
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel hafnium–graphite oxide catalyst for the Meerwein–Ponndorf–Verley reaction and the activation effect of the solvent†
Xiaomin Lia,
Zhengjiang Dua,
Yi Wua,
Yadong Zhena,
Rixin Shaoa,
Bingqi Lia,
Chengmeng Chen b,
Quansheng Liua and
Huacong Zhou*a
b,
Quansheng Liua and
Huacong Zhou*a
aCollege of Chemical Engineering, Inner Mongolia University of Technology, Inner Mongolia Key Laboratory of High-Value Functional Utilization of Low Rank Carbon Resources, Hohhot 010051, Inner Mongolia, China. E-mail: hczhou@imut.edu.cn
bCAS Key Laboratory of Carbon Materials, Institute of Coal Chemistry, Chinese Academy of Sciences, China
First published on 9th March 2020
Abstract
Construction and application of novel hydrogenation catalysts is important for the conversion of carbonyl or aldehyde compounds into alcohols in the field of biomass utilization. In this work, a novel, efficient, and easily prepared hafnium–graphite oxide (Hf–GO) catalyst was constructed via the coordination between Hf4+ and the carboxylic groups in GO. The catalyst was applied into the hydrogenation of biomass derived carbonyl compounds via the Meerwein–Ponndorf–Verley (MPV) reaction. The catalyst gave high efficiency under mild conditions. An interesting phenomenon was found whereby the activity of the catalyst increased gradually in the initial stage during reaction. The solvent, isopropanol, was proved to have an activation effect on the catalyst, and the activation effect varied with different alcohols and temperatures. Further characterizations showed that isopropanol played the activation effect via replacing the residual solvent (DMF) in micro- and mesopores during the preparation process, which was hard to be completely removed by common drying process.
Introduction
Catalytic conversion of biomass into value-added chemicals has attracted increasing attention due to the rapid depletion of fossil fuels and escalating consumption of energy.1,2 From the viewpoint of CO2 emission, biomass is well known as a carbon-neutral resource,3,4 which can be converted into various chemicals, such as alcohols or hydrocarbons,5,6 gluconic acid,7 5-hydroxymethylfurfural,8–10 lactic acid,11,12 dimethyl furan,13 levulinic acid (LA),14,15 and γ-valerolactone (GVL).16–18 The hydrogenation conversion of carbonyl derivatives from biomass into alcohols or their derivatives was an important step among the reaction chains of biomass utilization. For example, levulinic acid or its esters, key platforms in biomass conversion, can be converted into the famous derivative GVL via hydrogenation reaction, and GVL has broad applications such as fuel additives and as a raw material for the production of valuable hydrocarbons.19–25 Hydrogenation reactions and the corresponding catalysts were especially important for the catalytic conversion utilization of biomass. To date, various hydrogenation catalysts have been developed for the conversion of carbonyl platforms from biomass, such as palladium,26 ruthenium,27 platinum,28 iridium,29 nickel,30 cobalt,31 molybdenum,32 and copper.33 Though moderate or even high efficiency was achieved by using these catalysts, there are still some issues to be improved. One is the potential catalyst cost and metal reserves on earth for the precious metal catalysts during future large-scale applications, and thus it was still desirable to explore more potential candidates for hydrogenation reaction. On the other hand, these reported catalysts were often applied using gaseous H2 as hydrogen sources. Generally, a relatively high H2 pressure was often required to improve the catalytic performances of the catalysts, leading to potential safety issues in applications. Furthermore, many transition metal based catalysts often required relatively high temperatures to achieve satisfied performances, which increased the energy input and risk of accidents.Compared to hydrogenation under H2 atmosphere, catalytic transfer hydrogenation known as Meerwein–Ponndorf–Verley (MPV) reduction using secondary alcohols or formic acid as hydrogen source is often seen as milder hydrogenation process. Various catalysts were reported to be efficient to catalyze MPV reaction.26–35 Among the transition metal catalysts for MPV reactions, zirconium-based catalysts were comprehensively reported giving high efficiency.34,35 Very recently, hafnium-based catalysts were reported with higher activity and milder reaction conditions than the zirconium-based catalysts. Different organic ligands were used to prepare Hf catalysts, including porphyrins, benzoic acid, and phytate.36–41 Up to now, the kinds of ligands being reported are still very limited and some of them suffer from the high costs, inconvenience to obtain, and/or not satisfied thermal stability. Therefore, it was desirable to explore more potential materials to construct Hf based catalysts.
Besides constructing new and efficient catalysts, the effects of the solvents on catalyst activities were also important issues in catalytic conversion systems of biomass. For MPV reactions, the common solvent, isopropanol, played its role generally by acting as both solvent and hydrogen donor by donating hydrogen atoms. Among the reported catalysts for MPV reactions, more attentions were paid on the construction of novel catalysts.26–35 However, the effects of the solvents on the activity of the catalysts were not concerned enough. Therefore, disclosing the effects of the solvents when constructing novel MPV catalysts is relatively appreciated.
In this work, graphite oxide (GO) was used to construct a novel hafnium–graphite oxide (Hf–GO) hybrid catalyst. GO was chosen due to the abundant acidic carboxylic groups and phenolic hydroxyl groups connected with aromatic structures.42 Besides, GO is a readily purchasable and stable carbonaceous material with unique physicochemical properties and broad applications.43–48 This study was started by the preparation of Hf–GO catalyst (Scheme 1). The catalyst was prepared simply by coordination reaction of HfCl4 with commercial GO in DMF. The solvent, isopropanol, was found and proved to have an activation effect on the catalyst, and the possible reason was analyzed. As far as we know, this is the first report of constructing Hf-based catalyst using graphite oxide as the ligand for the catalytic transfer hydrogenation of ethyl levulinate and other carbonyl compounds. With the advantages of high efficiency, excellent stability, and facile preparation, the Hf–GO catalyst may have potential applications in the field of biomass conversion.
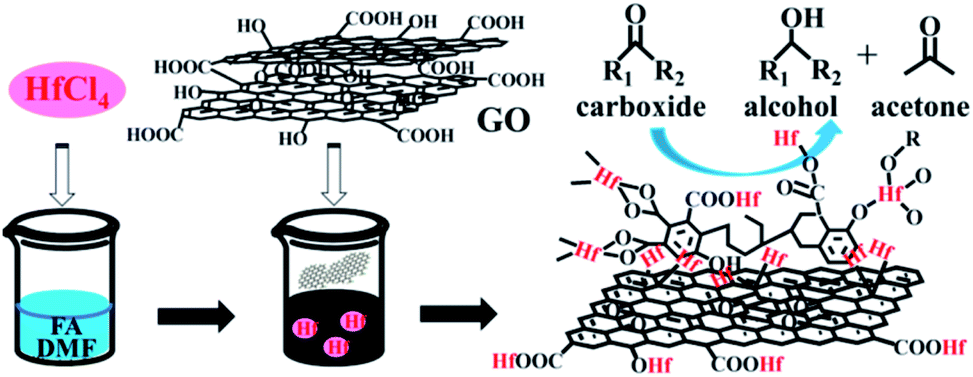 | ||
| Scheme 1 Schematic illustration of the Hf–GO catalyst preparation and its application in the conversion of carbonyl compounds into alcohols. | ||
Results and discussion
Studies of the preparation conditions of catalysts
In the preparation process of the catalyst, the effects of raw material ratio, aging temperature and aging duration for Hf–GO preparation were investigated (Fig. 1). The exact content of the oxygen-containing acidic groups were complex to determine precisely. Therefore, the effect of the amount of Hf precursor (HfCl4) on the catalytic activity was studied by varying the mass ratios of Hf precursor to GO. As shown in Fig. 1A, the conversion and product yield reached the highest values when the mass ratio of Hf precursor to GO was 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. With further increasing the Hf precursor dosage, the GVL yield had a slight decrease. The activity of the catalyst came to the optimal values when the aging temperature and aging duration came to 80 °C and 3 h, respectively (Fig. 1B and C), and further increasing the temperature or time the yield had no significant change. Many reports employed formic acid as a modulator to facilitate the formation of the crystalline structures during the preparation of catalysts.40,49,50 The effect of formic acid as well as other acidic modulators was studied for Hf–GO preparation, shown in Fig. 1D and Table S2.† Among the organic and inorganic acidic modulators, formic acid showed the promoting effect on the activity of the catalyst. The addition of suitable amount of formic acid could increase the activity of the catalyst, and both GVL yield (76.7%) and EL conversion (82.0%) reached the highest values when the molar ratio 21:1 of formic acid to Hf was used.
:1. With further increasing the Hf precursor dosage, the GVL yield had a slight decrease. The activity of the catalyst came to the optimal values when the aging temperature and aging duration came to 80 °C and 3 h, respectively (Fig. 1B and C), and further increasing the temperature or time the yield had no significant change. Many reports employed formic acid as a modulator to facilitate the formation of the crystalline structures during the preparation of catalysts.40,49,50 The effect of formic acid as well as other acidic modulators was studied for Hf–GO preparation, shown in Fig. 1D and Table S2.† Among the organic and inorganic acidic modulators, formic acid showed the promoting effect on the activity of the catalyst. The addition of suitable amount of formic acid could increase the activity of the catalyst, and both GVL yield (76.7%) and EL conversion (82.0%) reached the highest values when the molar ratio 21:1 of formic acid to Hf was used.
