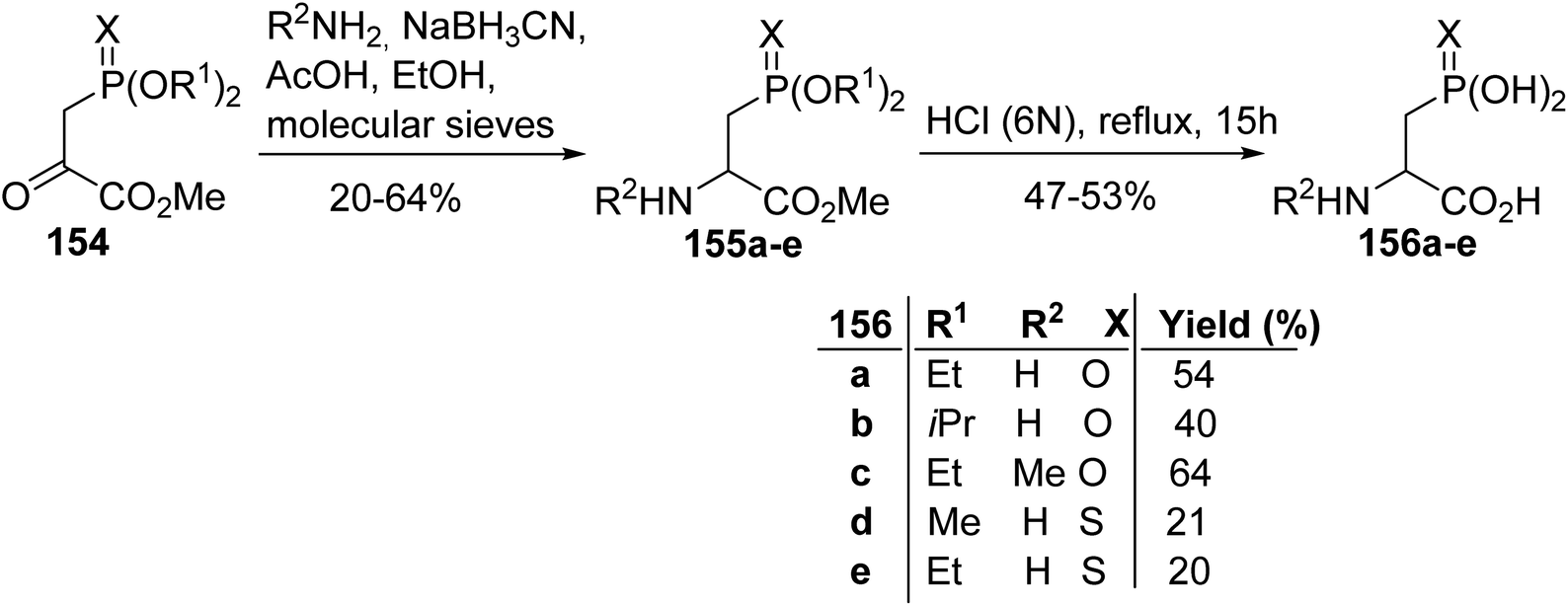
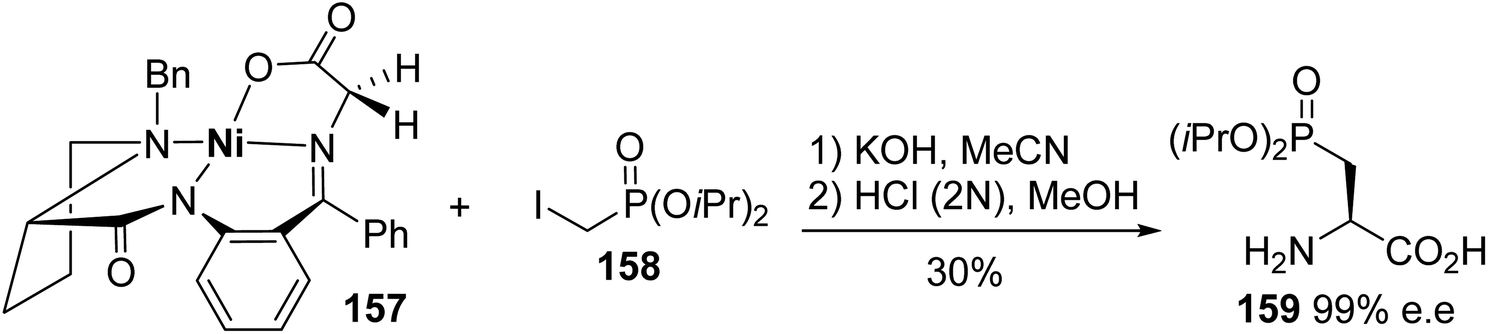
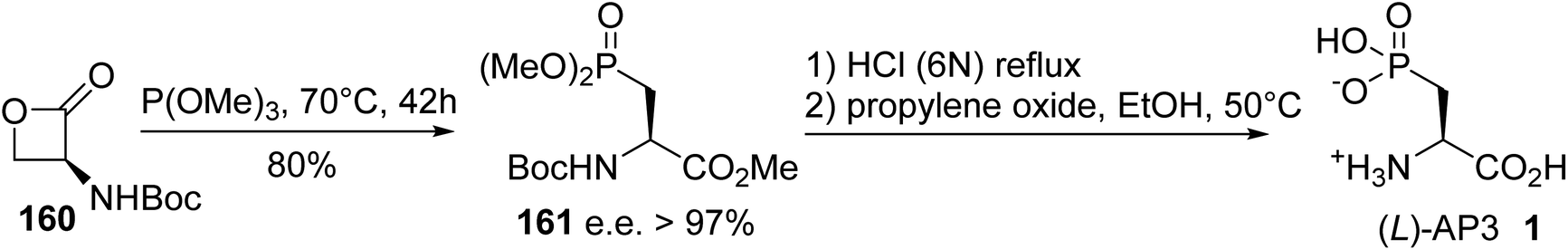
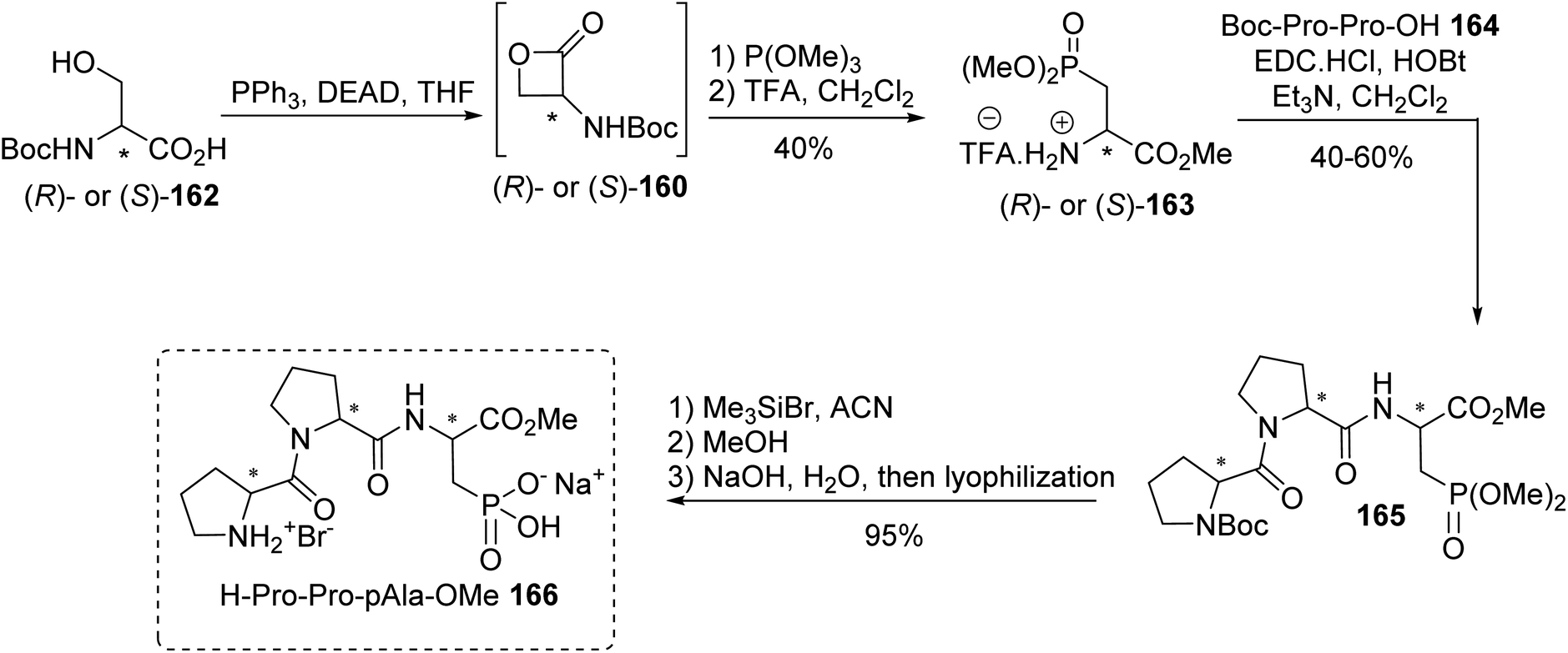
