
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D hierarchical rose-like Ni2P@rGO assembled from interconnected nanoflakes as anode for lithium ion batteries†
Gan Cai,
Zhenguo Wu ,
Tao Luo,
Yanjun Zhong*,
Xiaodong Guo,
Zhiye Zhang,
Xinlong Wang and
Benhe Zhong
,
Tao Luo,
Yanjun Zhong*,
Xiaodong Guo,
Zhiye Zhang,
Xinlong Wang and
Benhe Zhong
School of Chemical Engineering, Engineering Research Center of Comprehensive Utilization and Clean Processing of Phosphorus Resources of Ministry of Education, Sichuan University, Chengdu 610065, P. R. China. E-mail: yjzhong@scu.edu.cn; Fax: +86-28-85405235; Tel: +86-28-85405235
First published on 23rd January 2020
Abstract
In recent years, anode materials of transition metal phosphates (TMPs) for lithium ion batteries (LIBs) have drawn a vast amount of attention from researchers, due to their high theoretical capacity and comparatively low intercalation potentials vs. Li/Li+. However, in practice, their application remains constrained by poor electrical conductivity, and dramatic volume expansion and severe agglomeration during the lithium process, which leads to questionable kinetic issues and a prompt decline in capacity during cycling. Herein, through an elaborate design, we developed a novel three-dimensional (3D) hierarchical rose-like architecture self-assembled from two-dimensional (2D) Ni2P nanoflakes immobilized on reduced graphene oxide (rGO) via a combination of a hydrothermal process and phosphating treatment. Such a design provides unique superiority for Ni2P-based anode materials for LIBs. Paraphrasing, the 3D hierarchical structure of Ni2P distributes the stress on the anode material while cycling and provides more lithium storage space. The rGO not only enhances the conductivity of materials, but also serves as a flexible framework which immobilizes Ni2P so that it prevents it from pulverization. Therefore, the synergistic effect between them guarantees the integrity of the material structure after a long-term cycling Li+ intercalation and deintercalation process. When it acted as anode material for LIBs, the as-obtained 3D rose-like Ni2P@rGO electrode exhibited a noticeable electrochemical performance, which delivers a discharge capacity of 330.5 mA h g−1 at a current density of 100 mA g−1 after 100 cycles and retains 200.5 mA h g−1 at 1000 mA g−1.
1. Introduction
Throughout approximately 30 years of development, LIBs have been revolutionary and are dominant in modern electronics, electric vehicles, and permanent energy storage, owing to their superiority in high output voltage and energy density, no memory effect, long lifespan, low self-discharge rate and promising safety.1–3 For the past few years, along with the development of society, the demand for LIBs with high energy density has increased constantly, and the research and development of advanced materials with a considerably higher capacity than those in current use has attracted attention, in particular anode materials,4 because the graphite anode materials which have been widespread in commercial LIBs deliver only a capacity of 372 mA h g−1 in theory.5Transition metal phosphates (TMPs) have garnered increasing attention due to their unique crystal structure,6,7 thermodynamic stability8 and low operating potential for energy storage and conversion.9 Therein, Ni phosphate has become the research hot topic because of its rich natural resources10 and high theoretical specific capacity.11 Metal-rich phosphides, especially Ni2P, has been extensively studied on account of their lower complexity of synthesis methods12,13 and superior chemical stability than other Ni-based phosphorus-rich phosphide.14,15 Nevertheless, the large volume expansion and decreased electrical conductivity during circulation seriously limit their cycling stability.16 Researchers have been involved in intense efforts to overcome these shortcomings, mainly including transferring the material to nanoscale,17 embedding with carbon coating,18,19 and optimization of the morphology.20 Among various strategies, making Ni2P-based hybrids by introducing carbon matrix have been proved to be highly effective way. The adopted carbon matrix such as porous carbon,21,22 graphene,23,24 graphene oxide,25,26 reduced graphene oxide,27,28 etc. can often serve as the remarkable buffer reservoir of electrode material expansion and electrolyte so that significantly enhance the transport efficiency of electron and lithium ion.29,30 For example, Dong et al.31 synthesized a construction of sandwich-like Ni2P nanoarray through hydrothermal method and two-step high temperature annealing. Results demonstrate that the interaction between Ni2P and 2D graphene can reduce the mechanical strain and enhance material conductivity, which leads to an impressive reversible capacity of 417 mA h g−1 at 300 mA g−1 for LIBs. Zhang et al.32 accomplished a structure of hyperfine Ni2P nanoparticles inserted in planiform carbon skeleton (Ni2P@C). They indicated that the carbon skeleton can boost electronic conductivity LIBs and the rough surface can expand the contact area between the electrodes and the electrolyte. The specific capacity of the Ni2P@C electrode reaches up to 265.5 mA h g−1 after 400 cycles. Both 2D and 3D carbon substrates can not only well alleviate the volume deformation of TMPs anode, but also be beneficial in the aspects of electrical conductivity.33,34 The TMPs embedded with the carbon material are usually sphere,35 flake array36 or random particle,37 while there is still lots of interesting attempts worthy of conducting in designing the multi-channel 3D porous structure of Ni2P growing on carbon matrix.38,39 Porous structure has been extensively used in functional materials for its fantastic advantage on sufficient specific surface, fast mass transport, and volume expansion buffering.40,41 It is also noteworthy that the recent research on Ni2P anode mainly focus on the design and synthesis of nanosized Ni2P materials, while research on micron-Ni2P materials was ignored to some extent. Although electrodes fabricated by nanoparticles have higher electrochemical energy storage performance than those made of micronparticles, in most cases, the former one has lower energy density than that of the latter.42 Beyond that, micronparticles are easier to prepare than nanoparticles during electrode production. All the foregoing considerations offer enormous impetus for seeking out appropriate strategy that use to produce micronscale Ni2P materials with multi-channel 3D structure and load it on carbon substrates simultaneously.
Herein, we attempted to design and fabricate a micro-sized Ni2P@rGO with 3D construction by two-step process involving hydrothermal treatment and phosphating strategy (illustrated in Fig. 1). Consequently, micro-sized 3D rose-like architecture assembled by 2D interconnected Ni2P nanoflakes immobilized on rGO was obtained, and it demonstrates largely boosted electrochemical properties when used as anode for LIBs, with a capacity of 817.8 mA h g−1 under 100 mA g−1, and delivering satisfactory cycling stability with capacity conservation rate of nearly 67% after 100 cycles at 100 mA g−1. This is mainly contributed chiefly by the hierarchical porosity architecture which reduces the average deformation force of the material resulting from the increasing specific surface area and accompanying remission of material pulverization when lithium ion insertion/extraction from the anode.43 Furthermore, the rGO in composite not only serves as framework, but also provides a good conductive network and synergistic effect to prevent particle from aggregation, which lead to a large initial capacity, impressive rate capability and intact retention of morphology.
