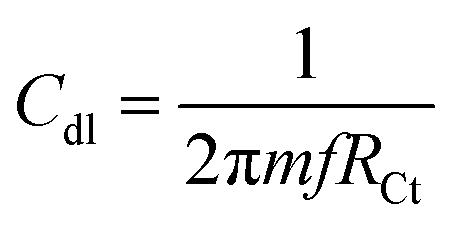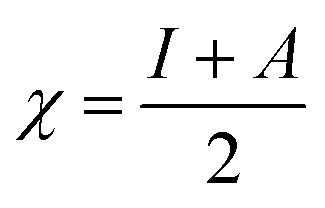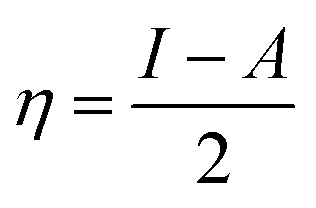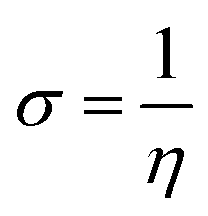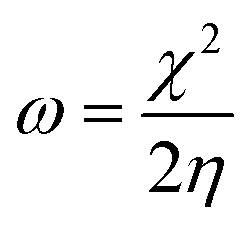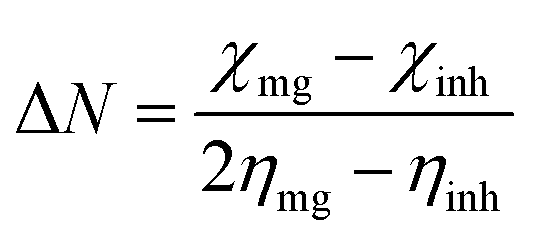DOI:
10.1039/C9RA10702A
(Paper)
RSC Adv., 2020,
10, 11426-11434
Experimental and computational studies of graphene oxide covalently functionalized by octylamine: electrochemical stability, hydrogen evolution, and corrosion inhibition of the AZ13 Mg alloy in 3.5% NaCl
Received
19th December 2019
, Accepted 27th February 2020
First published on 20th March 2020
Abstract
Recently, carbon allotropes were shown to play a key role in energy harvesting and as hydrophobic coatings on metal alloys. We have designed octylamine-functionalized graphene oxide materials for energy harvesting and as an anti-corrosion coating for metal alloy protection in a 3.5% NaCl medium. The material has been characterized by different techniques to confirm the structure and composition of the modified graphene oxide sheet: FTIR spectroscopy, XRD, Raman spectroscopy, FESEM and TEM. The electrochemical stability and corrosion inhibition efficiency were studied by electrochemical methods. The electrochemical stability increased with an increase in the applied voltage up to 500 mV, and the corrosion inhibition efficiency was shown to be 73%. The coating stability studies showed a long stability time in the corrosion medium.
1. Introduction
Graphene oxide and functionalized graphene oxide materials have been explored in the energy field and as hydrophobic coatings. Several authors investigated graphene oxide-functionalized materials for corrosion inhibition efficiency in different environments to check how the corrosion of metal alloys was controlled. Amino acid-functionalized graphene oxide was studied as a corrosion inhibition layer on magnesium alloys.1 The nanoparticle-incorporated graphene oxide behaved as a corrosion inhibition barrier on the mild steel alloy surface in an acidic medium.2,3 Furthermore, phenylenediamine-functionalized graphene oxide acted as a corrosion barrier layer on the copper surface.4 Virtanen discussed the biological importance of the Mg alloy and its interaction with the body and its fluids.5 Atrens et al. investigated how the addition of impurities such as Fe, Ni and Co improved the corrosion rate in a biological medium.6 Wen et al. showed that triethanolamine possessed excellent corrosion inhibition in a 3.5% NaCl environment. Their electrochemical results showed an inhibition efficiency of up to 91% due to the nonbonding electrons of the amine groups adsorbed onto the Mg alloy surface.7 Hoche et al. studied iron impurities in the form of ferric chloride in a corrosion medium of 3.5% NaCl environment. The results suggested that the iron impurities have a significant effect on the improvement of the corrosion inhibition efficiency of Mg alloys.8 Razavi et al. studied the recent research progress of Mg alloy implants. In the future, Mg alloys will play a significant role in implant materials due to their light weight and eco-friendliness in biological environments.9 Williams et al. examined the strong electrolyte FeCl2 as a corrosion inhibitor in a medium, and it controlled localized corrosion propagation.10 Miskovic et al. studied different compositions of magnesium alloys containing elements such as Zn and Ca to show how they improved the corrosion resistance of pure Mg alloys. They suggested that the hydrogen evolution was controlled by varying the composition of the alloy.11 Birbilis et al. investigated the corrosion inhibition of a lanthanum-coated Mg alloy in a 0.1 M NaCl solution. The results suggested that the La-coated alloy improved the corrosion resistance in a 1 M NaCl environment due to the high electron transfer capability of lanthanum.12 Chu et al. conducted studies of corrosion inhibition of a Mg alloy coated with carbon allotropes like the diamond-based carbon plasma in a bacterial culture.13 The carbon-coated Mg alloy showed excellent corrosion resistance. Wan et al. studied the corrosion inhibition of a phosphate-coated Mg alloy in a phosphate buffer solution. They found that the bisphosphate-coated Mg alloy showed strong corrosion resistance.14 Han et al. studied the corrosion inhibition in a hydroxyapatite (HA)-coated Mg alloy in a buffered solution, and found that the HA-coated Mg alloy improved the corrosion resistance in the buffer solution.15 The corrosion inhibition of an arginine-based polyester-coated Mg alloy was studied by Xi et al.16 They showed the arginine-decorated polyester to have excellent interaction with blood cells as compared with poly(glycolide)-co-lactide, and to have an inhibition efficiency that was higher as compared with the polyglycolide-coated Mg alloys. The corrosion inhibition of the magnesium alloy coated with graphene oxide by the plasma method was studied in a 3.5% NaCl solution by Wang.17 The results suggested that the corrosion inhibition of the Mg alloy increased in the NaCl medium with the coating by the plasma technique. The use of an aromatic amine covalently functionalized graphene oxide as a corrosion inhibitor was studied on mild steel in 3.5% NaCl solution by Bagherzadeh et al.18 They found that the naphthalene amine showed a higher inhibition efficiency. Also, R. Wang et al. investigated the corrosion inhibition of the plasma technique synthesised graphene oxide-coated Mg alloy in 3.5% NaCl solution. They concluded that the graphene oxide showed excellent corrosion inhibition efficiency in 3.5% NaCl medium.19 Also, the corrosion inhibition of the perfluorinated polysiloxane-functionalized graphene oxide coated Mg alloy was studied by Pyo et al. Their results showed excellent inhibition efficiency as compared with conventional epoxy-coated Mg alloys.20 The corrosion inhibition of the silane-incorporated graphene oxide-coated Mg alloy was studied in 3.5% NaCl medium by Zhang et al.21 The corrosion inhibition studies of the polyvinyl alcohol functionalized graphene oxide coated with 2024 aluminum alloy in 0.2 M NaCl medium were performed by Lutkenhaus.22 The results showed that with the increased percentage composition, the inhibition efficiency increased as well. Furthermore, the corrosion inhibition efficiency studies of the few-layer graphene oxide sheet-coated copper metal were carried out in 5% NaCl solution by Ham.23 The results showed that the alloy surface was smooth after being immersed in the corrosion medium for 4 days, and no pitting corrosion occurred on the alloy surface.24 Our previous work studied an amino acid-functionalized graphene oxide-coated magnesium alloy in 3.5% NaCl medium. After immersion for 5 days in the corrosion medium, the alloy did not form any corrosion product on the alloy surface due to the strong adhesion of the coating on the alloy surface.25,26 In the present work, we have completed studies of corrosion inhibition of the octylamine-functionalized graphene oxide-coated Mg alloy in a 3.5% NaCl medium. The novelty of this work is that the long-chain octylalkylamine enhances the graphene oxide hydrophobic surface repulsion, and thus decreases the corrosive ion penetration.