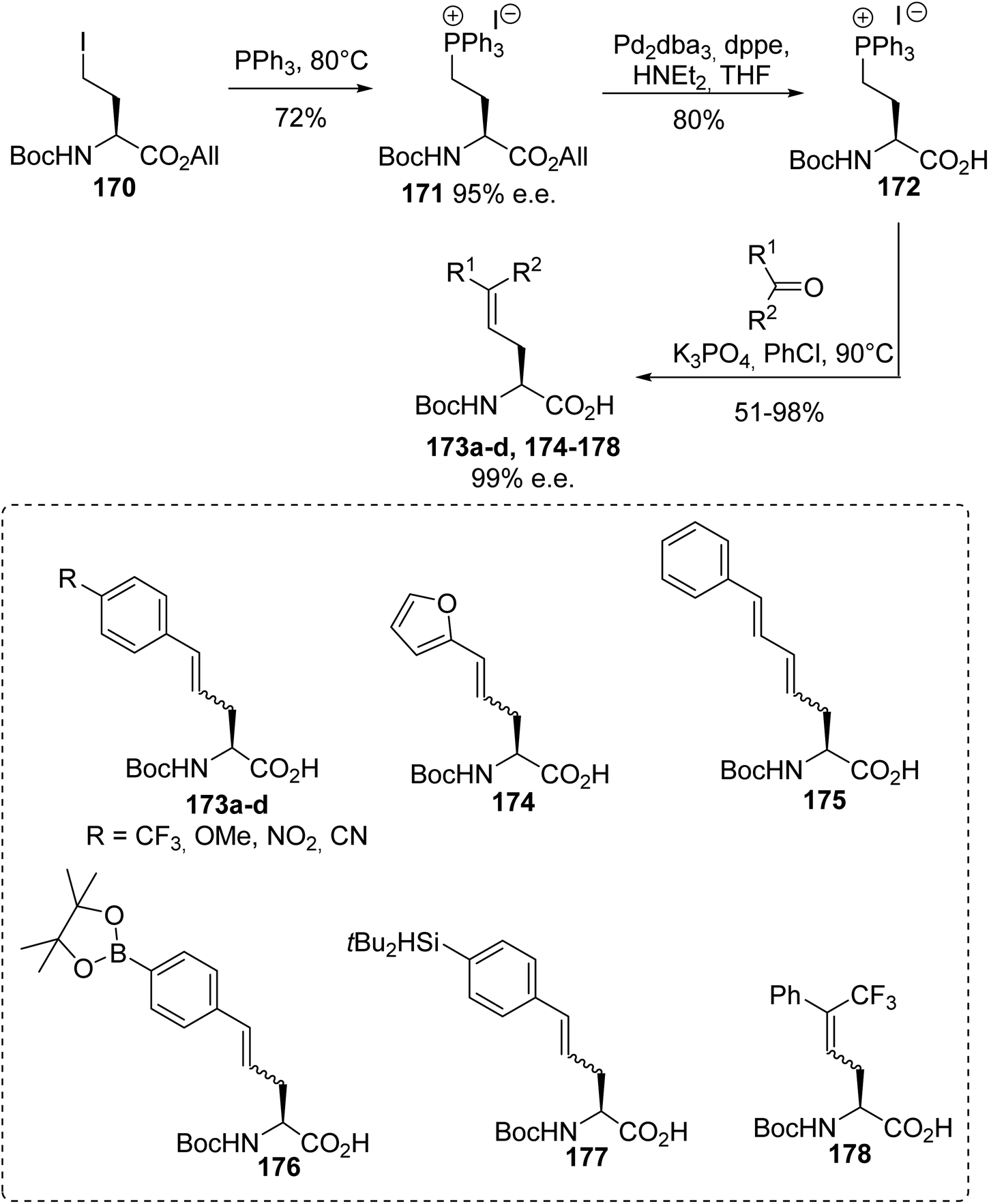
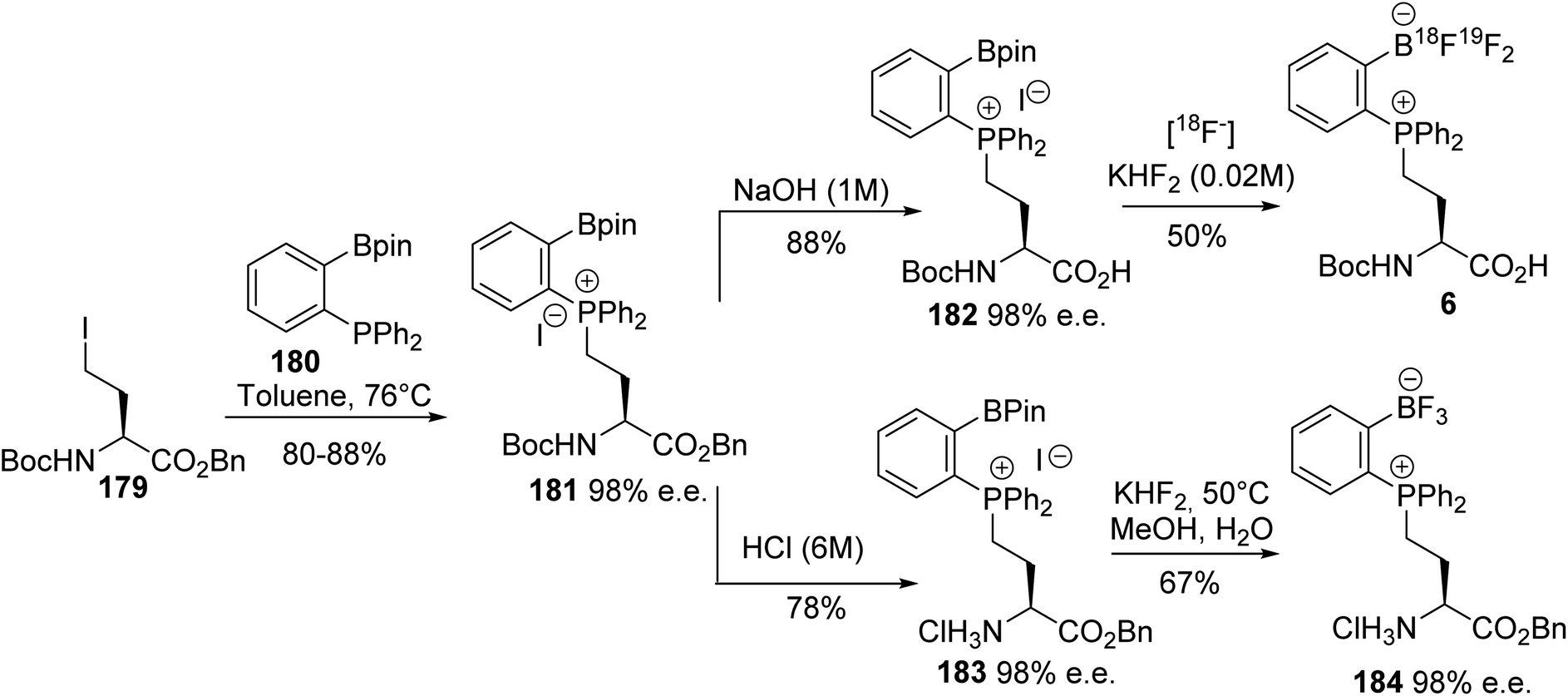
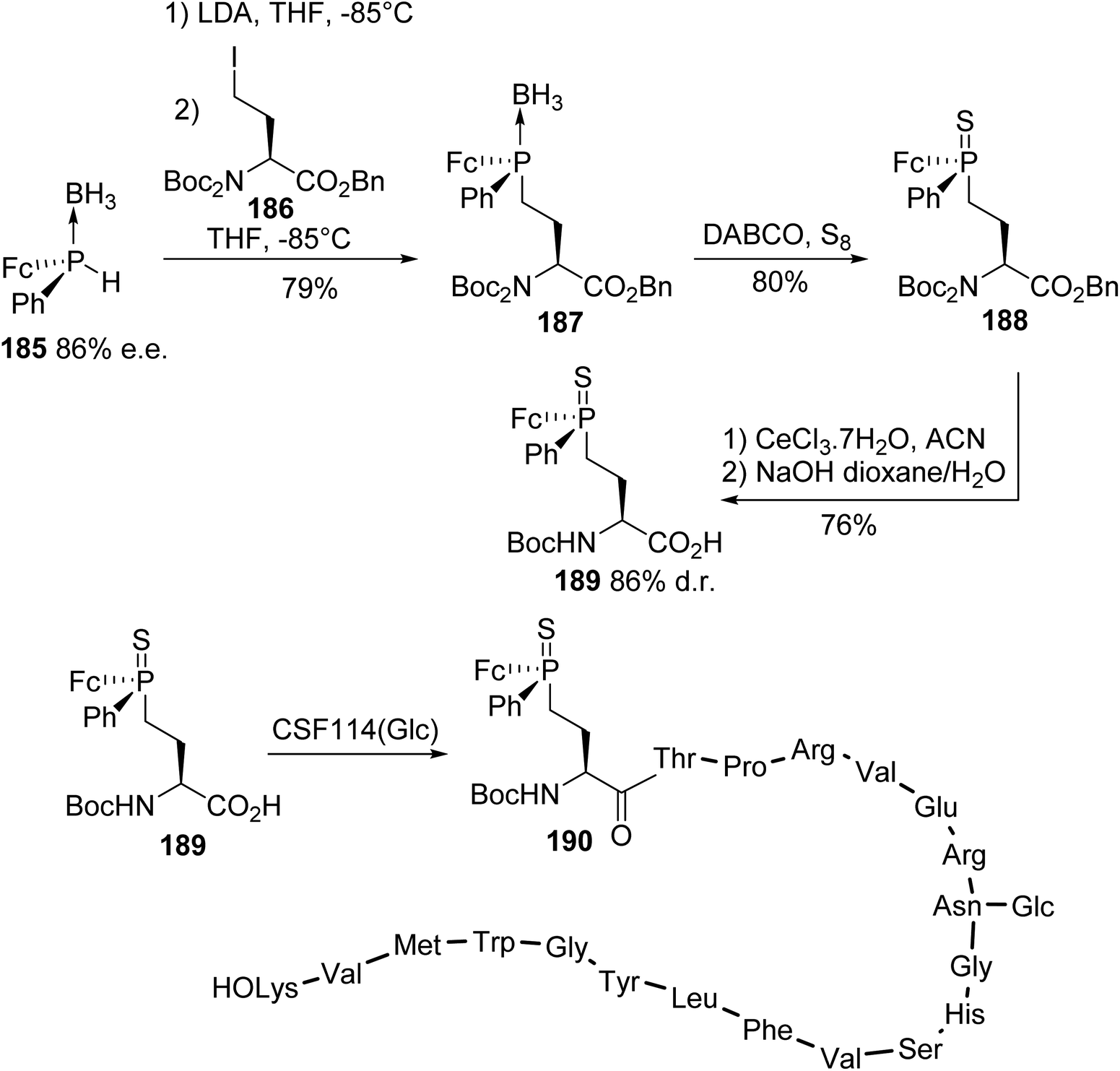
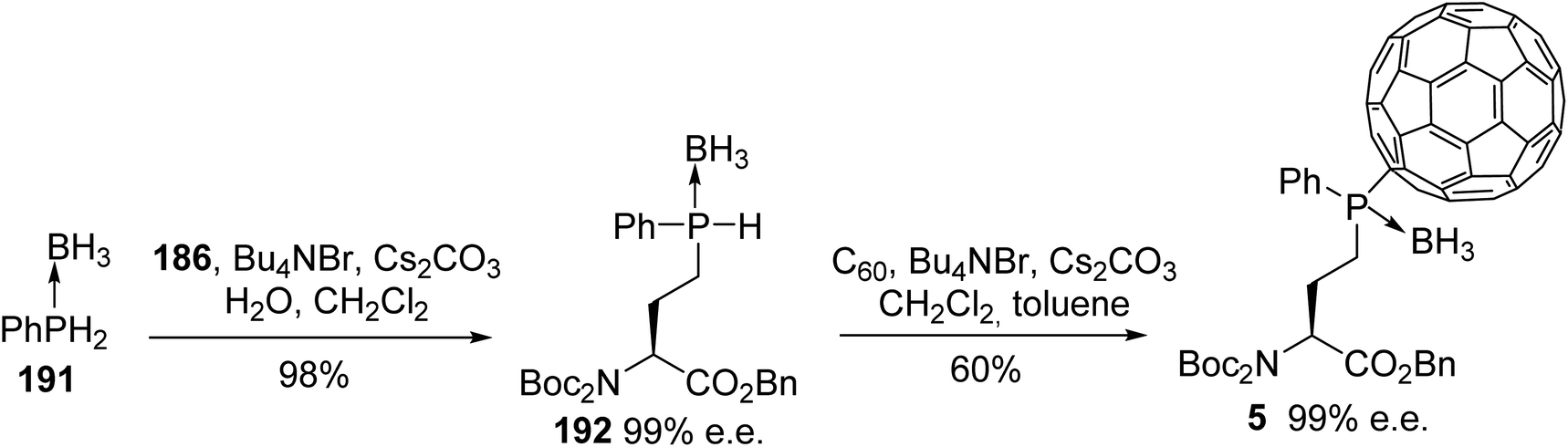