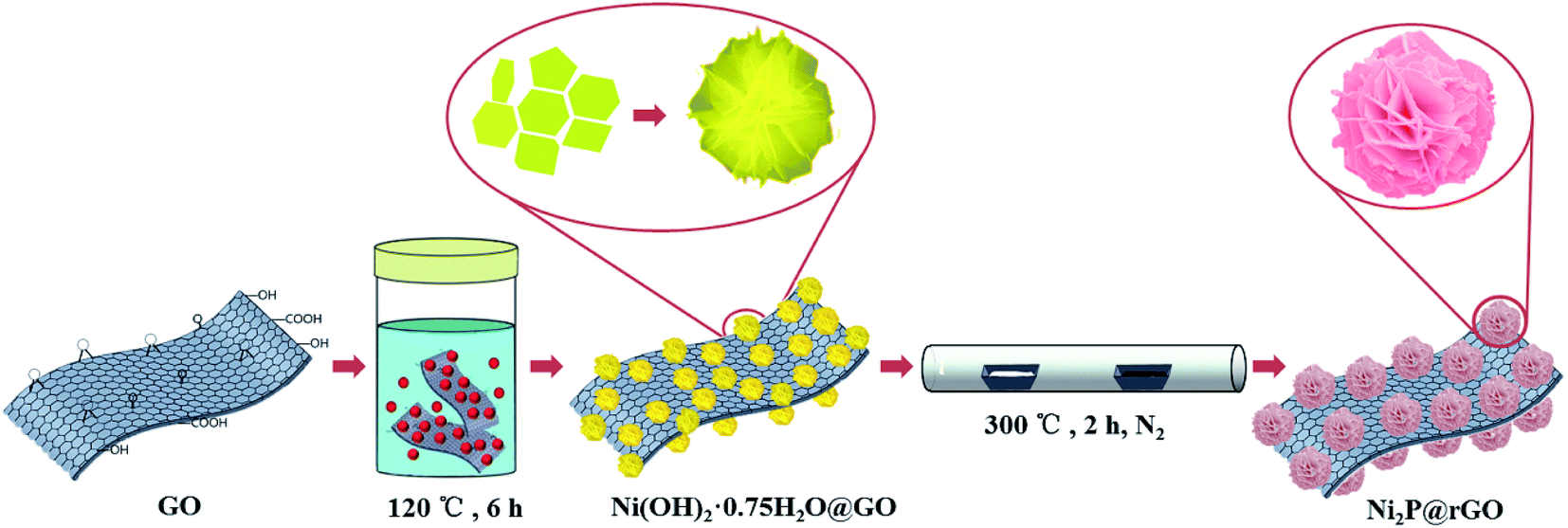 | ||
| Fig. 1 Schematic illustration of the formation of the 3D rose-like Ni2P@rGO composite. | ||
2. Experimental
2.1 Materials preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. Subsequently, the materials were heated at 300 °C for 2 h under a heating rate of 2 °C min−1 in N2 atmosphere. After naturally cooling, the black sample was collected and it was labeled as Ni2P@rGO.
:5. Subsequently, the materials were heated at 300 °C for 2 h under a heating rate of 2 °C min−1 in N2 atmosphere. After naturally cooling, the black sample was collected and it was labeled as Ni2P@rGO.2.2 Materials characterization
The morphologies structure of the as-obtained materials was acquired by a scanning electron microscope (SEM, FEI Inspect F50) and a transmission electron microscope (TEM, JEOL JEM-2100F). X-ray diffraction (XRD) patterns to analyze the phase structure of samples were detected by a X'Pert PRO with Cu Kα radiation, in a scan of 0.0262° s−1 at 40 mA, 40 kV. X-ray photoelectron spectroscopic (XPS) investigation was employed an Escalab 250Xi, Thermo Scientific spectrometer using an Al Kα X-ray source. Raman spectroscopy was obtained by HR EVOLUTION at a wavelength of 785 nm. The graphene content of Ni2P@rGO composite was characterized by thermal gravimetric analysis (TGA) on a STA 409 F3 at a heating rate of 10 °C from 40–900 °C in air atmosphere. Fourier Transform Infrared Spectroscopy (FTIR) were tested on a PerkinElmer Shelton, CT06484.2.3 Electrochemical measurements
The electrochemical measurements were performed on CR2032 coin cells. The anode electrode was prepared through mixing the active materials (as-prepared materials), conductive agent (acetylene black) and binder (polyvinylidene difluoride, PVDF) with a mass fraction of 80 wt%, 10 wt% and 10 wt%, respectively. The mixture was coated on a copper current collect and dried in vacuum for 12 hours. The active material loading electrode was controlled at 1.8 ± 0.2 mg cm−2. The adopted electrolyte for cells was 1 M LiPF6 dissolved in a 4:1 (v:v) ethylene carbonate (EC)–diethyl carbonate (DEC) mixture. Galvanostatic charge/discharge tests were executed in a voltage range of 0.01–3.0 V via battery test station (Neware, China). Zennium IM6 electrochemical workstation was employed to gather electrochemical impedance spectra (EIS) data from 100 kHz to 0.01 Hz at room temperature. Cyclic voltammetry (CV) profiles were investigated on an electrochemical workstation (LK9805) between 0.01 and 3.0 V with scan of 0.2 mV s−1.
3. Results and discussion
The in situ synthesis process of 3D rose-like Ni2P@rGO is schematically illustrated in Fig. 1. In the hydrothermal process, the oxygenic functional groups on the surface of GO provides abundant active sites for the adsorption of nickel ions (Ni2+), and the resulting chemical bonds enable the subsequent precursor to grow tightly on GO. Ni2+ is attracted by active sites which uniformly distributed on the GO surface and form a tight bond with the help of oxygen-containing functional group. NH4F and H2NCONH2 then irrigate these Ni2+ seeds to make the “rose” bloom. Addition of the ammonium fluoride leads the precursor to grow in situ with 3D hierarchical rose-like morphology.38 During low temperature phosphating, PH3 is generated by NaH2PO2·H2O thermal decomposition42 and reach the downstream and graphene oxide driven by the upstream airflow, and the 3D rose-like structure is well remained after the annealed phosphating. Accordingly, 3D rose-like Ni2P@rGO is obtained. Without graphene oxide or graphene, the precursor is assembled spontaneously to a 3D structure centered on Ni2+ dispersed in the solvent at high temperatures. When graphene is brought into system, 3D rose-like Ni2P can not incorporate well with graphene and reflect in only a fraction of crystal assembled with graphene and plentiful crystal present independently, as will be demonstrated in the following section.The representative photograph (Fig. 2a) of Ni(OH)2·0.75H2O@GO reveals a large number of microflowers with a diameter of about 2–4 μm growing on the GO sheets base closely, whereas without the attraction of strong bonds of GO substrate ultrathin 2D nanoflakes assembled to the shape of a rose, even under situation of graphene introduction, a large proportion of roses still grow independently (Fig. S1a and c†). It is interesting to find that 3D rose-like structure assembled from 2D interconnected nanoflakes further held after annealing processes in N2 regardless of mingling carbon material or not, as shown in Fig. 2b, c, S1b and S1d,† apart from the nanoflakes growing ever thicker. An intact and regular structure of rose petals can be further illustrated by TEM images in Fig. 2d. Simultaneously TEM image shown in Fig. 2e provides more evidence for the uniform distribution of 3D rose-like Ni2P on rGO basement. HRTEM image in Fig. 2f of Ni2P@rGO shows well-defined crystal lattice with the interplanar distance of 0.221 nm, matching well with the (111) crystalline face of the Ni2P phase. EDX element mapping result shown in Fig. 2g reveals an evenly distribution of C, P, and Ni elements in the 3D rose-like Ni2P@rGO composite. All the results suggest that 3D rose-like Ni2P@rGO was successfully constructed through a facile in situ two-step method.
