
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature controlled condensation of nitriles: efficient and convenient synthesis of β-enaminonitriles, 4-aminopyrimidines and 4-amidinopyrimidines in one system†
Yinghua Li,
Yingzu Zhu,
Shiqun Xiang ,
Weibin Fan,
Jiang Jin and
Deguang Huang*
,
Weibin Fan,
Jiang Jin and
Deguang Huang*
State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences Fuzhou, Fujian 350002, China. E-mail: dhuang@fjirsm.ac.cn
First published on 12th February 2020
Abstract
A series of β-enaminonitriles, 4-aminopyrimidines and 4-amidinopyrimidines were synthesized by condensation of organonitriles in one system. A wide variety of compounds were obtained in good to excellent yields by simply controlling the reaction temperature. The base-induced transformation process is easy for production. The scope and versatility of the method have been successfully demonstrated with 72 examples. The flexible and diversified characteristics of nitriles were introduced based on electronic effect, steric effect, position of substituted groups and intermolecular hydrogen bonding. An updated reaction mechanism is proposed based on the study of the stoichiometric reaction conditions at variable temperature, and on the investigation of products with cis–trans configuration transformation.
Introduction
As a versatile and designable building block, β-enaminonitrile plays an important role in the synthesis of pharmaceuticals, fungicides, dyes and biological pesticides.1,2 Many heterocyclic compounds, e.g. pyrimidine,3 pyridine,4 pyrrole,5 pyrazole,6 imidazole,7 isothiazole8 and pyrrolinone9 are proposed to be synthesized from β-enaminonitriles (Scheme 1). The great significance of them in synthetic chemistry has consistently stimulated the development of new methods for their preparation. Meanwhile, the base-facilitated alkylation of stabilized carbanions, directed by functional groups, has been extensively employed in the construction of small molecule precursors for organic synthesis.10 Among which, the alkylation of deprotonated nitrile anions occupies a unique position as being powerful nucleophiles, ideally suited for transformation into a variety of functional groups.11 The challenging issues lie ahead in controlling selectivity and efficiency of transformation, from the viewpoints of practicality in laboratory synthesis and industrial applications.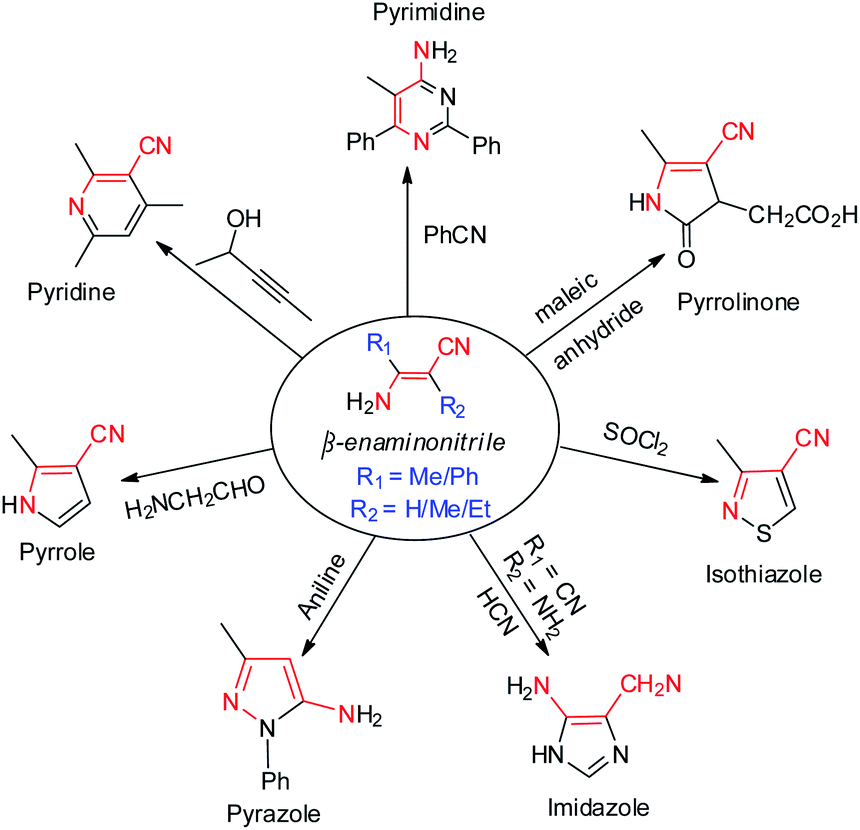 | ||
| Scheme 1 Synthesis of diverse heterocycles from β-enaminonitriles. | ||
β-Enaminonitriles were commonly synthesized by condensation of two functional amide/nitrile molecules under high temperature and/or strong base conditions (Scheme 2a and b).1,12,13 Other methods were also developed to obtain β-enaminonitriles, including the refluxing of methylenecarbonitriles with triethyl orthoformate and amine (Scheme 2c),14 the metal-catalysed condensation of active nitriles with isocyanides or cyclopropanes (Scheme 2d and e),15,16 the cyclization of chloromethyl p-tolyl sulfoxide with cyanomethyl lithium and (Scheme 2f),17 and the Thorpe–Ziegler condensation of aliphatic nitriles (Scheme 2g).18 However, all these reactions show limited tolerance for the synthesis of various enaminonitriles due to the rigorous reaction conditions and/or limited reaction substrates.1,12–18 Thus, the utilization of β-enaminonitriles for the synthesis of different substituent 4-aminopyrimidines is rarely described. 4-Aminopyrimidines and their derivatives are an important building blocks found in the molecules of natural products.19 The biochemical activity of them has been discovered and explored for the treatment of diseases such as T cell-mediated autoimmune and inflammatory disorders,20 external injuries21 and cancer.22 An efficient method is desired for the synthesis of various β-enaminonitriles, 4-aminopyrimidines and their derivatives, consistent with the development of heterocyclic chemistry and its related medical synthetic chemistry.
