
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic, selective, and stereocontrolled construction of C4 quaternary and homobenzylic dihydroisoquinolones by sp3 C–H benzylation†
Timothy K. Beng * and
Antonio Moreno
* and
Antonio Moreno
Department of Chemistry, Central Washington University, Ellensburg, WA 98926, USA. E-mail: Timothy.beng@cwu.edu
First published on 28th February 2020
Abstract
C1 benzylated isoquinoline derivatives constitute the core of benzylisoquinoline alkaloids (BIAs). However, their C4 congeners remain elusive. Here, we describe a diastereoselective, catalytic, and modular C(sp3)–C(sp3) coupling protocol wherein β-amino sp3 C–H bonds of readily affordable vicinally functionalized dihydroisoquinolones are replaced by sp3 C–benzyl bonds. The method provides expedient access to C4 quaternary and homobenzylic dihydroisoquinolones, which are attractive fragments for potential drug discovery.
Introduction
One of the most succinct approaches to C(sp3)–C(sp3) bond construction is α-enolate alkylation of carbonyl compounds.1 The approach provides the opportunity to modify ordinary and broadly available unactivated carboxylic acid derivatives such as esters. However, the strongly basic conditions that are frequently employed in enolate nucleophilic addition/substitution reactions often lead to a compromise in efficiency, selectivity, and functional group compatibility. Transition metal-catalyzed cross-coupling reactions continue to emerge as viable alternatives to these aforementioned environmentally unfriendly approaches.2 In particular, transition metal-catalyzed C(sp3)–C(sp3) coupling wherein sp3 C–H bonds are replaced by sp3 C–alkyl bonds, leading to vicinally functionalized N-heterocycles, is an attractive transformation given that alkyl-substituted cyclic amines/amides constitute the core of several fragrances, agrochemicals, ligands, alkaloid natural products and pharmaceuticals.3 Despite its poor departing ability, the hydrogen atom is still viewed as an ideal functional handle owing to its relative abundance in organic molecules, which increases its availability for manipulation in either de novo or late-stage functionalization strategies. The dehydrogenation of alcohols to the corresponding carbonyl compounds, which subsequently undergo condensation with a CH-acidic compound followed by hydrogen auto-transfer to form α-alkylated products, is a redox neutral strategy that is quite appealing. Fittingly, several transition metals are capable of catalyzing the dehydrative coupling of alcohols with carbonyl-containing compounds.4 However, no dehydrative coupling method exists for α-alkylation of α,α-substituted esters with alcohols to generate α-quaternary esters. This is quite understandable since all of the aforementioned methods rely on the formation of an aldol or Knoevenagel-type condensation product followed by concomitant reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. Benzylic and homobenzylic all-carbon quaternary stereocenters have shown the propensity to mitigate a spectrum of structural diversity and conformational constraint issues in medicinal chemistry.5 We have therefore identified the catalytic and stereocontrolled installation of a C4 quaternary homobenzylic stereocenter on the skeleton of dihydroisoquinolones (DHIQs) as an important research objective.
C bond. Benzylic and homobenzylic all-carbon quaternary stereocenters have shown the propensity to mitigate a spectrum of structural diversity and conformational constraint issues in medicinal chemistry.5 We have therefore identified the catalytic and stereocontrolled installation of a C4 quaternary homobenzylic stereocenter on the skeleton of dihydroisoquinolones (DHIQs) as an important research objective.