 | ||
| Fig. 2 (a) SEM images of Ni(OH)2·0.75H2O@GO precursor. (b and c) SEM images of Ni2P@rGO. (d and e) TEM images of Ni2P and Ni2P@rGO. (f) HRTEM images of Ni2P@rGO. (g) EDX mapping of Ni2P@rGO and the corresponding elemental mappings of C, P and Ni elements (as labeled). | ||
To notarize the phase structure, the XRD patterns of the precursor Ni(OH)2, Ni(OH)2@G, Ni(OH)2@GO are illustrated in Fig. S2a.† Diffraction peaks located at 11.3°, 22.7°, 34.4°, 38.7° match well with the (003), (006), (012) and (015) crystal planes of Ni(OH)2·0.75H2O (JCPDS: 38-0715), respectively.44 After the phosphating processes, Ni(OH)2, Ni(OH)2@G, Ni(OH)2@GO are transformed into Ni2P, Ni2P@G and Ni2P@rGO, respectively, as manifestly justified by XRD patterns in Fig. S2b† and 3a. For the Ni2P@rGO sample, the diffraction peaks at 40.8°, 44.6°, 47.3° and 54.2° can be indexed to (111), (201), (210) and (300) planes of Ni2P phase (JCPDS No. 74-1385).45 The broad peak at ∼22° was corresponding to the graphene frame structure.46 TGA result is shown in Fig. S3,† there is a weight loss of the material continuously decreased before ∼400 °C due to the burning of carbon in the Ni2P@rGO composite material. It figures out that the mass fraction of graphene oxide is about 8.89 wt%. The Raman spectra in Fig. S4a† of the 3D rose-like Ni2P@rGO sample displays the characteristic D bands corresponding to defective carbon on 1350 cm−1 and G bands of graphitic sp2− type carbon on 1590 cm−1, respectively. Compare to the individual GO, the peak positions of 3D rose-like Ni2P@rGO are shifted to lower wavelength, signifying that the oxygen functional groups of GO were reduced.47 The chemical bond between Ni2P and rGO was further explored by FTIR and displayed in the Fig. S4b.† For pure graphene oxide, there are three absorption peaks at 1066, 1635 and 3430 cm−1, corresponding to C–O stretching in C–O–C group, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carboxyl stretching, C–OH stretching, respectively. While in Ni2P@rGO, the strength of these three peaks is decreased, which is most probably caused by chemical interaction between graphene and Ni2P.24 Beyond that, other characteristic peaks in Ni2P@rGO are caused by the presence of Ni2P.
O carboxyl stretching, C–OH stretching, respectively. While in Ni2P@rGO, the strength of these three peaks is decreased, which is most probably caused by chemical interaction between graphene and Ni2P.24 Beyond that, other characteristic peaks in Ni2P@rGO are caused by the presence of Ni2P.
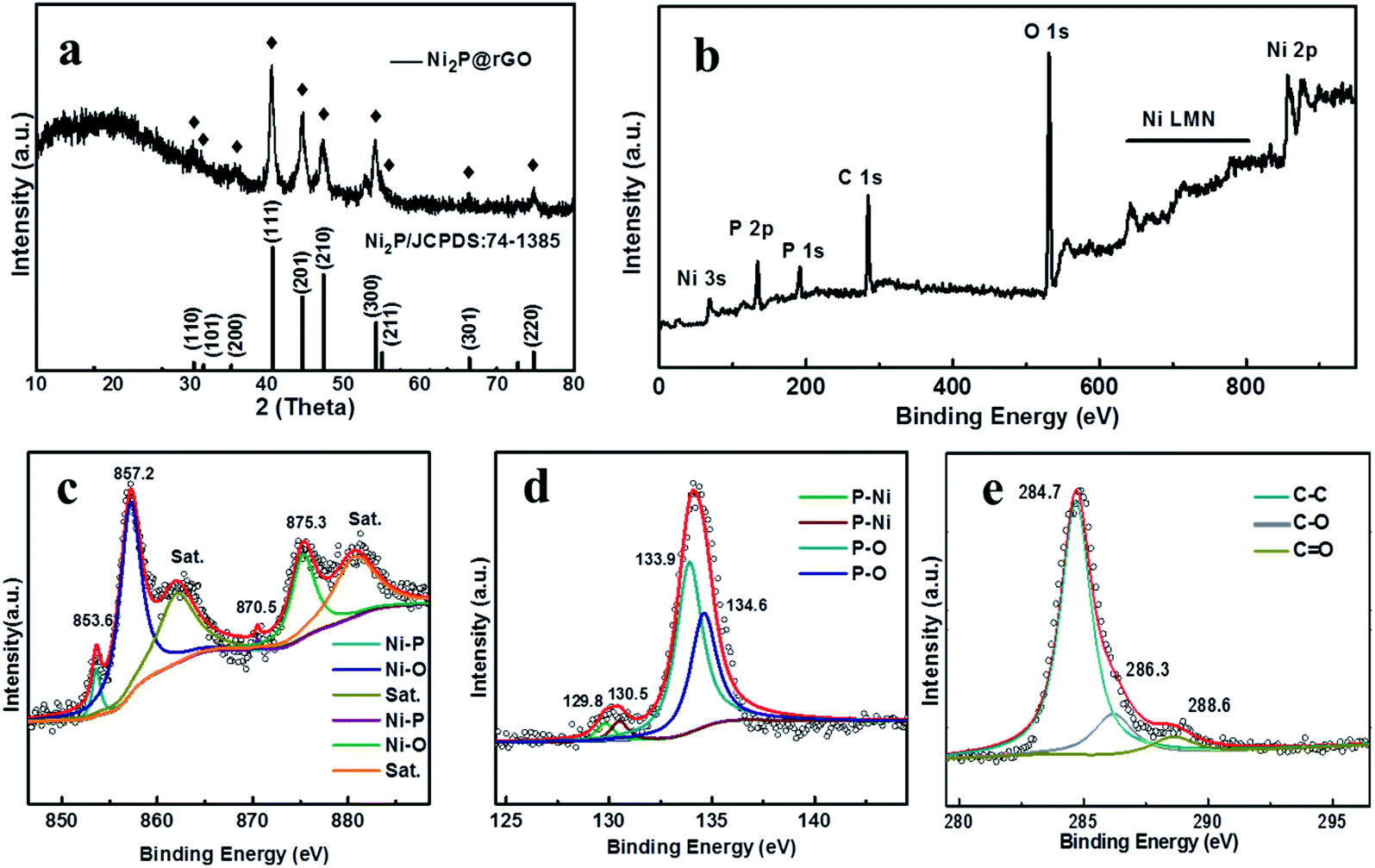 | ||
| Fig. 3 Characterizations of Ni2P@rGO: (a) XRD patterns. (b) Full XPS spectra of Ni2P@rGO. High resolution spectra of (c) Ni 2p (d) P 2p, and (e) C 1s. | ||
The chemical composition of Ni2P@rGO is further proven by XPS and discussed in Fig. 3b–e. Fig. 3b shows the full XPS spectrum of Ni2P@rGO composite, in which Ni, P, and C elements are detected. In the high-resolution Ni 2p spectrum (Fig. 3c), there are six prominent bands: two satellite peaks locate at binding energy of 861.4 and 879.3 eV, Ni 2p3/2 and Ni 2p1/2 of Ni–P bands are represented by the peak at 853.6, 870.5, and characteristic peak at 857.2 and 875.3 eV are corresponding to the Ni 2p3/2 and Ni 2p1/2 of Ni–O, respectively.31 Fig. 3d describes the high-resolution XPS spectrum of P 2p, The peaks located at 129.8 and 130.5 eV were matched for P 2p3/2 and P 2p1/2 of P–Ni, and the peaks at 133.9 and 134.5 eV were matched for P 2p3/2 and P 2p1/2 of P–O, and this result is in accordance with the existing results in the literature.48 In the C 1s XPS spectrum as shown in Fig. 3e, three peaks can be found, which correspond to sp2-hybridized C–C and oxygen containing functional groups of C–O and CO respectively.49 Peaks of oxygenated functional groups is significantly decreased associated with successful reducing reaction on GO. Moreover, the XPS spectra of Ni 2p and P 2p of the as-prepared Ni2P sample (Fig. S5a and b†) exhibit the same spectra with Ni2P@rGO sample, indicating that the introduction of rGO would not affect the formation of Ni2P.