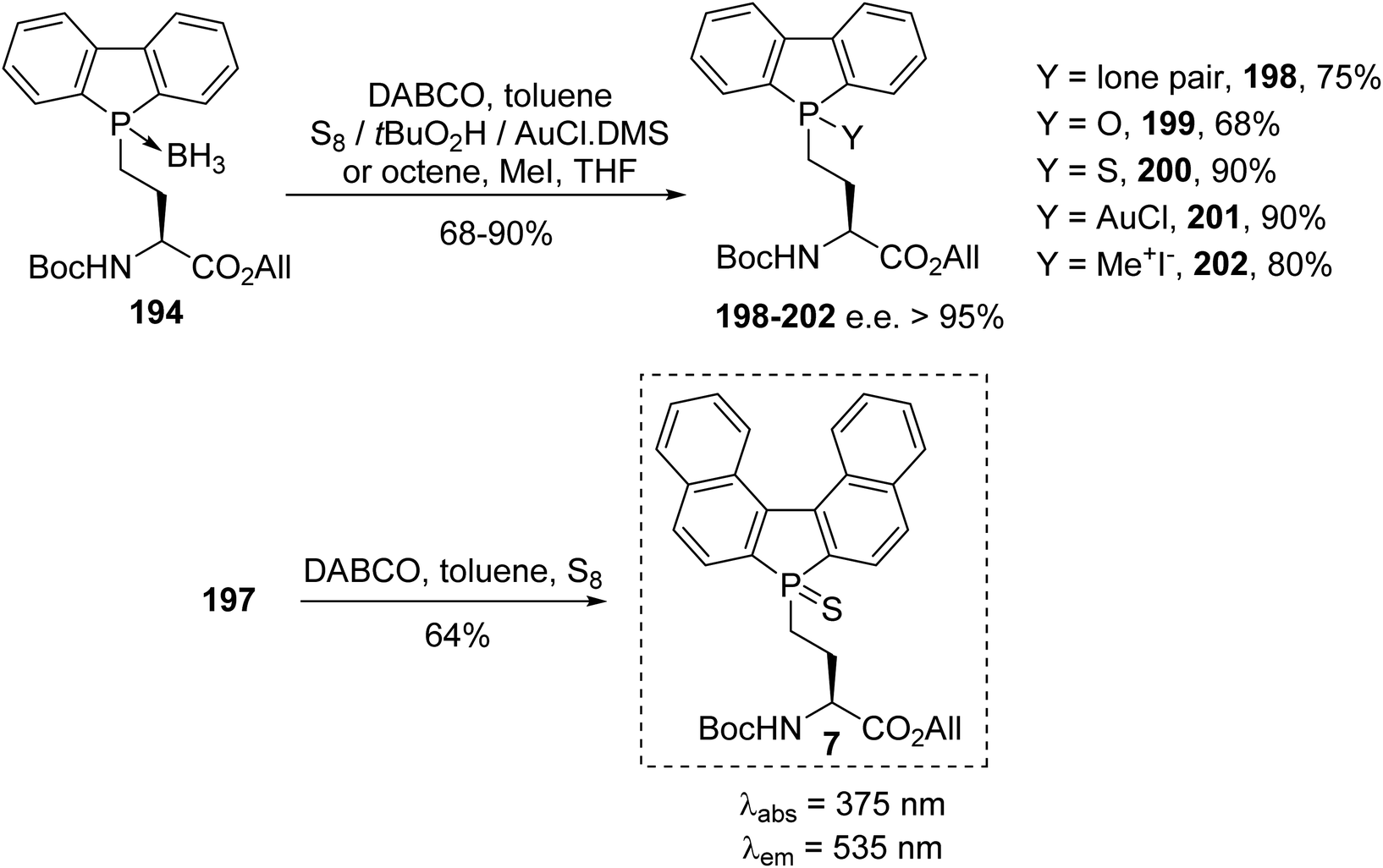
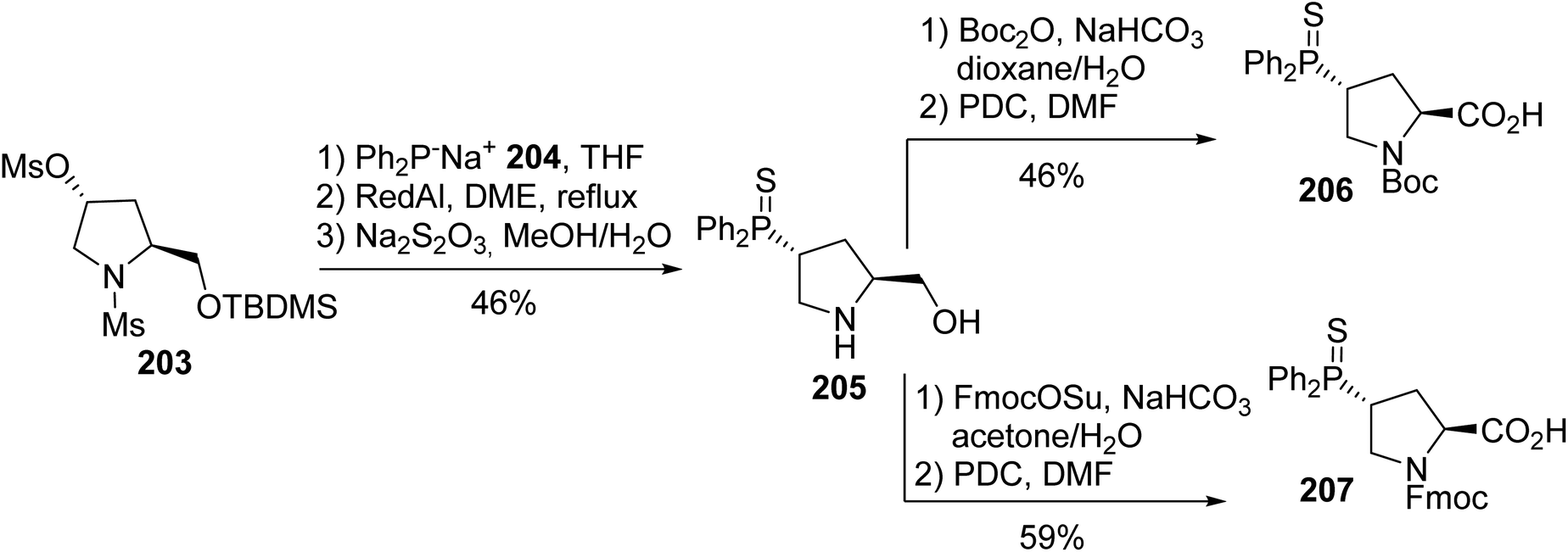
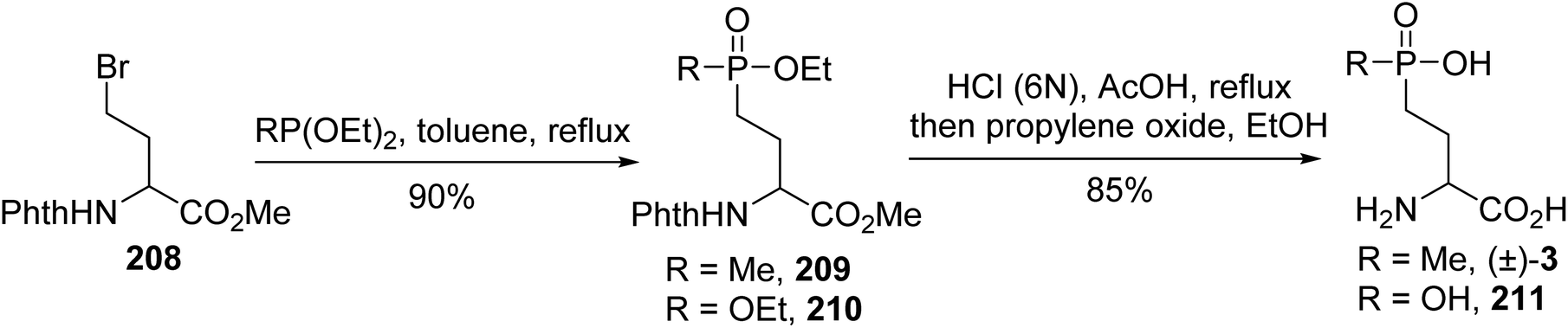
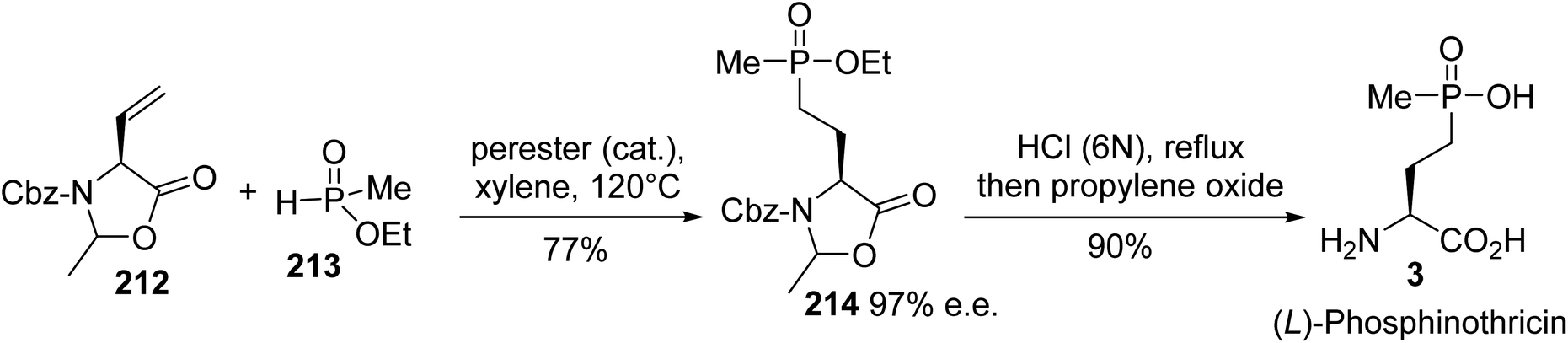