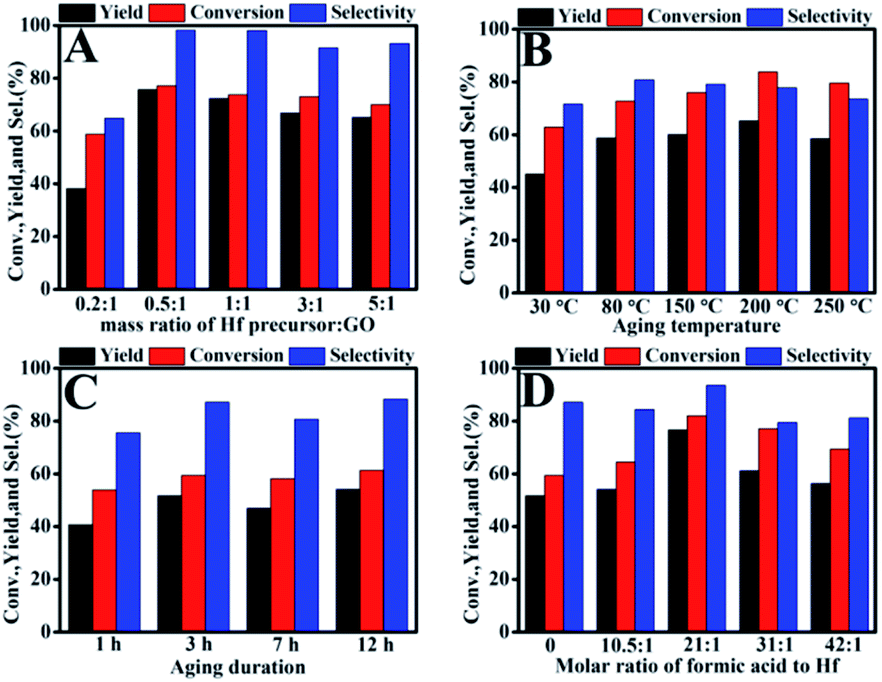 | ||
| Fig. 1 Effects of preparation conditions on the performance of Hf–GO catalyst for the conversion of EL to GVL. (A) Effects of the mass ratio of Hf precursor:GO, (B) aging temperature, (C) aging duration, and (D) formic acid dosage. | ||
As seen in Fig. 1A, the mass ratio of Hf precursor to GO was the key factor influencing the activity of the catalyst, and thus we characterized the Hf–GO catalysts prepared under different raw material ratios. The textural properties of the catalysts with various mass ratios were first characterized using N2 adsorption–desorption isotherms (Fig. S1†). The surface area and pore volume first dropped for mass ratios of Hf precursor to GO from 0.2 to 1, and then slightly increased with larger mass ratios. Compared with the surface area and pore volume, the change in average pore diameter was reversed (Table S1†). The N2 adsorption–desorption isotherms revealed the typical mesoporous character (Fig. S1†). The Hf content in Hf–GO-0.5:1 was 10.01 wt%, which was higher than that of Hf–GO-0.2:1 (6.64 wt%) as confirmed by inductively coupled plasma atomic emission spectrometry (ICP-AES Table S1†). The Hf contents in other catalysts almost maintained constant despite further increasing the mass ratios of Hf precursor to GO. Therefore, 0.5:1 was selected as the optimal ratios for the preparation of Hf–GO.
The effects of the modulators were analyzed in detail (Fig. 1D and Table S2†). As seen, the GVL yield and EL conversion reached 51.7% and 59.4% at 150 °C in 3 h without formic acid. The activity of the catalyst gradually increased with the formic acid dosage increased, and the GVL yield (76.7%) and EL conversion (82.0%) reached the highest values when the molar ratio 21:1 of formic acid to Hf was used. Further increasing the formic acid dosage, the activity gradually decreased. It was reported that the monocarboxylic acids were usually added in the synthesis of MOFs as modulators.40,49–51 Seemingly, the monocarboxylic acid is not necessarily incorporated in the structure but can strongly affect the connectivity of the inorganic node by coordinates to metal ions (Hf4+).52–54 The carboxylate ligands have lower pKa and possess stronger coordination ability, leading to the partial replacement of the modulators and the formation of structural defects.55–63 It was also reported that the addition of formic acid could increase the acidity.64 We also studied the effect of different modulators for Hf–GO preparation shown in Table S2.† We chose five different modulators with identical mole ratio to Hf. The results show that the catalyst using formic acid as modulator gave the highest catalytic activity.
Based on the above results, the preparation conditions were investigated, including the mass ratio of hafnium precursor (HfCl4) to GO, aging temperature, aging duration, modulators and their dosage. The catalyst showed higher activity for the conversion of ethyl levulinate into GVL under the preparation conditions of the mass ratio of HfCl4 to GO 0.5:1, aging temperature 80 °C, aging duration 3 h, formic acid as modulator with the dosage of the molar ratio of formic acid to Hf 21:1. The Hf–GO catalyst prepared under this condition was further characterized and studied.
Catalyst characterization
The structures of the catalyst obtained under the above optimized conditions were characterized (Fig. 2). SEM and TEM showed that Hf–GO maintained the typical dimensional sheet structures of GO (Fig. 2A and B). EDS mapping results showed that Hf element was introduced into GO successfully and dispersed on GO uniformly (Fig. 2C and D). The FTIR spectrum of GO and Hf–GO catalyst in Fig. 2E exhibited the asymmetric (GO, 1731 cm−1; Hf–GO, 1723 cm−1) and symmetric (GO, 1616 cm−1; Hf–GO, 1655 cm−1) stretching vibration of carboxylate groups. FTIR showed that the wavenumber difference of the asymmetric and symmetric vibrations of carboxylate anions was narrowed from 115 cm−1 for GO to 68 cm−1 for Hf–GO, indicating that Hf4+ was coordinated with carboxylate groups.65–67 It has reported the bands at around 520 and 760 cm−1 were characteristic of Hf–O bonds.68 The new absorption band at 690 cm−1 could be assigned to the Hf–O bond vibration (Fig. 2E).68 As shown in XRD patterns (Fig. 2F), no obvious characteristic peaks of HfO2 or Hf(OH)4 appeared, indicating that Hf dispersed uniformly and did not form crystal aggregates. The 001 peak of GO shifted from 12° to 10°, corresponding to the layer distance enlargement from 0.74 nm to 0.80 nm, indicating that some of the Hf4+ were inserted into the layers of GO. The Raman spectra revealed that the G band for Hf–GO had a red shift to 1589 cm−1 and D band had a blue shift to 1350 cm−1 as compared to GO (1596 cm−1, 1345 cm−1) (Fig. S2†). Furthermore, the intensity ratio of D and G peaks (ID/IG) increased slightly from 0.94 to 1.00 (Table S4†) after assembly of GO with Hf4+. XPS full spectra, fitted spectra of Hf 4d and Hf 4f again proved the successful introduction of Hf4+ in Hf–GO (Fig. 3). The XPS curve-fitted spectrum revealed the Hf 4d5/2 and Hf 4d3/2, Hf 4f7/2 and Hf 4f5/2 could be detected corresponding to the hafnium–oxygen bonds at 213.8 eV and 224.4 eV, 17.4 eV and 19.1 eV, respectively (Fig. 3C and D). And these spectra were calibrated by C 1s peak at 284.8 eV. In Fig. 3B and C 1s peaks were deconvoluted into four bands based on the literatures.69–72 The contents of C–C or C–H, C–OH or C–O–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and OC–O from Fig. 3B were summarized in Table S5.