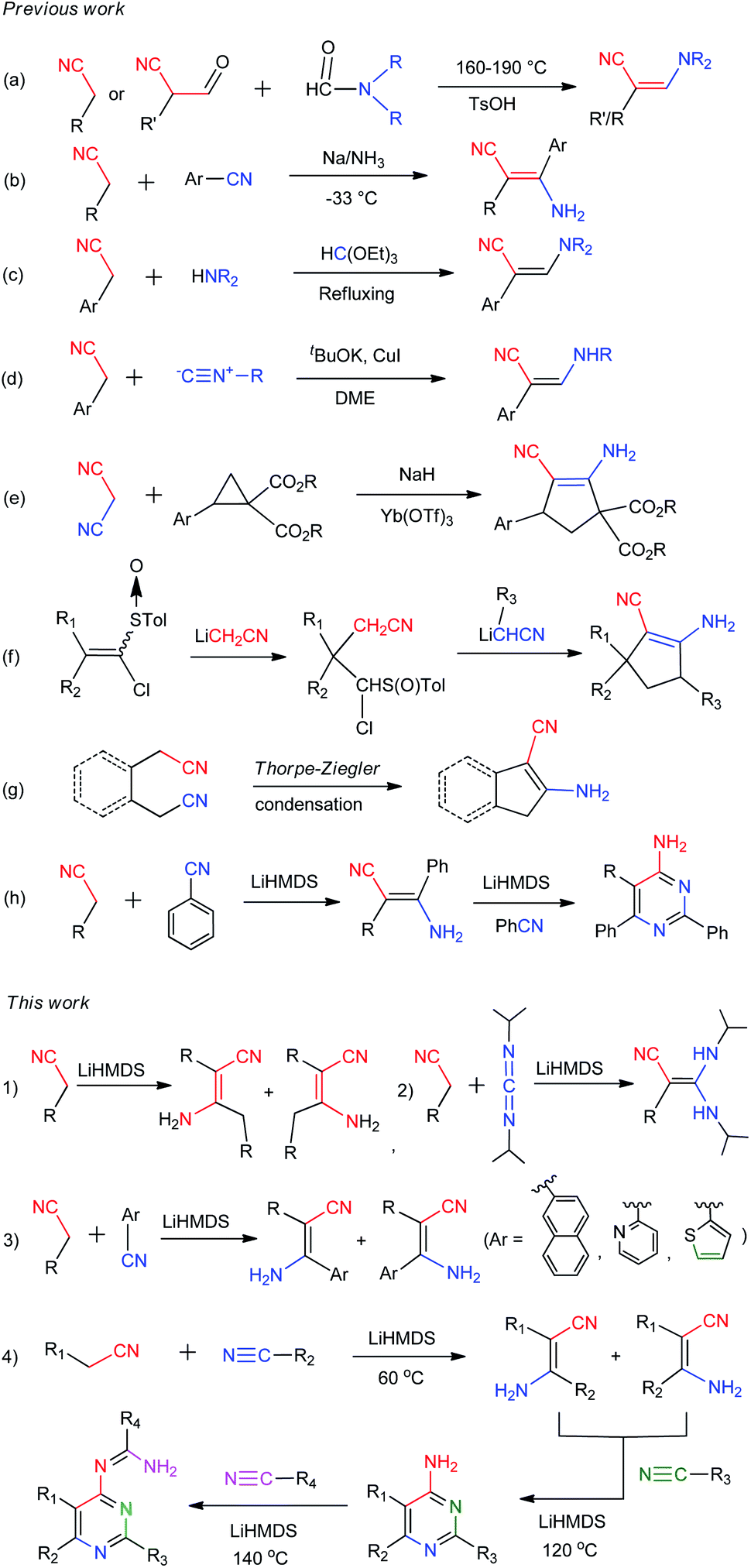 | ||
| Scheme 2 Methods and routines for the synthesis of β-enaminonitriles and its related 4-aminopyrimidines, 4-amidinopyrimidines. | ||
Organonitriles possess versatile chemical properties,23 and the nitrile group can be easily transformed into a variety of heterocyclic groups such as amines, amides, amidines, tetrazoles, triazoles, oxazoles and thiazoles.24 The employment of nitriles as nitrogen source has become a scientific interest in the construction of nitrogenous substances and heterocyclic compounds.1,25 We recently reported a simple and catalyst-free method for the synthesis of β-enaminonitriles and 4-aminopyrimidines by using aliphatic nitriles as electron donors and benzonitriles as electron acceptor (Scheme 2h).26 However, the application of aliphatic nitriles and other types of aromatic nitriles (e.g. naphthyl, pyridyl, thienyl) as electron acceptors has not been concerned. Also, the condensation of aliphatic nitriles only and the discussion of the cis–trans isomerization of β-enaminonitriles was not involved in that paper. Herein, we introduce the method for the synthesis of β-enaminonitriles by self-condensation of aliphatic nitriles, as well as reaction of aliphatic nitriles with N,N′-diisopropylcarbodiimide (DIC) and various aromatic nitriles. The discussion of cis–trans isomerization of β-enaminonitriles is concluded based on the results of condensation of various nitriles. Furthermore, the formation of 4-aminopyrimidines with various types of nitriles is examined, and a new type condensation of four-component nitriles is developed for the construction of novel multi-substituted 4-amidinopyrimidines.
Results and discussion
All the reactions were carried out under N2 atmosphere in Teflon screw-cap sealed tubes. The condensation of phenylacetonitrile (1a) was selected as a model reaction and optimized by changing reagents or reaction conditions to find out the most suitable condition (Table 1). Based on the experimental results obtained, it is concluded that the reaction of substrates 1a (0.4 mmol) and lithium hexamethyldisilazide (LiHMDS, 0.2 mmol) in dimethoxyethane (DME) at 120 °C for 24 h gives the best result with the production of compound 2a in yield up to 86% (entry 6). The LiHMDS salt works as a highly efficient base in deprotonation of the α-H of 1a, and it was added in stoichiometric ratio for a completed reaction. Other alkali salts and/or organic bases of tBuOK, EtONa, Cs2CO3, 4-dimethylaminopyridine (DMAP) and 1,8-diazabicyclo [5.4.0]-undec-7-ene (DBU) afforded the product of 2a in lower yields (entry 1–2) or without yield (entry 3–5). Meanwhile, the type of solvent was important for the formation of 2a (entry 6–11). Less polar solvents such as DME, anisole and THF would help to increase the reaction yield, among which the DME is the best choice (entry 6). In contrast, more polar solvents would be unfavourable for the generation of product with the decline of yield to trace in DMF (entry 11). The existence of hydrogen bonding interaction between solvent and inter mediate imine/amine is suggested to be responsible for the inaction of the self-condensation reaction in more polar solvent system.
|
|||||
|---|---|---|---|---|---|
| En | Base | Solvent | Temp (°C) | T (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), base (0.20 mmol), solvent (1 mL), N2.b Isolated yield. DME = dimethoxyethane, DMAP = 4-dimethylaminopyridine, DBU = 1,8-diazabicyclo[5,4,0]undec-7-ene, THF = tetrahydrofuran, DCE = dichloroethane, DMF = N,N-dimethylformamide. | |||||
| 1 | tBuOK | DME | 120 | 24 | 12 |
| 2 | EtONa | DME | 120 | 24 | 26 |
| 3 | Cs2CO3 | DME | 120 | 24 | None |
| 4 | DMAP | DME | 120 | 24 | None |
| 5 | DBU | DME | 120 | 24 | None |
| 6 | LiHMDS | DME | 120 | 24 | 86 |
| 7 | LiHMDS | Anisole | 120 | 24 | 55 |
| 8 | LiHMDS | THF | 120 | 24 | 45 |
| 9 | LiHMDS | Dioxane | 120 | 24 | None |
| 10 | LiHMDS | DCE | 120 | 24 | None |
| 11 | LiHMDS | DMF | 120 | 24 | None |
| 12 | LiHMDS | DME | 60 | 24 | 3 |
| 13 | LiHMDS | DME | 100 | 24 | 22 |
| 14 | LiHMDS | DME | 140 | 24 | 61 |
| 15 | LiHMDS | DME | 120 | 12 | 67 |
| 16 | LiHMDS | DME | 120 | 36 | 71 |
The percent conversion of nitrile was examined in a temperature range of 60–140 °C (entry 6, 12–14), and the temperature of 120 °C was determined as standard condition according to the transformation efficiency of compound 2a. Lower temperature would decrease the yield dramatically down to 3% at 60 °C (entry 12), and higher temperature might not help to improve the efficiency of production (entry 14). In addition, the influence of reaction time on the production of 2a was monitored at points of 12 h, 24 h and 36 h (entry 6, 15–16). It was found that 24 h would be a right time for the achievement of reaction. Shorter or longer time would be disadvantageous for the transformation of 1a to 2a with yields decreased to 67% at 12 h and 71% at 36 h, respectively.