Intrinsic to our design of selecting DHIQs as substrates for sp3 C–H benzylation is the recognition that a subset of natural products, which display a broad spectrum of biological activities,6 harbor benzyl groups on the carbon skeleton of the azaheterocycle. Examples of these benzylisoquinoline alkaloids (BIAs) are depicted in Fig. 1. Although the literature is inundated with examples of C1 benzylated isoquinoline derivatives, general and selective strategies for C4 benzylation leading to all-carbon quaternary stereocenters remain elusive.7 We have sought to bridge this gap and herein report a C(sp3)–C(sp3) site- and diastereoselective benzylation methodology wherein sp3 C–H bonds of DHIQs are replaced by sp3 C–benzyl bonds, in sterically congested environments (Fig. 2A). Conceptually, the approach is closely related to the well-heeled Pd-catalyzed allylic alkylation of carbonyl compounds.8 While this manuscript was in preparation, we became aware of Yang's annulation-based approach to C4 quaternary and homobenzylic DHIQs (Fig. 2B).9
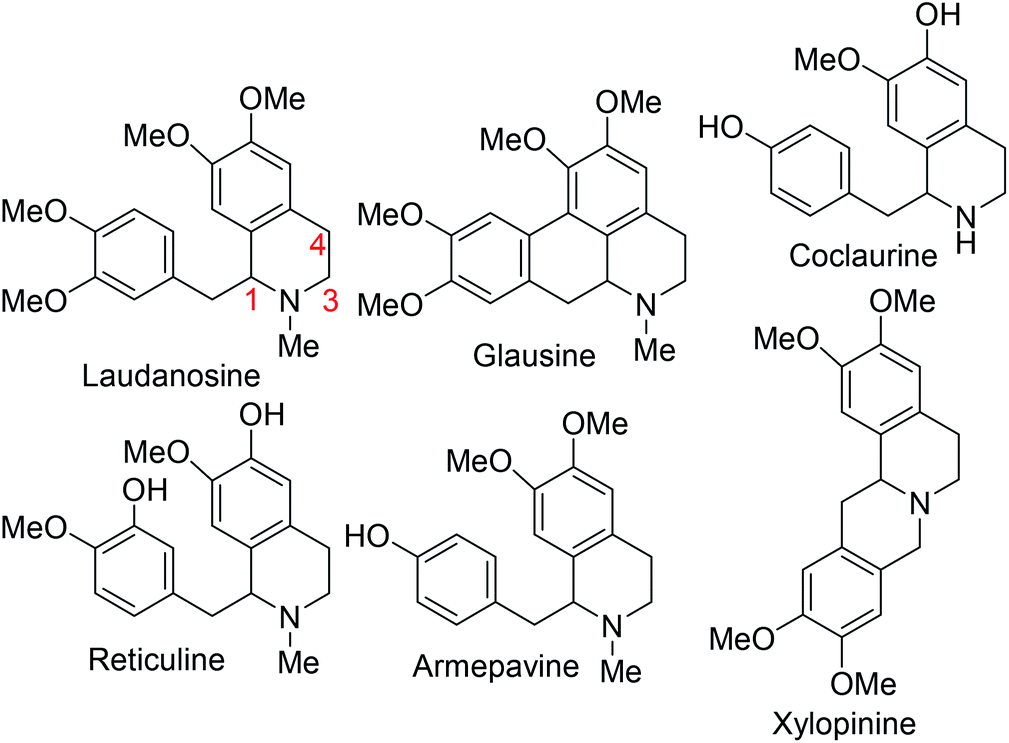 | ||
| Fig. 1 Examples of biologically active benzylisoquinoline alkaloids (BIAs). | ||
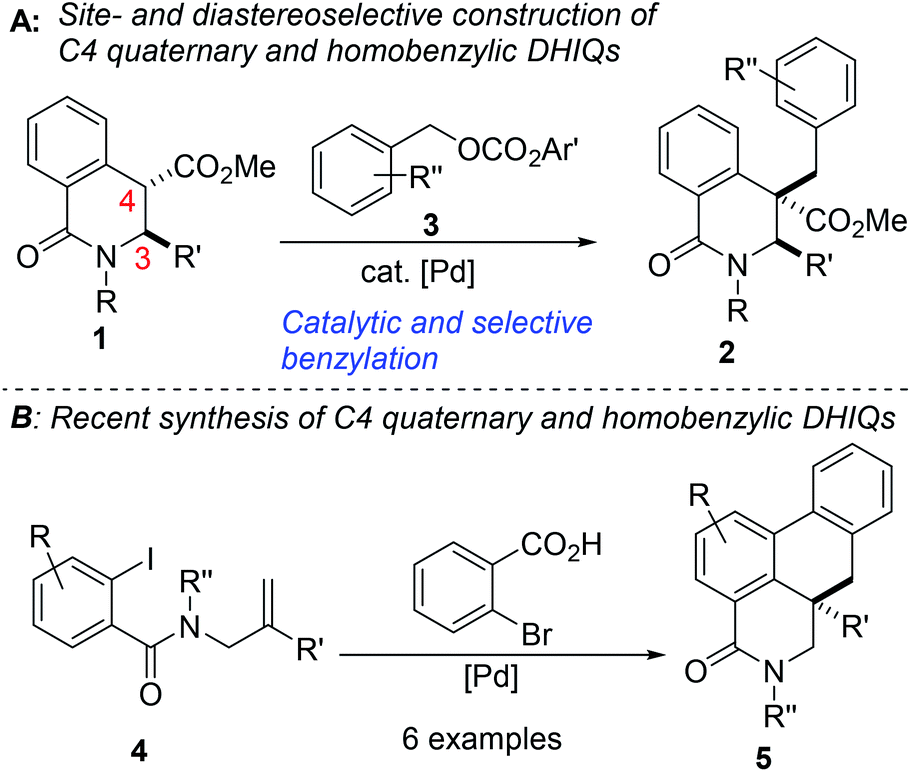 | ||
| Fig. 2 (A) Proposed plan for the selective construction of C4 quaternary and homobenzylic dihydroisoquinolones (B) Yang's recent synthesis of C4 quaternary and homobenzylic polycyclic dihydroisoquinolones. | ||
Results and discussion
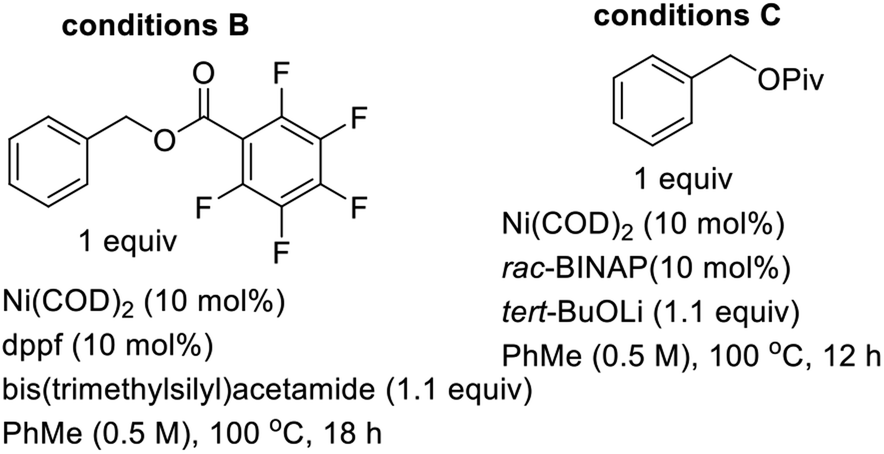
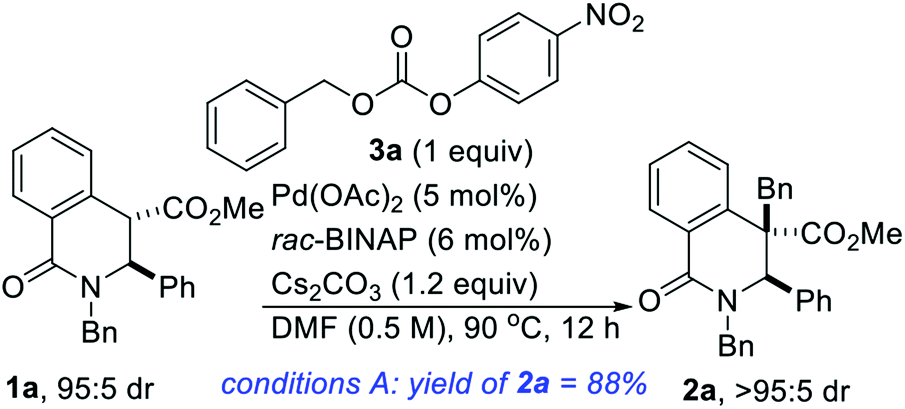
