
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ru(II)-catalyzed C6-selective C–H acylmethylation of pyridones using sulfoxonium ylides as carbene precursors†
Yangjie Fuab,
Zhaohui Wangab,
Qiyu Zhangab,
Zhiyu Lia,
Hong Liu ab,
Xiaoling Bi*a and
Jiang Wang*b
ab,
Xiaoling Bi*a and
Jiang Wang*b
aJiangsu Key Laboratory of Drug Design and Optimization, Department of Medicinal Chemistry, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China. E-mail: bxl@163.com
bState Key Laboratory of Drug Research, Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu Chong Zhi Road, Shanghai 201203, China. E-mail: jwang@simm.ac.cn
First published on 11th February 2020
Abstract
In this study, we describe a method using sulfoxonium ylides as carbene precursors to achieve C6-selective acylmethylation of pyridones catalyzed by a ruthenium(II) complex. This approach featured mild reaction conditions, moderate to excellent yields, high step economy, and had excellent functional group tolerance with good site selectivity. Besides, gram-scale preparation, synthetic utility, and mechanistic studies were conducted. It offers a direct and efficient way to synthesize pyridone derivatives.
1 Introduction
Pyridone is exhibited as a privilege scaffold in a large range of biological active agents, attracting much attention from medicinal chemists (Fig. 1).1 Consequently, how to achieve the late stage functionalization of pyridone has attracted intensive attention.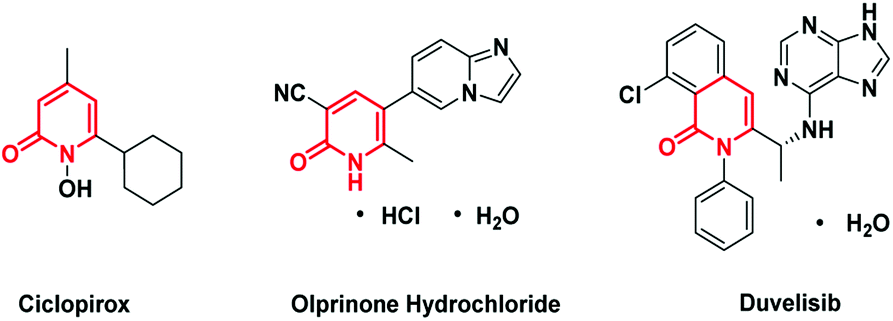 | ||
| Fig. 1 Pharmaceuticals containing a C6-alkylated pyridone core structure. | ||
Traditionally, the direct alkylation of pyridone was usually afforded by pre-functionalization with a halogen followed by transition-metal catalyzed coupling reactions. Recently, the direct C–H functionalization strategy to form C–C or C–X bonds has become a more effective and reliable synthetic route.2 Transition-metal-promoted C3 (ref. 3) and C5 (ref. 4) positions of 2-pyridones have been probed exhaustively owing to the sufficient electron density of C–H bonds in these positions. However, only limited examples have been reported on the direct C–H bond functionalization on C6 position of pyridone.5 For instance, Cramer and collaborators described the synthesis