Encouraged by the above results, we started to examine the scope of the reaction system as to other nitrile substrates (Table 2). A total of 29 organonitriles were chosen for analysis of the implication of reaction, including aliphatic nitriles, aromatic nitriles, heteroaromatic nitriles, dinitriles. The reactions produced 37 β-enaminonitrile compounds in the same way as phenylacetonitrile (1a) worked. The yield distribution and production efficiency of β-enaminonitriles were discussed based on the electronic effect, steric effect and the synergic effect of functional groups on the nitriles. It was found that all the aliphatic nitriles could work both as electron donors and electron acceptors in the above reaction system, by means of deprotonation of the α-H of nitriles and polarization of the carbon atom on the CN triple bond. By contrast, the aromatic nitriles could only work as electron acceptors owing to the absence of α-H. Firstly, 25 alkyl organonitriles (2a–2y) were deprotonated and performed to study the electronic effect of functional groups in manner of self-condensation. The experimental results showed that electronic effect of substituted groups on the aliphatic nitriles had a significant impact on the production of β-enaminonitriles. The reaction of phenylacetonitrile derivatives with electron-donating groups and weak electron-withdrawing substituted groups on the para-position of phenyl ring afforded the product of β-enaminonitriles in high yields (2a–2g, 70–86%). The electron-withdrawing fluoro group would offer the product in lower yield (2h, 63%) compared to its analogous bromo and chloro groups (2f, 2g). No product of β-enaminonitrile could be isolated when the strong electron-withdrawing groups (nitro and/or trifluoromethyl) substituted phenylacetonitrile derivatives were used for reaction. This point of view was consistent with the results presented in the formation of compounds 2p–2w, in which the reaction of naphthyl groups and various alkyl groups substituted nitriles generated the products in yields 62–78%. Secondly, the steric hindrance to the reaction was studied by employment of methoxyl group (2e, 2j), fluoro group (2h, 2k, 2m) and methyl group (2c, 2i, 2l) in the para-, meta- and ortho-positions, respectively. The result showed that the yields of reaction decreased dramatically from the substitution of para-position (2c, 2h) to ortho-position (2l, 2m), and no self-condensation product was found when the methoxyl group was employed in the ortho-position of the phenyl ring of phenylacetonitrile. This conclusion was in accordance with the fact that the dimethyl groups substituted nitriles (2n, 2o) provided the products in much lower yields (47% and 34%). Finally, the condensation reaction was examined with mixed nitriles by using the deprotonated n-butyronitrile, 3-phenylpropionitrile, 2-pyridylacetonitrile as electron donors and benzonitrile, 2-cyanopyridine, 2-thiophenecarbonitrile, 2-naphthonitrile as electron acceptors (3a–3l). All the reactions were carried out at 60 °C and the products were obtained in good to excellent yields, except for those reactions using 2-pyridylacetonitrile as electron donor. The experiments of 2-pyridylacetonitrile were run at 140 °C for a complete transformation of starting materials to compounds (3c, 3f, 3i, 3l) with the yields above 70%. It is supposed that the interaction of hydrogen bonding between the pyridine group of starting material and the amine group of product decreased the free degree of molecules, which would lead to the difficulty in the molecular collision at lower temperature.
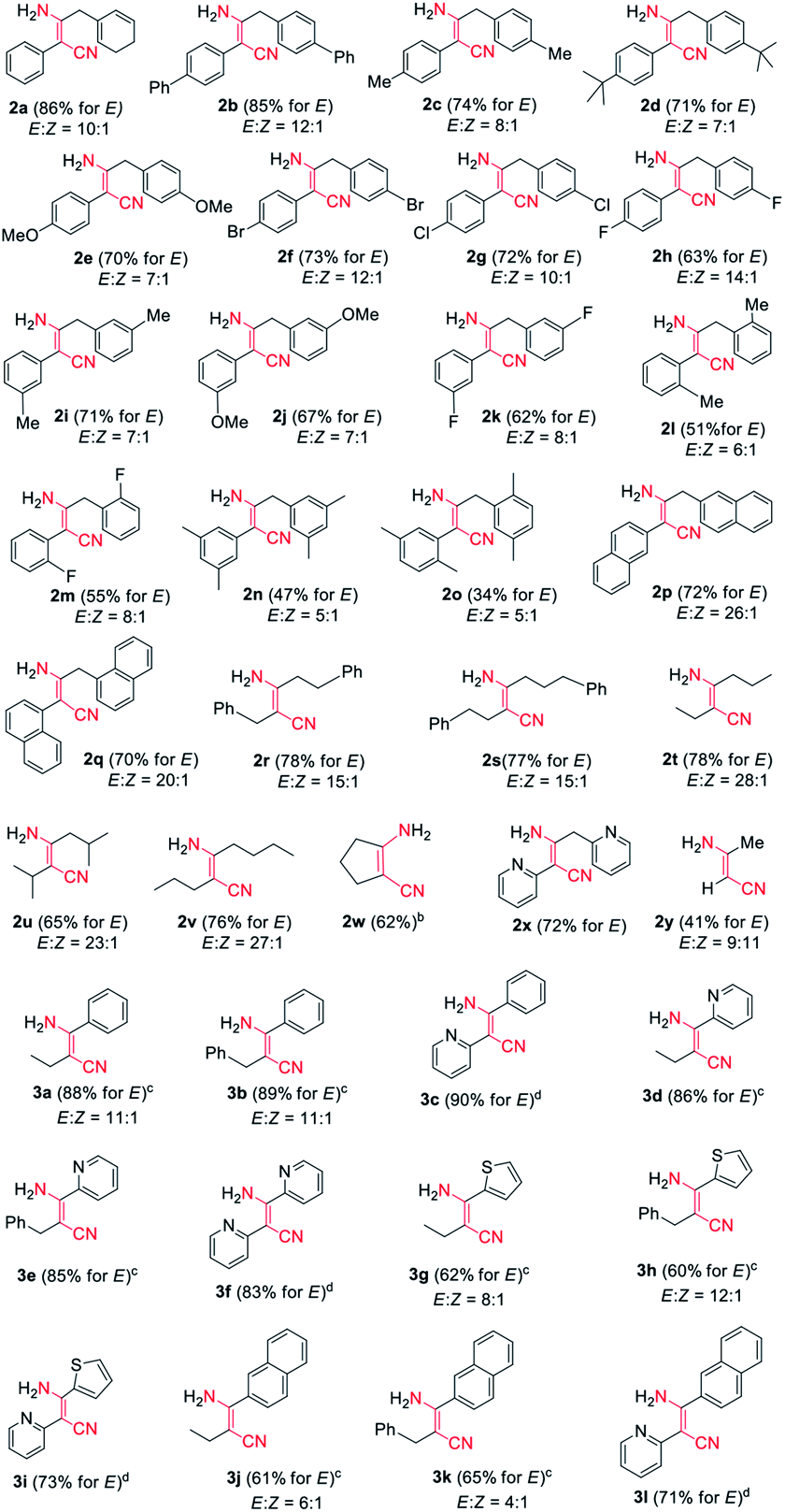
In addition, it was found that the cis–trans isomerization was less evident in the formation of β-enaminonitriles in our reaction, since all the reactions produced trans isomers as main products. The yields of (Z)-β-enaminonitriles was less than 10% in the condensation of aliphatic nitriles only (worked as both electron donor and electron acceptor). The proportion of cis isomers turned out to be higher in the reaction of aliphatic nitriles (electron donors) with aromatic nitriles (electron acceptors). The yields could reach 16% in the formation of compound 3k. It is thought that the aryl group on the aromatic nitriles decreased the energy barrier of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond cleavage, which would lead to decreasing energy consumption for C–C bonding again steric hindrance for the generation of cis-form products. Beyond that, no cis-form compounds was found in the formation of pyridyl group contained β-enaminonitriles. The strong intramolecular hydrogen bonding would bring the reaction to the generation of (E)-β-enaminonitriles during this process (Fig. 1). There was a special case: the condensation of acetonitrile in our conditions afforded cis–trans isomers of 3-aminocrotononitrile (2y) in a ratio of about 1
N bond cleavage, which would lead to decreasing energy consumption for C–C bonding again steric hindrance for the generation of cis-form products. Beyond that, no cis-form compounds was found in the formation of pyridyl group contained β-enaminonitriles. The strong intramolecular hydrogen bonding would bring the reaction to the generation of (E)-β-enaminonitriles during this process (Fig. 1). There was a special case: the condensation of acetonitrile in our conditions afforded cis–trans isomers of 3-aminocrotononitrile (2y) in a ratio of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which could be due to the similar steric effect of methyl group and amine group to the cyanide group on the other carbon atom.