Gleaning from prior reports on Pd-catalyzed benzylation of azlactones, simple unbranched esters, and 3-aryl oxindoles, using benzyl electrophiles,10 we commenced studies on C4 benzylation of DHIQs using model substrate 1a. Hindered and unactivated esters such as 1a generally have an unfavorable ester–enolate equilibrium owing to their low α-CH acidities. Additionally, it can be difficult to control the steric course of alkylation of acyclic esters since the enolate geometry is not predictable a priori. Another inherent challenge of these studies involves the difficulty associated with generating the presumed π-benzyl–palladium intermediate, which requires dearomatization of the electrophilic component.10a After surveying different reaction conditions (Table 1), we were pleased to find that 1a undergoes efficient benzylation to furnish product 2a in 88% isolated yield when carbonate 3a is employed. The optimization studies further revealed that DMF out-performs other solvents such as 2-methyltetrahydrofuran (2-MeTHF), N,N-dimethylacetamide (DMA), and 1,4-dioxane. Cs2CO3 is a more suitable base than NaOAc and Hunig's base, but the reasons are unclear at this point. Performing the reaction at an elevated temperature for a shorter duration has no beneficial effect on the efficacy of the transformation (entry 6). No coupling occurs when the BINAP ligand is replaced by triphenylphosphine (entry 7) or tri-tert-butylphosphine (entry 8). Although the diastereoselectivity of benzylation is impeccable, the use of enantiopure versions of BINAP does not lead to any asymmetric induction (entries 9 & 10). Benzyl carbonate 3a appears to be best suited for this site-selective benzylation compared to the other benzyl electrophiles that we have surveyed (entries 11–13). Nickel-catalyzed conditions11 were also evaluated but they did not perform as well as the optimized Pd-catalyzed conditions (entries 14 & 15).