2. Experiment
Natural graphite powder, KMnO4, H2O2, octylamine, and THF (tetrahydrofuran) were purchased from Alfa Aesar. The graphene oxide (GO) was synthesized by the modified Hummers' method. In brief: 50 ml of H2SO4 and 40 ml of H2PO4 were put into a 250 ml round bottom flask; then, 1 g of graphite powder was added to the above mixture.27 The mixture was kept in a 1000 ml beaker containing ice, and then 3 g of KMnO4 was added slowly to the mixture and the mixture temperature was increased to above room temperature up to 90 °C. After cooling down to room temperature, the mixture was kept at 70 °C and refluxed for 24 h. The mixture was cooled down to room temperature, and then centrifuged at 7000 rpm, and washed with deionized water until the mixture was neutral. The final product was dried at 80 °C for 24 h. The octylamine functionalization was a one-step method. In brief, 100 mg of GO was dispersed in THF by sonication and 5 ml of octylamine was added to the above dispersed mixture. The mixture was refluxed at 60 °C for 24 h, and then centrifuged at 5000 rpm to isolate the final product. The black product was washed with ethanol and deionized water, and finally dried at 80 °C for 24 h.
2.1. Material characterization
The functionalized material was characterized by different techniques to confirm the octylamine was covalently functionalized on the graphene oxide matrix. The functional groups were characterized by FTIR spectroscopy using a PerkinElmer Spectrum 65 FT-IR spectrometer. Microstructures were studied by a Raman WITec microscope at 530 nm laser excitation (Germany), powder XRD D8 ADVANCE (Bruker), Carl Zeiss microscope equipped with EDX, 3.0 keV FESEM, and TEM by FEI Model Tecnai G2S Twin (200 kV) GATAN image software to confirm the crystallinity of the materials.
2.2. Corrosion inhibition studies
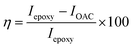
The Mg alloy was cut into 1 cm × 1 cm squares and mounted with cold-cure materials, and polished with different size grade silicon papers for the active alloy surface. The as-prepared Mg alloy was kept in a desiccator before use. The coating materials were prepared in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio of graphene oxide and epoxy for coating the Mg alloys. The polyamine was used as a binder. The alloys were coated by a 0.5 μm brush. After coating, the Mg alloy was dried at 80 °C for 24 h. The as-prepared alloy was used as a working electrode, with a platinum mesh as the counter electrode and Ag/AgCl2 as a reference electrode to carry out the electrochemical reaction. The working electrode was immersed in a 3.5% NaCl medium for 5 days. The potentiodynamic polarization technique was carried out from OCP ±250 at scan rate of 10 mV s−1. The impedance studies were carried out from 0.01 mHz to 100 kHz by a 920D CHI electrochemical workstation. The corrosion inhibition efficiency was evaluated by the following eqn (1) and (2). Hence the octylamine functionalized graphene oxide redox stability was studied by three cell electrochemical system, and working electrode was used platinum disk electrode. The electrochemical cell size was square 5 cm × 5 cm (H × L), and 1 M NaOH was used as for the water electrolyte. The applied voltage from the open circuit potential ± at a scan rate of 10 mV s−1.
:1 weight ratio of graphene oxide and epoxy for coating the Mg alloys. The polyamine was used as a binder. The alloys were coated by a 0.5 μm brush. After coating, the Mg alloy was dried at 80 °C for 24 h. The as-prepared alloy was used as a working electrode, with a platinum mesh as the counter electrode and Ag/AgCl2 as a reference electrode to carry out the electrochemical reaction. The working electrode was immersed in a 3.5% NaCl medium for 5 days. The potentiodynamic polarization technique was carried out from OCP ±250 at scan rate of 10 mV s−1. The impedance studies were carried out from 0.01 mHz to 100 kHz by a 920D CHI electrochemical workstation. The corrosion inhibition efficiency was evaluated by the following eqn (1) and (2). Hence the octylamine functionalized graphene oxide redox stability was studied by three cell electrochemical system, and working electrode was used platinum disk electrode. The electrochemical cell size was square 5 cm × 5 cm (H × L), and 1 M NaOH was used as for the water electrolyte. The applied voltage from the open circuit potential ± at a scan rate of 10 mV s−1.| |
 | (1) |
| |
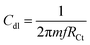 | (2) |
2.3. Computational studies
We used the C30H10(CO2)(OH)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) GO model, with one –CO2H, one –OH, and one O groups. The octylamine molecule was coordinated to this GO model through the NC group, which was formed by removing two hydrogens from the NH2-group of the octylamine and the O-atom from the CO group of the GO, and then coordinating the N atom of the octylamine to the C atom of the GO model. We performed DFT studies of this model, both neutral and protonated on the NC group, using the Gaussian 09 package, revision B.01.28 Calculations were done using the hybrid DFT functional B3LYP29 and split-valence basis set 6-31+G* having one set of polarization and one set of diffuse functions, both on the heavier atoms (C, N, O).30,31 It should be emphasized that the introduction of the diffuse function set made the total number of wavefunctions quite significant, and thus the calculations became noticeably time- and resource-demanding. This was the reason why a smaller GO model was chosen. We optimized the structures, calculated frequencies, and performed NBO and MO analysis of our models with implicit effects from water taken into account (dielectric constant ε = 78.3553). Implicit solvent calculations were done using the self-consistent reaction field IEF-PCM method (the UFF default model as employed in the Gaussian 09 package, with the electrostatic scaling factor α set to 1.0). For the global reactivity analysis, the electrophilicity (χ), global hardness (η), global softness (σ), and nucleophilicity (ω) values were calculated according to the equations below:
O) GO model, with one –CO2H, one –OH, and one O groups. The octylamine molecule was coordinated to this GO model through the NC group, which was formed by removing two hydrogens from the NH2-group of the octylamine and the O-atom from the CO group of the GO, and then coordinating the N atom of the octylamine to the C atom of the GO model. We performed DFT studies of this model, both neutral and protonated on the NC group, using the Gaussian 09 package, revision B.01.28 Calculations were done using the hybrid DFT functional B3LYP29 and split-valence basis set 6-31+G* having one set of polarization and one set of diffuse functions, both on the heavier atoms (C, N, O).30,31 It should be emphasized that the introduction of the diffuse function set made the total number of wavefunctions quite significant, and thus the calculations became noticeably time- and resource-demanding. This was the reason why a smaller GO model was chosen. We optimized the structures, calculated frequencies, and performed NBO and MO analysis of our models with implicit effects from water taken into account (dielectric constant ε = 78.3553). Implicit solvent calculations were done using the self-consistent reaction field IEF-PCM method (the UFF default model as employed in the Gaussian 09 package, with the electrostatic scaling factor α set to 1.0). For the global reactivity analysis, the electrophilicity (χ), global hardness (η), global softness (σ), and nucleophilicity (ω) values were calculated according to the equations below:| |
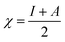 | (3) |
| |
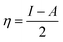 | (4) |
| |
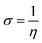 | (5) |
| |
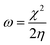 | (6) |
| |
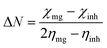 | (7) |
3. Result and discussion
3.1. Spectroscopy studies
As shown in Fig. 1, the octylamine-functionalized OH group stretching frequency appeared at 3500 cm−1. Hence, the GO CO stretching frequency appeared at 1713 cm−1, and the stretching frequency appeared at 2878 cm−1 due to the CC bond in the GO matrix. Furthermore, the octylamine-functionalized GO stretching frequency appeared at 3300 cm−1 due to the alkyl nitrogen bond. The stretching frequency appeared at 2200 cm−1 due to the long alkyl chain.32 The alkyl chain C–H bond vibration appeared at 738 cm−1. The bond vibration appearing at 1035 cm−1 was associated with an alkyl chain, and the bond appeared at 1052 cm−1 due to the C–O–C stretching frequency. Furthermore, the stretching frequency appeared at 1537 cm−1 due to the sp2 carbon modification on the graphene oxide matrix. The stretching frequency at 2169 cm−1 was due to the presence of the graphene oxide carboxylate. As shown in Fig. 1C, the characteristic D peak from GO graphitic carbon appearing at 1200 cm−1 was associated with a carbon disorder of the graphene oxide matrix, and the G peak appearing at 1600 cm−1 was associated with an sp2 carbon C–C bond vibration.33 Furthermore, the octylamine-functionalized GO peak appeared at 200 cm−1 and 400 cm−1 due to the presence of an alkyl chain on the graphitic carbon matrix, and the graphitic carbon peaks appeared at 1200 cm−1 and 1600 cm−1 for the D and G peaks, respectively. Furthermore, Fig. 1D showed the GO Raman mapping of a layer from the few-layer GO, and does not illuminate due to the absence of octylamine. In Fig. 1E, the GO layer was blinking due to the presence of octylamine, and the layer thickness was shown to be around 500 nm. In another case, the crystalline GO sheet showed new peaks at 2θ = 10°, 15°, 20°, 25° and 30° due to the presence of the octylamine-functionalized GO sheet.34 Hence, the GO graphitic carbon peak appeared at 2θ = 10°.