:1, which could be due to the similar steric effect of methyl group and amine group to the cyanide group on the other carbon atom.
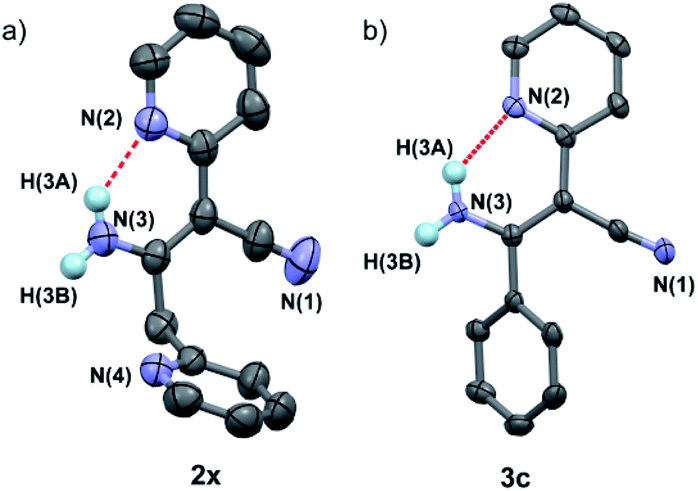 | ||
| Fig. 1 Crystal structures of compounds 2x (CCDC number: 1969352) (a) and 3c (1956321) (b), showing the strong intermolecular hydrogen bondings between amine and pyridine groups (N2⋯H–N3 = 1.998 Å for compound 2x and 2.010 Å for compound 3c). | ||
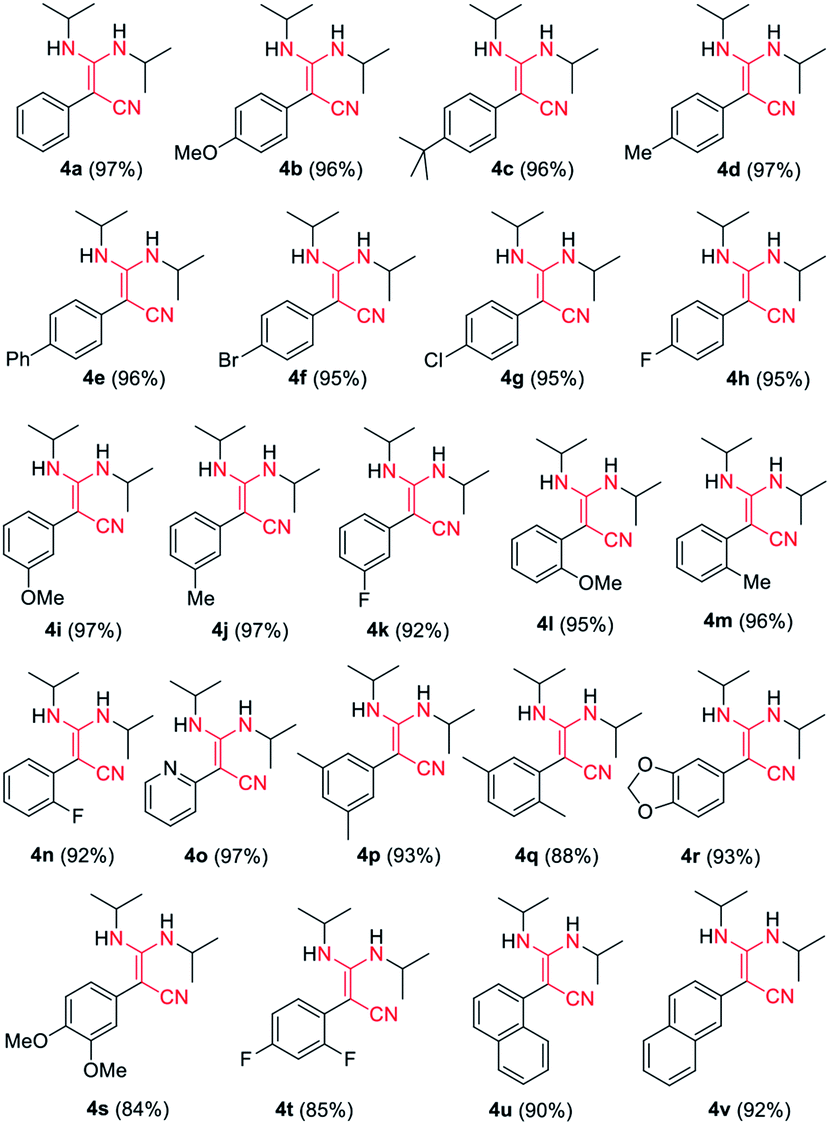
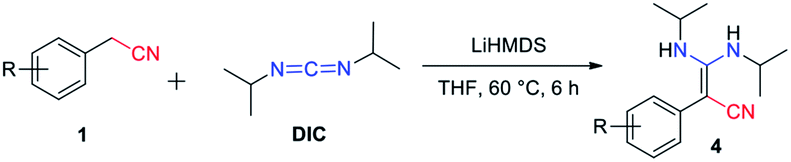
Our condensation reaction also demonstrates good reactivity and stability towards the formation of functional β-enaminonitriles with N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN type substrates (Table 3 and Fig. S1†). The reaction of phenylacetonitrile with N,N′-diisopropylcarbodiimide (DIC) in THF (60 °C, 6 h) afforded compound 4a in yield 97% (Table 3). The scope of phenylacetonitriles substrates was explored by changing functional groups on the phenyl ring, including electron donating groups (MeO−, tBu−, Me−; 4b–4d), electron withdrawing groups (Ph−, Br−, Cl−, F−; 4e–4h), difunctional groups (dimethyl, dimethoxyl, dioxolane, difluoro; 4p–4t) and aromatic groups (pyridyl, naphthyl; 4o, 4u and 4v). All the reactions showed high production efficiency with the yields obtained from 84% to 97%, this result indicated that the above electron groups had weak impacts on the yield of the reaction. The difunctional substituted groups such as 3,5-dimethyl (4p), 2,5-dimethyl (4q), 2,4-difluoro (4t) and 3,4-dimethoxyl (4s) could reduce the generation of product by ca. 10%. Larger substituted group might be disadvantageous for the production of compound 4 and its derivative. Meanwhile, the reaction of DIC with 2-pyridylacetonitrile (4o), 1-naphthyl-acetonitrile (4u) and 2-naphthylacetonitrile (4v) gave the target products in good yields of 97%, 90% and 92%, respectively. It shows that our reaction have a good tolerance in the transformation of various aryl acetonitriles to β-enaminonitriles. The reactivity of nitrile in our system was also examined by reaction of phenylacetonitrile with benzophenone. The reaction afforded the product of 2,3,3-triphenylacrylonitrile in yield 80% after dehydroxylation (Fig. S2†). In addition, the synthetic utility of our reaction was also examined by running the experiments on gram scale. The self-condensation of phenylacetonitrile (1a) or cross-coupling of 1a with DIC on 5 gram scale yielded the products of 2a and 4a in high overall yields of 80% and 95% (Scheme 3), respectively.