|
||
|---|---|---|
| Entry | Deviation from conditions A | % yield |
| 1 | 2-Me THF as solvent | 32 |
| 2 | N,N-Dimethylacetamide (DMA) as solvent | 72 |
| 3 | 1,4-Dioxane as solvent | 36 |
| 4 | NaOAc in place of Cs2CO3 | 23 |
| 5 | Hunig's base in place of Cs2CO3 | 28 |
| 6 | Performed at 110 °C for 4 h | 76 |
| 7 | 10 mol% PPh3 in place of rac-BINAP | 0 |
| 8 | 10 mol% tert-Bu3P in place of rac-BINAP | 0 |
| 9 | S-BINAP in place of rac-BINAP | 83(50:50 er) |
| 10 | R-BINAP in place of rac-BINAP | 81(50:50 er) |
| 11 | BnOCO2tBu in place of BnOCO2Me | 79 |
| 12 | BnOP(O)(OEt)2 in place of BnOCO2Me | 63 |
| 13 | BnOP(O)(OPh)2 in place of BnOCO2Me | 80 |
| 14 | Conditions B in place of conditions A | 77 |
| 15 | Conditions C in place of conditions A | 53 |
 |
||
With satisfactory conditions for site-selective and catalytic and diastereoselective benzylation of DHIQs in hand, the scope of the transformation with respect to the stereoelectronics of the nucleophile (i.e., the DHIQ) was next explored (Scheme 1). DHIQs bearing N-alkyl, allyl, and benzyl substituents were surveyed. Pleasingly, the quaternary DHIQs are obtained in synthetically attractive yields when benzyl carbonate 3a is added to a wide range of diversely functionalized lactamoyl esters (see 2a–m). C3-arylated DHIQs harboring alkoxy, allyloxy, benzyloxy, and halogen substituents all react satisfactorily (entries 2a–i). Furthermore, a DHIQ bearing an alkynyl substituent at C3 is amenable to this diastereoselective benzylation protocol (see 2j). The transformation fully tolerates C3 alkenyl substituents (entries 2k–m), which bodes well for late-stage diversification. Although speculative, the diastereoselectivity of the transformation is presumably governed by substituent effects, especially the vicinal C3 substituent.
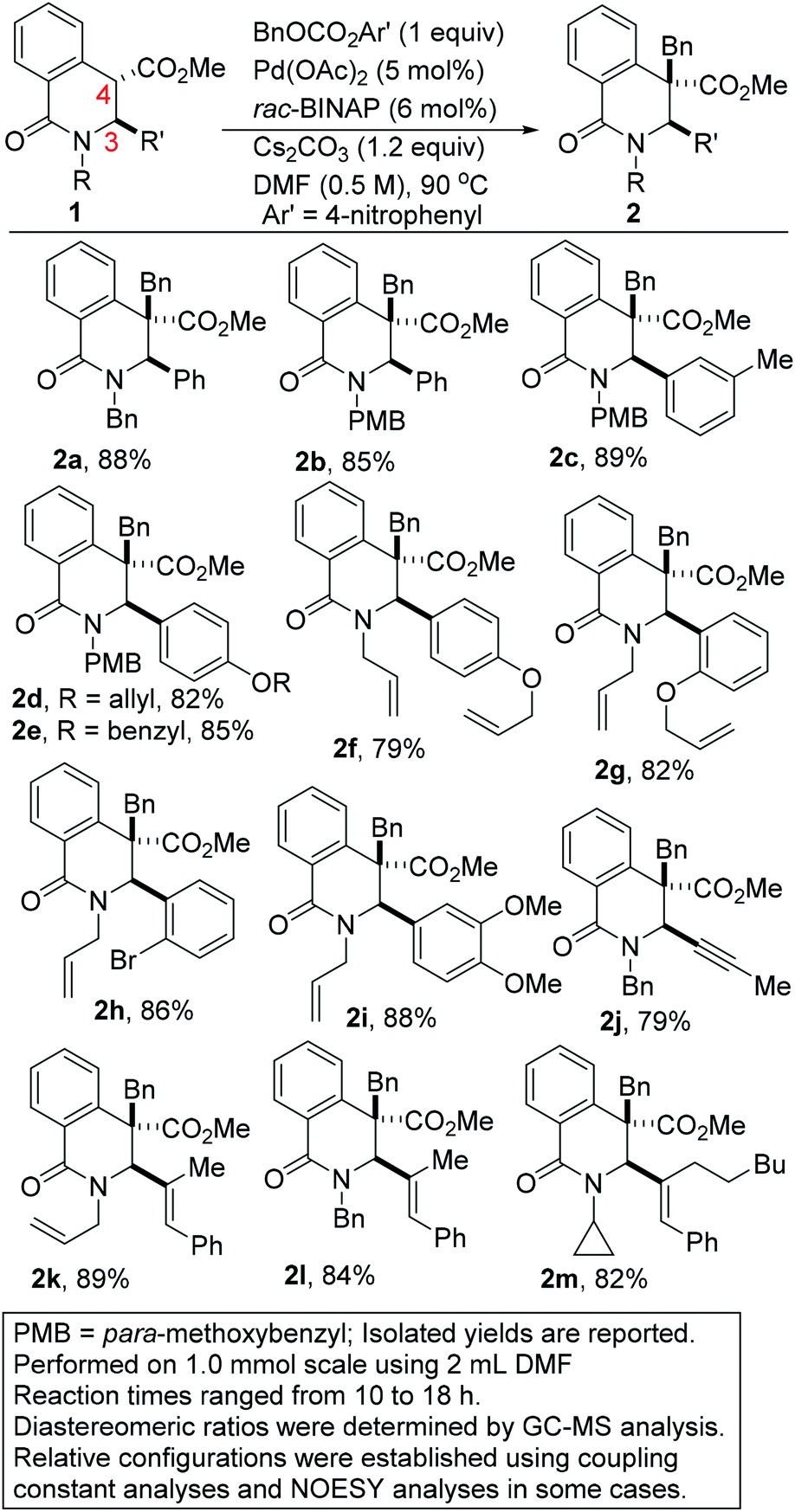 | ||
| Scheme 1 Scope of nucleophile in the selective benzylation of DHIQs. | ||
The scope of the transformation with respect to the electrophilic benzyl carbonate coupling partner has been briefly explored (Scheme 2). In the event, we find that electron-rich benzyl carbonates out-perform their electron-deficient counterparts (see 2o vs. 2r). As has been previously articulated,10a ionization of the benzyl electrophile to the η3-benzyl-palladium cationic