The galvanostatic discharge–charge curves of initial two cycles for the three electrodes at 100 mA g−1 are shown in Fig. 4a and b. It further demonstrates difference in charge and discharge mechanisms which lead to the capacity gaps among them. As displayed in Fig. 4a, the formation of SEI film mainly occurs at about 1.4 V in the first cycle and then vanish in the subsequent cycles. Along with the advancement of discharge/charge process, we can find that the discharge platform of Ni2P@rGO in the second discharge lap is lower than that of the other two materials, which illustrates that rGO is beneficial to alleviate the polarization phenomenon, thereby preserves a stable electrochemical kinetics. The galvanostatic discharge/charge curves of Ni2P@rGO at different current densities is displayed in Fig. 4c. It should be noted that the discharge/charge curves in the second cycle of each current density were selected for comparison, in order to exclude the unstable factors in the current switching process and better demonstrate the real rate performance of the material. The results demonstrate almost coincided trend illustrating reversible Li+ insertion/extraction among the 3D channels and splendid stability of material structure. To give a further insight into the electrochemical reaction mechanism to Ni2P@rGO electrode, Fig. 4d give the CV curves of initial three cycles for Ni2P@rGO at scan rate of 0.2 mV s−1 in the voltage range of 0.01 to 3 V. The cathodic and anodic peaks are concerned with the reaction of Ni2P + 3Li+ + 3e− ↔ Li3P + 2Ni.50 Obviously, the CV curve of the initial cycle was different from those of the next cycles, the broad peak at ∼0.5 V corresponds to the Li+ imbedding in Ni2P and the peak lay on ∼1.4 V could be inferred as the formation of SEI film. In the subsequent cycles, the peak of ∼1.4 V fade away and the peak of ∼0.5 V is moved to ∼1.6 V, testifying that the formation of SEI film mainly happens in first cycle. As for anodic scan, namely the charge process, there are three peaks, which represent the transformations from Ni to Ni12P5, Ni5P2 and Ni2P, respectively.15,51 In addition, starting from the second cycle, the CV curves tend to be stable, which is indicative of the reversible Li-ion exchange in the electrochemical system.
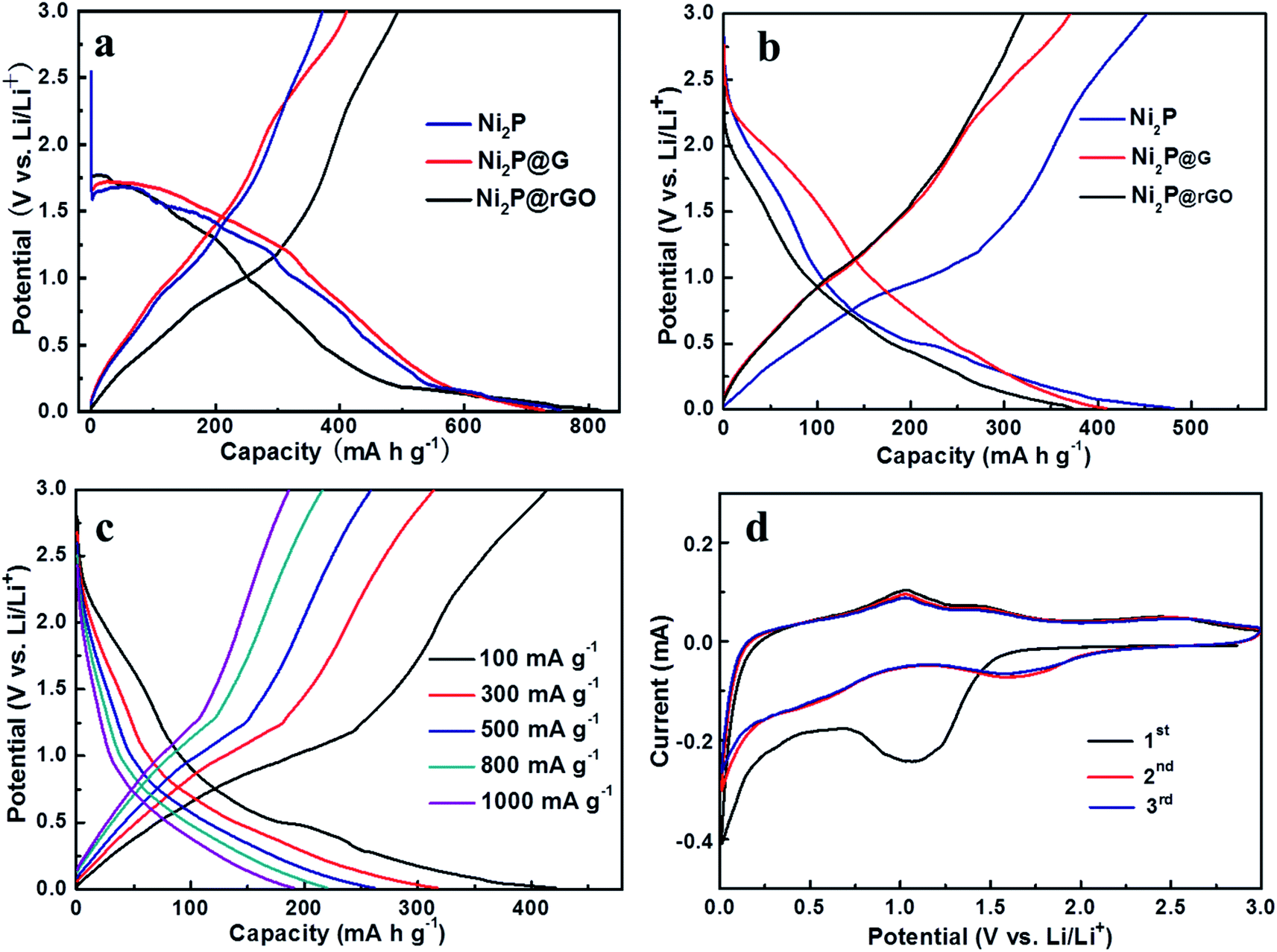 | ||
| Fig. 4 Galvanostatic discharge–charge profiles of Ni2P, Ni2P@G and Ni2P@rGO at 100 mA g−1 in different cycle of 1st cycle (a) and 2nd cycle (b), and Ni2P@rGO at different current densities (c), and (d) CV curves of Ni2P@rGO at a constant scan rate of 0.2 mV s−1 from 0.01 to 3 V. | ||
The rate performance comparison of the as-obtained three electrodes is shown in Fig. 5a. Apparently, the Ni2P@rGO electrode exhibits the optimum rate capability, with reversible capacity of 388.7, 304.6, 255.2 and 214.5 mA h g−1 when the current density increases gradually from 100 to 300, 500 and 800 mA h g−1, respectively. And even the current density reach 1000 mA g−1, the reversible capacity is still maintained 200.5 mA h g−1. The specific discharge capacity can recover to 288.2 mA h g−1 when the current density reset to 100 mA g−1, implying good lithium storage kinetics of Ni2P@rGO. In comparison, the Ni2P@G electrode delivers 330.2, 239.7, 191.6, 152.6 and 131.9 mA h g−1 at the identical current density, and for pure Ni2P the corresponding capacity is 341.3, 207.5, 147.3, 103.2 and 83.2 mA h g−1, respectively. Remarkable rate performance of Ni2P@rGO can be ascribed to the enhanced electrical conductivity enhanced by virtuous mutual promotion between rGO and 3D hierarchical structure. Furthermore, carbon base framework plays a key role of maintaining the 3D rose-like hierarchical structure stability while resisting impact of tremendous quantity of ions under a large current.52 It also provides a firm conducting network, reducing the charge transfer resistance so as to facilitate electron transfer, which is also proved in the impedance spectroscopy delivered below.