† The main peak at 284.8 eV originates in sp2 (CC), sp3 (C–C) carbon and C–H. The deconvoluted peak centered at the binding energies of 286.7, 287.2, and 288.8 eV were considered as the C–O (hydroxyl and ethers groups), CO (carbonyl groups), and OC–OH (carboxyl groups) oxygen-containing carbonaceous bands, respectively. The intensity of the peak assigned to oxygen-containing carbonaceous bands of the C–O, CO and OC–O for GO decreased after the introduction of Hf4+. These results indicated the successful covalent coordination of Hf4+ with COOH in GO. ICP-AES showed that the content of Hf in the catalyst was around 9.04 wt%. The results of elemental analysis of different catalysts were given in Table S6.† N2 adsorption–desorption isotherms showed that the porous structure and the surface area, pore volume, and average pore size of the catalyst were 61.8 m2 g−1, 0.08 cm3 g−1, and 6.1 nm, respectively (Fig. S3C and Table S7†). TG analysis showed that the thermal stability after 300 °C of the Hf–GO catalyst was better than that of GO due to the coordination of Hf4+ with the carboxylic groups, and the catalyst was stable enough under reaction temperature range (<200 °C) (Fig. S3D†). The detailed discussions of catalyst characterization were given in ESI.†
O, and OC–O from Fig. 3B were summarized in Table S5.† The main peak at 284.8 eV originates in sp2 (CC), sp3 (C–C) carbon and C–H. The deconvoluted peak centered at the binding energies of 286.7, 287.2, and 288.8 eV were considered as the C–O (hydroxyl and ethers groups), CO (carbonyl groups), and OC–OH (carboxyl groups) oxygen-containing carbonaceous bands, respectively. The intensity of the peak assigned to oxygen-containing carbonaceous bands of the C–O, CO and OC–O for GO decreased after the introduction of Hf4+. These results indicated the successful covalent coordination of Hf4+ with COOH in GO. ICP-AES showed that the content of Hf in the catalyst was around 9.04 wt%. The results of elemental analysis of different catalysts were given in Table S6.† N2 adsorption–desorption isotherms showed that the porous structure and the surface area, pore volume, and average pore size of the catalyst were 61.8 m2 g−1, 0.08 cm3 g−1, and 6.1 nm, respectively (Fig. S3C and Table S7†). TG analysis showed that the thermal stability after 300 °C of the Hf–GO catalyst was better than that of GO due to the coordination of Hf4+ with the carboxylic groups, and the catalyst was stable enough under reaction temperature range (<200 °C) (Fig. S3D†). The detailed discussions of catalyst characterization were given in ESI.†
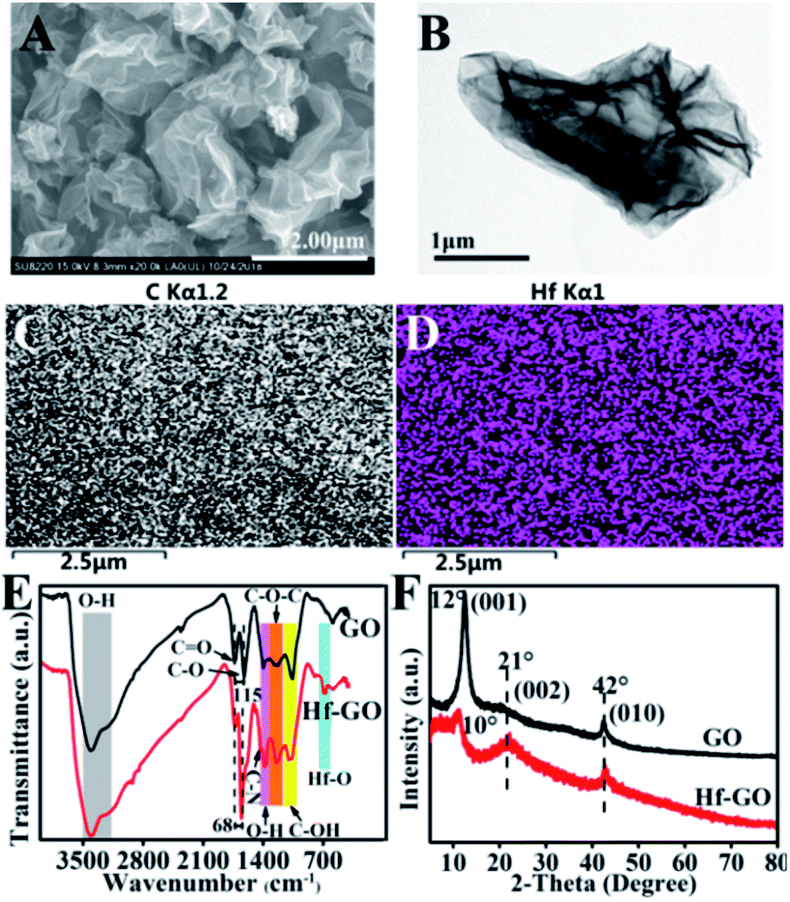 | ||
| Fig. 2 (A) SEM image, (B) TEM image, (C and D) C and Hf EDS mappings of Hf–GO, (E) FTIR spectra, (F) XRD patterns of Hf–GO and GO. | ||
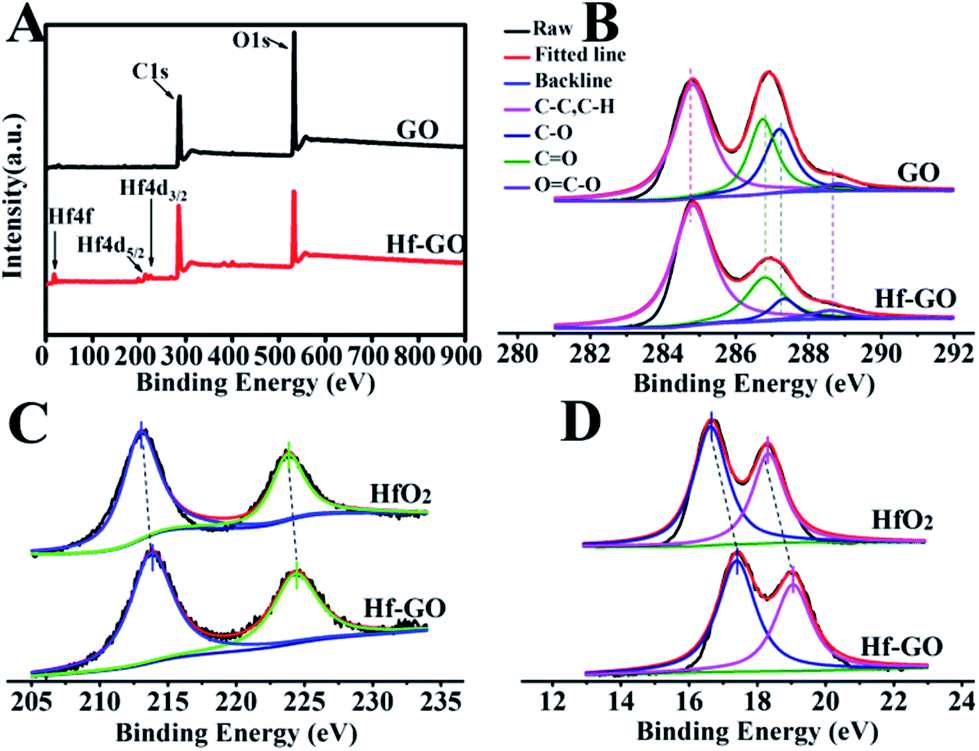 | ||
| Fig. 3 (A) XPS full spectra, (B) the fitted spectra of C 1s peak of GO and Hf–GO, (C) the fitted spectra of Hf 4d, and (D) Hf 4f of HfO2 and Hf–GO. | ||
Effect of reaction conditions on transfer hydrogenation
Next, the effects of the reaction conditions on transfer hydrogenation were investigated with isopropanol as the hydrogen source, including the catalyst dosage (Fig. S4A†), reaction temperature (Fig. S4B†) and reaction time (Fig. S4C†). The discussions were given in ESI.† The catalyst gave satisfied reaction results and rate under the conditions of catalyst dosage >0.05 g and reaction temperature >150 °C for the studied reaction system. The EL conversion, GVL yield, and GVL selectivity could reach to 96.5%, 88.8%, and 92.0%, respectively, under the conditions of catalyst dosage 0.1 g, reaction temperature 150 °C, and reaction time 9 h. The GVL yield did not increase when removing the solid catalyst during reaction, indicating Hf–GO was a heterogeneous catalyst (Fig. S4D†).Comparison with other catalysts