intermediate can either proceed through coordination of Pd(0) to the π-system and concomitant ionization to the η3-benzyl-palladium cation or through SN2 displacement of the carbonate leaving group by palladium leading to the σ-bound cation, which then isomerizes to the π-benzyl cation. In these studies, we surmise that substitution followed by isomerization is operative given that less electron-rich benzyl carbonates react slowly and less efficiently under identical conditions; consistent with a mechanism involving SN2 displacement as opposed to pre-coordination. Tsuno and co-workers have previously rationalized that increased electron density would stabilize the transition state in an SN2-type reaction, leading to charged intermediates.12 Additionally, an SN2 displacement with palladium has been proposed in studies on oxidative addition to benzyl chloride.13
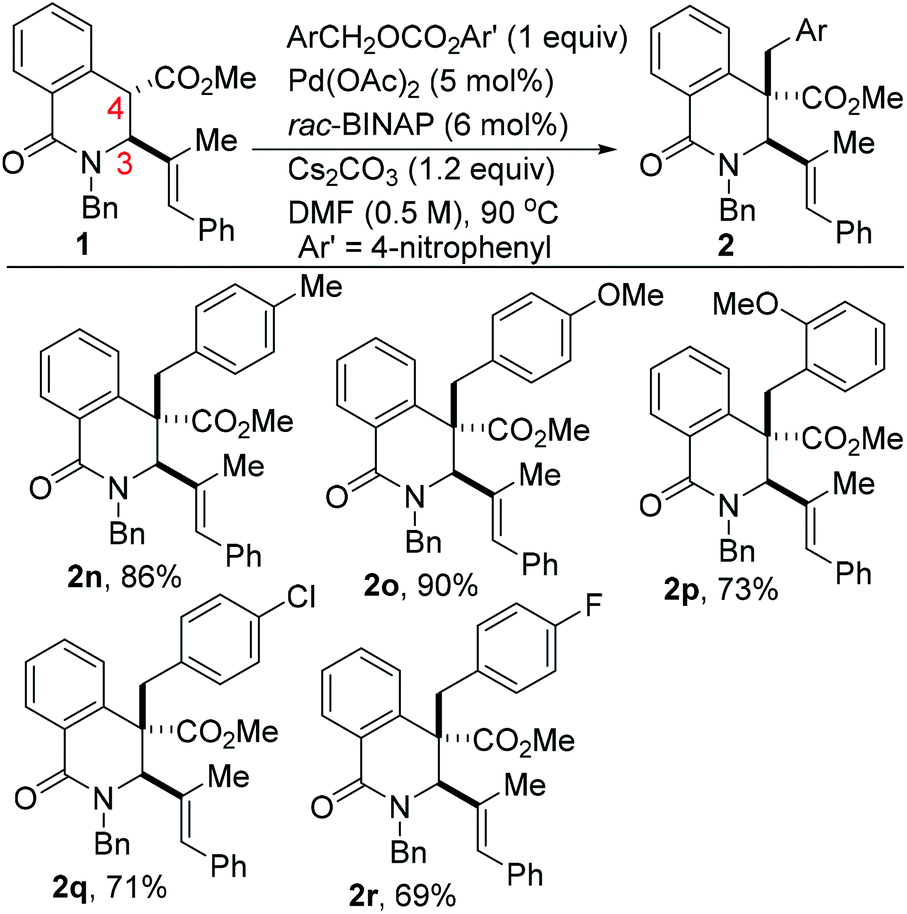 | ||
| Scheme 2 Scope of benzyl carbonate electrophile in the catalytic and selective benzylation of DHIQs. | ||
A potentially beneficial aspect of these studies is the scalable nature of the reaction without any compromise in efficiency and selectivity. This has set the stage for post-diversification studies, leading to the construction of attractive fragments for potential drug discovery. For example, lithium borohydride-assisted chemoselective reduction of the ester group affords isoquinolone alkenols of type 6 (Scheme 3). The practical and reliable synthesis of saturated azaheterocycles bearing a hydroxymethyl group such as 6, is noteworthy given that this substructure constitutes the core of several alkaloids, including calycotomine.14
 | ||
| Scheme 3 Chemoselective reduction of C4 quaternary and homobenzylic dihydroisoquinolones. | ||
Conclusions
In summary, the successful implementation of a Pd-catalyzed α-ester functionalization strategy wherein remote sp3 C–H bonds of vicinally functionalized dihydroisoquinolones are replaced by sp3 C–benzyl bonds has led to the diastereoselective synthesis C4 quaternary and homobenzylic dihydroisoquinolones. The transformation tolerates a variety of synthetically useful functional groups such as haloarenes, alkenes, and alkynes. Electron-rich benzyl carbonates are more amenable to this sp3 C–H benzylation than their electron-deficient