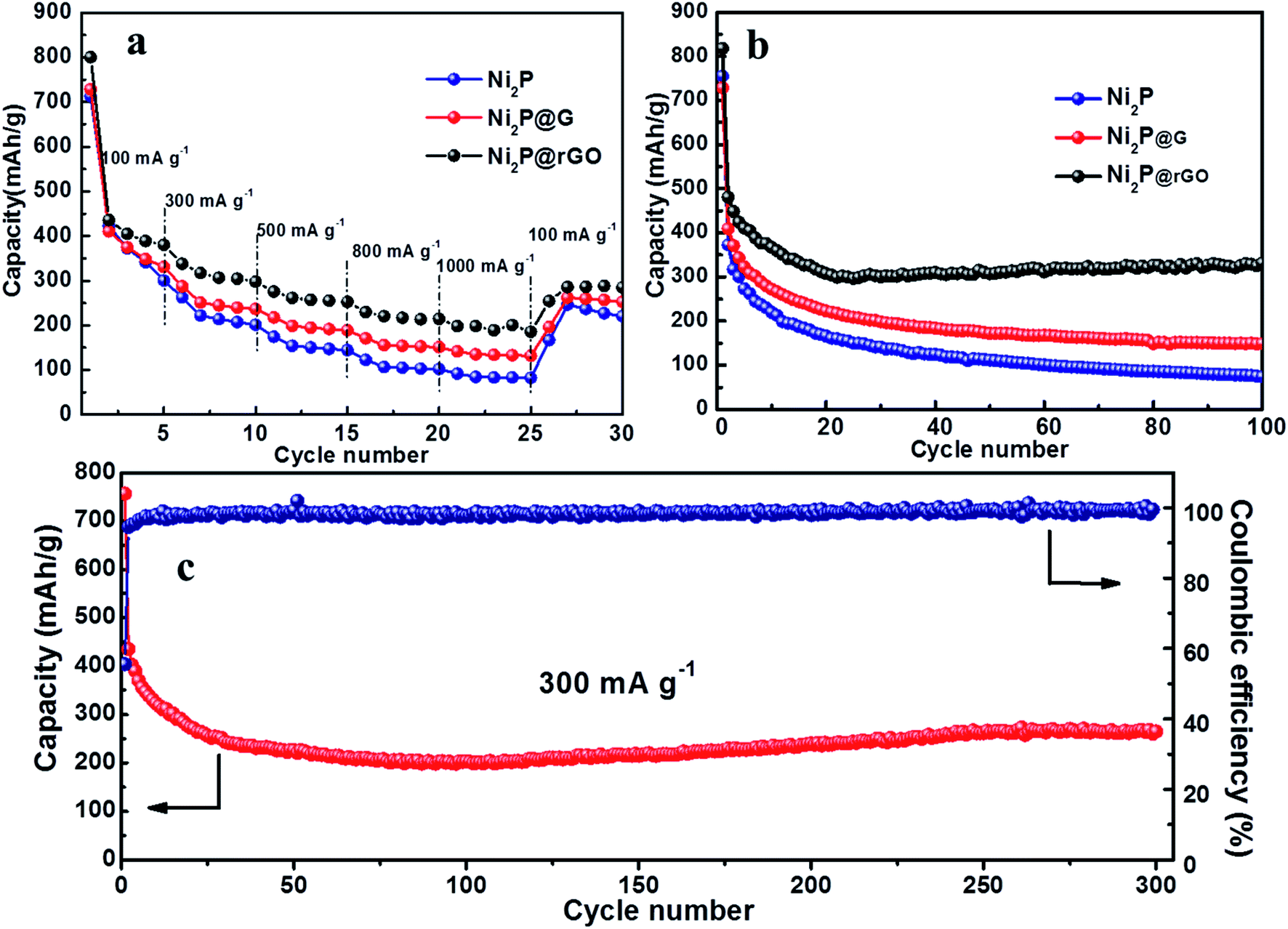 | ||
| Fig. 5 (a) Rate performance of Ni2P, Ni2P@G and Ni2P@rGO at different current densities from 100 mA g−1 to 1 A g−1. (b) Cycling performance of Ni2P, Ni2P@G and Ni2P@rGO at 100 mA g−1. (c) Long cycle performance of Ni2P@rGO at 300 mA g−1. | ||
Cycling stability of these three electrodes were further studied at a current density of 100 mA g−1, as shown in Fig. 5b. For the 3D rose-like Ni2P@rGO electrode, it exhibits a discharge and charge capacity of 817.8 and 493.8 mA h g−1, respectively, achieving a coulombic efficiency (CE) of ∼60.4%. The irreversible capacity derives from the formation of a solid electrolyte interface (SEI).53 In the subsequent cycles, the CE increases to nearly 100%, which is indicative of favorable invertibility of the electrode material. After 100 cycles, it still exhibits a superior electrochemical performance with a reversible capacity of 330.5 mA h g−1. Aware that graphene is also active for Li+ intercalation/deintercalation, the reversible capacity contribution of graphene in Ni2P@rGO can't be ignored. According to our TGA results mentioned above and the electrochemical performance of pure graphene materials collected in the literatures,54–56 the reversible capacity contribution of graphene is presumably 40 mA h g−1. It shows that the specific capacity of Ni2P@rGO is much higher than the algebraic sum of graphene and pure Ni2P under the same test condition, revealing that the excellent capacity performance thanks mainly to the synergistic effect.