of 1,6-annulated 2-pyridones by selective intramolecular nickel catalyzed cyclization.5c Afterwards, more C–H functionalization at C6 position of pyridone mediated by transition-metal have been reported.6 Miura and colleagues exploited selective C6 borylation of pyridone with bis(pinacolato)diboron via rhodium catalyzed C–H bond activation. The synthetic utility has been extended by subsequent Suzuki–Miyaura cross-coupling to form new C–C bonds and after removal of the directing group, the C6-arylated NH-pyridone has been afforded.6c At the same time, our group has successively reported the rhodium or cobalt-catalyzed, C6-selective C–H alkylation, arylation, and amidation of pyridones by using potassium trifluoroborates or oxazolones (Scheme 1a and b).6d,6h
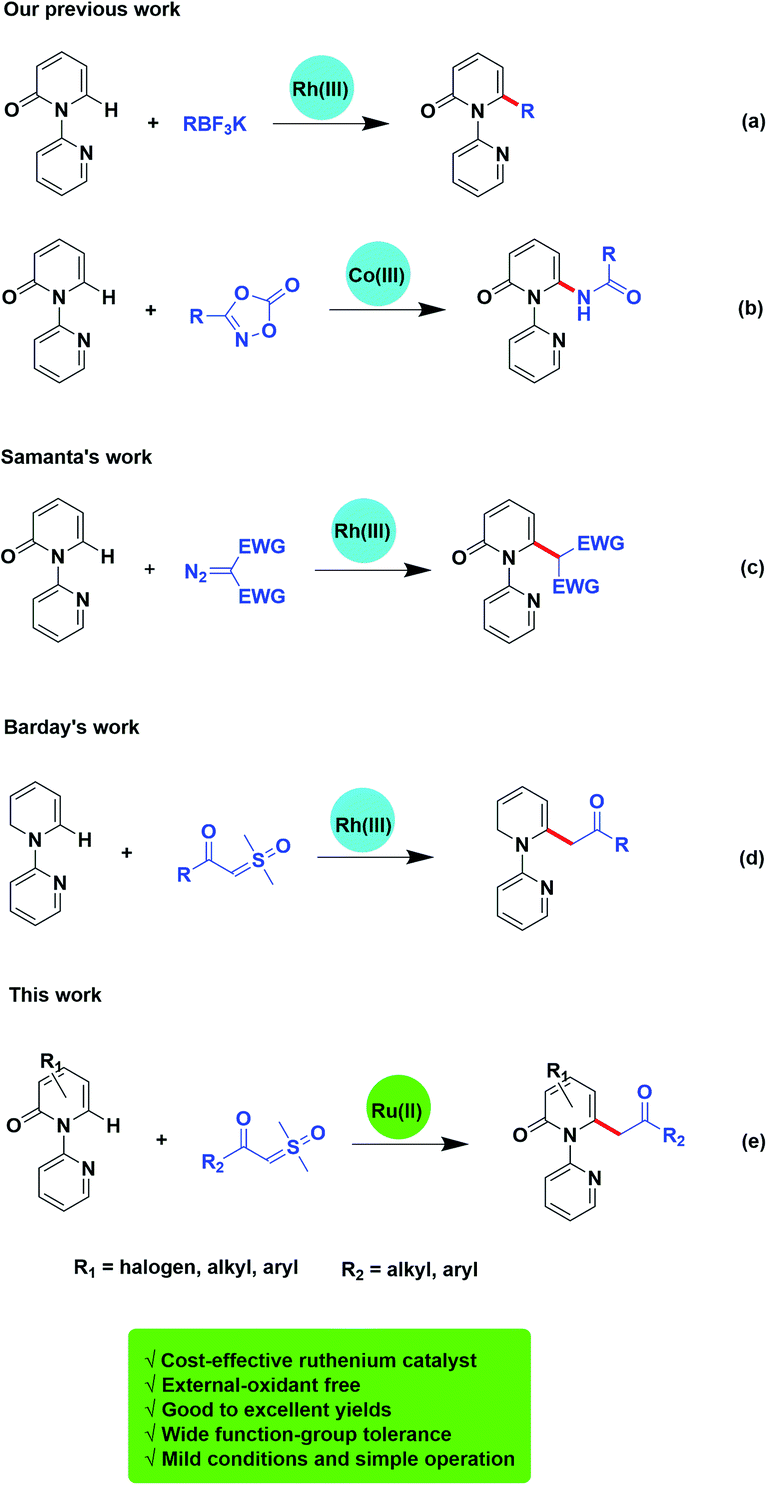 | ||
| Scheme 1 C6-selective C–H functionalization of pyridones. | ||
Transition-metal-catalyzed C–H functionalization is based on carbene migratory insertion to achieve the transformation. In transition-metal-catalyzed C–H functionalization, α-diazo carbonyls are commonly used as carbene precursor.7,8 Samanta and colleagues disclosed a rhodium-mediated C6-selective alkylation of 2-pyridones employing α-diazocarbonyl derivatives (Scheme 1c).7c However, there are still some limitations of diazo compounds serving as a carbene precursor, such as the potential explosiveness due to the evolution of nitrogen gas. To overcome these problems, other carbene surrogates were explored, such as cyclopropenes,9 hydrazones,10 ketone-functionalized enynes,11 triazoles,12 and sulfoxonium ylides. Sulfoxonium ylides have been reported to be employed in industry, and are more safe alternatives to diazo compounds.13 And recently, Barday and co-workers developed the cross-coupling reactions of α-carbonyl sulfoxonium ylides with arenes and heteroarenes using (Cp*RhCl2)2 as the catalyst (Scheme 1d).13d
Instead of using the noble metals such as rhodium and iridium, to date, more examples on direct C–H bond functionalization catalysed by ruthenium, a cost-effective transition-metal, has attracted attention and been developed. Herein, we reveal ruthenium(II)-catalyzed C6-selective direct acylmethylation of pyridones using sulfoxonium ylides (Scheme 1e).