 |
| | Fig. 1 Octylamine-functionalized GO and GO spectroscopy and XRD studies. (A) GO FTIR, (B) octyl amine functionalized graphene oxide FTIR, (C) Raman spectroscopy of GO, octylamine functionalized graphene oxide and (D and E) GO and octylamine functionalized Raman mapping, (F) crystalling studies of GO and octylamine functionalized GO. | |
3.2. Microstructure of OAC functionalized GO sheet
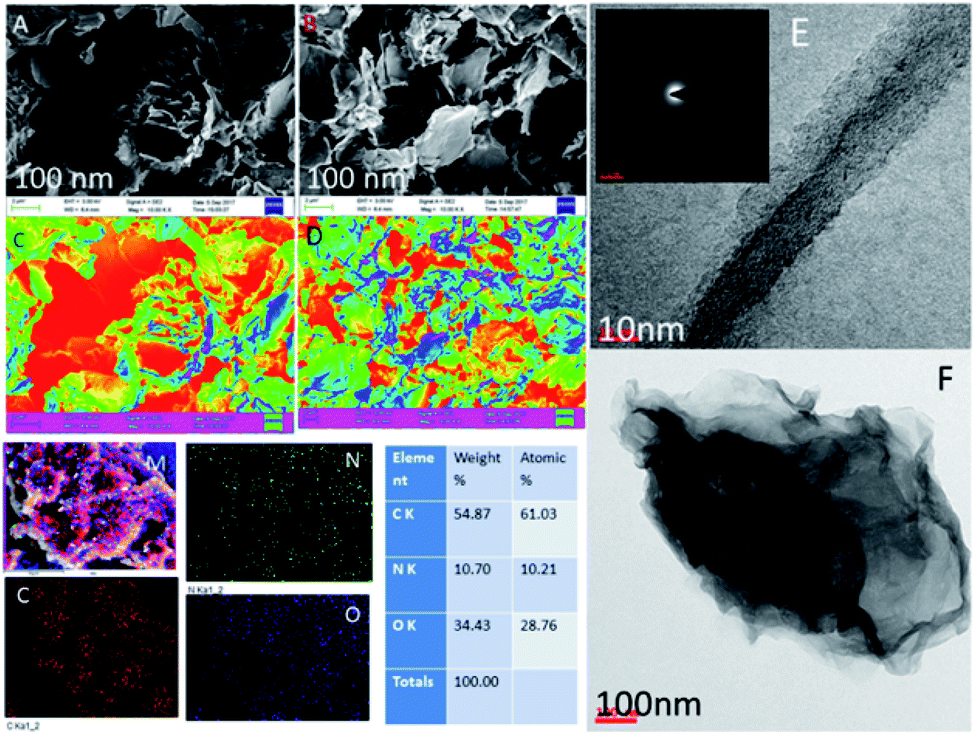
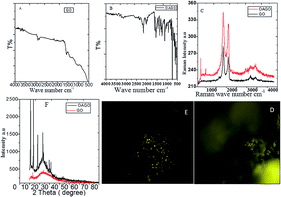
Fig. 2 shows the GO and octylamine-functionalized GO microstructure. In Fig. 2A, the GO sheet shows a flower-shaped GO sheet and a few layers due to the 3 h sonication. Furthermore, there was no GO layer crack. Hence, Fig. 2B suggested that the octylamine-functionalized GO sheet showed porous and ribbon shapes due to the octylamine covalently bound with the GO layer. Furthermore, Fig. 2C showed the GO few layer confirmation with coloring. In another case, Fig. 2D showed that nitrogen covalently interacted with the graphene oxide matrix in the octylamine-functionalized graphene oxide mapping images. Fig. 2E shows the TEM image of the octylamine-functionalized GO sheet. GO sheets are regularly formed at high densities due to the alkyl chain π–π crosslinked over the GO layer.35,36 The polycrystalline morphology was also indexed (Fig. 2E), and it was confirmed from the XRD result. Further elemental results showed that the octylamine covalently functionalized on the graphene oxide via π–π bond. Furthermore, the features shown in Fig. 2F suggested that the dark zigzag graphene oxide single layers formed.