In order to see the performances of the state-of-the-art catalyst for the MPV reaction, the performance of Hf–GO was compared various other catalysts, including Hf-, Zr-, Pd-, Ru-, Pt-, Ir-, Co-, Ni-, Mo-, and Cu-based catalysts (Table 1). The catalysts with the same reaction and similar reaction conditions were chosen and compared. The performances of around 37 kinds of catalysts were compared. When using H2 as hydrogen source, Mo2C/CNT (entry 37), Pd/AC (entry 21), Ni/γ-Al2O3 (entry 36) gave higher activities than other catalysts, with TOF values of 10.3 h−1 (150 °C), 7.94 h−1 (100 °C), and 7.67 h−1 (200 °C), respectively. When using isopropanol as hydrogen source, GO–Hf prepared in this work gave the highest activity with TOF values of 3.51 h−1 (150 °C). Among all the 37 catalysts, Hf–GO ranks basically the forth in viewpoint of TOF value. The above comparison analysis should that Hf–GO was efficient for the MPV reaction of ethyl levulinate.| Entry | Catalyst | Reaction conditions | EL conv. (%) | LA conv. (%) | GVL yield (%) | GVL sel. (%) | TOFb (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Preparation condition: 1 mL formic acid was dissolved in DMF (400 mL), 1.59 mmol metal chloride was added into DMF solution with continuously stirred and completely dissolved. After that, 1.0 g of GO was directly added to the HfCl4 solution and the obtained mixture was stirred for 3 h at 30 °C, then aged at 80 °C under static conditions for 3 h. The suspended solution was separated by filtration to give black precipitate, and successively washed with DMF, ethanol for 4 times, dried under vacuum conditions at 80 °C for 24 h, and crowded into powders. Reaction conditions: 1 mmol EL, 0.1 g catalyst (5 mol% Hf), 5 mL 2-PrOH.b TOF (turnover frequency) = (mole of GVL)/(mole of active metal × reaction time).c Data obtained from ref. 64, FDCA: furan dicarboxylic acid, 16 mol% Hf.d Data obtained from ref. 65, HA: humic acid.e Data obtained from ref. 73, the mole of Hf was 52 mol%.f Data obtained from ref. 73, 95 mol% Hf.g Data obtained from ref. 73, 54 mol% Hf (ICP determined).h Data obtained from ref. 49, MOF-808: 1,3,5-benzenetricarboxylic acid, 10 mol% Hf.i Data obtained from ref. 74, DUT67: 2,5-thiophenedicarboxylic acid.j Data obtained from ref. 74, UiO-66: terephthalic acid.k Data obtained from ref. 75, PPOA: phenylphosphonic acid.l AC: active carbon.m PhyA: phytic acid.n RSL: raw Shengli lignite.o HAs: humic acids.p Humic acids (HA) extracted from lignite and the solid residues (SR).q HBA: 4-hydroxybenzoic acid dipotassium salt.r CA: cyanuric acid.s NG: not given. | ||||||||
| 1 | Blank | IPA, 150 °C, 5 h | <1 | 0 | 0 | — | This work | |
| 2 | GO | IPA, 150 °C, 3 h | 36.8 | 0 | 0 | — | This work | |
| 3 | Hf–GOa | IPA, 150 °C, 5 h | 95.5 | 87.7 | 91.8 | 3.51 | This work | |
| 4 | Hf–GOa | IPA, 130 °C, 15 h | 88.4 | 82.6 | 93.5 | 1.10 | This work | |
| 5 | Zr–GOa | IPA, 150 °C, 5 h | 54.8 | 54.7 | 99.8 | 2.18 | This work | |
| 6 | Al–GOa | IPA, 150 °C, 5 h | 12.2 | 7.5 | 62.0 | 0.3 | This work | |
| 7 | Cr–GOa | IPA, 150 °C, 5 h | 3.5 | 0.9 | 25.8 | 0.04 | This work | |
| 8 | Fe–GOa | IPA, 150 °C, 5 h | 7.1 | 1.3 | 17.7 | 0.05 | This work | |
| 9 | Cu–GOa | IPA, 150 °C, 5 h | 5.0 | 1.1 | 23.1 | 0.04 | This work | |
| 10 | Sn–GOa | IPA, 150 °C, 5 h | 22.3 |
2.4 | 10.6 | 0.10 | This work | |
| 11 | FDCA–Hfc | IPA, 160 °C, 4 h | >99 | 98 | 98 | 1.53 | 64 | |
| 12 | HfO2c | IPA, 160 °C, 4 h | 28 | 22 | 79 | 0.34 | 64 | |
| 13 | Zr–HAd | IPA, 150 °C, 11 h | 99.5 | 84.2 | 84.6 | 0.1 | 65 | |
| 14 | Hf–ATMPe | IPA, 150 °C, 4 h | 95 | 86 | 91 | 0.41 | 73 | |
| 15 | HfO2f | IPA, 150 °C, 4 h | 49 | 35 | 71 | 0.09 | 73 | |
| 16 | Hf–EDPAg | IPA, 150 °C, 4 h | 87 | 74 | 85 | 0.34 | 73 | |
| 17 | Hf–MOF-808h | IPA, 120 °C, 8 h | NGs | 94 | NG | 1.18 | 49 | |
| 18 | DUT67(Hf)i | IPA, 160 °C, 4 h | 98.9 | 90.5 | 91.5 | — | 74 | |
| 19 | UiO-66(Hf)j | IPA, 160 °C, 4 h | 73.9 | 58.5 | 79.1 | — | 74 | |
| 20 | PPOA–Hfk | IPA, 160 °C, 6 h | 100 | 85 | 85 | — | 75 | |
| 21 | Pd/ACl | H2 0.5 MPa, 100 °C, 5 h, H2O | 38 | 28 | 74 | 7.94 | 26 | |
| 22 | Co | H2 3.3 MPa, 130 °C, 3 h | 99 | 94 | 95 | 0.19 | 31 | |
| 23 | Zr–PhyAm | IPA, 150 °C, 6 h | 100 | 96.7 | 96.7 | 0.25 | 35 | |
| 24 | Zr–RSLn | IPA, 160 °C, 12 h | 92.4 | 81.1 | 87.8 | 0.48 | 76 | |
| 25 | Zr–HAso | IPA, 150 °C, 15 h | >99 | 85.0 | >85.9 | 0.09 | 34 | |
| 26 | Zr–HAfp | IPA, 150 °C, 9 h | 92.7 | 90.1 | 97.2 | 0.09 | 77 | |
| 27 | Zr–HAtp | IPA, 150 °C, 7 h | 92.8 | 92.6 | 99.8 | 0.09 | 77 | |
| 28 | Zr–SRfp | IPA, 150 °C, 7 h | 92.4 | 92.0 | 99.6 | 0.43 | 77 | |
| 29 | Zr–SRtp | IPA, 150 °C, 9 h | 95.4 | 92.0 | 96.3 | 0.31 | 77 | |
| 30 | Zr–HBAq | IPA, 150 °C, 4 h | 100 | 94.4 | 94.4 | — | 78 | |
| 31 | Zr–CAr | IPA, 150 °C, 4 h | 100 | 96.9 | 96.9 | — | 79 | |
| 32 | Ru/OMC-P | H2 7 bar, 70 °C, 6 h, H2O | 98 | 92 | 94 | 1.61 | 27 | |
| 33 | Ru/carbon | H2 1 bar, 265 °C, 50 h, H2O | 100 | 98.6 | 98.6 | 0.04 | 28 | |
| 34 | Pd/carbon | H2 1 bar, 265 °C, 50 h, H2O | 100 | 90 | 90 | 0.04 | 28 | |
| 35 | Pt/carbon | H2 1 bar, 265 °C, 50 h, H2O | 100 | 30 | 30 | 0.02 | 28 | |
| 36 | Ni/γ-Al2O3 | H2 50 bar, 200 °C, 4 h, H2O | 92 | 92 | 100 | 7.67 | 30 | |
| 37 | Mo2C/CNT | H2 30 bar, 150 °C, 1 h, H2O | 83 | 75 | 90.3 | 10.3 | 32 | |
| 38 | Cu-catalyst | H2 70 bar, 200 °C, 10 h, H2O | >99 | 91 | >91 | — | 80 | |
| 39 | Ni/MgAlO2.5 | H2 3 MPa, 160 °C, 1 h, dioxane | 100 | 99.7 | 99.7 | 1.46 | 81 | |
Substrate scope expansion