2 Results and discussion
Based on the precedent reported research, 2-pyridone (1a) and α-benzoyl sulfoxonium ylide (2a) were selected to probe the reaction conditions for transition-metal catalyzed acylmethylation of pyridone (Table 1). Initially, the coupling reaction between substrate 1a (0.4 mmol) and 2a (0.8 mmol) was triggered by a screen of various transition metal complexes. Ruthenium(II) (5 mol%), cobalt(III) (5 mol%), and rhodium(III) (5 mol%) were independently investigated in the presence of AgSbF6 (10 mol%) in hexafluoroisopropanol (HFIP) and the mixture was stirred at 60 °C under an argon atmosphere for 24 h. The results indicated that [Ru(p-cymene)Cl2]2 was the optimal catalyst (Table 1, entries 1–3). Additionally, if replacing the [Ru(p-cymene)Cl2]2 with [RuCl(p-cymene)((S)-binap)]Cl, Ru(PPh3)3Cl2, or RuCl[(R,R)-Tsdpen](p-cymene), the yield of 3aa was decreased (Table 1, entries 4–6). Solvent was subsequently examined and results demonstrated that 3aa could be obtained in a higher yield in HFIP than in others including 1,2-dichloorethaan (DCE), acetonitrile, dioxane, methanol, and ethanol (Table 1, entries 7–11). Changing the additive from AgSbF6 to AgNTf2, AgOTf, or Ag(OAc)2 could diminish the yield of 3aa (Table 1, entries 12–14). The yield slightly decreased caused by the reduction of [Ru(p-cymene)Cl2]2 and AgSbF6 (Table 1, entries 15 and 16). Whilst when the reaction was conducted at 90 °C, 3aa could also be attained in 91% yield which was no more discrepancy with conducting at 60 °C (Table 1, entry 15). However, decreasing the temperature to 40 °C, the yield was reduced to 67% (Table 1, entry 16). The reaction could also be carried out in air with 76% yield (Table 1, entry 17), but without ruthenium(II) complex or Ag(I) additive, the reaction was no longer proceeded (Table 1, entries 18 and 19).
|
||||
|---|---|---|---|---|
| Entry | Cat.b | Ag salt | Solvent | Yieldc (%) |
| a Reaction conditions: compound 1a (0.4 mmol), compound 2a (0.8 mmol), cat. (5 mol%) and Ag salt (10 mol%) in solvent (3 mL) at 60 °C for 24 h, under Ar atmosphere. N.R. = no reaction.b Catalyst A = [Cp*Co(CO)I2], catalyst B = (Cp*RhCl2)2, catalyst C = [Ru(p-cymene)Cl2]2, catalyst D = [RuCl(p-cymene)((S)-binap)]Cl, catalyst E = Ru(PPh3)3Cl2, catalyst F = RuCl[(R,R)-Tsdpen](p-cymene).c Isolated yield.d Cat. (2.5 mol%).e Ag salt (5 mol%).f At 90 °C.g At 40 °C.h At air condition. | ||||
| 1 | A | AgSbF6 | HFIP | 13 |
| 2 | B | AgSbF6 | HFIP | 49 |
| 3 | C | AgSbF6 | HFIP | 91 |
| 4 | D | AgSbF6 | HFIP | 31 |
| 5 | E | AgSbF6 | HFIP | 16 |
| 6 | F | AgSbF6 | HFIP | 63 |
| 7 | C | AgSbF6 | DCE | 22 |
| 8 | C | AgSbF6 | MeCN | 13 |
| 9 | C | AgSbF6 | Dioxane | 21 |
| 10 | C | AgSbF6 | CH3OH | 12 |
| 11 | C | AgSbF6 | CH3CH2OH | 64 |
| 12 | C | AgNTf2 | HFIP | 84 |
| 13 | C | AgOTf | HFIP | 78 |
| 14 | C | Ag(OAc)2 | HFIP | Trace |
| 15d | C | AgSbF6 | HFIP | 84 |
| 16e | C | AgSbF6 | HFIP | 82 |
| 17f | C | AgSbF6 | HFIP | 91 |
| 18g | C | AgSbF6 | HFIP | 67 |
| 19h | C | AgSbF6 | HFIP | 76 |
| 20 | — | AgSbF6 | HFIP | N.R. |
| 21 | C | — | HFIP | N.R. |

With the optimized reaction conditions obtained, we investigated the substrate scope of pyridones 1a–1r (Scheme 2). The results showed that C3 substituted of 2-pyridones can sustain multiple functional groups, including electron-withdrawing groups or electron-donating groups, and even halogens to afford the desirable products in good to moderate yields (3ba–3ea, 72–87%). Substituents installed on the C4 position of pyridones can be processed smoothly by obtaining the desired products in good to moderate yields (3fa–3ia, 72–84%). Satisfyingly, although suffering from steric hindrance for the C5-substituted 2-pyridones, the desired compounds could be afforded in considerable yields (3ja–3ma, 77–85%). Moreover, this transformation was also compatible to isoquinolinones by attaining target molecules in good to excellent yields (3na–3ra, 72–81%).