 |
| | Fig. 2 FESEM and TEM microscopy studies of the octylamine-functionalized GO and GO. (A and B) FESEM microstructure of GO and octylamine funtionalized GO, and (C and D) GO and octyl amine functionalized GO few layer structure, (E) TEM microstructure of GO and (F) octylamine functionalized graphene oxide ribbon structure. | |
3.3. Electrochemical stability studies
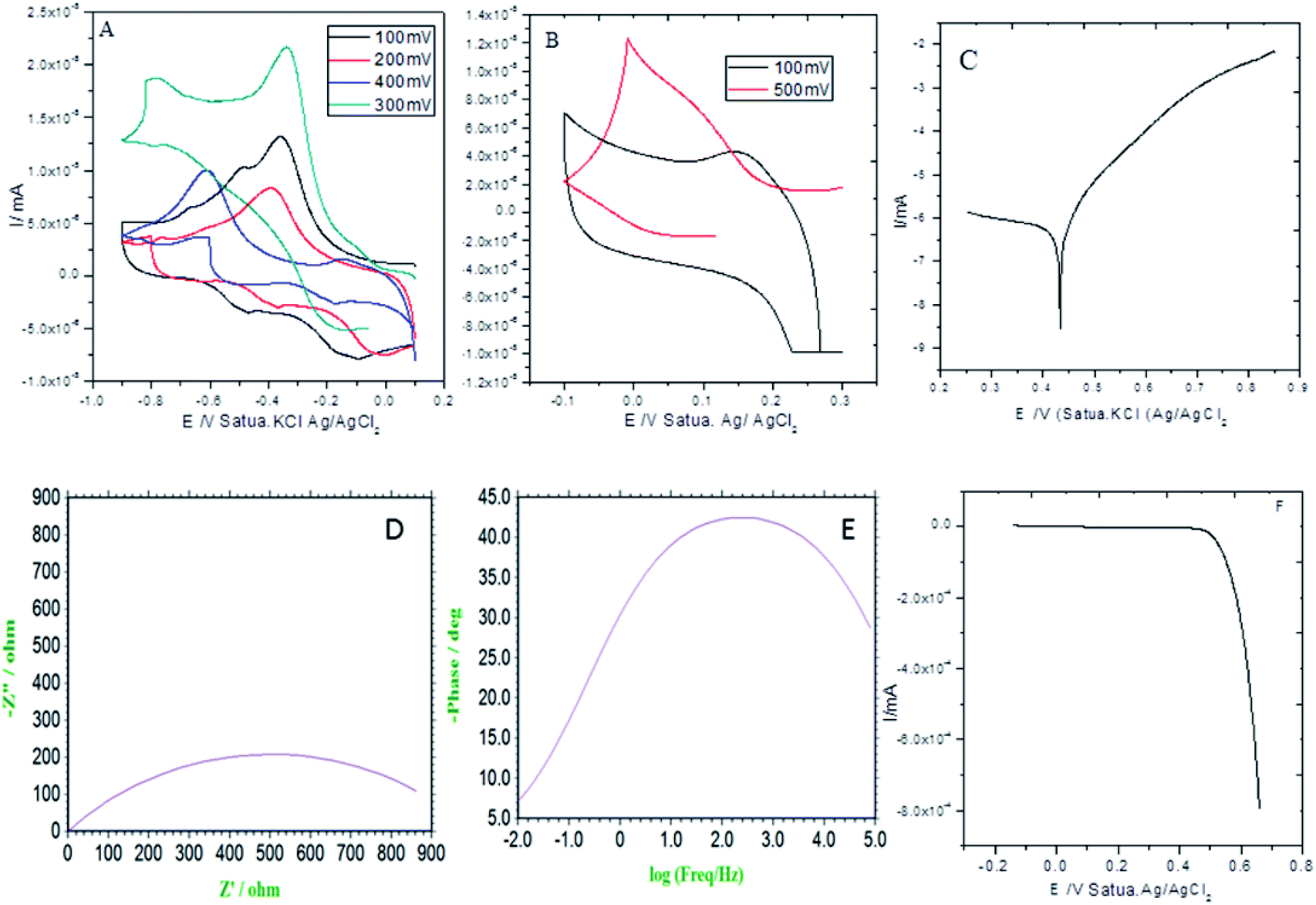
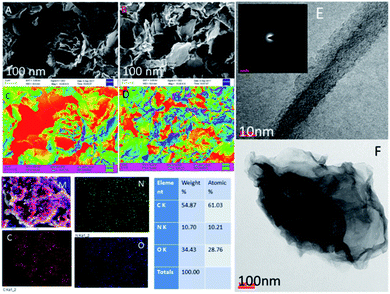
As shown in Fig. 3A (CV: the applied voltage from 300 mV to 400 mV at 10 mV scan rates), the current is increased with the increase of the applied voltage due to the presence of the alkyl chain inductive effect. At the applied voltage of 300 mV, redox behavior appeared at 0.2 mV to 0.6 mV due to the presence of non-bonding electrons of the pyridine ring. Furthermore, the redox behavior was increased at the applied voltage of 400 mV by the effect of the five-membered ring electron delocalization within the GO matrix. The oxidation peak appeared from 0.2 mV to 0.7 mV due to the alkyl chain electron continued exchange with the GO matrix.37,38 Next, at the applied voltage of 500 mV, the redox behavior was increased from 0.3 mV to 0.5 mV due to the presence of the graphene oxide carbonyl delocalized electrons. Hence, the GO graphitic carbon non-bonding electrons pushed electrons to the alkyl chain. Furthermore, Fig. 3B shows that at an applied voltage from 100 mV to 300 mV at 20 mV scan rates, the current was increased due to intercalation of the GO epoxy non-bonding electrons.39 However, according to the Tafel curve shown in Fig. 3C, the anodic oxygen evolution is less as compared to cathodic hydrogen evolution due to the long alkyl chain inductive effect on the graphene oxide surface. The hydrogen evolution was initiated from 0.5 mV to 0.7 mV due to the alkyl chain electron intercalation on the GO substrate. The current density was increased due to the free non-bonding electrons of the carbonyl groups and carboxylate anions. In Fig. 3D, the Nyquist plot from impedance spectroscopy is shown for the applied frequency range from 0.01 kHz to 100 kHz frequencies to find out the zero resistance. The semicircle diameter was increased due to the increased hydrogen evolution. The charge transfer was increased at 970 Ω due to the alkyl chain electron intercalation.40–42 As shown in Fig. 3E, the impedance phase was increased due to the non-bonding electron intercalation of the GO carbonyl and carboxylate groups. Six-membered pyridine ring electrons were delocalized within the GO matrix. Furthermore, we studied hydrogen evolution from 0.4 mV to 6 mV using the LSV technique. The LSV result supported the observation that octylamine functionalized the GO sheet, acting as an excellent 2D carbon material for hydrogen evolution in conventional methods.