CN type substrates (Table 3 and Fig. S1†). The reaction of phenylacetonitrile with N,N′-diisopropylcarbodiimide (DIC) in THF (60 °C, 6 h) afforded compound 4a in yield 97% (Table 3). The scope of phenylacetonitriles substrates was explored by changing functional groups on the phenyl ring, including electron donating groups (MeO−, tBu−, Me−; 4b–4d), electron withdrawing groups (Ph−, Br−, Cl−, F−; 4e–4h), difunctional groups (dimethyl, dimethoxyl, dioxolane, difluoro; 4p–4t) and aromatic groups (pyridyl, naphthyl; 4o, 4u and 4v). All the reactions showed high production efficiency with the yields obtained from 84% to 97%, this result indicated that the above electron groups had weak impacts on the yield of the reaction. The difunctional substituted groups such as 3,5-dimethyl (4p), 2,5-dimethyl (4q), 2,4-difluoro (4t) and 3,4-dimethoxyl (4s) could reduce the generation of product by ca. 10%. Larger substituted group might be disadvantageous for the production of compound 4 and its derivative. Meanwhile, the reaction of DIC with 2-pyridylacetonitrile (4o), 1-naphthyl-acetonitrile (4u) and 2-naphthylacetonitrile (4v) gave the target products in good yields of 97%, 90% and 92%, respectively. It shows that our reaction have a good tolerance in the transformation of various aryl acetonitriles to β-enaminonitriles. The reactivity of nitrile in our system was also examined by reaction of phenylacetonitrile with benzophenone. The reaction afforded the product of 2,3,3-triphenylacrylonitrile in yield 80% after dehydroxylation (Fig. S2†). In addition, the synthetic utility of our reaction was also examined by running the experiments on gram scale. The self-condensation of phenylacetonitrile (1a) or cross-coupling of 1a with DIC on 5 gram scale yielded the products of 2a and 4a in high overall yields of 80% and 95% (Scheme 3), respectively.
|
|---|
| a Reaction conditions: 1 (0.20 mmol), DIC (0.20 mmol), LiHMDS (0.20 mmol), 60 °C, 6 h, N2, isolated yield. DIC = N,N′-diisopropylcarbodiimide. |
 |
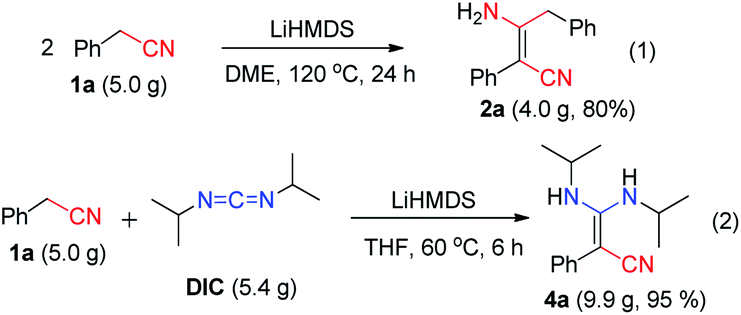 | ||
| Scheme 3 Studies of the synthetic utility of β-enaminonitriles. | ||
In order to explore the application of β-enaminonitriles for the construction of various substituted 4-aminopyrimidines, the condensation of β-enaminonitrile with another portion of nitrile was examined under base condition (Table 4). The reactions were carried out at 120 °C and 140 °C for the condensation of two-species (5b–5d, 5f) and three-species organonitriles (5e, 5g–5i) respectively. The self-condensation of acetonitrile was also completed at a temperature of 140 °C (5a). Nine types of 4-aminopyrimidines were obtained in yields 63–77%, which reflected that the reaction had a wide range of application in the synthesis of nitrogen containing heterocycles. Higher temperature was required for the achievement of multi-substituted 4-aminopyrimidines. It showed that the condensation of multi-components of nitriles was more difficult than that of fewer-components. Also, the eletrophilicity of nitrile has an impact on the formation of 4-aminopyrimidine. The CN groups of aliphatic nitriles posses less electrophilicity to be attacked than aromatic nitriles, and the condensation of aliphatic nitriles for the synthesis of 4-aminopyrimidines needs higher temperature (≥140 °C). Furthermore, it was found that the component of 2-pyridylacetonitrile might not be suitable for the building of pyrimidine core, since the reactions of pyridylacetonitrile-contained β-enaminonitriles (2w, 3c, 3f, 3i, 3l) with aromatic nitriles did not yield the target products under the same conditions. It is thought that the strong hydrogen bonding between the pyridine and amine group in the molecule limits the change of conformation from ‘trans’ to ‘cis’ (Fig. 1). The inversion of configuration is critical for the construction of pyrimidine core through cycloaddition with another portion of nitrile.
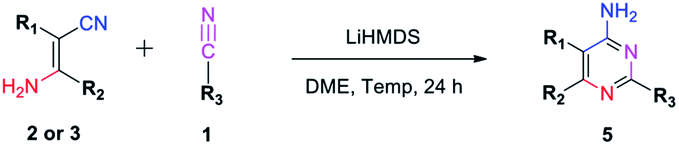
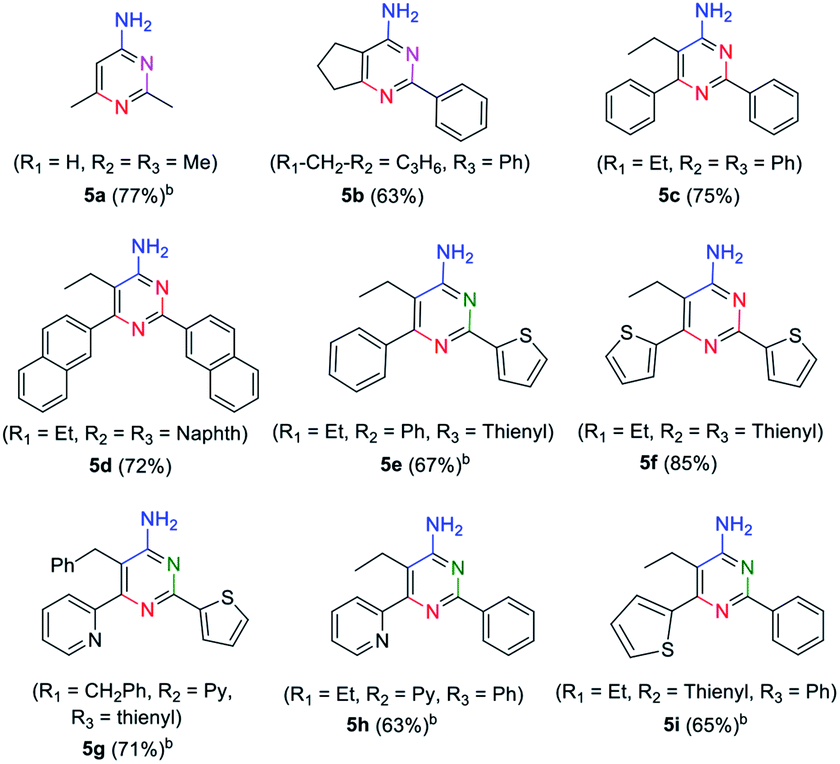
Interestingly, our method also provides an efficient way to synthesize 4-amidinopyrimidines by further condensation of 4-aminopyrimidines with a fourth portion of nitriles (Scheme 4).