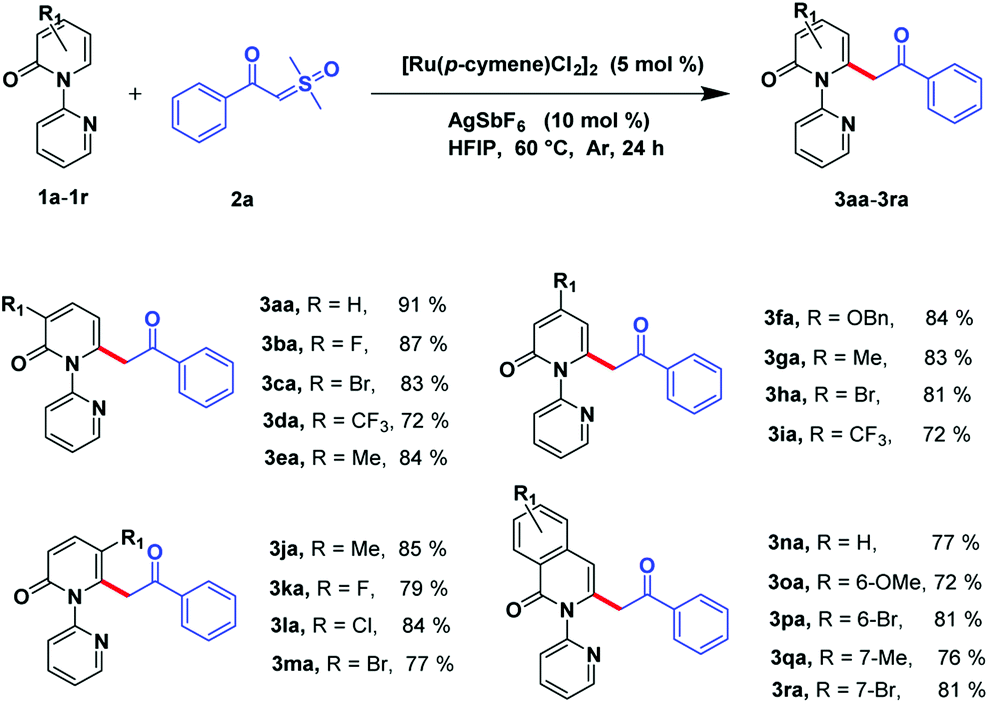 | ||
| Scheme 2 Substrate scope of pyridions.a,b a Reaction conditions: compound 1a–1r (0.4 mmol), compound 2a (0.8 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), and AgSbF6 (10 mol%) in HFIP (3 mL) at 60 °C, under Ar in 24 h. b Isolated yield. | ||
Next, we investigated the scope of sulfoxonium ylides. The acylmethylation proposal was suitable for various kinds of α-benzoyl sulfoxonium ylides (Scheme 3). It can be tolerated by electron-donating groups, such as CH3 and OMe, and can be processed smoothly even if electron-withdrawing groups, such as CF3, or halogens (F, Cl, and Br), are incorporated in the derivatives. Different positions such as the ortho-, meta-, and para-of the phenyl ring can favorably afford the relevant products (3ab–3an) in high yields (76–86%). Gratifyingly, this reaction could also be carried out with heterocyclic compounds such as thiophene and the corresponding product (3ao) was detected in 75% yield. The sulfoxonium ylides can also bear some alkyl substrates and the relevant products could be detected in acceptable yields (3ap–3aq, 63–68%).
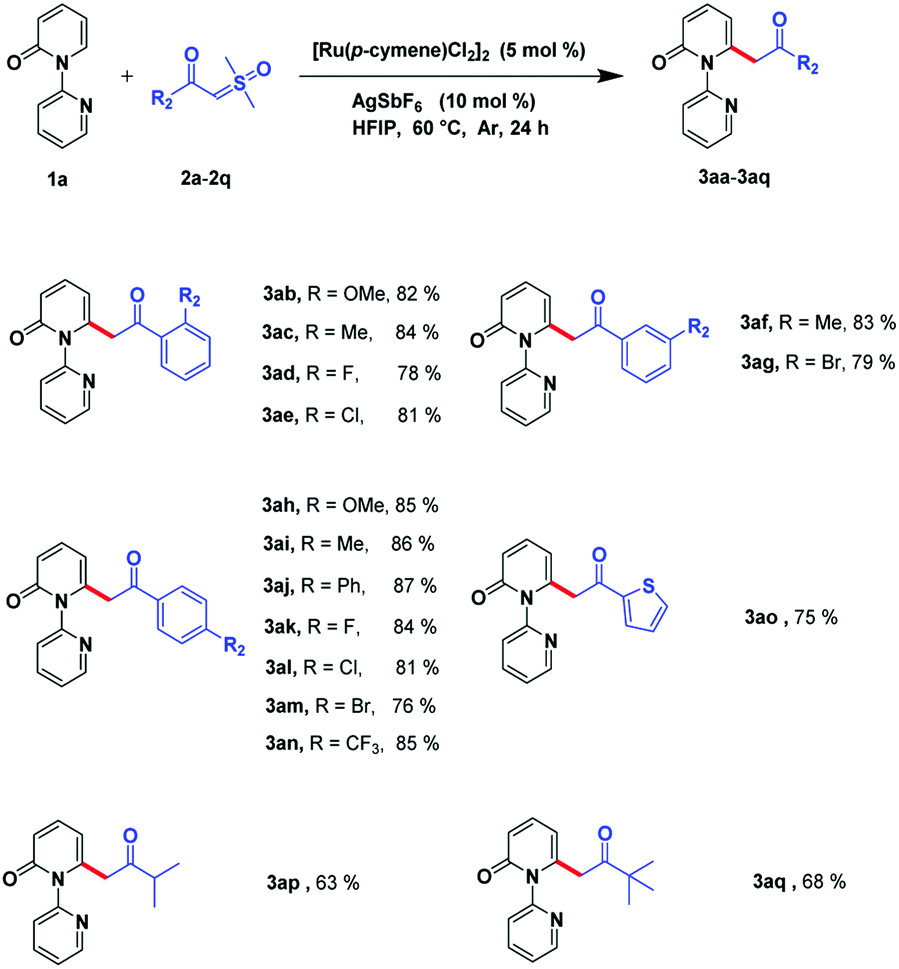 | ||
| Scheme 3 Substrate scope of sulfoxonium ylides.a,b a Reaction conditions: compound 1a (0.4 mmol), compound 2a–2q (0.8 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), and AgSbF6 (10 mol%) in HFIP (3 mL) at 60 °C, under Ar in 24 h. b Isolated yield. | ||