counterparts. Post-diversification has led to the synthesis of C4 quaternary dihydroisoquinolones bearing a hydroxymethyl group, a substructure that constitutes the core of several isoquinoline alkaloids, including calycotomine. The development of an enantioselective version of the transformation is ongoing as well as efforts to discover potentially potent antileishmania compounds.Experimental
All experiments involving air and moisture sensitive reagents such as organolithium reagents were carried out under an inert atmosphere of nitrogen and using freshly distilled solvents. Column chromatography was performed on silica gel (230–400 mesh). Thin-layer chromatography (TLC) was performed using Silicycle Siliaplate™ glass backed plates (250 μm thickness, 60 Å porosity, F-254 indicator) and visualized using UV (254 nm) or KMnO4 stain. Unless otherwise indicated, 1H, 13C, and DEPT-135 NMR, and NOESY spectra were acquired using CDCl3 solvent at room temperature. Chemical shifts are quoted in parts per million (ppm). HRMS-EI+ data were obtained using either electronspray ionization (ESI) or electron impact (EI) techniques. High-resolution ESI was obtained on an LTQ-FT (ion trap; analyzed using Excalibur). High resolution EI was obtained on an Autospec (magnetic sector; analyzed using MassLynx). Representative GC-MS traces are provided to substantiate the diastereomeric ratios.General procedure A
General procedure B
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20). Yield = 406 mg, 88%. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 1.5 Hz, 1H), 7.44–7.33 (m, 6H), 7.32–7.24 (m, 5H), 7.28–7.15 (m, 4H), 6.51 (dd, J = 8.0, 1.1 Hz, 1H), 6.44 (dt, J = 7.1, 1.4 Hz, 2H), 5.77 (d, J = 14.5 Hz, 1H), 4.62 (s, 1H), 3.49 (d, J = 14.5 Hz, 1H), 3.31 (d, J = 13.1 Hz, 1H), 3.28 (s, 3H), 2.88 (d, J = 13.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 171.12, 163.15, 136.79, 136.61, 135.93, 135.10, 131.26, 130.75, 130.63, 129.40, 128.96, 128.90, 128.69, 128.49, 128.17, 128.09, 128.05, 127.86, 127.83, 126.93, 67.12, 58.19, 51.81, 48.43, 45.99. FTIR (KBr): 2976.0754, 2927.2335, 1721.7979, 1650.1792, 1492.0415, 1438.4625, 1362.2698, 1320.5399, 1290.1484, 1206.364, 1180.3512, 1146.7618, 1132.397, 995.8166, 918.8793, 700.1334. HRMS calc. for C31H27NO3 461.1991, found 461.1208.