 |
| | Fig. 3 Electrochemical stability studies of the octylamine-functionalized GO sheet. (A and B) CV studies, (C) potentiodynamic polarization, (D) Nyquist plot, (E) frequency impedance and (F) linear sweep voltammetry. | |
3.4. Corrosion inhibition studies
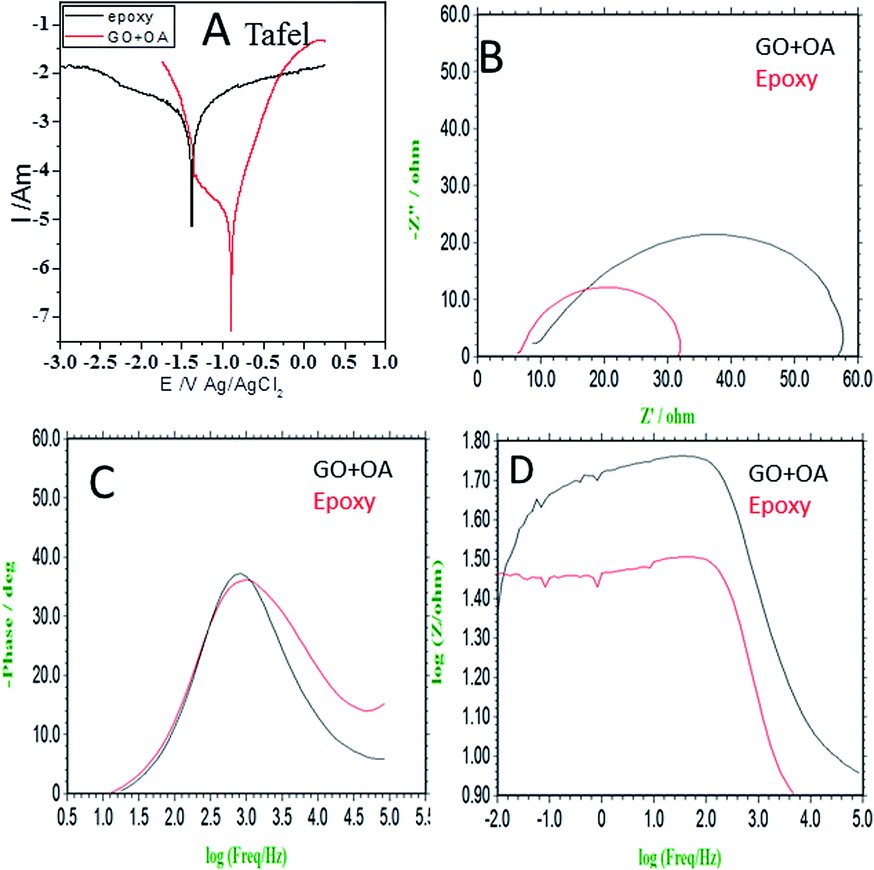
In Fig. 4a, the results of the potentiodynamic polarized studies of the epoxy and octylamine (OA)-coated Mg alloys are shown. The epoxy-coated Mg alloy corrosion current density was increased by the effect of the increased chloride ion diffusion rate. Furthermore, the alloy-pitting corrosion propagation was increased by the poor epoxy coating adhesion efficiency. The corrosion potential was decreased by the effect of the alloy having localized corrosion. The epoxy coating physical adsorption was poor. Due to this, the alloy was prone to corrosion in a 3.5% NaCl environment. For the octylamine-functionalized graphene oxide-coated magnesium, the corrosion current was decreased by the effect of the octylamine hydrophobic surface and prevention of aggressive ion penetration to the surface.43 Furthermore, the octylamine-coated Mg alloy corrosion potential was increased by the long alkyl chains blocking the corrosive ion diffusion to the coating. The corrosion inhibition efficiency values are presented in Table 1. In Fig. 4b, the impedance spectrum of the epoxy and octylamine-decorated GO-coated Mg alloy is shown. The conventional epoxy-coated Mg alloy was corroded due to the poor physical adhesion of the epoxy coating on the Mg alloy.44 Furthermore, the Nyquist plot was decreased by the effect of chloride ion diffusion to the traditional epoxy coating, and the Cdl value was 7.231 μF. In Fig. 4c the impedance phase was decreased, and in Fig. 4d the impedance frequency was also suppressed because of the poor efficiency of the conventional epoxy coating adhesion.
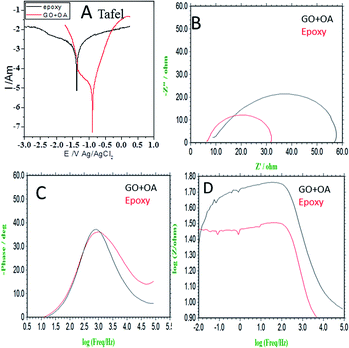 |
| | Fig. 4 Electrochemical studies of the epoxy and octylamine-decorated GO-coated magnesium alloy immersed in 3.5% NaCl (A) Tafel, (B) Nyquist, (C) impedance phase, (D) impedance frequency. | |
Table 1 Corrosion inhibition studies of epoxy and octylamine coated magnesium alloy
| S. no. |
−ECorr/mV |
−ICorr/mA |
η% |
RCt (Ω) |
Cdl (μF) |
η% |
| Epoxy |
1.383 |
4.937 |
|
22 |
7.231 |
|
| GO + OA |
2.461 |
1.307 |
73 |
52 |
1.529 |
78 |
However, the octylamine-functionalized graphene oxide impedance Nyquist plot showed an excellent physical barrier layer on the Mg alloy surface. Furthermore, in Fig. 4c the impedance phase angle was increased as compared with conventional epoxy coating due to the strong physical adsorption on the alloy surface. The in Fig. 4d impedance frequency was increased due to the octylamine hydrophobic surface. The impedance values are presented in Table 1. From the electrochemical studies, the octylamine showed excellent physical barrier layer formation on the Mg alloy surface in the 3.5% NaCl solution.
3.5. AFM alloy microstructure
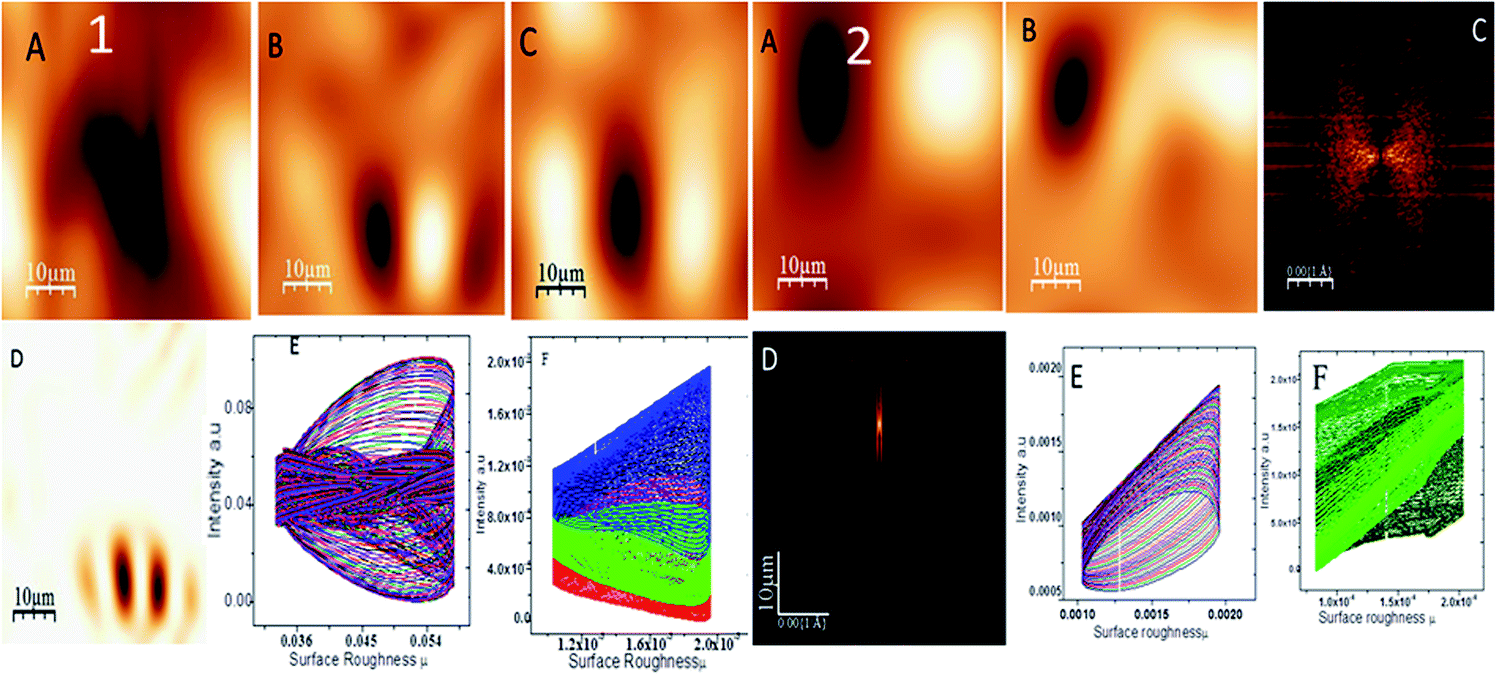
Fig. 5 shows the topography of the epoxy and octylamine (OAC)-functionalized graphene oxide-coated magnesium alloy in 3.5% NaCl corrosion medium. The epoxy-coated Mg alloy propagated pitting corrosion on the alloy surface due to the increased corrosive ion diffusion to the epoxy coating. Fig. 5(1A) indicates pitting propagation by attack of corrosive ions. Fig. 5(1B) shows the results of investigation of the corrosive ion penetration to different places of the alloy surface to explore the pitting size. Fig. 5(1B and C) shows that the pitting size is increased due to the heavy electrolyte chloride ions. Fig. 5(1D) indicates the pitting size propagation. Fig. 5(1E and F) shows the pitting size at around 0.8–0.9 μm due to the heavy electrolyte penetration to the barrier coating. Furthermore, Fig. 5(2A) shows the octylamine-functionalized GO-coated Mg alloy topography. It is suggested that the pitting propagation was controlled by the octylamine-functionalized GO sheet due to the highly hydrophobic surface.45 For the alkyl amine-functionalized GO-coated Mg alloy surface, different areas for pitting propagation were studied but no pitting initiation was found due to the hydrophobic surface. Fig. 5(2C and D) suggest that the pitting propagation is controlled by the alkyl amine-functionalized GO sheet. Fig. 5(2E and F) show the pitting size of different areas of the Mg alloy surface, indicating that the pitting initiation was controlled by the long alkyl chain GO materials.