To indicate the synthetic utility of this strategy for the approach to C6-acylmethylation piperidin-2-one, gram-scale synthesis of compound 3aa was conducted and the product was obtained in 89% yield (Scheme 4a). Furthermore, hydrogenation of 3aa was examined to form 4aa in 69% yield (Scheme 4b).
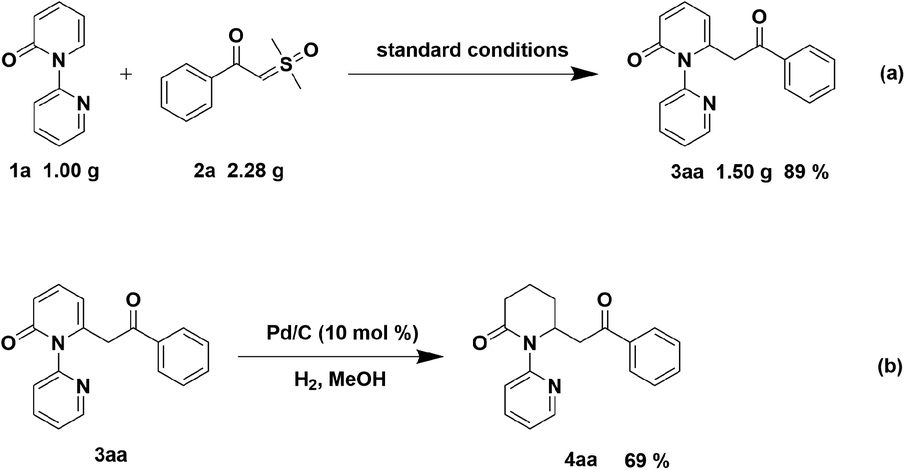 | ||
| Scheme 4 Gram-scale synthesis and synthetic transformation of compound 3aa. | ||
In order to investigate the preliminary mechanism, a series of experiments were designed and performed. Firstly, a hydrogen–deuterium (H/D) exchange experiment was conducted to gain insight into the C–H cleavage, when 2-pyridone (1a) was examined in the optimized condition with the presence of CD3OD and no deuterium exchange was observed. It demonstrate the irreversible of C–H bond cleavage catalyzed by ruthenium. Furthermore, the kinetic isotope effect (KIE) experiment was conducted, employing [D1]-1a as substrate, illustrated a KIE of 1.3, indicated that the rate-limited step was not the division of the C–H bond. Additionally, an intermolecular competition reaction between 3-(trifluoromethyl)-2H-[1,2′-bipyridin]-2-one (1d) and 3-methyl-2H-[1,2′-bipyridin]-2-one (1e) with compound 2a were carried out in one sealed tube. Finally, it gave a higher yield of 3ea than 3da, revealing that the electron-donating substrate has faster reaction rate (Scheme 5).