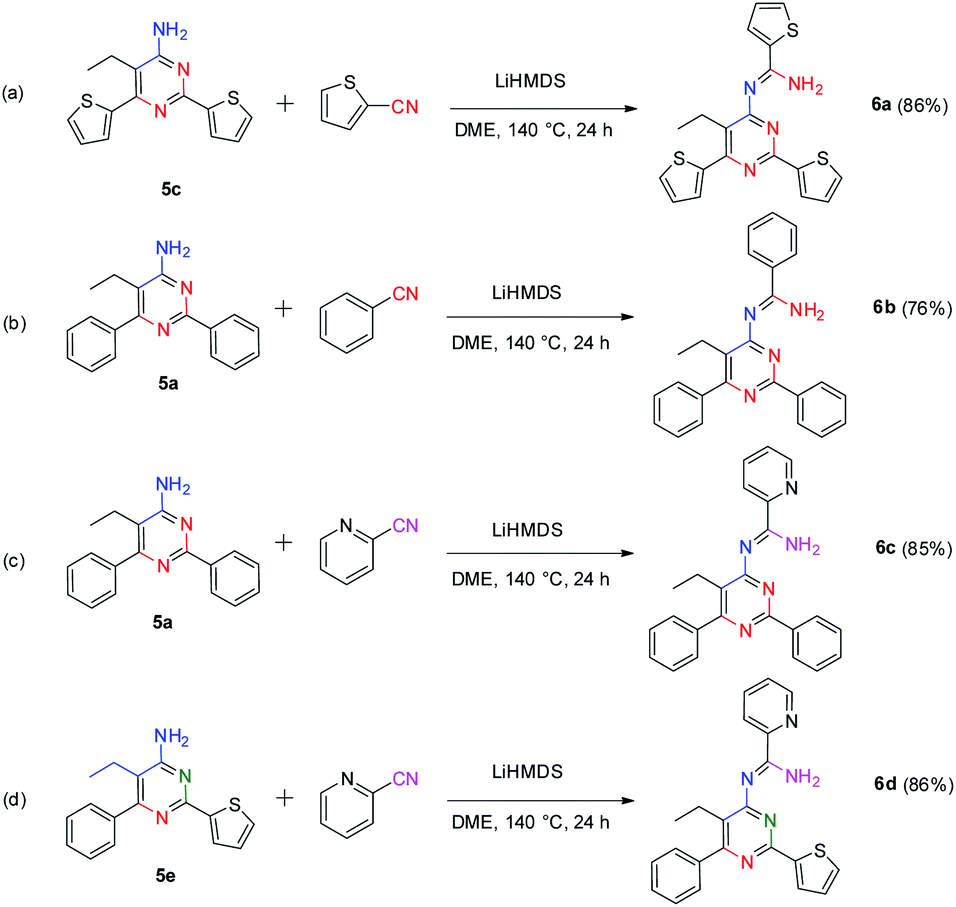 | ||
| Scheme 4 Studies of the synthetic utility of β-enaminonitriles. | ||
To our best knowledge, the synthesis of 4-amidinopyrimidine was rarely reported, and the only example of (2,6-dimethyl-4-acetamidine)-pyrimidine was obtained as a by product in the condensation of acetonitrile under pressure of a few thousand atmosphere.27 The preparation of different component 4-amidinopyrimidine has not been reported so far. In our lab, the reactions run smoothly with four typical type of 4-amidinopyrimidines and the products were obtained in yields over 76%. All the compounds were characterized by 1H NMR, 13C NMR, mass spectra and X-ray crystallography (Fig. 2).
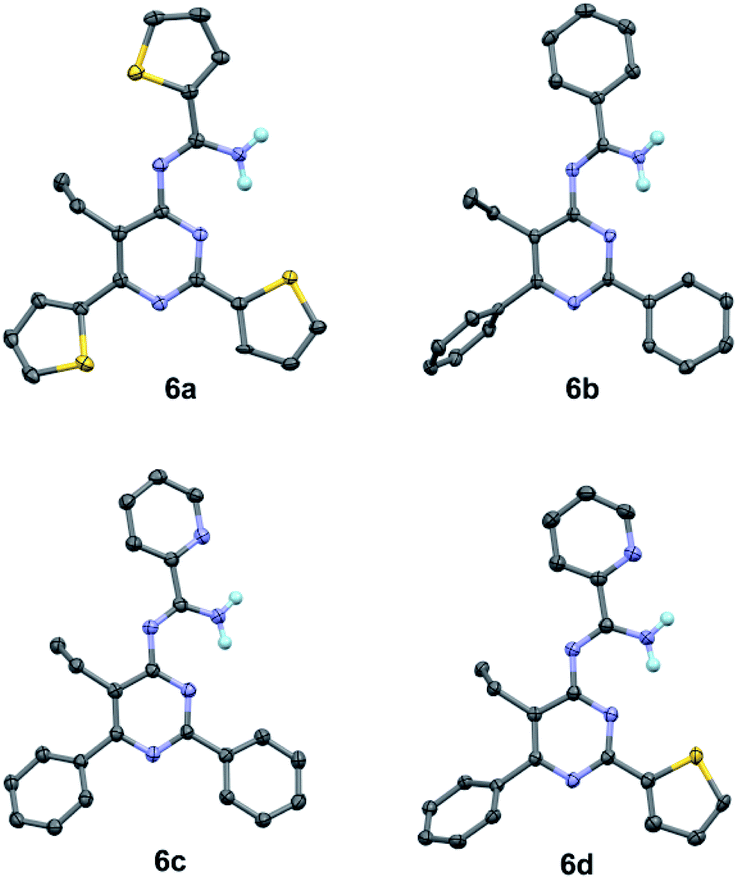 | ||
| Fig. 2 Crystal structures of compounds 6a–6d (CCDC number: 1956324–1956327). | ||
The bond lengths and bond angles of heterocyclic rings are comparable to those presented in the analogous compounds,26 indicating the formation of pyrimidine cores and peripheral amidine function groups.
Based on the above experimental results, an updated mechanism is proposed in Scheme 5. One of hydrogen atoms on the methylene group of aliphatic nitrile is deprotonated by LiHMDS to form a cyanoalkylide anion (a). The anion attacks an adjacent nitrile molecule on the carbon atom of cyanide group to generate an intermediate imine species (b) and (c). The species (c) attacks another nitrile molecule to produce the imine species (d), following by an intramolecular cycloaddition reaction to form the intermediate (e). The imine species (e) attacks a fourth nitrile molecule to form the four-component intermediate (f). The chemical shift of hydrogen atom (H1) and the electron transfer of C–H bond to the terminal imine nitrogen affords the product of 6 spontaneously. In addition, the intermediates (b), (c) and (e) would transform to compounds 2, 2′ and 5 by controlling the addition of nitriles and the reaction temperature.