Encouraged by the excellent performance of Hf–GO in the conversion of EL to GVL, we investigated the possibility of the MPV reactions of other carbonyl compounds with different structures (Table 2). The results showed that aliphatic and aromatic aldehydes could be effectively hydrogenated to corresponding alcohols at 70–100 °C within 2–4 h (entries 2–8). While ketones, especially with long aliphatic chains, needed a higher reaction temperature or longer reaction time of the harsh conditions to achieve satisfied conversions and yields as compared to aldehydes (entries 10 and 11). Hf–GO displayed high efficiency for cyclohexanone (conversion, yield, selectivity all >99%) (entry 9), under not yet optimized conditions. These results substantiated the universality of Hf–GO catalyst in the conversion of different carbonyl compounds with various structures.| Entry | Substrate | Product | T (°C) | T (h) | Conv. (%) | Yield (%) | Sel. (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: substrate 1 mmol, isopropanol 5 mL, and Hf–GO 0.1 g (5 mol% Hf). | |||||||
| 1 | 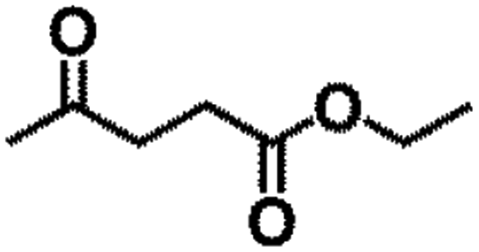 |
 |
150 | 9 | 96.5 | 88.8 | 92.0 |
| 2 |  |
 |
100 | 3 | 95.9 | 87.8 | 91.5 |
| 3 | 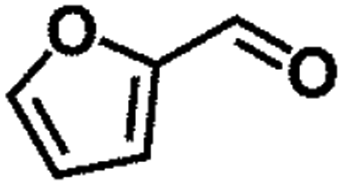 |
 |
70 | 4 | 89.7 | 85.5 | 95.4 |
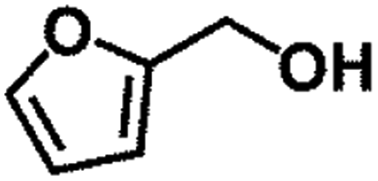
| 4 |  |
 |
90 | 3 | 97.9 | 95.9 | 98.0 |
| 5 |  |
 |
100 | 3 | 89.7 | 81.0 | 90.3 |
| 6 | 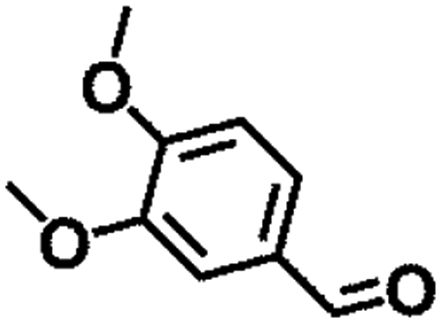 |
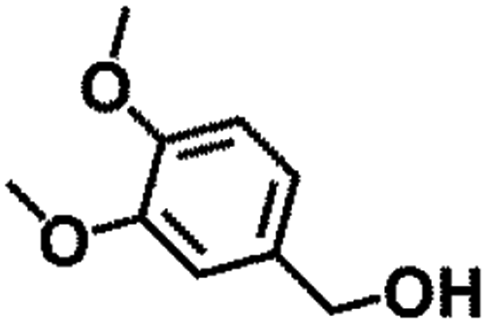 |
80 | 3 | 93.0 | 91.7 | 98.6 |
| 7 | 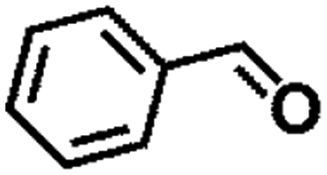 |
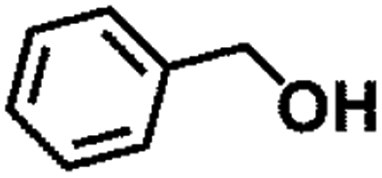 |
90 | 2 | 96.4 | 87.2 | 90.5 |
| 8 |  |
 |
80 | 4 | 93.7 | 88.7 | 94.6 |
| 9 |  |
 |
100 | 5 | >99 | >99 | >99 |
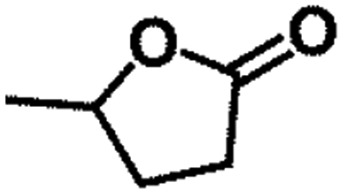
| 10 | 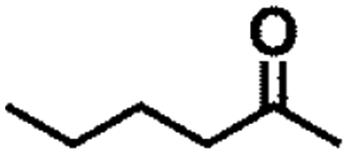 |
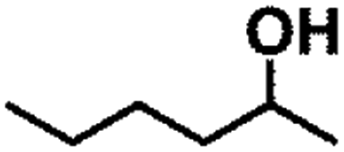 |
150 | 8 | >99 | >99 | >99 |
| 11 | 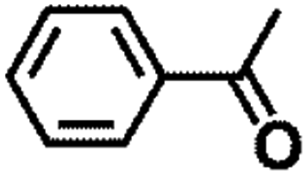 |
 |
120 | 7 | 99.3 | 98.8 | 99.5 |
Recycle of the Hf–GO catalyst
During the recycling process, an interesting and unexpected phenomenon was observed that the activity of Hf–GO increased with the initial 3 uses and then stabilized at a similar level without significant decreasing after 11 cycles compared to the first use (Fig. 4). The slight decreasing trend from the 6th use to the 11th use was due to the loss of the catalyst during the centrifugation recycling process. After the catalyst dosage was replenished to the initial level, the performance could be recovered (12th use). The activity increasing of Hf–GO during the initial uses draw us much attention because few reports mentioned this phenomenon for the analogue catalysts. We speculated that Hf–GO was activated by certain components in the reaction system. Mainly four components in the reaction system existed, including EL (the reactant), GVL (the product), isopropanol (the solvent), and acetone (the by-product). In order to disclose the “activator”, the catalyst was pretreated by the above components, respectively, under 150 °C to simulate the reaction conditions. As shown in Fig. 5, only the pretreatment by isopropanol (Run 1 in Fig. 5A) could increase the activity of Hf–GO compared with no pretreatment (Run 0 in Fig. 5A). The pretreatment by EL, GVL, and acetone could not activate Hf–GO (Run 0 vs. Run 1) and the activity only increased after the first catalytic reaction (Run 2 and 3 vs. Run 1). Thus, it was isopropanol that played the role of activation effects. Subsequently, temperature was proved to be a key factor for the activation effect (Fig. S5†). The activation effect became more and more obvious with the increasing of temperature in the range of room temperature to 150 °C (Fig. S6†). Further increasing the pretreatment temperature led to the decreasing of the activating trend, which might be due to the gradual decomposition of the carboxylate groups under higher temperatures. Temperatures in the range of 120–150 °C were optimal activation temperature, which just fell into the optimal reaction temperature of EL to GVL.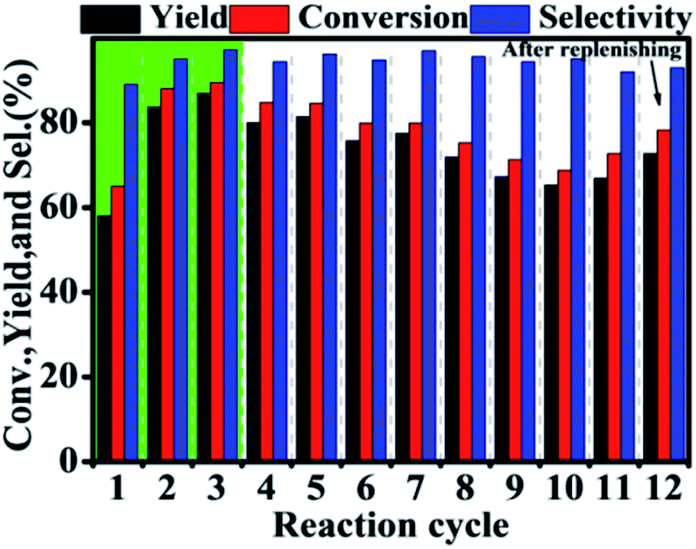 | ||
| Fig. 4 Recycle of the Hf–GO catalyst. Reaction conditions: 1 mmol EL, 0.1 g catalyst (5 mol% Hf), 5 mL 2-PrOH, 150 °C, 2 h. | ||
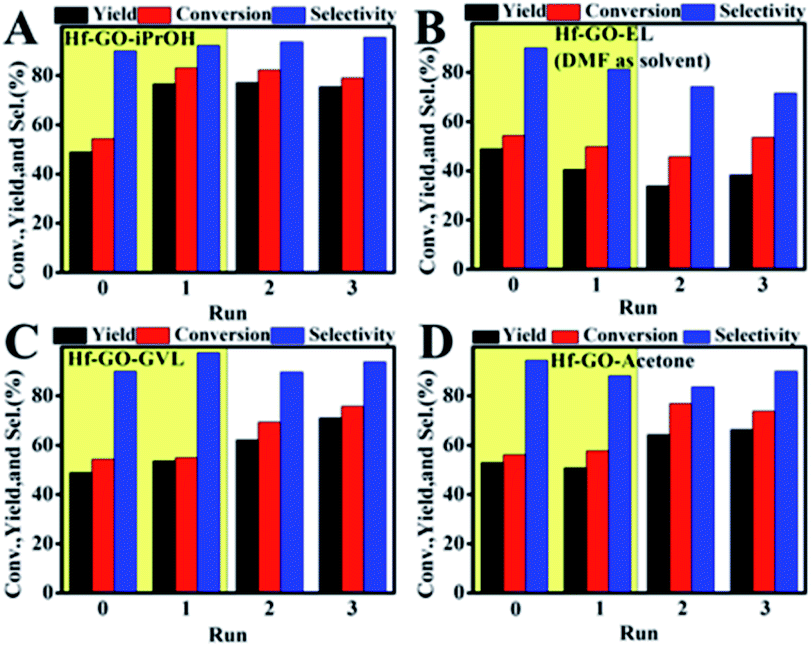 | ||
| Fig. 5 Performance of the Hf–GO–X catalyst (Hf–GO pretreated by solvent X). (A) X = iPrOH; (B) X = ethyl levulinate (EL); (C) X = GVL; (D) X = acetone. Run 0: the performance of the fresh Hf–GO catalyst without pretreatment. Run 1, 2 and 3: the performance of the Hf–GO–X catalyst for subsequent three times use. | ||
Besides isopropanol, other alcohols were also studied to check if they had activation effects on Hf–GO (Fig. S7 and S8†). It could be seen that all the alcohols studied had activation effects on Hf–GO but the extents varied. The activation effects of the secondary alcohols were generally higher than that of the primary alcohols, with isopropanol and isobutanol giving the most significant activation effect. Other solvents such as decane, acetone, and 2-hexanone were also used to pretreat Hf–GO, and it was found that these solvents could not activate the catalyst (Fig. S9†). The reasons for this would be discussed below.