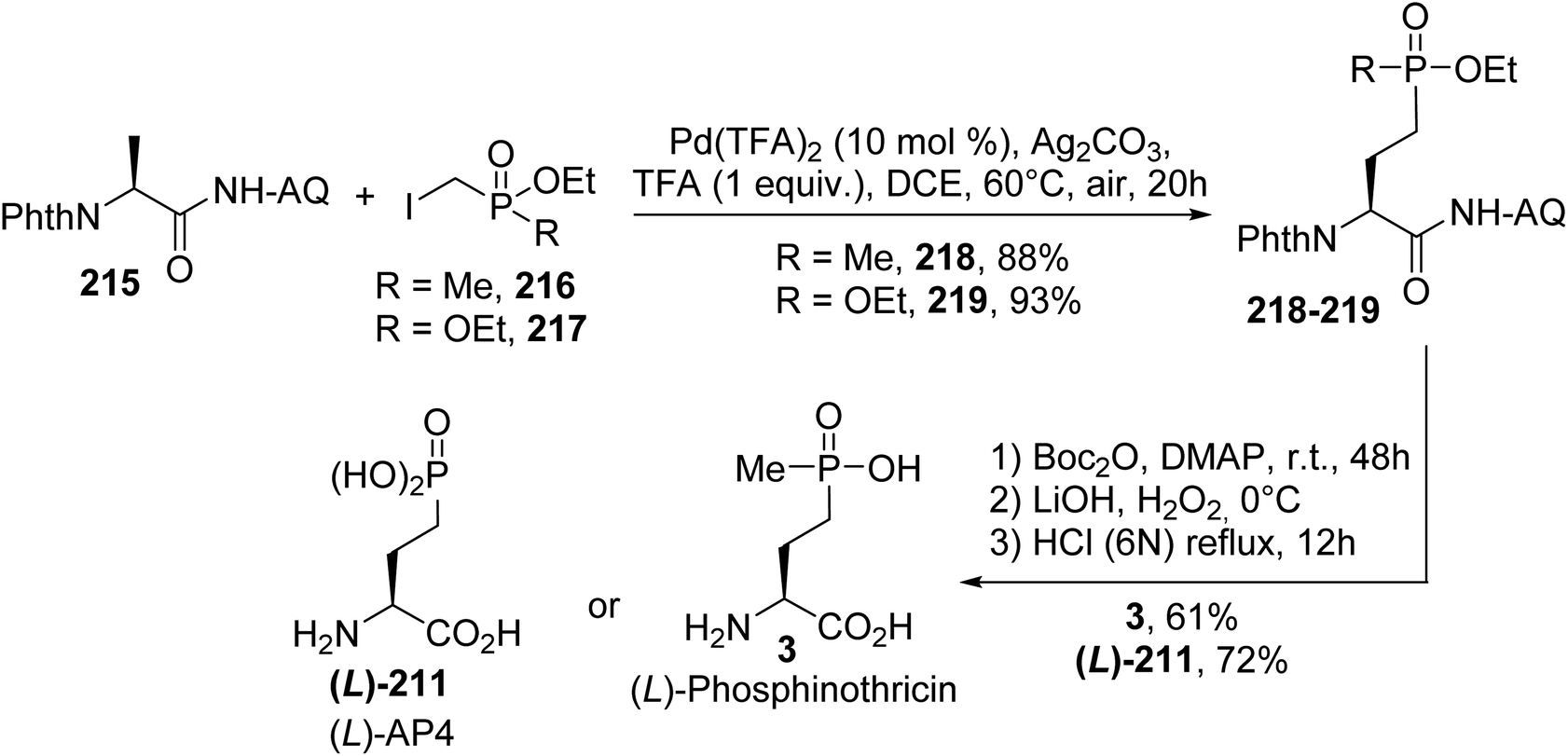

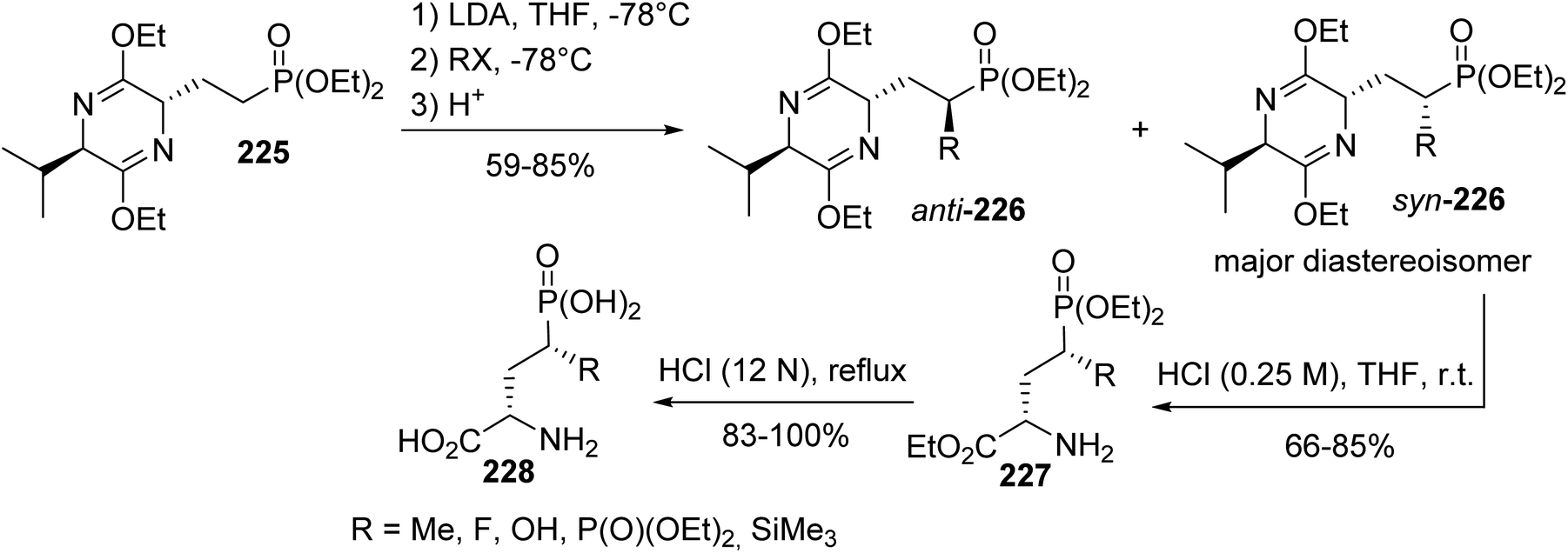
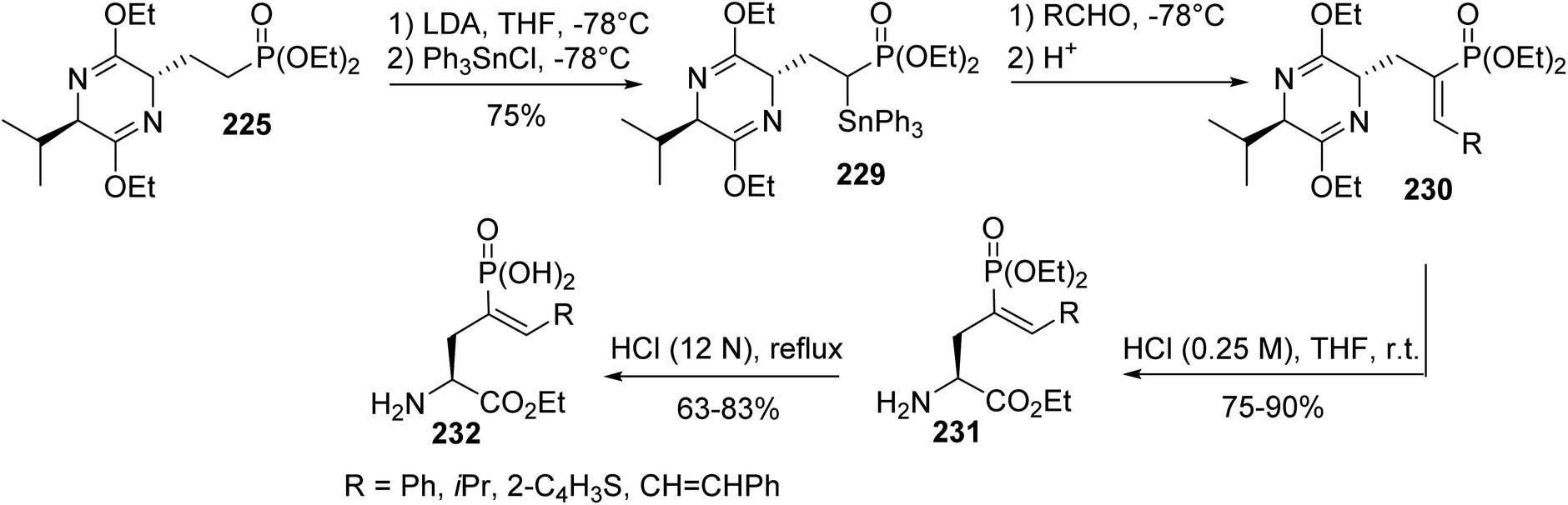
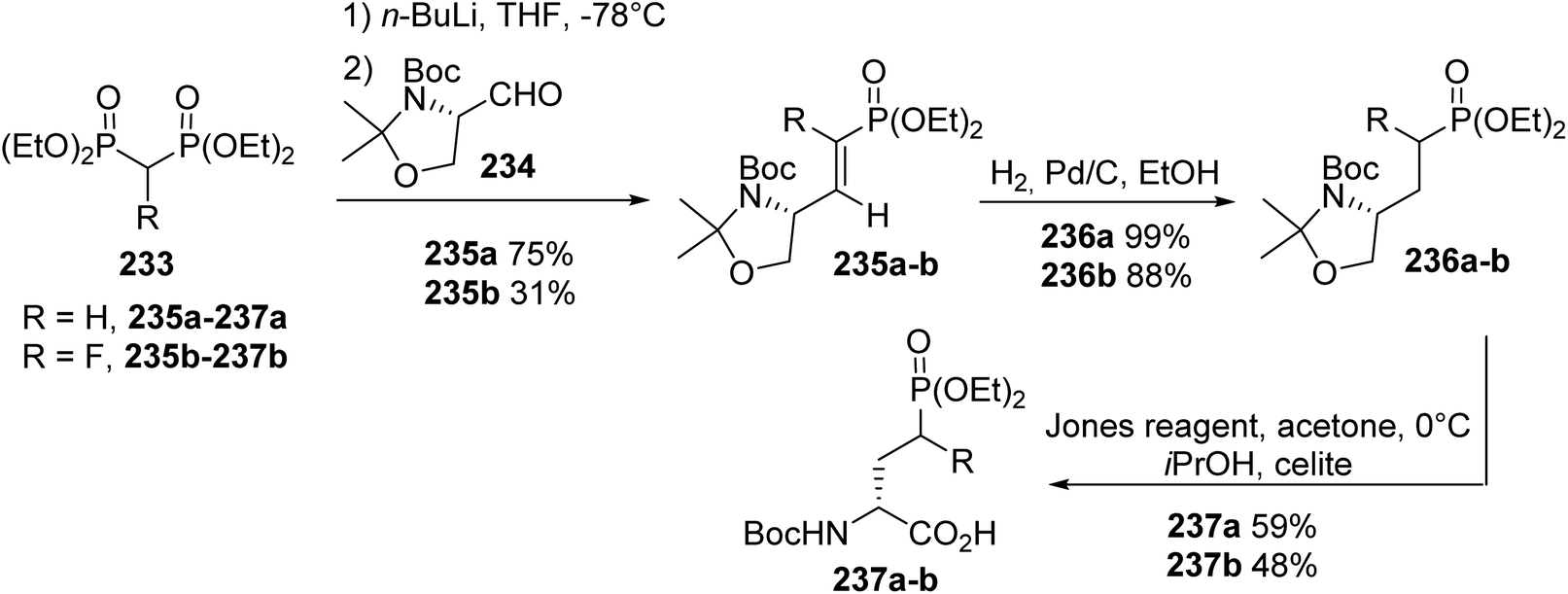