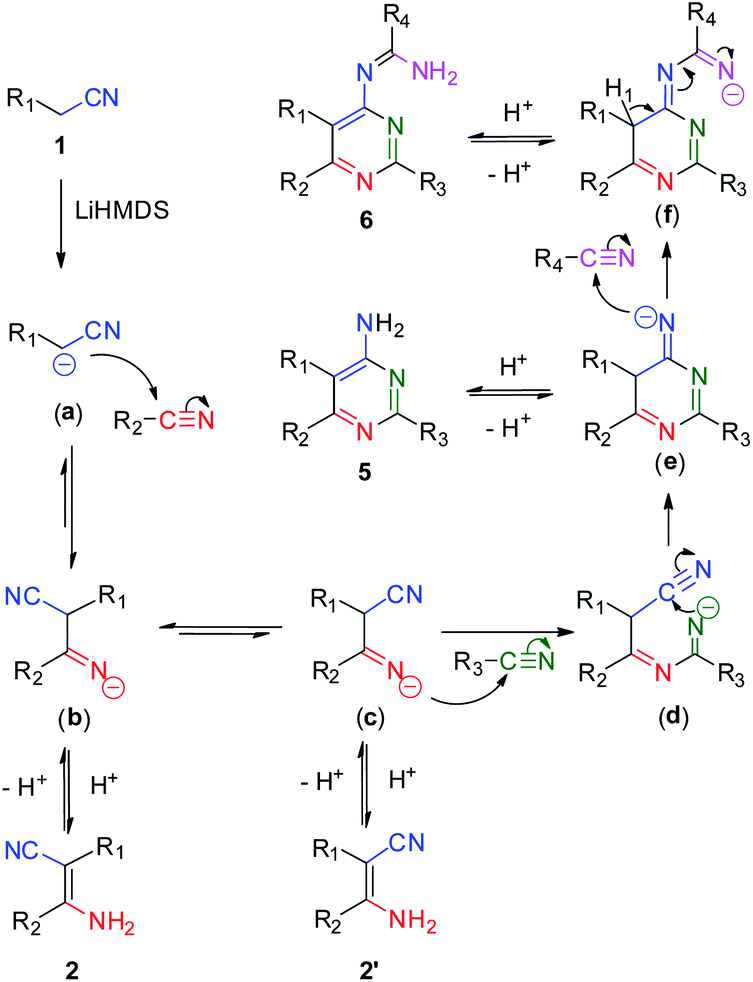 | ||
| Scheme 5 Plausible mechanism of the condensation of various nitriles under base conditions. | ||
Conclusions
In summary, we have described a simple and efficient method for the synthesis of β-enaminonitriles, 4-aminopyrimidines and 4-amidinopyrimidines in a total of 72 samples by means of condensation of organonitriles. The products can be obtained gradually by simply controlling the reaction temperature and the addition of nitrile substrates. The presence of various products in one system indicates the flexible and diversified characteristics of nitriles as electron donor and/or electron acceptor under base conditions. The extra reactivity and stability were studied based on electronic effect, steric effect, position of substituted groups and intermolecular hydrogen bonding. The employment of easily available reagents and mild reaction condition makes this method useful for synthesis chemistry. This work is superior in simple reaction condition, low-cost materials, broad substrate applicability and wide adaptability of production. It might provide a clue to synthesize various multi-substituted 4-aminopyrimidines and/or 4-amidinopyrimidines in favour of the development of biological chemistry, pharmaceutical chemistry and pesticide chemistry.Experimental section
General information
Unless otherwise stated, the loading of starting materials were completed in a glove box. The sealed Teflon screw-cap tube (50 mL) was moved out of glove box, and heated to react under N2 atmosphere. All the liquid nitriles were distilled before use. Other commercial-grade chemicals were used without further purification. DME was distilled over Na and degassed before use. Flash chromatography was performed on silica gel (200–300 mesh). The single crystal data of compounds were collected by a Cu-Kα rotating anode source at 100 K, using a Supernova diffractometer with the ω-scan method. ESI-MS were obtained using a Bruker Impact II quardrupole time-of-flight mass spectrometer. The 1H NMR and 13C NMR spectra were recorded on Bruker Avance III (400 MHz) and chemical shifts were expressed in δ ppm values with reference to tetramethylsilane (TMS) as internal standard. The NMR spectra were recorded in solvent of CDCl3 (Safety Note: along with standard practice (eye protection, fume hood, laboratory coat) the use of LiHMDS requires no water, far away from the environment of fire source and training for the operator.).General experimental procedures
:1 v/v) to afford β-enaminonitriles in yields up to 90% (3c).:1 v/v) to afford compound 4 in yields up to 97% (4a).:1 v/v) to afford compound 5 in yields up to 85% (5f).:1 v/v) to afford compound 6 in yields up to 86% (6a).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant No. 21371171) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000).Notes and references
- (a) A. W. Erian, Chem. Rev., 1993, 93, 1991–2005 CrossRef CAS; (b) S. Bondock, A. E. Tarhoni and A. A. Fadda, Curr. Org. Chem., 2011, 15, 753–781 CrossRef CAS.
- (a) M. A. A. Radwan and M. El-Sherbiny, Bioorg. Med. Chem., 2007, 15, 1206–1211 CrossRef CAS PubMed; (b) S. Bondock, W. Fadaly and M. A. Metwally, Eur. J. Med. Chem., 2009, 44, 4813–4818 CrossRef CAS PubMed; (c) O. Fedorov, K. Huber, A. Eisenreich, P. Filippakopoulos, O. King, A. N. Bullock, D. Szklarczyk, L. J. Jensen, D. Fabbro, J. Trappe, U. Rauch, F. Bracher and S. Knapp, Chem. Biol., 2011, 18, 67–76 CrossRef CAS PubMed; (d) J. Liu, Z. Liu, P. Liao, L. Zhang, T. Tu and X. Bi, Angew. Chem., Int. Ed., 2015, 54, 10618–10622 CrossRef CAS PubMed; (e) H. Wang, H. Xu, B. Li and B. Wang, Org. Lett., 2018, 20, 5640–5643 CrossRef CAS PubMed.
- (a) H. J. Kabbe, Justus Liebigs Ann. Chem., 1967, 704, 144–149 CrossRef CAS; (b) Y. Han, K. Ebinger, L. E. Vandevier, J. W. Maloney, D. S. Nirschl and H. N. Weller, Tetrahedron Lett., 2010, 51, 629–632 CrossRef CAS; (c) Y. Loidreau, P. Marchand, C. Dubouilh-Benard, M.-R. Nourrisson, M. Duflos, O. Lozach, N. Loaëc, L. Meijer and T. Besson, Eur. J. Med. Chem., 2012, 58, 171–183 CrossRef CAS PubMed; (d) S. Schenone, M. Radi, F. Musumeci, C. Brullo and M. Botta, Chem. Rev., 2014, 114, 7189–7238 CrossRef CAS PubMed.
- (a) F. Bohlmann and D. Rahtz, Chem. Ber./Recl., 1957, 90, 2265–2272 CrossRef CAS; (b) A. R. Katritzky, A. Denisenko and M. Arend, J. Org. Chem., 1999, 64, 6076–6079 CrossRef CAS; (c) M. C. Bagley, Z. Lin and S. J. Pope, Chem. Commun., 2009, 5165–5167 RSC; (d) D. Sarkar, N. Rout, M. K. Ghosh, S. Giri, K. Neue and H. Reuter, J. Org. Chem., 2017, 82, 9012–9022 CrossRef CAS PubMed.
- A. Alberola, A. G. Ortega, M. L. Sidaba and C. Sañudo, Tetrahedron, 1999, 55, 6555–6566 CrossRef CAS.
- M. Marinozzi, G. Marcelli, A. Carotti and B. Natalini, RSC Adv., 2014, 4, 7019–7023 RSC.
- (a) J. P. Ferris, P. C. Joshi, E. H. Edelson and J. G. Lawless, J. Mol. Euol., 1978, 11, 293–311 CrossRef CAS PubMed; (b) M. J. Alves, B. L. Booth and M. F. J. R. P. Proenc, J. Chem. Soc., Perkin Trans. 1, 1990, 6, 1705–1712 RSC.
- (a) T. Naito, S. Nakagawa, J. Okumura, K. Takahashi, K. Masuko and Y. Narita, Bull. Chem. Soc. Jpn., 1968, 41, 965–971 CrossRef CAS; (b) R. E. Hackler, K. W. Burow, S. V. Kaster and D. I. Wickiser, J. Heterocycl. Chem., 1989, 26, 1575–1578 CrossRef CAS.