Characterization of the recycled and captivated Hf–GO
The intrinsic mechanism for the activation effect of alcohols was analyzed by characterizing the fresh and pretreated/used Hf–GO catalysts using various techniques, including XPS, elemental analysis, XRD, Raman, TG analysis, and N2 adsorption–desorption. No significant morphology changes and Hf chemical state changes were found for the pretreated and recycled Hf–GO (Fig. S10–S13†). The N contents decreased after solvent pretreatment except for pretreatment by DMF (Table S6†). TG analysis also showed that the weight losses of Hf–GO pretreated by solvents were much lower than that of the fresh Hf–GO (Fig. S14†). FTIR showed that The C–N stretching vibration at 1424 cm−1 of DMF was obvious for fresh Hf–GO,82 but it was found to be weakened after solvent treatment (Fig. S15†). Base on the above analysis, it could be deduced that solvent pretreatment removed residual DMF in Hf–GO. The residual DMF in the pore structures of Hf–GO was hard to remove even washed by ethanol and then dried under vacuum and 150 °C (boiling point of DMF under standard atmosphere) (Table S6,† entry 9). As shown in Table S6,† the N content in GO was only 0.04 wt%, but it increased to 1.55 wt% for fresh Hf–GO. The N content decreased to different extents after pretreatment by different solvents (except for Hf–GO pretreated by DMF), indicating the solvent pretreatment could indeed remove DMF residual in the catalyst. The Hf content had no significant changes, and the changes of C, H, and O contents could be attributed to the removal of DMF. It should be noted that the pretreatment by decane and acetone could also remove DMF (Table S6†), but these solvents could not activate the catalyst only if Hf–GO was allowed to contact with isopropanol (as mentioned above, Fig. S9†). Therefore, the structures of the pretreated Hf–GO were further characterized by BET and pore structure analysis (Fig. 6 and Table S7†). It could be seen that the surface area, the volumes of total pores and mesopores of Hf–GO–decane and Hf–GO–acetone were all much lower than those of Hf–GO–isopropanol, which may result in no obvious activation effect for these solvents. Isopropanol pretreatment mainly affected the structures of micro- and mesopores, indicating DMF in these pore structures was removed (Fig. 6A and B). Another possible reason was that although these solvents could remove DMF in mesopores of Hf–GO, they could not interact with the active Hf–O sites and provide hydrogen to promote the occurrence of the reaction. The surface contents of Hf in Hf–GO pretreated by different solvents were analyzed by XPS, and no obvious difference were observed (Table S8†). The influence of mass transfer on the performance of the catalysts were detected by prolonging the reaction time, and both conversion and yield for Hf–GO–iPrOH were higher than those for Hf–GO-fresh throughout the whole time range, further indicating isopropanol pretreatment had an activation effects on Hf–GO (Fig. S16†). In conclusion, it could be postulated that isopropanol played its activation effects on Hf–GO by first replacing DMF in micro- and mesopores and then interacting with active Hf–O sites to promote the proceeding of the reaction.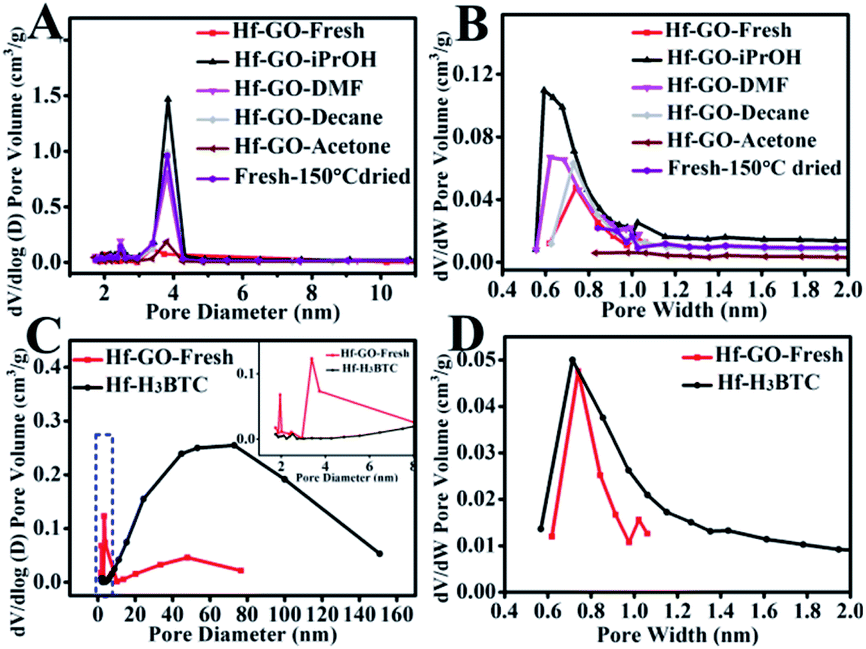 | ||
| Fig. 6 Mesopore distributions of different catalysts (A and C) and micropore distributions of different catalysts (B and D). The mesopore size distribution was calculated based on the DFT method. The micropore size distribution was calculated based on the H–K method. | ||
Besides Hf–GO catalyst, the above discussed activation effects of isopropanol also existed for Zr–GO catalyst, but was not obvious for Hf–H3BTC constructed by 1.3.5-benzene tricarboxylic acid (Fig. S17†). This was related to the different pore structures of the catalysts (Fig. 6 and S18†). Hf–GO had obvious micro- and mesoporous structures. For Hf–H3BTC, there were abundant pores larger than 10 nm and the distribution of pore diameters were broad, leading to the easy removal of DMF during preparation by washing and much less DMF residue in the catalyst compared to Hf–GO (Fig. 6C and D and Table S6†). Therefore, isopropanol could easily contact with the active sites in the first use, and no obvious increasing of activity was observed during the subsequent use. Comparatively, Hf–GO had abundant micro- and mesopores (Fig. 6A and B), leading to the residue of DMF, and thus isopropanol could activate the catalyst via removing DMF as discussed above.
Experimental
Materials
Ethyl levulinate (EL, 98%), γ-valerolactone (GVL, 98%), zircomiun tetrachloride (ZrCl4, 98%), 1,3,5-benzenetricarboxylic acid (H3BTC, 99%), furfural (FF, 99%) and furfuryl alcohol (FA, 98%) were provided by J&K Scientific Ltd. Hafnium tetrachloride (HfCl4, 98+%) and chromium(III) chloride anhydrous (CrCl3, 98%) were obtained from Alfa. Aluminum chloride (AlCl3, 99%), iron chloride (FeCl3, ≥99.9%), copper chloride (CuCl2, 98%) and tin chloride (SnCl4, AR) were provided by Aladdin Industrial Corporation. Graphite oxide (GO) were provided by Institute of Coal Chemistry, Chinese Academy of Sciences. Isopropanol (iPrOH, 99.9%) was provided by Innochem. N,N-Dimethylformamide (DMF, >99.9%), n-butanol (nBuOH, ≥99.7%), n-propanol (nPrOH, ≥99.9%), isobutanol (sBuOH, ≥99.5%), methanol (MeOH, ≥99.9%), ethanol (EtOH, 100%), 1-hexanol (nHeOH, >99.5%), n-octanol (nOcOH, ≥99%) and 2-hexanol (sHeOH, 99%) were provided by Aladdin Industrial Corporation. Decane (AR) and other chemicals were obtained from Beijing Institute of Chemical Reagent.Preparation of different M–GO catalysts
The M–GO catalysts (M = Hf, Zr, Al, Cr, Fe, Cu, Sn) were obtained by the simple synthesis of GO with an equivalent mole of metal chloride (i.e., HfCl4, ZrCl4, AlCl3, CrCl3, FeCl3, CuCl2 or SnCl4) and 0–2.0 mL of formic acid in DMF under stirring conditions. In a typical preparation process for Hf–GO, 1.359 g (1 mL) formic acid (FA) was dissolved in DMF (400 mL), 0.5 g HfCl4 was added into DMF solution with continuously stirred and completely dissolved. After that, 1.0 g of GO was directly added to the HfCl4 solution and the obtained mixture was stirred for 3 h at 30 °C, then aged at 80 °C under static conditions for 3 h. The suspended solution was separated by filtration to give black precipitate, and successively washed with DMF, ethanol for 4 times, dried under vacuum conditions at 80 °C for 24 h, and crowded into powders for use. For comparison, the Hf–H3BTC organic–inorganic hybrid was prepared by solvothermal treatment of HfCl4 and equimolecular 1,3,5-benzenetricarboxylic acid in DMF at 120 °C under static conditions for 48 h, referencing to a previously reported synthetic methods.51,83 All the solid samples were after dried at 80 °C for 4 h for the catalyst characterization and analysis of catalytic performance.The preparation parameters were studied, including effects of the mass ratio of Hf precursor: GO, aging temperature, aging duration, and formic acid dosage. The details were described as follows. (1) Effects of the mass ratio of Hf precursor to GO: 1 mL formic acid was dissolved in DMF (400 mL, five copies), HfCl4 of 0.2 g, 0.5 g, 1.0 g, 3.0 g, 5.0 g, respectively, was added into DMF solution with continuously stirred and completely dissolved. After that, 1.0 g of GO was directly added to the HfCl4 solution and the obtained mixture was stirred for 3 h at 30 °C, then aged at 80 °C under static conditions for 3 h. The suspended solution was separated by filtration to give black precipitate, and successively washed with DMF, ethanol for 4 times, dried under vacuum conditions at 80 °C for 24 h, and crowded into powders. (2) Aging temperature: 1 mL formic acid was dissolved in DMF (400 mL), 0.5 g HfCl4 was added into DMF solution with continuously stirred and completely dissolved. After that, 1.0 g of GO was directly added to the HfCl4 solution and the obtained mixture was stirred for 3 h at 30 °C, then aged at the required temperature under static conditions for 3 h. The suspended solution was separated by filtration to give black precipitate, and successively washed with DMF, ethanol for 4 times, dried under vacuum conditions at 80 °C for 24 h, and crowded into powders. (3) Aging duration: 1 mL formic acid was dissolved in DMF (400 mL), 0.5 g HfCl4 was added into DMF solution with continuously stirred and completely dissolved. After that, 1.0 g of GO was directly added to the HfCl4 solution and the obtained mixture was stirred for 3 h at 30 °C, then aged at 80 °C under static conditions for the required duration. The suspended solution was separated by filtration to give black precipitate, and successively washed with DMF, ethanol for 4 times, dried under vacuum conditions at 80 °C for 24 h, and crowded into powders. (4) Formic acid dosage: formic acid of 0 mL, 0.5 mL, 1.0 mL, 1.5 mL, 2.0 mL, respectively, was dissolved in DMF (400 mL, five copies), 0.5 g HfCl4 was added into DMF solution with continuously stirred and completely dissolved. After that, 1.0 g of GO was directly added to the HfCl4 solution and the obtained mixture was stirred for 3 h at 30 °C, then aged at 80 °C under static conditions for 3 h. The suspended solution was separated by filtration to give black precipitate, and successively washed with DMF, ethanol for 4 times, dried under vacuum conditions at 80 °C for 24 h, and crowded into powders. Reaction conditions: 1 mmol EL, 0.1 g catalyst, 5 mL 2-PrOH, reaction temperature 150 °C, and reaction time 3 h.