:20). Yield = 406 mg, 88%. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 1.5 Hz, 1H), 7.44–7.33 (m, 6H), 7.32–7.24 (m, 5H), 7.28–7.15 (m, 4H), 6.51 (dd, J = 8.0, 1.1 Hz, 1H), 6.44 (dt, J = 7.1, 1.4 Hz, 2H), 5.77 (d, J = 14.5 Hz, 1H), 4.62 (s, 1H), 3.49 (d, J = 14.5 Hz, 1H), 3.31 (d, J = 13.1 Hz, 1H), 3.28 (s, 3H), 2.88 (d, J = 13.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 171.12, 163.15, 136.79, 136.61, 135.93, 135.10, 131.26, 130.75, 130.63, 129.40, 128.96, 128.90, 128.69, 128.49, 128.17, 128.09, 128.05, 127.86, 127.83, 126.93, 67.12, 58.19, 51.81, 48.43, 45.99. FTIR (KBr): 2976.0754, 2927.2335, 1721.7979, 1650.1792, 1492.0415, 1438.4625, 1362.2698, 1320.5399, 1290.1484, 1206.364, 1180.3512, 1146.7618, 1132.397, 995.8166, 918.8793, 700.1334. HRMS calc. for C31H27NO3 461.1991, found 461.1208.Note: all other benzylated products depicted in Schemes 1 and 2 were prepared as described above. Spectroscopic data can be found in the ESI.†
:50). Yield = 114.6 mg, 91%. 1H NMR (400 MHz, CDCl3) δ 8.24 (dd, J = 7.7, 1.5 Hz, 1H), 7.54 (dd, J = 7.5, 1.8 Hz, 1H), 7.37–6.94 (m, 11H), 6.98–6.82 (m, 2H), 6.61 (s, 1H), 6.51–6.38 (m, 2H), 6.24 (dd, J = 7.9, 1.1 Hz, 1H), 5.46 (d, J = 14.2 Hz, 1H), 4.27 (d, J = 14.2 Hz, 1H), 4.22 (s, 1H), 3.91 (d, J = 11.3 Hz, 1H), 3.83 (s, 3H), 3.64 (d, J = 11.3 Hz, 1H), 2.88–2.73 (m, 2H), 1.78 (s, 1H) 1.30 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 163.87, 157.85, 139.53, 136.80, 136.45, 135.93, 131.97, 131.55, 131.26, 130.95, 129.21, 129.10, 128.68, 128.37, 127.59, 127.24, 127.16, 126.35, 125.25, 124.76, 120.99, 110.61, 70.54, 61.60, 55.47, 44.92, 42.64, 42.50, 14.48. FTIR (KBr): 3384.5368, 2972.9933, 2932.8937, 1638.2038, 1449.1308, 1364.7192, 1290.2159, 1270.3054, 1247.8533, 1206.5967, 1179.918, 1131.1074, 1071.4274, 994.4373, 924.8386, 881.7598, 797.4882, 700.0535. HRMS calc. for C34H33NO3 503.2460, found 503.2463.Note: all other alcohols depicted in Scheme 3 were prepared as described above. Spectroscopic data can be found in the ESI.†
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Central Washington University for financial support to T. K. B. A. M. is a STEP fellow. The School of Graduate Studies and Research is thanked for partial support of this work through a Faculty Research Award to T. K. B. as well as a Summer Fellowship to A.M.Notes and references
- B. M. Stoltz, N. B. Bennett, D. C. Duquette, A. F. G. Goldberg, Y. Liu, M. M. Loewinger and C. M. Reeves, Compr. Org. Synth., 2014, 3, 1–55 CAS.