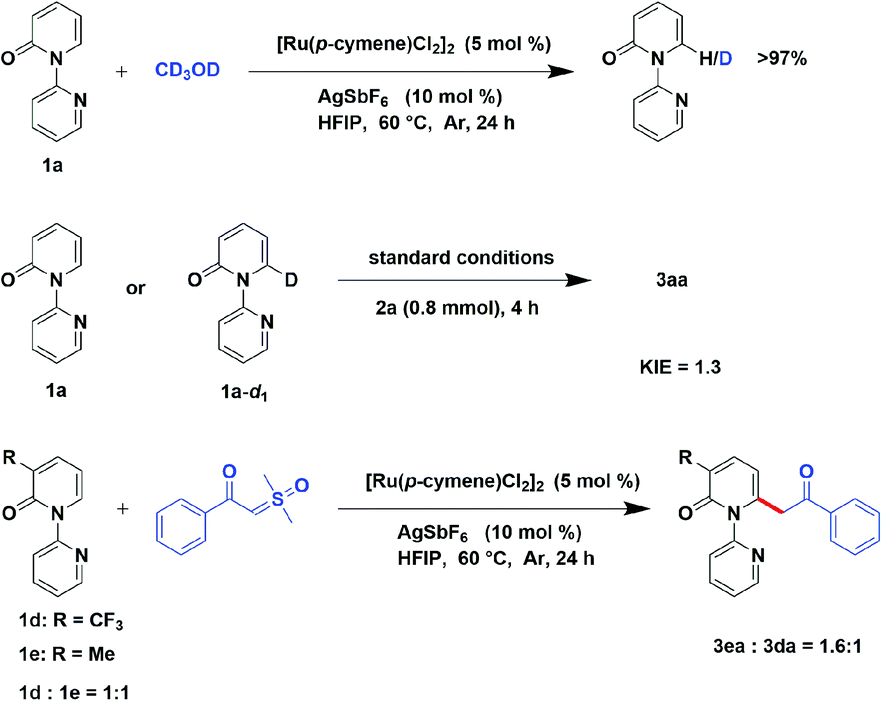 | ||
| Scheme 5 Mechanism study experiments. | ||
On the basis of the preliminary experimental results, a plausible acylmethylation catalytic cycle is proposed (Scheme 6). The reactive Ru(II) complex was first formed after ligand exchange of [Ru(p-cymene)Cl2]2 with AgSbF6, followed by a ortho C–H bond activation of pyridone. This process is assisted by the DG, pyridine motif and generate intermediate A. There is a ligand exchange among 2a and intermediate A, which affords the intermediate B. With the leaving of DMSO, ruthenium carbene intermediate C is produced. Migratory insertion of ruthenium-carbene generate intermediate D. Eventually, the intermediate D transfer the protonation, produce the product 3aa and liberate the active Ru-catalyst.
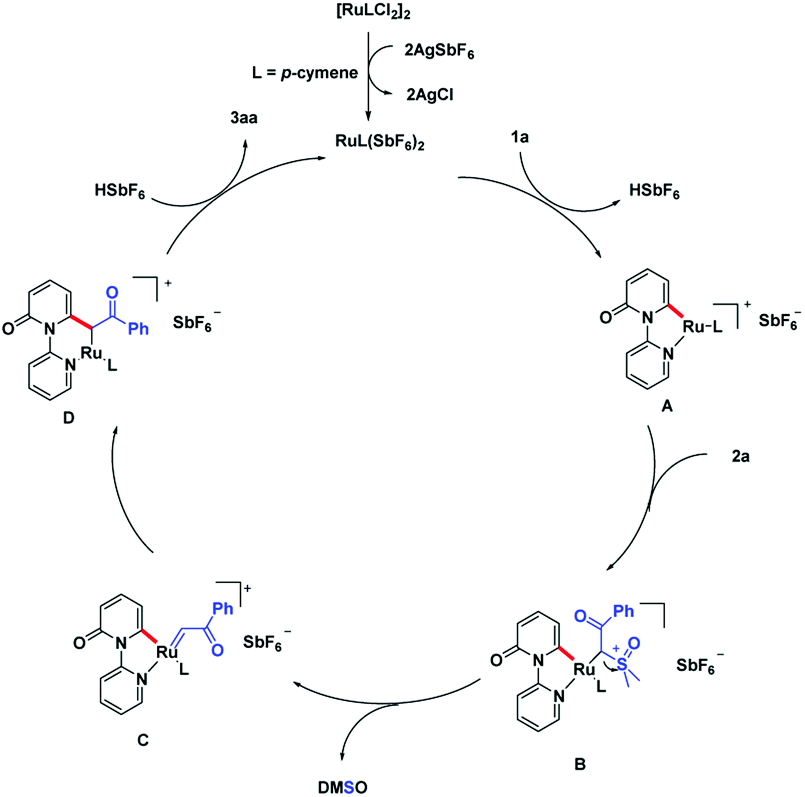 | ||
| Scheme 6 Proposed reaction mechanism. | ||
3 Conclusions
In summary, we achieved the ruthenium(II)-catalyzed C6-selective C–H acylmethylation of pyridones employing sulfoxonium ylides. This new transformation is achieved using the excellent role of the Ru(II) catalyst ([Ru(p-cymene)Cl2]2), and allows the synthesis of various C6-acylmethylated 2-pyridone derivatives. Besides, this approach features mild reaction conditions, moderate to excellent yields and high step economy. Furthermore, mechanistic study experiments were conducted to reveal the catalytic transformation cycle. It offers a direct and efficient way to synthesize pyridone derivatives and will be important to medicinal chemists.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation (81620108027, 21632008, and 21877118) and supported by grants from Science and Technology Commission of Shanghai Municipality (17431903100 and 18431907100).References
- (a) A. Hueber, P. Esser and K. Heimann, Exp. Eye Res., 1998, 67, 530–533 CrossRef PubMed; (b) T. Brunin, L. Legentil and H. Pierre, Tetrahedron, 2006, 62, 3959–3968 CrossRef CAS; (c) H. Zhou, T. Shen, Y. Luo, L. Liu, W. Chen and B. Xu, Int. J. Cancer, 2010, 127, 2467–2477 CrossRef CAS PubMed; (d) R. Paola, E. Mazzon and I. Paterniti, Eur. J. Pharmacol., 2011, 650, 612–620 CrossRef PubMed; (e) P. Blier and J. Pharmacol, Exp. Ther., 2014, 351, 585–595 CrossRef PubMed; (f) M. Ma, Q. Ren and C. Yang, Psychopharmacology, 2017, 234, 525–533 CrossRef CAS PubMed.