The as-obtained Hf–GO was treated with different organic solvents at 150 °C for 2 h to give Hf–GO–X (X = DMF, EL, GVL, iPrOH, other various alcohols, acetone, 2-hexanone, decane). 0.1 g of the fresh catalyst was stirred in the different organic solvents at 150 °C for 2 h, recovered by centrifugation, and washed three times with fresh isopropanol to give Hf–GO–X. For the effects of pretreating temperatures in isopropanol, the fresh Hf–GO was treated with isopropanol at various temperatures for 2 h to give Hf–GO-Y (Y = 25, 50, 70, 100, 120, 150, 170, 200 °C).
Catalyst characterization
Scanning electron microscopy (SEM) measurements were performed using a Hitachi SU8220 scanning electron microscope operated at 20 kV with an energy dispersive spectrometer (EDS) apparatus. Transmission electron microscopy (TEM) images were obtained using a TEM JEM-2010 with an accelerating voltage of 120 kV. The samples were dispersed in absolute ethanol, and the suspension was sonicated for 15 min to make the sample disperse well. Then the sample solution was dropped onto the copper network containing the carbon support film, and was allowed to volatilize at room temperature for 2 h to be tested. Fourier transform-infrared spectra (FTIR) were obtained using a PerkinElmer spectrometer. X-ray diffraction (XRD) was carried out via an Produce-Rigaku SmartLab 9 KW X-ray diffractometer with a Cu target configuration and a highly sensitive D/teX Ultra 250 inspection system at 20° per minute ranging from 5° to 90°. The tube voltage was 45 kW, and the current was 200 mA. Raman spectrum was collected on a Renishaw® inVia microscope in the range of 120 cm−1 to 4000 cm−1 with 532 nm laser for excitation with power of 0.3 mW, 50× objective, data acquisition time 3 s, and scanning 50 cycles for each spectrum. The thermogravimetric (TG) analysis of Hf–GO was performed using a thermogravimetric analysis system (Diamond TG/DTA6300, PerkinElmer Instruments) under an N2 atmosphere at the heating rate of 10 °C min−1. The surface area and pore diameters and pore volumes were determined via the nitrogen adsorption–desorption method using an American Mike Micromeritics ASAP 2020 sorption analyzer. The XPS measurements were carried out via an ESCALAB 250Xi spectrometer (Thermo Fisher Scientific) at a pressure of ∼3 × 10−9 mbar using Al Kα as the excitation source (hν = 1486.6 eV) and operating at 15 kV and 150 W, and C 1s orbital (284.8 eV) was used to correct the binding energy. The XPSpeak 4.1 software was used to perform manual peak splitting, establishing a baseline and add peaks, adjust parameters such as peak area and half-peak width to make the fitted data coincide with the original data, keep the Lorentzian–Gaussian ratio consistent (default 80). C 1s peaks were deconvoluted into four bands.69–72 Hf fitted spectra in which the peak area ratio was fixed (d5/2:d3/2 = 3:2, f7/2:f5/2 = 4:3) and the half-peak width was kept consistent. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) using a Agilent 720 system. Elemental analysis (EA) using a Thermo Fisher Flash 2000 system.
The hydrogenation reduction reactions
The hydrogenation reduction of various aldehydes and ketones using isopropanol as the solvent and hydrogen-donor was performed in a 10 mL Teflon-lined stainless steel autoclave equipped with a magnetic stirrer. In a typical reaction procedure, EL or other carbonyl compounds (1 mmol), isopropanol (5 mL) and the catalyst (100 mg) were added into the reactor and tightly sealed, then placed into a preheated oil-bath at a known temperature of 130–170 °C for a reaction time of 1–9 h. After reaction, the reactor was cooled in cold water to quench the reaction and the organic phase was diluted by isopropanol. The liquid samples were analyzed quantitatively by gas chromatography (TECHCOMP GC7900) using decane as the internal standard, and identification of the products was done by GC-MS (SHIMADZU-QP 2010).Catalyst recycle and heterogeneity
In the experiments to investigate the reusability of the Hf–GO catalyst, the catalyst was recovered by centrifugation, washed three times with fresh isopropanol, and then the catalyst was directly used for the next run of reaction. For heterogeneity, the solid catalyst was separated from the reaction mixture by centrifugation and the supernatant was allowed to react to determine if the product yield further increased in the absence of the solid catalyst.Conclusions
In summary, a novel and efficient Hf–GO catalyst was constructed using graphite oxide as the ligand for MPV reaction of carbonyl compounds. Both the preparation conditions and the reaction conditions were systematically studied. The prepared Hf–GO catalyst showed similar or even higher efficiency among the analogues. It was found that isopropanol had an activation effect on the catalyst by first removing residual DMF in micro- and mesopores and then interacting with the active Hf–O sites. With the advantages of good efficiency, excellent stability, and facility to prepare, the novel Hf–GO catalyst was promising in biomass conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21968021, 21606134, and 21566029), CAS “Light of West China” Program, and the Innovative and Entrepreneurial Talents Grassland Talents Engineering of Inner Mongolia.Notes and references
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- Q. Xu, X. L. Li, T. Pan, C. G. Yu, J. Deng, Q. X. Guo and Y. Fu, Green Chem., 2016, 18, 1287–1294 RSC.
- H. L. Wang, Y. Q. Pu, A. Ragauskas and B. Yang, Bioresour. Technol., 2019, 271, 449–461 CrossRef CAS PubMed.
- A. Q. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS PubMed.
- H. L. Wang, H. M. Wang, E. Kuhn, M. P. Tucker and B. Yang, ChemSusChem, 2018, 11, 285–291 CrossRef CAS PubMed.
- S. Wang, J. Wang, Q. F. Zhao, D. D. Li, J. Q. Wang, M. Cho, H. Cho, O. Terasaki, S. J. Chen and Y. Wan, ACS Catal., 2015, 5, 797–802 CrossRef CAS.
- M. Dashtban, A. Gilbert and P. Fatehi, RSC Adv., 2014, 4, 2037–2050 RSC.
- M. E. Zakrzewska, E. Bogel-Lukasik and R. Bogel-Lukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS PubMed.
- H. M. Mirzaei and B. Karimi, Green Chem., 2016, 18, 2282–2286 RSC.
- Z. C. Tang, W. P. Deng, Y. L. Wang, E. Z. Zhu, X. Y. Wan, Q. H. Zhang and Y. Wang, ChemSusChem, 2014, 7, 1557–1567 CrossRef CAS PubMed.
- D. Esposito and M. Antonietti, ChemSusChem, 2013, 6, 989–992 CrossRef CAS PubMed.
- G. H. Wang, J. Hilgert, F. H. Richter, F. Wang, H. J. Bongard, B. Spliethoff, C. Weidenthaler and F. Schuth, Nat. Mater., 2014, 13, 294–301 Search PubMed.
- A. Szabolcs, M. Molnar, G. Dibo and L. T. Mika, Green Chem., 2013, 15, 439–445 RSC.
- F. D. Pileidis and M. M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed.
- J. Yuan, S. S. Li, L. Yu, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Energy Environ. Sci., 2013, 6, 3308–3313 RSC.
- L. Deng, J. Li, D. M. Lai, Y. Fu and Q. X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529–6532 CrossRef CAS PubMed.
- S. H. Zhu, Y. F. Xue, J. Guo, Y. L. Cen, J. G. Wang and W. B. Fan, ACS Catal., 2016, 6, 2035–2042 CrossRef CAS.
- K. Yan, Y. Y. Yang, J. J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292–304 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- J. P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- M. G. Al-Shaal, A. Dzierbinski and R. Palkovits, Green Chem., 2014, 16, 1358–1364 RSC.
- L. Xin, Z. Y. Zhang, J. Qi, D. J. Chadderdon, Y. Qiu, K. M. Warsko and W. Z. Li, ChemSusChem, 2013, 6, 674–686 CrossRef CAS PubMed.
- F. Y. Ye, D. M. Zhang, T. Xue, Y. M. Wang and Y. J. Guan, Green Chem., 2014, 16, 3951–3957 RSC.
- A. Villa, M. Schiavoni, C. E. Chan-Thaw, P. F. Fulvio, R. T. Mayes, S. Dai, K. L. More, G. M. Veith and L. Prati, ChemSusChem, 2015, 8, 2520–2528 CrossRef CAS PubMed.
- P. P. Upare, J. M. Lee, D. W. Hwang, S. B. Halligudi, Y. K. Hwang and J. S. Chang, J. Ind. Eng. Chem., 2011, 17, 287–292 CrossRef CAS.