- H. V. Dobeneck, E. Brunner, H. Bunke, G. Metzner, R. Schmidt, E. Weil and J. Sonnenbichler, Liebigs Ann. Chem., 1981, 410–424 CrossRef.
- X. Zhang, Z. Zhang, S. Xiang, Z. Zhu, C. Chen and D. Huang, Inorg. Chem. Front., 2019, 6, 1135–1140 RSC and the reference therein.
- (a) R. C. Larock, Comprehensive Organic Transforma-tions: A Guide to Functional Group Preparations, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed; (b) M. Movassaghi and M. D. Hill, Nat. Protoc., 2007, 2, 2018–2023 CrossRef CAS PubMed; (c) R. Adepu, R. Sunke, C. L. T. Meda, D. Rambabu, G. R. Krishna, C. M. Reddy, G. S. Deora, K. V. L. Parsa and M. Pal, Chem. Commun., 2013, 49, 190–192 RSC; (d) R. Sunke, R. Adepu, R. Kapavarapu, S. Chintala, C. L. T. Meda, K. V. L. Parsa and M. Pal, Chem. Commun., 2013, 49, 3570–3572 RSC; (e) R. P. Frutos, X. Wei, N. D. Patel, T. G. Tampone, J. A. Mulder, C. A. Busacca and C. H. Senanayake, J. Org. Chem., 2013, 78, 5800–5803 CrossRef CAS PubMed; (f) D. Liu, Q. Nie and M. Cai, Tetrahedron, 2018, 74, 3020–3029 CrossRef CAS.
- (a) A. Novelli, A. P. G. V. de Varela and J. D. Bonafede, Tetrahedron, 1968, 24, 2481–2484 CrossRef CAS; (b) B. Stanovnik, J. Heterocycl. Chem., 1999, 36, 1581–1593 CrossRef CAS.
- S. A. Lang and E. Cohen, J. Med. Chem., 1975, 18, 441–443 CrossRef CAS PubMed.
- (a) O. S. Wolfbeis, Monatsh. Chem., 1981, 112, 875–877 CrossRef CAS; (b) H. M. Al-Matar, S. M. Riyadh and M. H. Elnagdi, Arkivoc, 2007, xiii, 53–62 Search PubMed.
- S. Kim and S. H. Hong, Adv. Synth. Catal., 2015, 357, 1004–1012 CrossRef CAS.
- M. K. Ghorai, R. Talukdar and D. P. Tiwari, Chem. Commun., 2013, 49, 8205–8207 RSC.
- (a) T. Satoh and D. Wakasugi, Tetrahedron Lett., 2003, 44, 7517–7520 CrossRef CAS; (b) D. Wakasugi and T. Satoh, Tetrahedron, 2005, 61, 1245–1256 CrossRef CAS.
- (a) H. Baron, F. G. P. Remfry and J. F. Thorpe, J. Chem. Soc., 1904, 85, 1726–1761 RSC; (b) K. Ziegler, H. Eberle and H. Ohlinger, Justus Liebigs Ann. Chem., 1933, 504, 94–130 CrossRef CAS; (c) F. F. Fleming and B. C. Shook, Tetrahedron, 2002, 58, 1–23 CrossRef CAS; (d) K. Yoshizawa, S. Toyota and F. Toda, Green Chem., 2002, 4, 68–70 RSC; (e) T. Paulis, K. Hemstapat, Y. Chen, Y. Zhang, S. Saleh, D. Alagille, R. M. Baldwin, G. D. Tamagnan and P. J. Conn, J. Med. Chem., 2006, 49, 3332–3344 CrossRef PubMed; (f) S. R. Jagtap, M. J. Bhanushali, N. S. Nandurkar and B. M. Bhanage, Synth. Commun., 2007, 37, 2253–2258 CrossRef CAS.
- (a) M. E. Webb, A. Marquet, R. R. Mendel, F. Rebeille and A. G. Smith, Nat. Prod. Rep., 2007, 24, 988–1008 RSC; (b) S. R. Walker, E. J. Carter, B. C. Huff and J. C. Morris, Chem. Rev., 2009, 109, 3080–3098 CrossRef CAS PubMed.
- P. Chen, D. Norris, J. Das, S. H. Spergel, J. Wityak, L. Leith, R. Zhao, B.-C. Chen, S. Pitt, S. Pang, D. R. Shen, R. Zhang, H. F. De Fex, A. M. Doweyko, K. W. Mclntyre, D. J. Shuster, K. Behnia, G. L. Schieven and J. C. Barrish, Bioorg. Med. Chem. Lett., 2004, 14, 6061–6066 CrossRef CAS PubMed.
- W. M. Zweers-Zeilmarker, G. J. Horbach and R. F. Witkamp, Xenobiotica, 1997, 27, 769–780 CrossRef PubMed.
- (a) D. L. W. Kwong, J. S. T. Sham, G. K. H. Au, D. T. T. Chua, P. W. K. Kwong, A. C. K. Cheng, P. M. Wu, M. W. M. Law, C. C. H. Kwok, C. C. Yau, K. Y. Wan, R. T. T. Chan and D. D. K. Choy, J. Clin. Oncol., 2004, 22, 2643–2653 CrossRef CAS PubMed; (b) H. R. Li, E. I. Shaagisultanova, K. Yamashita, Z. Piao, M. Perucho and S. R. Malkhosyan, Cancer Res., 2004, 64, 4760–4767 CrossRef CAS PubMed; (c) M. A. Walker, H. D. King, R. A. Dalterio, P. Trail, R. Firestone and G. M. Dubowchik, Bioorg. Med. Chem. Lett., 2004, 14, 4323–4327 CrossRef CAS.
- (a) Z. Rappoport, The Chemistry of the Cyano Group, Interscience Publishers, London, 1970 Search PubMed; (b) A. J. Fatiadi, In Preparation and Synthetic Applications of Cyano Compounds, Wiley, New York, 1983 Search PubMed; (c) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300–307 RSC.
- P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 RSC.
- (a) R. R. Williams and J. K. Cline, J. Am. Chem. Soc., 1936, 58, 1504–1505 CrossRef CAS; (b) I. R. Baxendale and S. V. Ley, J. Comb. Chem., 2005, 7, 483–489 CrossRef CAS PubMed; (c) S. N. Karad and R. S. Liu, Angew. Chem., Int. Ed., 2014, 53, 9072–9076 CrossRef CAS PubMed; (d) P. Chen, C.-X. Song, W.-S. Wang, X.-L. Yu and Y. Tang, RSC Adv., 2016, 6, 80055–80058 RSC; (e) R. A. J. Tinson, D. L. Hughes, L. Ward and G. R. Stephenson, ACS Omega, 2018, 3, 8937–8944 CrossRef CAS PubMed.
- Y. Zhu, Y. Li, S. Xiang, W. Fan, J. Jin and D. Huang, Org. Chem. Front., 2019, 6, 3071–3077 RSC.
- (a) T. L. Cairns, A. W. Larchar and B. C. McKusick, J. Am. Chem. Soc., 1952, 74, 5633–5636 CrossRef CAS; (b) K. Yanagiya, M. Yasumoto and M. Kurabayashi, Bull. Chem. Soc. Jpn., 1973, 46, 2804–2808 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of compounds; X-ray crystal structures of compounds 2j, 4a and 5e; synthesis and characterization of 2,3,3-triphenylacrylonitrile. CCDC 1956320–1956327, 1969352. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10866a |
| This journal is © The Royal Society of Chemistry 2020 |