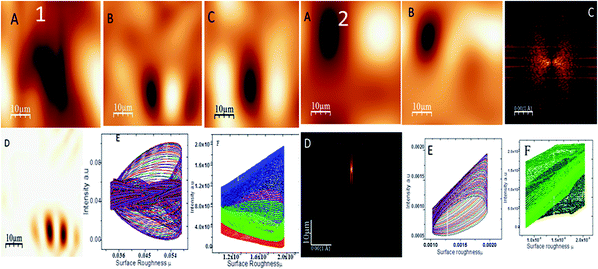 |
| | Fig. 5 Epoxy-coated Mg alloy surface roughness (1) and octylamine-decorated GO-coated Mg alloy roughness (2). | |
3.6. AZ13 Mg alloy microstructure
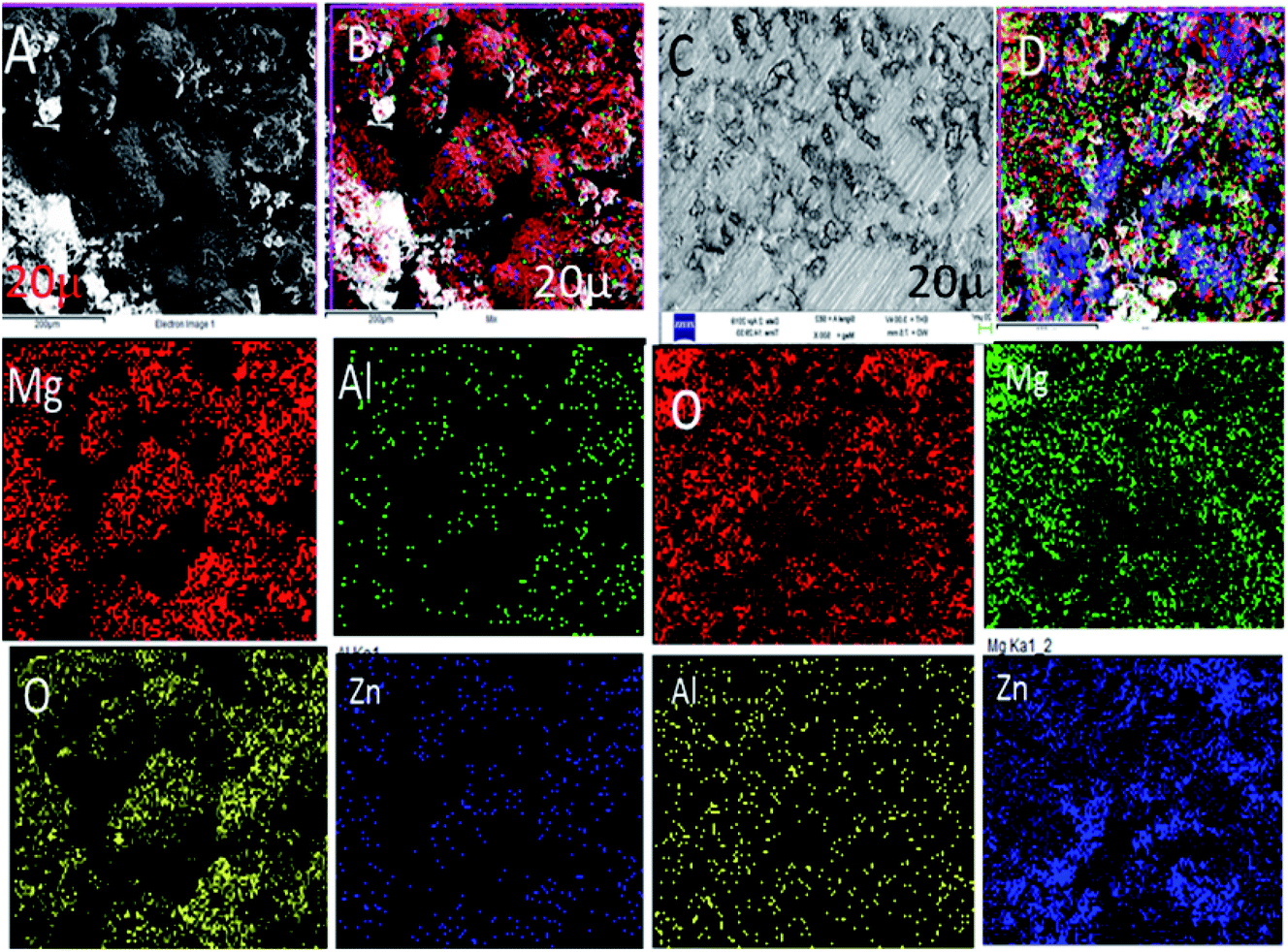
Fig. 6A shows the epoxy-coated Mg alloy microstructure immersed in 3.5% NaCl solution after 5 days. The results of being harshly affected by the heavy chloride ion penetration to the epoxy coating and damages of the grain boundary of the Mg alloy can be seen. The Mg alloy surface looks like hills due to the corrosive ion diffusion to the coating layer and damages at the alloy grain boundary. Fig. 6B suggests that the alloy undergoes uniform pitting corrosion by attack of the chloride ions. Furthermore, the epoxy-coated alloy elemental mapping results suggested that the oxygen concentration increased due to the Mg alloy oxide layer formed by the corrosive ion reacting with the Mg alloy surface.46 Hence, the elemental compositions were also decreased due to the large epoxy pore size. The octylamine-functionalized GO-coated Mg alloy surface controlled pitting initiation, as shown in Fig. 6C. The pitting propagation was stopped by the alkyl amine-functionalized graphene oxide hydrophobic surface. Fig. 6C shows pitting initiation healing by the alkyl amine-functionalized GO on the Mg alloys.47–50 Furthermore, the elemental compositions suggested that the oxygen concentration was less, as compared with the traditional epoxy-coated Mg alloys. From the microstructure, we concluded that the octylamine-functionalized GO materials have significant corrosion resistance in a 3.5% NaCl solution.