- W. Li, J. H. Xie, H. Lin and Q. L. Zhou, Green Chem., 2012, 14, 2388–2390 RSC.
- K. Hengst, M. Schubert, H. W. P. Carvalho, C. B. Lu, W. Kleist and J. D. Grunwaldt, Appl. Catal., A, 2015, 502, 18–26 CrossRef CAS.
- H. C. Zhou, J. L. Song, H. L. Fan, B. B. Zhang, Y. Y. Yang, J. Y. Hu, Q. G. Zhu and B. X. Han, Green Chem., 2014, 16, 3870–3875 RSC.
- E. F. Mai, M. A. Machado, T. E. Davies, J. A. Lopez-Sanchez and V. T. da Silva, Green Chem., 2014, 16, 4092–4097 RSC.
- Y. Y. Liu, P. Ghimire and M. Jaroniec, J. Colloid Interface Sci., 2019, 535, 122–132 CrossRef CAS PubMed.
- Y. F. Sha, Z. H. Xiao, H. C. Zhou, K. L. Yang, Y. M. Song, N. Li, R. X. He, K. D. Zhi and Q. S. Liu, Green Chem., 2017, 19, 4829–4837 RSC.
- J. L. Song, B. W. Zhou, H. C. Zhou, L. Q. Wu, Q. L. Meng, Z. M. Liu and B. X. Han, Angew. Chem., Int. Ed. Engl., 2015, 54, 9399–9403 CrossRef CAS PubMed.
- M. H. Beyzavi, N. A. Vermeulen, A. J. Howarth, S. Tussupbayev, A. B. League, N. M. Schweitzer, J. R. Gallagher, A. E. Platero-Prats, N. Hafezi, A. A. Sarjeant, J. T. Miller, K. W. Chapman, J. F. Stoddart, C. J. Cramer, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 13624–13631 CrossRef CAS PubMed.
- J. Zheng, M. Y. Wu, F. L. Jiang, W. P. Su and M. C. Hong, Chem. Sci., 2015, 6, 3466–3470 RSC.
- M. H. Beyzavi, N. A. Vermeulen, A. J. Howarth, S. Tussupbayev, A. B. League, N. M. Schweitzer, J. R. Gallagher, A. E. Platero-Prats, N. Hafezi, A. A. Sarjeant, J. T. Miller, K. W. Chapman, J. F. Stoddart, C. J. Cramer, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 3251 CrossRef CAS PubMed.
- X. X. Cai, J. P. Pan, G. M. Tu, Y. H. Fu, F. M. Zhang and W. D. Zhu, Catal. Commun., 2018, 113, 23–26 CrossRef CAS.
- S. Rojas-Buzo, P. García-García and A. Corma, Green Chem., 2018, 20, 3081–3091 RSC.
- J. L. Song, Z. M. Xue, C. Xie, H. R. Wu, S. S. Liu, L. Zhang and B. X. Han, ChemCatChem, 2018, 10, 725–730 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- H. L. Zhou, Y. C. Liu, L. Zhang, H. D. Li, H. Liu and W. J. Li, J. Colloid Interface Sci., 2019, 533, 287–296 CrossRef CAS PubMed.
- O. Metin, H. Can, K. Sendil and M. S. Gultekin, J. Colloid Interface Sci., 2017, 498, 378–386 CrossRef CAS PubMed.
- M. Nasrollahzadeh, S. M. Sajadi, A. Rostami-Vartooni, M. Alizadeh and M. Bagherzadeh, J. Colloid Interface Sci., 2016, 466, 360–368 CrossRef CAS PubMed.
- S. Rojas-Buzo, P. Garcia-Garcia and A. Corma, ChemSusChem, 2018, 11, 432–438 CrossRef CAS PubMed.
- Z. Hu, I. Castano, S. Wang, Y. Wang, Y. Peng, Y. Qian, C. Chi, X. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CrossRef CAS.
- A. H. Valekar, K.-H. Cho, S. K. Chitale, D.-Y. Hong, G.-Y. Cha, U. H. Lee, D. W. Hwang, C. Serre, J.-S. Chang and Y. K. Hwang, Green Chem., 2016, 18, 4542–4552 RSC.
- H. Reinsch, B. Bueken, F. Vermoortele, I. Stassen, A. Lieb, K.-P. Lillerud and D. De Vos, CrystEngComm, 2015, 17, 4070–4074 RSC.
- V. Bon, V. Senkovskyy, I. Senkovska and S. Kaskel, Chem. Commun., 2012, 48, 8407–8409 RSC.
- V. Bon, I. Senkovska, I. A. Baburin and S. Kaskel, Cryst. Growth Des., 2013, 13, 1231–1237 CrossRef CAS.
- G. R. Cai and H. L. Jiang, Angew. Chem., Int. Ed., 2017, 56, 563–567 CrossRef CAS PubMed.
- H. Reinsch, B. Bueken, F. Vermoortele, I. Stassen, A. Lieb, K. P. Lillerud and D. De Vos, Crystengcomm, 2015, 17, 4070–4074 RSC.
- Z. G. Hu, I. Castano, S. N. Wang, Y. X. Wang, Y. W. Peng, Y. H. Qan, C. L. Chi, X. R. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- Z. L. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gandara, H. B. Burgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CrossRef CAS PubMed.
- K. M. Choi, H. J. Jeon, J. K. Kang and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11920–11923 CrossRef CAS PubMed.
- S. He, Y. F. Chen, Z. C. Zhang, B. Ni, W. He and X. Wang, Chem. Sci., 2016, 7, 7101–7105 RSC.
- H. Li, T. Yang and Z. Fang, Appl. Catal., B, 2018, 227, 79–89 CrossRef CAS.
- Z. H. Xiao, H. C. Zhou, J. M. Hao, H. L. Hong, Y. M. Song, R. X. He, K. D. Zhi and Q. S. Liu, Fuel, 2017, 193, 322–330 CrossRef CAS.
- J. L. Song, L. Q. Wu, B. W. Zhou, H. C. Zhou, H. L. Fan, Y. Y. Yang, Q. L. Meng and B. X. Han, Green Chem., 2015, 17, 1626–1632 RSC.
- L. Peng, J. L. Zhang, J. S. Li, B. X. Han, Z. M. Xue and G. Y. Yang, Chem. Commun., 2012, 48, 8688–8690 RSC.
- H. Li, Y. Li, Z. Fang and R. L. Smith, Catal. Today, 2019, 319, 84–92 CrossRef CAS.
- G. Lv, H. Wang, Y. Yang, T. Deng, C. Chen, Y. Zhu and X. Hou, ACS Catal., 2015, 5, 5636–5646 CrossRef CAS.
- P. P. Upare, J.-W. Yoon, M. Y. Kim, H.-Y. Kang, D. W. Hwang, Y. K. Hwang, H. H. Kung and J.-S. Chang, Green Chem., 2013, 15, 2935 RSC.
- Y. Wang, Z. M. Rong, Y. Wang, T. Wang, Q. Q. Du, Y. Wang and J. Qu, ACS Sustainable Chem. Eng., 2016, 5, 1538–1548 CrossRef.
- O. Akhavan, ACS Nano, 2010, 4, 4174–4180 CrossRef CAS PubMed.
- C. Xie, J. L. Song, B. W. Zhou, J. Y. Hu, Z. R. Zhang, P. Zhang, Z. Jiang and B. X. Han, ACS Sustainable Chem. Eng., 2016, 4, 6231–6236 CrossRef CAS.
- Z. Cai, W. K. Li, F. M. Wang and X. B. Zhang, J. Taiwan Inst. Chem. Eng., 2018, 93, 374–378 CrossRef CAS.
- W. B. Wu, Y. Li, H. Li, W. F. Zhao and S. Yang, Catalysts, 2018, 8, 264 CrossRef.
- J. X. Hao, L. M. Han, Y. F. Sha, X. X. Yu, H. Y. Liu, X. Y. Ma, Y. Z. Yang, H. C. Zhou and Q. S. Liu, Fuel, 2019, 239, 1304–1314 CrossRef CAS.
- B. B. Zhang, J. X. Hao, Y. F. Sha, H. C. Zhou, K. L. Yang, Y. M. Song, Y. P. Ban, R. X. He and Q. S. Liu, Fuel, 2018, 217, 122–130 CrossRef CAS.
- J. L. Song, L. Q. Wu, B. W. Zhou, H. C. Zhou, H. L. Fan, Y. Y. Yang, Q. Meng and B. X. Han, Green Chem., 2015, 17, 1626–1632 RSC.
- Z. M. Xue, J. Y. Jiang, G. F. Li, W. C. Zhao, J. F. Wang and T. C. Mu, Catal. Sci. Technol., 2016, 6, 5374–5379 RSC.
- K. Yan, J. Y. Liao, X. Wu and X. M. Xie, RSC Adv., 2013, 3, 3853–3856 RSC.
- K. Jiang, D. Sheng, Z. H. Zhang, J. Fu, Z. Y. Hou and X. Y. Liu, Catal. Today, 2016, 274, 55–59 CrossRef CAS.
- J. Cheng, J. Wang, X. Z. Wang and H. Wang, Ceram. Int., 2017, 43, 7159–7165 CrossRef CAS.
- H. Furukawa, F. Gandara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10795a |
| This journal is © The Royal Society of Chemistry 2020 |