On the contrary, Ni2P and Ni2P@G electrodes show much poorer specific capacity capability and a relatively rapid attenuation in the subsequent process. The initial discharge capacities of Ni2P and Ni2P@G can reach up to 754.9 and 728.3 mA h g−1, however, 100 cycles later, only deliver 74.3 and 149.5 mA h g−1, respectively. Splendid cyclic stability is closely related to material structure conservation during cycling process which is figured out by SEM images of three materials after 100 cycles, as shown in Fig. S6.† It can be seen that there are almost no distinct changes in the overall appearance of 3D rose-like architecture for Ni2P@rGO, as a result of increasing compressive strength lifted by harmonious symbiosis between 3D rose and flexile rGO. As a comparison, the structure of Ni2P is completely pulverized and without any complete rose-like structure remained after long-term lithium ion constantly embedded in and out. Meanwhile, by reason of insufficient and weak binding between Ni2P and graphene, the petal structure is ruined and only the vague outline can be seen. Furthermore, long cycle life at 300 mA g−1 for Ni2P@rGO electrode was also explored and shown in Fig. 5c. The specific discharge capacity decreases gradually during the first 80 cycles, then shows a rising tendency and stabilizes at 245.8 mA h g−1 until 300 cycles. The gradual increase in reversible capacity during the cycling can be attributed to the continuous motivation of the electrode material during the cycle, where the electrolyte invades into the electrode material stage by stage, which is a common phenomenon in TMP@C materials.57 By comparing with some reported Ni2P-based composites (Table 1), we can find that the Ni2P@rGO performs competitive cycling capacity to some extent. Observed from SEM image (Fig. S6d†) for Ni2P@rGO electrode cycling after 300 times, it still retains the original morphology characteristics of 3D hierarchical rose-like structure assembled from interconnected nanoflakes although some degree of pulverization and collapse occurs, which explains why it keeps a high reversible capacity after 300 cycles.
EIS measurement was carried out to analyze the electrode reaction impedance and the diffusion of the three electrodes. Fig. 6a gives the Nyquist plots, which are mainly composed of a semicircle and an oblique line in the high-frequency region and low-frequency region, respectively. With the simplified equivalent circuit (Fig. 6b), the impedances the experimental impedance data were simulated and shown in Fig. 6c. Rs is related to the electrolyte resistance. The semicircle in high-frequency is related to the SEI film impedance (Rf) and charge transfer impedance (Rct) at the electrode–electrolyte interface. Sloping line means lithium ion diffusion impedance ascribed to the Warburg impedance. The constant-phase element (CPE) is double layer capacitance. It is clear that the semicircle diameter of Ni2P@rGO is much smaller than those of Ni2P@G and Ni2P electrodes, implying much lower SEI film impedance and charge transfer impedance. The specific resistance values of each part are also calculated by fitting and listed in Table 2. Results show that the Rs values of the three materials are very similar, resulting from the same test conditions and battery package methods. The Rf value for Ni2P, Ni2P@G and Ni2P@rGO has a trend to decline which are 44.28 ohm, 11.17 ohm and 9.79 ohm, respectively. It can also find the Rct value of Ni2P@rGO (30.53 ohm) was only a quarter of that in Ni2P (121.60 ohm). This phenomenon can be explained by the rGO skeleton helped electrons migrate to active materials which reduces the charge transfer impedance. Eqn (1) and (2) were employed to acquire the lithium ion diffusion coefficient in the three battery systems to further explore the electrode performance.51
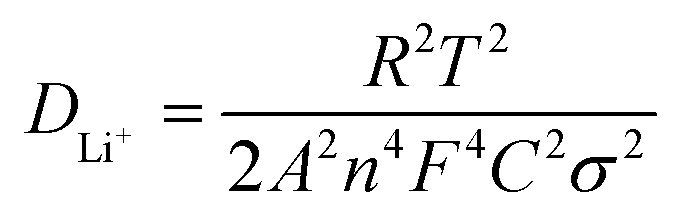 | (1) |
| Zre = Re + Rct + σω−1/2 | (2) |
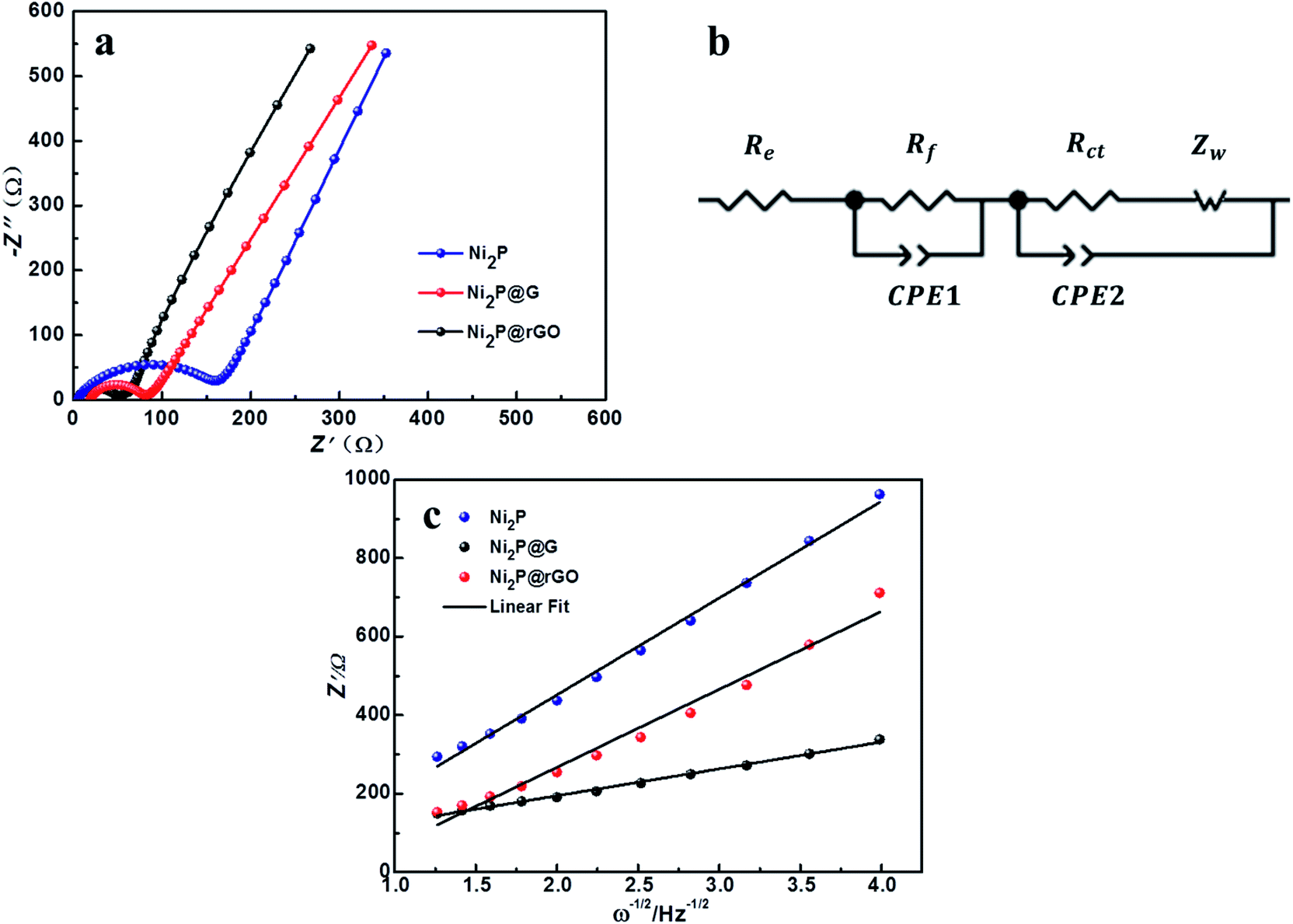 | ||
| Fig. 6 (a) EIS profiles of Ni2P, Ni2P@G, Ni2P@rGO. (b) Equivalent circuit diagram. (c) The relationship between Z′ and ω−1/2 at low frequency. | ||
| Re (Ω) | Rf (Ω) | Rct (Ω) | DLi+ (cm−2 s−1) | |
|---|---|---|---|---|
| Ni2P | 5.11 | 44.28 | 121.60 | 1.09 × 10−13 |
| Ni2P@G | 5.01 | 11.17 | 65.49 | 1.68 × 10−13 |
| Ni2P@rGO | 3.84 | 9.79 | 30.53 | 1.41 × 10−12 |
The diffusion coefficients lithium ions of three materials were calculated and listed in Table 2. A significantly larger DLi+ of Ni2P@rGO with 1.41 × 10−12 cm−2 s−1 can be found than others, which is in good agreement with its enhanced electrochemical performance, indicating rGO formed a better bond with the Ni2P leading to increase its electrical conductivity and the transport rate of Li+.