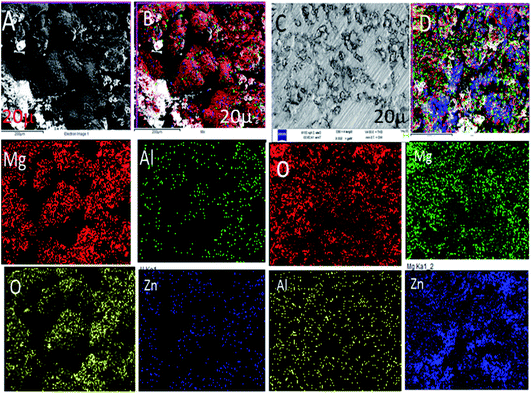 |
| | Fig. 6 FESEM microstructures of the epoxy and octylamine-functionalized GO-coated Mg alloys after immersion in the 3.5% NaCl environment for 5 days (A) epoxy coated Mg alloy, (B) uniform oxide layer formed on surface, (C and D) alkyl amine functionalized GO coated magnesium alloy surface. | |
3.7. Mechanism of alkyl amine on GO matrix
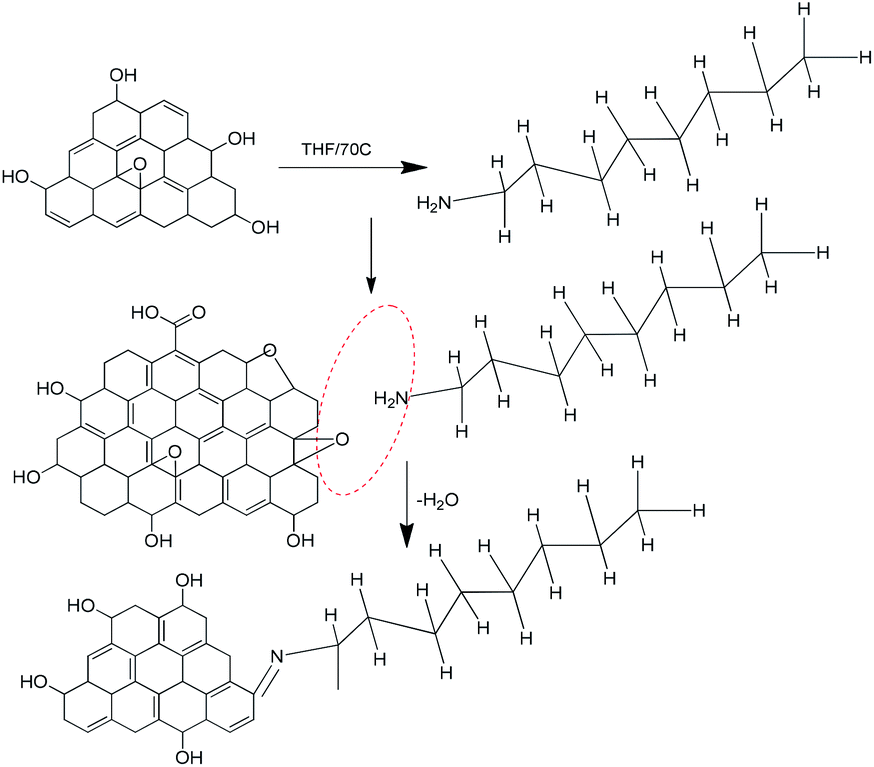
Fig. 7 shows the octylamine covalent bond formation on GO.
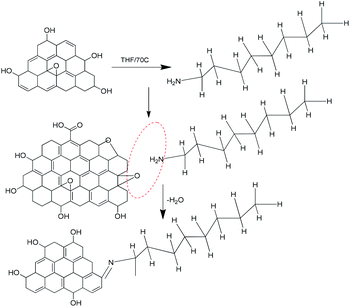 |
| | Fig. 7 Proposed octylamine functionalization of the GO matrix. | |
The GO epoxy group was involved in a ring opening reaction, and octylamine was deprotonated by the effect of the strong base NaH.
The octylamine nucleophile reacted with epoxy to give water as the leaving product, and form a new bond CN– and then the aminated graphene oxide sheet.45,46 The graphene oxide epoxy functional groups stretching frequency.
Disappear, it indicates that –NC new carbon bond formed and confirmed by FTIR techniques.
3.8. Computational studies
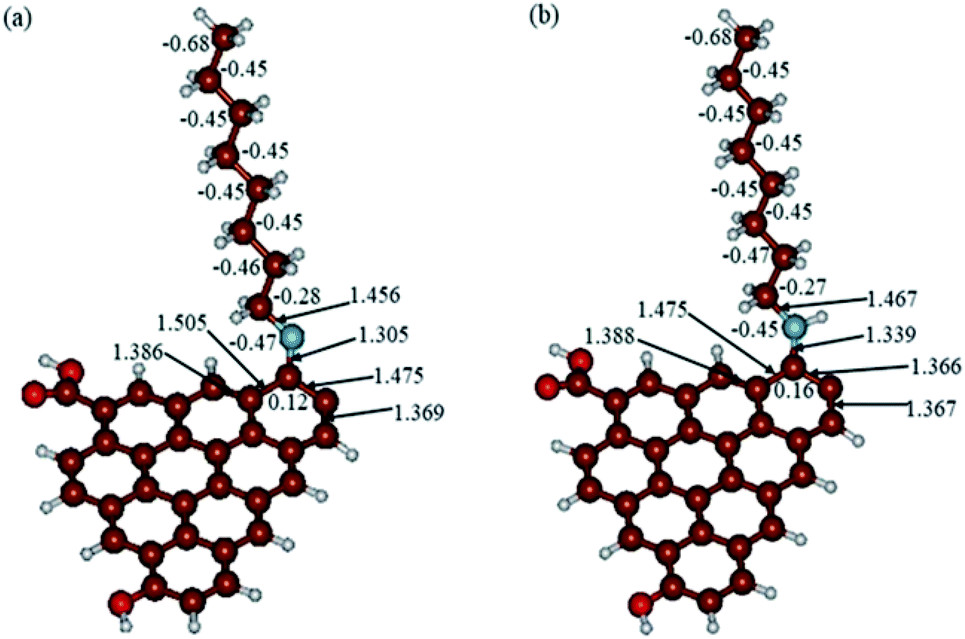
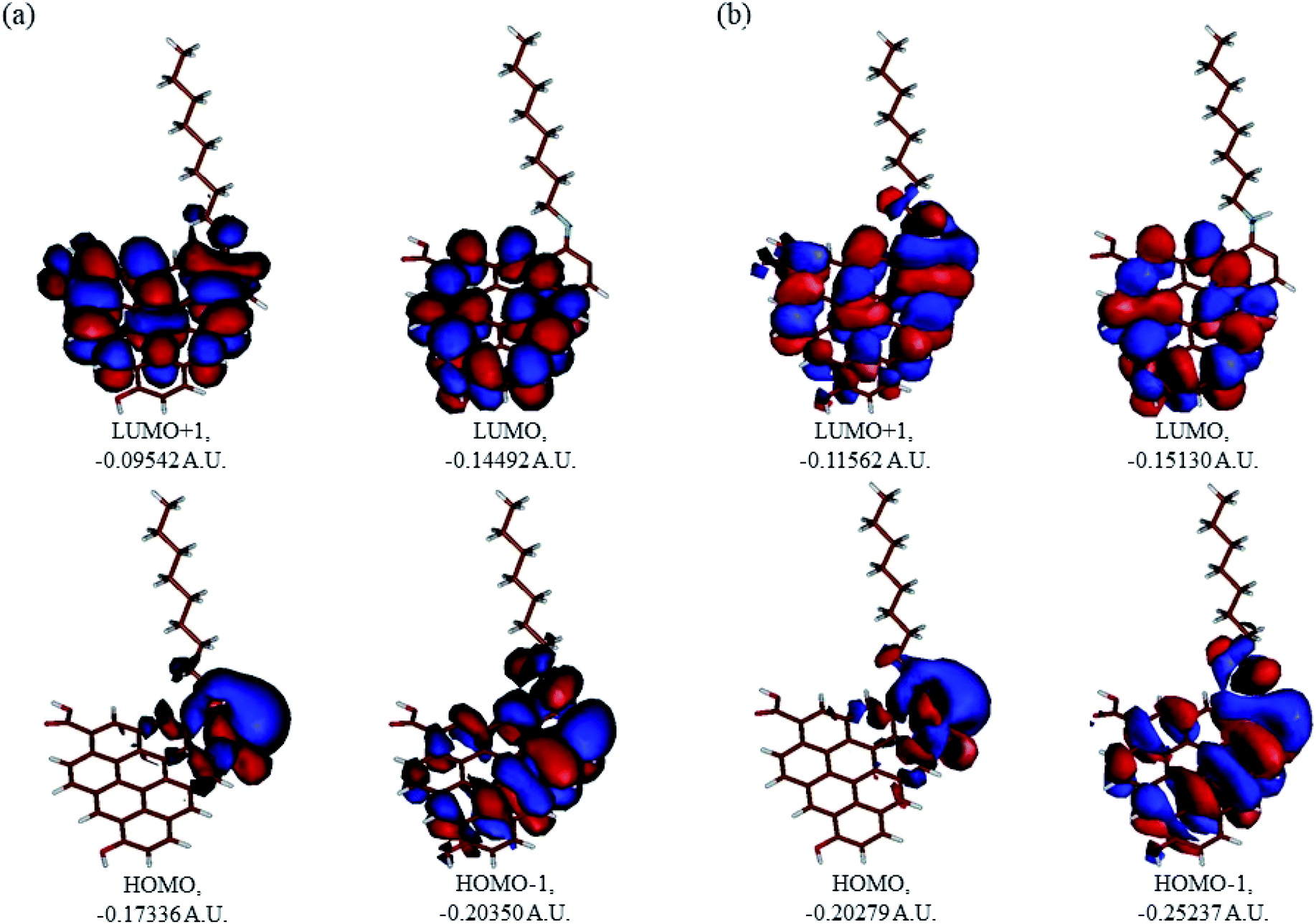
In Fig. 9, the implicit water optimized structures of the octylamine-functionalized GO model are given for the neutral (a) and protonated (b) species, along with the calculated NBO charges. Because only one layer of GO was used in the model, it became somewhat distorted upon optimization, as expected. As can be seen, upon protonation on the NC group, the C(octylamine)–N bond became elongated by ca. 0.011 Å, and the N–C(graphene oxide) bond distance became elongated even more by 0.035 Å. However, the NBO analysis results show that, upon protonation, the charges of the octylamine moiety remained essentially the same, and only the charge on the carbon of GO to which the N-atom was bound increased slightly by 0.04e. In general, as can be seen from the NBO results, the octylamine moiety atoms bore quite noticeable negative charges, −0.25 to −0.68e. Thus, the octylamine-functionalized GO surface could act as a nucleophilic agent and could interact with various oxidizers in solution. In Fig. 8, four frontier orbitals of the octylamine-functionalized GO model are given for the neutral (a) and protonated (b) structures, HOMO and HOMO−1, and LUMO and LUMO+1. Analysis of the MOs shows the following: (i) mostly graphene oxide and N-atom of the octylamine contribute in the frontier orbitals under consideration. (ii) The energy differences between HOMO and HOMO−1 are relatively small, ca. 0.81–1.33 