- (a) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (c) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed; (d) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed; (e) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412–424 CrossRef CAS PubMed; (f) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (g) L. Yang and H. M. Huang, Chem. Rev., 2015, 115, 3468–3517 CrossRef CAS PubMed; (h) C. Bruneau and P. H. Dixneuf, Top. Organomet. Chem., 2016, 55, 137–188 CrossRef CAS; (i) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachaland and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (j) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- (a) A. Nakatani, K. Hirano, T. Satoh and M. Miura, Chem.–Eur. J., 2013, 19, 7691–7695 CrossRef CAS PubMed; (b) E. E. Anagnostaki, A. D. Fotiadou, V. Demertzidou and A. L. Zografos, Chem. Commun., 2014, 50, 6879–6882 RSC; (c) A. Nakatani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 1377–1385 CrossRef CAS PubMed; (d) A. Modak, S. Rana and D. Maiti, J. Org. Chem., 2015, 80, 296–303 CrossRef CAS PubMed; (e) A. Najib, S. Tabuchi, K. Hirano and M. Miura, Heterocycles, 2016, 92, 1187–1203 CrossRef CAS; (f) P. Chauhan, M. Ravi, S. Singh, P. Prajapati and P. P. Yadav, RSC Adv., 2016, 6, 109–118 RSC.
- (a) T. Itahara and F. Ouseto, Synthesis, 1984, 1984, 488–489 CrossRef; (b) Y. Y. Chen, F. Wang, A. Q. Jia and X. W. Li, Chem. Sci., 2012, 3, 3231–3236 RSC; (c) Y. Li, F. Xie and X. Li, J. Org. Chem., 2016, 81, 715–722 CrossRef CAS PubMed; (d) S. Maity, D. Das, S. Sarkar and R. Samanta, Org. Lett., 2018, 20, 5167–5171 CrossRef CAS PubMed.
- (a) R. Tamura, Y. Yamada, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2012, 51, 5679–5682 CrossRef CAS PubMed; (b) Y. Cao, H. Xing, Q. Yang, Z. Li, T. Chen, Z. Bao and Q. Ren, Ind. Eng. Chem. Res., 2014, 53, 10784–10790 CrossRef CAS; (c) P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2015, 54, 633–637 CAS; (d) A. Biswas, D. Giri, D. Das, A. De, S. K. Patra and R. Samanta, J. Org. Chem., 2017, 82, 10989–10996 CrossRef CAS PubMed; (e) K. Hirano and M. Miura, Chem. Sci., 2018, 9, 22–32 RSC.
- (a) R. Odani, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2014, 53, 10784–10788 CrossRef CAS PubMed; (b) Y. Li, F. Xie and X. Li, J. Org. Chem., 2016, 81, 715–722 CrossRef CAS PubMed; (c) W. Miura, K. Hirano and M. Miura, Org. Lett., 2016, 18, 3742–3745 CrossRef CAS PubMed; (d) P. F. Peng, J. Wang, H. L. Jiang and H. Liu, Org. Lett., 2016, 18, 5376–5379 CrossRef CAS PubMed; (e) K. A. Kumar, P. Kannaboinaa and P. Das, Org. Biomol. Chem., 2017, 15, 5457–5461 RSC; (f) D. Das, P. Poddar, S. Maity and R. Samanta, J. Org. Chem., 2017, 82, 3612–3621 CrossRef CAS PubMed; (g) W. Miura, K. Hirano and M. Miura, J. Org. Chem., 2017, 82, 5337–5344 CrossRef CAS PubMed; (h) F. Gao, X. Han, C. P. Li, L. J. Liu, Z. Q. Cong and H. Liu, RSC Adv., 2018, 8, 32659–32663 RSC; (i) D. Das and R. Samanta, Adv. Synth. Catal., 2018, 360, 379–384 CrossRef CAS.