Based on the above analyses, the boosted electrochemical performance of Ni2P@rGO mainly thanks to its microstructure features, which combines the advantages of 3D hierarchical rose-like Ni2P nanoflake and deformation-resistant structure of rGO. The 3D hierarchical structure raises the lithium storage ability and the rGO skeleton increases the contact area between electrode and electrolyte so that reduces the resistance and distributes the stress. Besides, the rGO matrix who acts as a flexible chassis can regulate the volume expansion of 3D rose-like Ni2P during the cycle and provide a conductive framework for rapid electronic transfer.
4. Conclusion
In summary, we designed and fabricated a micro-sized 3D rose-like Ni2P@rGO composites via a mild two-step method involving hydrothermal process and phosphating treatment. This hierarchical structure is assembled from 2D interconnected Ni2P nanoflakes immobilized on rGO matrix. When served as anode material in LIBs, the composite demonstrates enhanced electrochemical performance in respect of rate capability, reversibility, and cycle stability, which is endowed with a reversible capacity of 200.5 mA h g−1 at 1000 mA g−1, and 245.8 mA h g−1 at 300 mA g−1 during 300 times cycling measurement. The remarkable electrochemical performance is ascribed to the synergistic effect between 3D hierarchical rose-like Ni2P and rGO in which rGO provides a conductive framework for Ni2P, and avoids its structure from pulverize in long cycles. This study provides a new strategy to develop high-performance electrode materials of 3D hierarchical structured metal phosphides immobilized on rGO matrix, and can be extended to other functional material systems for electrochemical energy storage and conversion applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work acknowledges the support of the Research Foundation for the Postdoctoral Program of Sichuan University (No. 2017SCU12018), National Key Research Projects (No. 2017YFB0307504) and Sichuan Science and Technology Planning Project (No. 2019YFH0149).References
- R. Kumar, S. Sahoo, E. Joanni, R. K. Singh, W. K. Tan, K. K. Kar and A. Matsuda, Prog. Energy Combust. Sci., 2019, 75, 100786 CrossRef.
- S. J. Yu, V. M. H. Ng, F. J. Wang, Z. H. Xiao, C. Y. Li, L. B. Kong, W. X. Que and K. Zhou, J. Mater. Chem. A, 2018, 6, 9332–9367 RSC.
- X. Wang, H. M. Kim, Y. Xiao and Y. K. Sun, J. Mater. Chem. A, 2016, 4, 14915–14931 RSC.
- M. S. Kim, B. Z. Fang, J. H. Kim, D. Yang, Y. K. Kim, T. S. Bae and J. S. Yu, J. Mater. Chem., 2011, 21, 19362–19367 RSC.
- W. Y. Long, B. Z. Fang, A. Ignaszak, Z. Z. Wu, Y. J. Wang and D. Wilkinson, Chem. Soc. Rev., 2017, 46, 7176–7190 RSC.
- H. N. Guo, C. C. Chen, K. Chen, H. C. Cai, X. Y. Chang, S. Liu, W. Q. Li, Y. J. Wang and C. Y. Wang, J. Mater. Chem. A, 2017, 5, 22316–22324 RSC.
- H. T. Lei, M. X. Chen, Z. Z. Liang, C. Y. Liu, W. Zhang and R. Cao, Catal. Sci. Technol., 2018, 8, 2289–2293 RSC.
- Y. Pei, Y. Cheng, J. Y. Chen, W. Smith, P. Dong, P. M. Ajayan, M. X. Ye and J. F. Shen, J. Mater. Chem. A, 2018, 6, 23220–23243 RSC.
- X. B. Liu, W. X. Li, X. D. Zhao, Y. C. Liu, C. W. Nan and L. Z. Fan, Adv. Funct. Mater., 2019, 29, 1–9 Search PubMed.
- P. Jiang, Q. Liu and X. P. Sun, Nanoscale, 2014, 6, 13440–13445 RSC.
- Y. Y. Feng, Y. OuYang, L. Peng, H. J. Qiu, H. L. Wang and Y. Wang, J. Mater. Chem. A, 2015, 3, 9587–9594 RSC.
- J. Yang, N. Yang, Q. Xu, L. S. Pearlie, Y. Z. Zhang, Y. Hong, Q. Wang, W. J. Wang, Q. Y. Yan and X. C. Dong, ACS Sustainable Chem. Eng., 2019, 7, 13217–13225 CrossRef CAS.
- J. Fullenwarth, A. Darwiche, A. Soares, B. Donnadieu and L. Monconduit, J. Mater. Chem. A, 2014, 2, 2050–2059 RSC.
- P. Lou, Z. Cui, Z. Jia, J. Sun, Y. Tan and X. Guo, ACS Nano, 2017, 11, 3705–3715 CrossRef CAS PubMed.
- Y. Feng, H. Zhang, Y. Mu, W. Li, J. Sun, K. Wu and Y. Wang, Chem.–Eur. J., 2015, 21, 9229–9235 CrossRef CAS PubMed.
- Y. Lu, X. L. Wang, Y. J. Mai, J. Y. Xiang, H. Zhang, L. Li, C. D. Gu, J. P. Tu and S. X. Mao, J. Phys. Chem. C, 2012, 116, 22217–22225 CrossRef CAS.
- J. Song, S. Park, V. Mathew, J. Gim, S. Kim, J. Jo, S. Kim, M. H. Alfaruqi, J. P. Baboo, I.-H. Kim, S.-J. Song and J. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 35235–35242 CrossRef CAS PubMed.
- Y. Wang, Q. Pan, K. Jia, H. B. Wang, J. J. Gao, C. L. Xu, Y. J. Zhong, A. A. Alshehri, K. A. Alzahrani, X. D. Guo and X. P. Sun, Inorg. Chem., 2019, 58, 6579–6583 CrossRef CAS PubMed.
- X. Miao, R. Yin, X. Ge, Z. Li and L. Yin, Small, 2017, 13, 1702138 CrossRef PubMed.
- J. Zheng, X. Huang, X. Pan, C. Teng and N. Wang, Appl. Surf. Sci., 2019, 473, 699–705 CrossRef CAS.
- Y. L. Xing, S. B. Wang, B. Z. Fang, G. Song, D. P. Wilkinson and S. C. Zhang, J. Power Sources, 2018, 385, 10–17 CrossRef CAS.