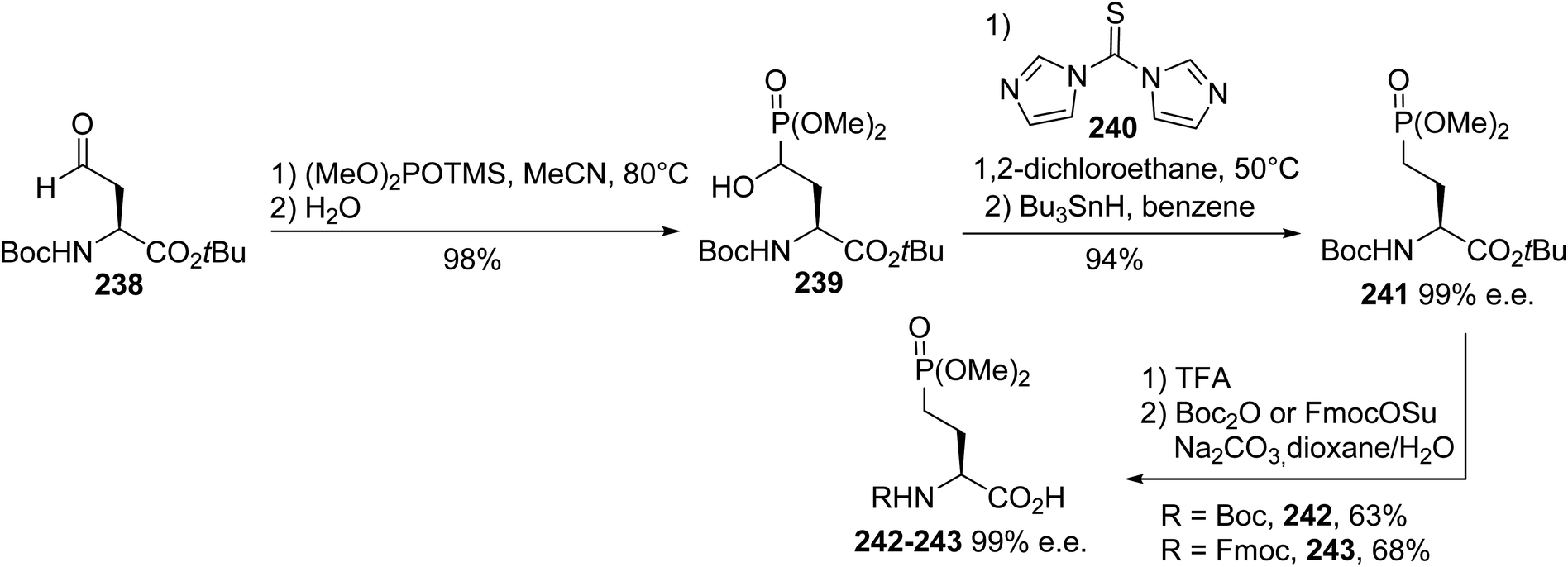
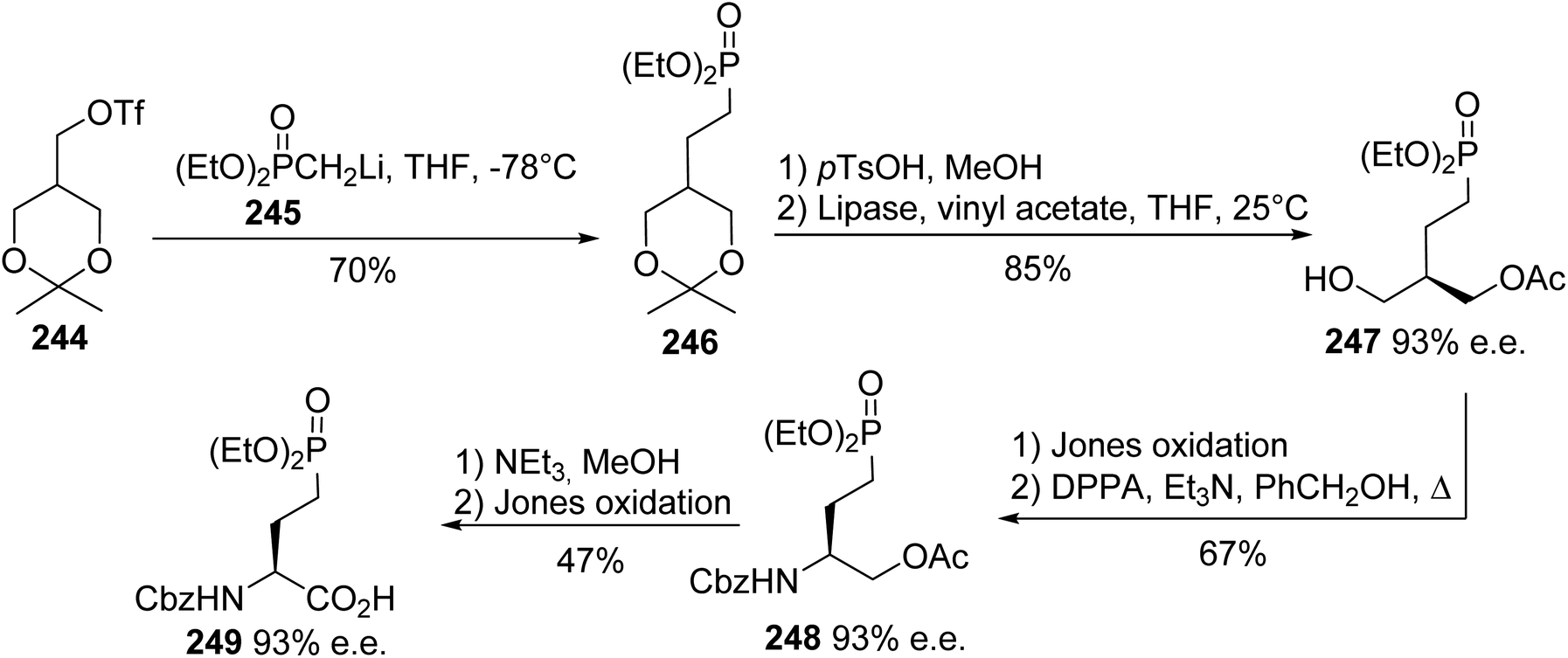

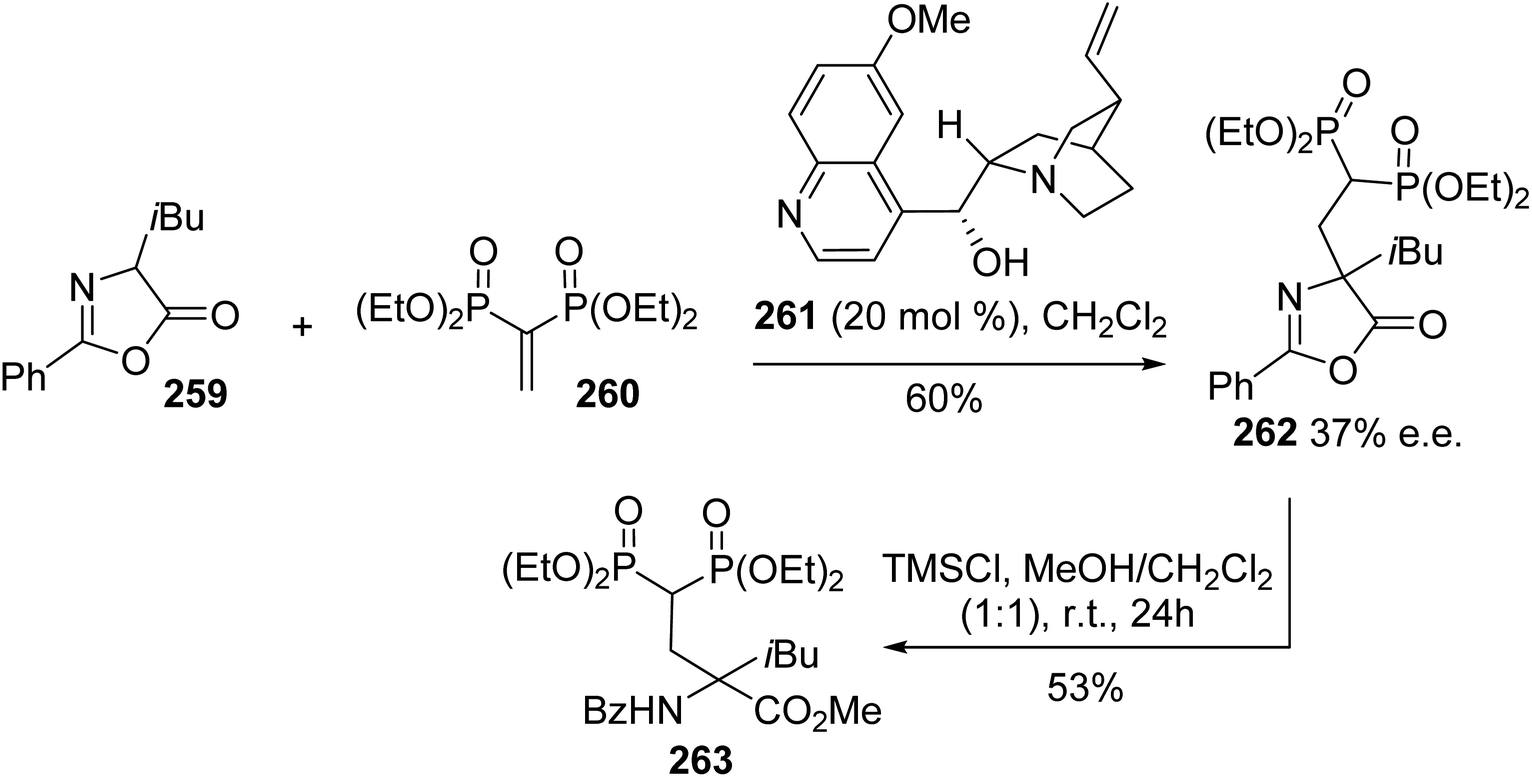
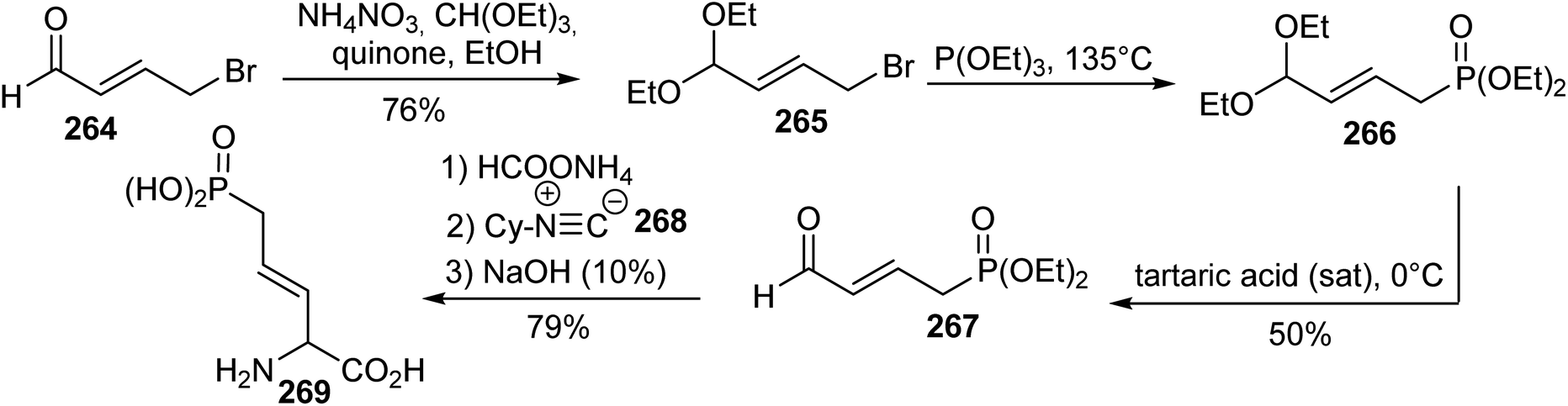