- (a) J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587–5590 CrossRef CAS PubMed; (b) M. Roche, A. Hamze, O. Provot, J.-D. Brion and M. Alami, J. Org. Chem., 2013, 78, 445–454 CrossRef CAS PubMed; (c) D. Das, A. Biswas, U. Karmakar, S. Chand and R. Samanta, J. Org. Chem., 2016, 81, 842–848 CrossRef CAS PubMed; (d) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13828 CrossRef CAS PubMed; (e) M. Paraja, R. Barroso, M. P. Cabal and C. Valdés, Adv. Synth. Catal., 2017, 359, 1058–1062 CrossRef CAS.
- (a) E. Brachet, A. Hamze, J.-F. Peyrat, J.-D. Brion and M. Alami, Org. Lett., 2010, 12, 4042–4045 CrossRef CAS PubMed; (b) B. Tréguier, M. Lawson, G. Bernadat, J. Bignon, J. Dubois, J. D. Brion, M. Alami and A. Hamze, ACS Comb. Sci., 2014, 16, 702–710 CrossRef PubMed; (c) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508–4511 CrossRef CAS PubMed; (d) P. K. Patel, J. P. Dalvadi and K. H. Chikhalia, Tetrahedron Lett., 2015, 56, 6585–6589 CrossRef CAS; (e) Y. Zhou, F. Ye, X. Wang, S. Xu, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 6109–6118 CrossRef CAS PubMed; (f) Y. Xia, F. Hu, Y. Xia, Z. Liu, F. Ye, Y. Zhang and J. Wang, Synthesis, 2017, 49, 1073–1086 CAS; (g) S. Qu and C. J. Cramer, J. Org. Chem., 2017, 82, 1195–1204 CrossRef CAS PubMed; (h) X. Hu, X. Chen, Y. Shao, H. Xie, Y. Deng, Z. Ke, H. Jiang and W. Zeng, ACS Catal., 2018, 8, 1308–1312 CrossRef CAS.
- (a) H. Zhang, K. Wang, B. Wang, H. Yi, F. Hu, C. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234–13238 CrossRef CAS PubMed; (b) L. Dian and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 3682–3686 CrossRef CAS PubMed.
- (a) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236–247 CrossRef CAS PubMed; (b) A. P. Jadhav, D. Ray, V. U. B. Rao and R. P. Singh, Eur. J. Org. Chem., 2016, 2016, 2369–2394 CrossRef CAS.
- S. Y. Hong, J. Jeong and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 2408–2412 CrossRef CAS PubMed.
- (a) N. Selander, B. T. Worrell, S. Chuprakov, S. Velaparthi and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 14670–14673 CrossRef CAS PubMed; (b) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC; (c) Y. Wang, X. Lei and Y. Tang, Synlett, 2015, 26, 2051–2059 CrossRef; (d) V. Helan, A. V. Gulevich and V. Gevorgyan, Chem. Sci., 2015, 6, 1928–1931 RSC; (e) W. H. Jeon, J.-Y. Son, J. E. Kim and P. H. Lee, Org. Lett., 2016, 18, 3498–3501 CrossRef CAS PubMed; (f) Y. Jiang, R. Sun, X.-Y. Tang and M. Shi, Chem.–Eur. J., 2016, 22, 17910–17924 CrossRef CAS PubMed.
- (a) F. Wang, L. Jin, L. Kong and X. Li, Org. Lett., 2017, 19, 1812–1815 CrossRef CAS PubMed; (b) Y. Xu, X. Zhou, G. Zheng and X. Li, Org. Lett., 2017, 56, 13117–13121 Search PubMed; (c) J. Vaitla, A. Bayer and K. H. Hopmann, Angew. Chem., Int. Ed., 2017, 56, 4277–4281 CrossRef CAS PubMed; (d) M. Barday and C. Janot, Angew. Chem., Int. Ed., 2017, 56, 13117–13121 CrossRef CAS PubMed; (e) J. Zhu, S. Sun and J. Cheng, Tetrahedron Lett., 2018, 59, 2284–2287 CrossRef CAS; (f) Y. Xu, G. Zheng, X. Yang and X. Li, Chem. Commun., 2018, 54, 670–673 RSC; (g) Y. Wu, Z. Chen, Y. Yang, W. Zhu and B. Zhou, J. Am. Chem. Soc., 2018, 140, 42–45 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10749e |
| This journal is © The Royal Society of Chemistry 2020 |