- M. S. Kim, D. Bhattacharjya, B. Z. Fang, D. S. Yang, T. S. Bae and J. S. Yu, Langmuir, 2013, 29, 6754–6761 CrossRef CAS PubMed.
- H. J. Li, S. Y. Hao, Z. Tian, Z. X. Zhao and X. M. Wang, Electrochim. Acta, 2019, 321, 134624 CrossRef CAS.
- Y. X. Zhang, L. Sun, L. Q. Bai, H. C. Si, Y. Zhang and Y. H. Zhang, Nano Res., 2019, 12, 607–618 CrossRef CAS.
- C. Wu, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1604015 CrossRef.
- M. Sun, H. J. Liu, J. H. Qu and J. H. Li, Adv. Energy Mater., 2016, 6, 1600087 CrossRef.
- C. An, Y. Wang, Y. Wang, G. Liu, L. Li, F. Qiu, Y. Xu, L. Jiao and H. Yuan, RSC Adv., 2013, 3, 4628–4633 RSC.
- Y. L. Shih, C. L. Wu, T. Y. Wu and D. H. Chen, Nanotechnology, 2019, 30, 115601 CrossRef.
- B. Fang, J. H. Kim, M. S. Kim, A. Bonakdarpour, A. Lam, D. P. Wilkinson and J. S. Yu, J. Mater. Chem., 2012, 22, 19031–19038 RSC.
- B. Fang, J. H. Kim, C. Lee and J. S. Yu, J. Phys. Chem. C, 2008, 112, 639–645 CrossRef CAS.
- C. F. Dong, L. J. Guo, Y. Y. He, C. J. Chen, Y. T. Qian, Y. N. Chen and L. Q. Xu, Energy Storage Mater., 2018, 15, 234–241 CrossRef.
- R. Z. Zhang, K. J. Zhu, J. D. Huang, L. Y. Yang, S. T. Li, Z. Y. Wang, J. R. Xie, H. Wang and J. Liu, J. Alloys Compd., 2019, 775, 490–497 CrossRef CAS.
- B. Wang, Q. Ru, C. Su, S. Cheng, P. Liu, Q. Guo, X. Hou, S. Su and F. C.-C. Ling, Chemelectrochem, 2018, 5, 1467–1473 CrossRef CAS.
- B. Z. Fang, S. Q. Fan, J. H. Kim, M. S. Kim, M. Kim, N. K. Chaudhari, J. Ko and J. S. Yu, Langmuir, 2010, 26, 11238–11243 CrossRef CAS PubMed.
- Z. X. Guang, Y. Huang, X. F. Chen, X. Sun, M. Y. Wang, X. S. Feng, C. Chen and X. D. Liu, Electrochim. Acta, 2019, 307, 260–268 CrossRef CAS.
- H. Li, X. Wang, Z. Zhao, Z. Tian, D. Zhang and Y. Wu, Chemelectrochem, 2019, 6, 404–412 CrossRef CAS.
- J. Jiang, C. Wang, W. Li and Q. Yang, J. Mater. Chem. A, 2015, 3, 23345–23351 RSC.
- H. Zheng, X. Huang, Z. Wu, H. Gao, W. Dong and G. Wang, Chem. - Asian J., 2017, 12, 2956–2961 CrossRef CAS PubMed.
- W. B. Hua, X. D. Guo, Z. Zheng, Y. J. Wang, B. H. Zhong, B. Fang, J. Z. Wang, S. L. Chou and H. Liu, J. Power Sources, 2015, 275, 200–206 CrossRef CAS.
- Y. L. Xing, Y. J. Wang, C. G. Zhou, S. C. Zhang and B. Z. Fang, ACS Appl. Mater. Interfaces, 2014, 6, 2561–2567 CrossRef CAS PubMed.
- B. Fang, M. S. Kim, J. H. Kim, S. Lim and J. S. Yu, J. Mater. Chem., 2010, 20, 10253–10259 RSC.
- H. N. Guo, H. C. Cai, W. Q. Li, C. C. Chen, K. Chen, Y. Zhang, Y. W. Li, M. Y. Wang and Y. J. Wang, Inorg. Chem. Front., 2019, 6, 1881–1889 RSC.
- B. Fang, M. Kim, S. Q. Fan, J. H. Kim, D. P. Wilkinson, J. Ko and J. S. Yu, J. Mater. Chem., 2011, 21, 8742–8748 RSC.
- K. Yao, M. Zhai and Y. Ni, Electrochim. Acta, 2019, 301, 87–96 CrossRef CAS.
- G. Chen, S. Tang, Y. Song, X. Meng, J. Yin, Y. Xia and Z. Liu, Chem. Eng. J., 2019, 361, 387–397 CrossRef CAS.
- D. F. Yang, B. H. Xu, Q. L. Zhao and X. S. Zhao, J. Mater. Chem. A, 2019, 7, 363–371 RSC.
- R. Kumar, E. Joanni, R. K. Singh, D. P. Singh and S. A. Moshkalev, Prog. Energy Combust. Sci., 2018, 67, 115–157 CrossRef.
- J. G. Tu, M. Y. Wang, X. Xiao, H. P. Lei and S. Q. Jiao, ACS Sustainable Chem. Eng., 2019, 7, 6004–6012 CrossRef CAS.
- L. G. Guex, B. Sacchi, K. F. Peuvot, R. L. Andersson, A. M. Pourrahimi, V. Strom, S. Farris and R. T. Olsson, Nanoscale, 2017, 9, 9562–9571 RSC.
- Q. Li, J. J. Ma, H. J. Wang, X. Yang, R. Yuan and Y. Q. Chai, Electrochim. Acta, 2016, 213, 201–206 CrossRef CAS.
- J. M. Wang, B. B. Wang, X. J. Liu, G. Wang, H. Wang and J. T. Bai, J. Colloid Interface Sci., 2019, 538, 187–198 CrossRef CAS PubMed.
- B. Fang, J. H. Kim, M. S. Kim and J. S. Yu, Acc. Chem. Res., 2013, 46, 1397–1406 CrossRef CAS PubMed.
- Y. Ma, J. Huang, L. Lin, Q. Xie, M. Yan, B. Qu, L. Wang, L. Mai and D.-L. Peng, J. Power Sources, 2017, 365, 98–108 CrossRef CAS.
- Y. Qian, L. Jiang, Z. Ullah, Z. Guan, C. Yu, S. Zhu, M. Chen, W. Li, Q. Li and L. Liu, Nanotechnology, 2019, 30, 225401 CrossRef CAS PubMed.
- K. H. Park, B. G. Kim and S. H. Song, Nanomaterials, 2019, 10, 9 CrossRef PubMed.
- X. X. Liu, D. L. Chao, D. P. Su, S. K. Liu, L. Chen, C. X. Chi, J. Y. Lin, Z. X. Shen, J. P. Zhao, L. Q. Mai and Y. Li, Nano Energy, 2017, 37, 108–117 CrossRef CAS.
- S. Shi, Z. Li, Y. Sun, B. Wang, Q. Liu, Y. Hou, S. Huang, J. Huang and Y. Zhao, Nano Energy, 2018, 48, 510–517 CrossRef CAS.
- Y. Lu, J. P. Tu, J. Y. Xiang, X. L. Wang, J. Zhang, Y. J. Mai and S. X. Mao, J. Phys. Chem. C, 2011, 115, 23760–23767 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10729k |
| This journal is © The Royal Society of Chemistry 2020 |