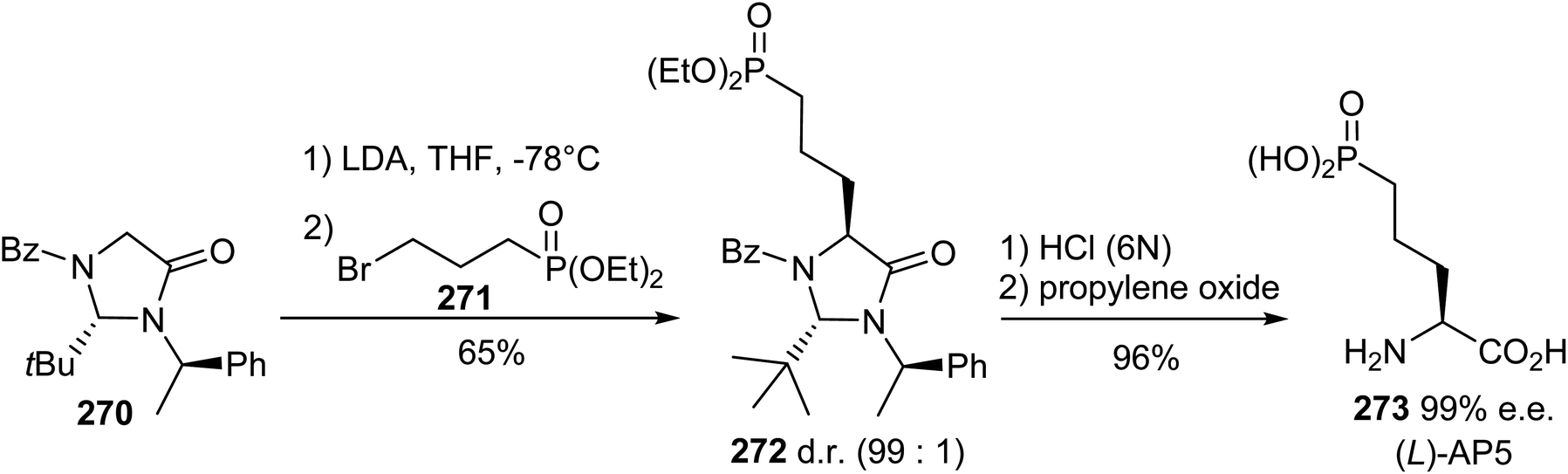
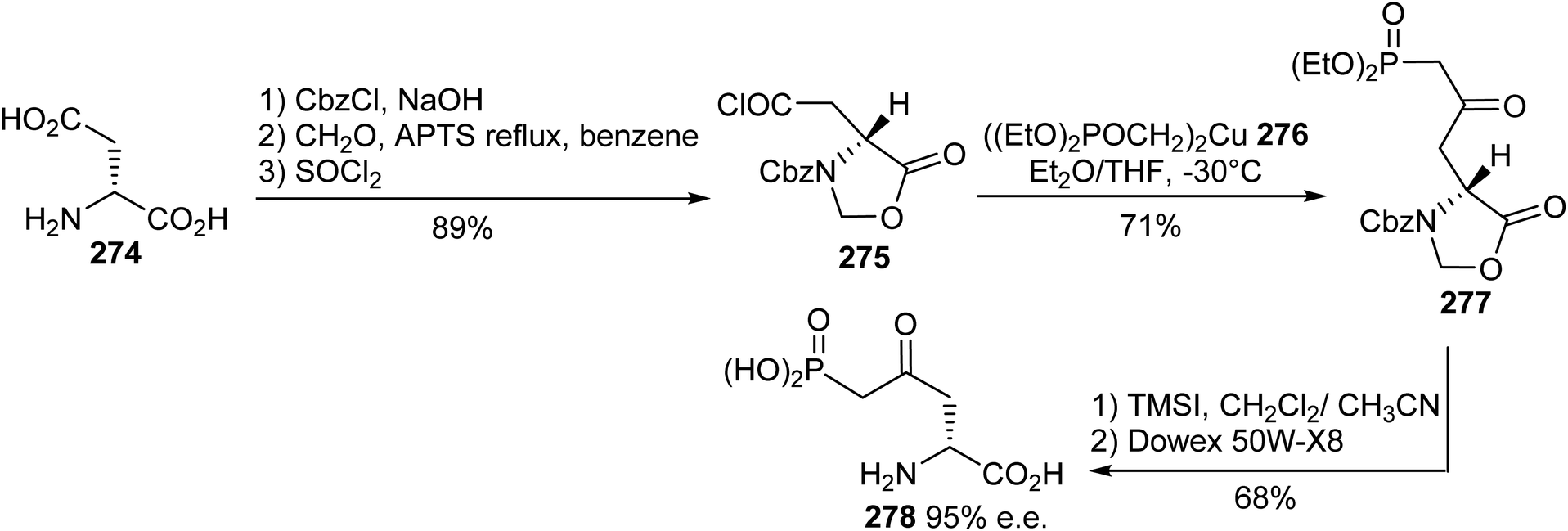
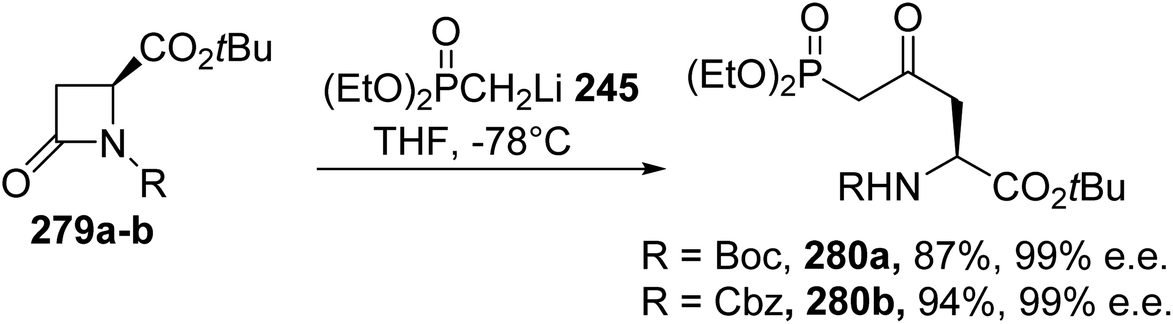
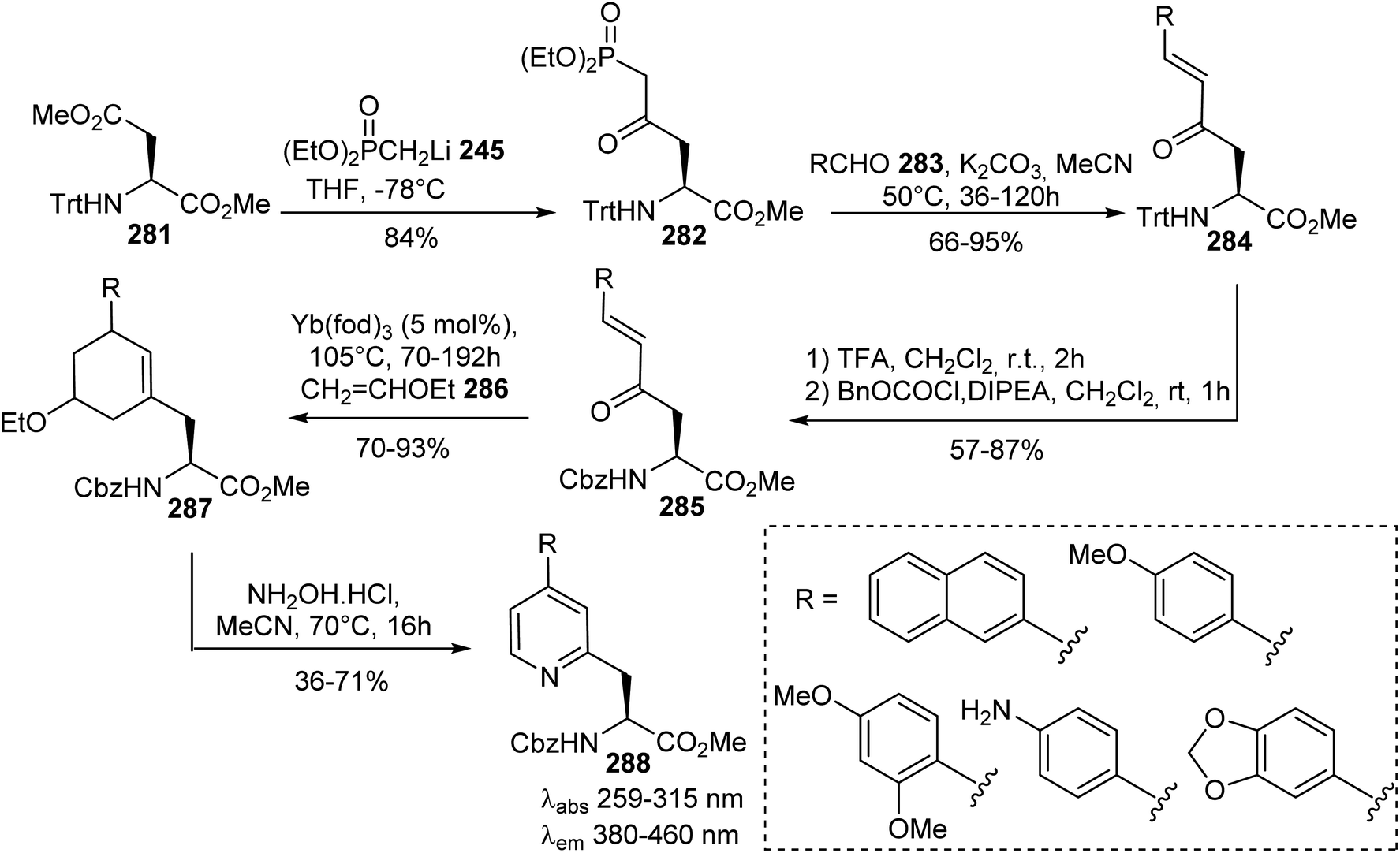