- (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed; (b) M. E. Welsch, S. A Snyder and B. R Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed; (c) N. Palmer, T. M. Peakman, D. Norton and D. C. Rees, Org. Biomol. Chem., 2016, 14, 1599–1610 RSC; (d) C. W. Murray and D. C. Rees, Angew. Chem., Int. Ed., 2016, 55, 488–492 CrossRef CAS PubMed; (e) I. P. Singh and P. Shah, Expert Opin. Ther. Pat., 2017, 27, 17–36 CrossRef CAS PubMed.
- (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (b) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826–839 CrossRef PubMed; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed. Engl., 2012, 51, 8960–9009 CrossRef CAS PubMed; (d) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (e) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- (a) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (b) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46 CrossRef CAS PubMed.
- (a) J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473 CrossRef CAS; (b) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 13, 2745 CrossRef PubMed; (c) A. C. B. Burtoloso, Synlett, 2009, 20, 320 CrossRef; (d) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC; (e) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 20, 5969 CrossRef.
- (a) M. Perez, Z. Wu, M. Scalone, T. Ayad and V. Ratovelomanana-Vidal, Eur. J. Org. Chem., 2015, 6503–6514 CrossRef CAS; (b) I. P. Singh and P. Shah, Expert Opin. Ther. Pat., 2017, 27, 17–36 CrossRef CAS PubMed.
- For an isolated example of directing group-assisted C4 benzylation using a lithium amide base, see M. M. Amer, A. C. Carrasco, D. J. Leonard, J. W. Ward and J. Clayden, Org. Lett., 2018, 20, 7977–7981 CrossRef CAS PubMed.
- For reviews, see: (a) J. Tsuji, Tetrahedron, 2015, 71, 6330–6348 CrossRef CAS; (b) B. M. Trost, Tetrahedron, 2015, 71, 5708–5733 CrossRef CAS PubMed; (c) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed; (d) B. M. Trost, M. R. Machacek and A. P. Aponick, Acc. Chem. Res., 2006, 39, 747–760 CrossRef CAS PubMed; (e) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed; (f) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS.
- For an annulation-based approach to C4 quaternary and homobenzylic dihydroisoquinolones, see X. Luo, L. Zhou, H. Lu, G. Deng, Y. Liang, C. Yang and Y. Yang, Org. Lett., 2019, 21, 9960–9964 CrossRef CAS PubMed.
- (a) B. M. Trost and L. C. Czabaniuk, Angew. Chem., Int. Ed., 2014, 53, 2826–2851 CrossRef CAS PubMed; (b) B. M. Trost and L. C. Czabaniuk, Chem.–Eur. J., 2013, 19, 15210–15218 CrossRef CAS PubMed; (c) B. M. Trost and L. C. Czabaniuk, J. Am. Chem. Soc., 2012, 134, 5778–5781 CrossRef CAS PubMed; (d) B. M. Trost and L. C. Czabaniuk, J. Am. Chem. Soc., 2010, 132, 15534–15536 CrossRef CAS PubMed; (e) M. Assié, J.-Y. Legros and J.-C. Fiaud, Tetrahedron: Asymmetry, 2005, 16, 1183–1187 CrossRef; (f) S. Tabuchi, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2016, 55, 6973–6977 CrossRef CAS PubMed; (g) K. J. Schwarz, C. Yang, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2018, 57, 12102–12105 CrossRef CAS PubMed.
- E. J. Tollefson, L. E. Hanna and E. R. Jarvo, Acc. Chem. Res., 2015, 48, 2344–2353 CrossRef CAS PubMed.
- S. H. Kim, S.-D. Yoh, C. Lim, M. Mishima, M. Fujio and Y. Tsuno, J. Phys. Org. Chem., 1998, 11, 254 CrossRef CAS.
- P. K. Wong, K. S. Y. Lau and J. K. Stille, J. Am. Chem. Soc., 1974, 96, 5956 CrossRef CAS.
- A. Chatterjee and N. Adityachaudhury, J. Org. Chem., 1962, 27, 309–310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/c9ra10888b |
| This journal is © The Royal Society of Chemistry 2020 |