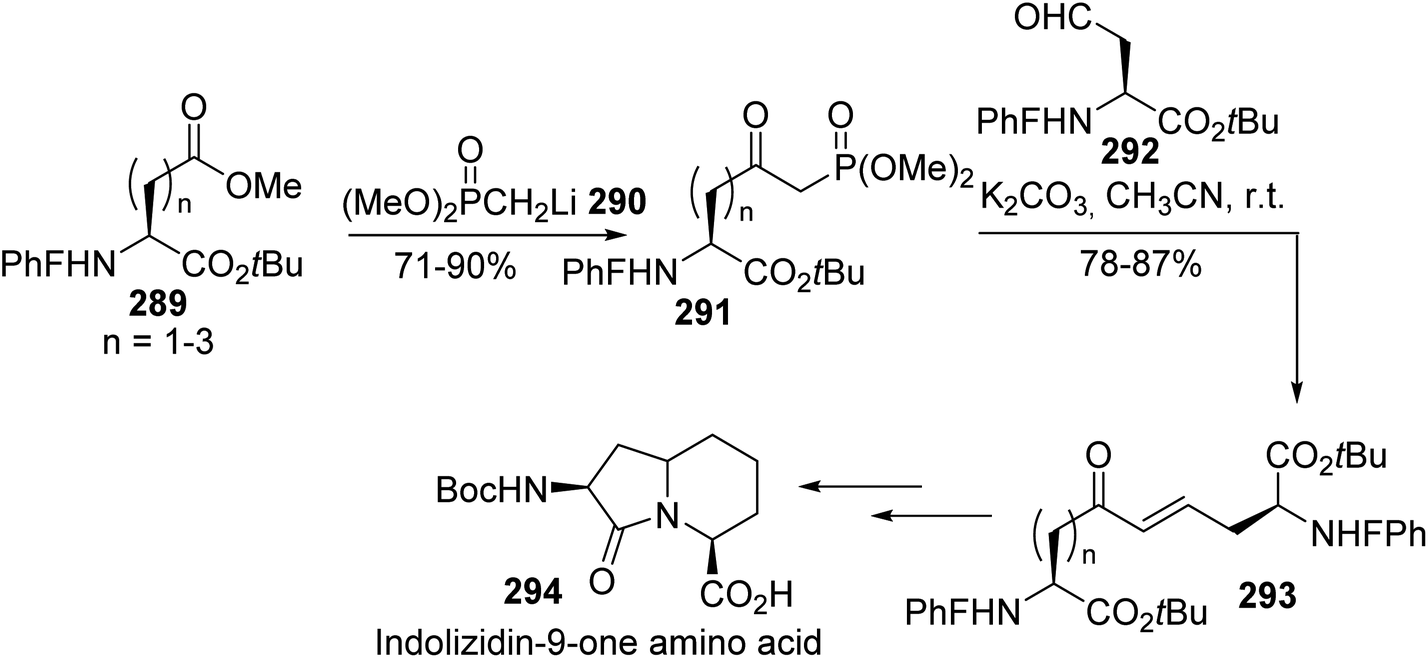
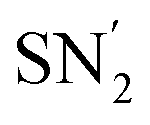 ring-opening of vinyl aziridines to afford the δ-phosphinoamino esters 297 with 70% yield and 97% e.e. at room temperature (Scheme 68).
ring-opening of vinyl aziridines to afford the δ-phosphinoamino esters 297 with 70% yield and 97% e.e. at room temperature (Scheme 68).