eV. Thus, participation of these orbitals in (electro)chemical processes can be considered possible. (iii) The HOMO–LUMO gaps for the neutral and protonated systems were calculated to be relatively small, 0.028 and 0.051 Ha and or ca. 0.76 and 1.39 eV, respectively, which means that the octylamine-functionalized GO should be quite reactive in the solution media. (iv) For the protonated structure, the MO energies were computed to be lower than for the neutral structure due to the extra stabilization of the cation MOs in the polar solvent. Also, the HOMO–LUMO gap of the protonated structures was computed to be higher by ca. 0.63 eV than that of the neutral structure, which might mean that in general, the protonated species might be somewhat less reactive towards oxidants in solution. (v) The computed global hardness values of the two models were quite small, ca. 0.014–0.026 A.U., and the global softness σ values were quite noticeable (see Table 2). This suggests that both neutral and protonated systems would be highly reactive towards oxidizing agents attacking the surface.47 This is also supported by the noticeably small energy HOMO–LUMO gap value (ΔE) (Table 2). The small calculated values of the global electronegativity (χ) and electrophilicity (ω) also suggest a high potential reactivity of the octylamine covalently functionalized GO towards the oxidizing electrophilic agents.
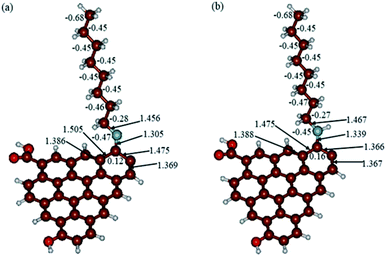 |
| | Fig. 8 Calculated structures of the GO model functionalized with octylamine: neutral (a) and protonated (b). Distances are given in Å. NBO charges are provided next to the atoms. The color code: brown for C, white for H, red for O, and blue for N. | |
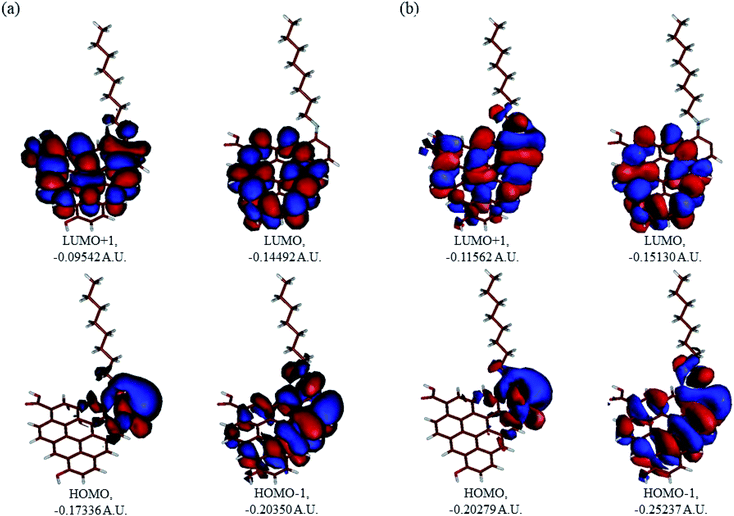 |
| | Fig. 9 Molecular orbitals of the GO model functionalized with neutral octylamine (a) and protonated (b). | |
Table 2 Global reactivity parameters computed for the GO model functionalized with octylamine with implicit effects from water. Values in A.U. (A) for the neutral structure, (B) for the protonated structure
| |
HOMO |
LUMO |
ΔE |
I |
A |
χ |
η |
σ |
ω |
ΔE |
| A |
−0.17336 |
−0.14492 |
0.02844 |
0.17336 |
0.14492 |
0.15914 |
0.01422 |
70.3235 |
0.89049 |
0.29537 |
| B |
−0.20279 |
−0.15130 |
0.05149 |
0.20279 |
0.15130 |
0.17704 |
0.02575 |
38.835 |
0.60864 |
0.29330 |
4. Conclusions
The octylamine-functionalized GO materials have been shown to work as a corrosion inhibition barrier on the Mg alloy surface. The spectroscopy results reveal that octylamine was covalently connected with graphene oxide. The presence of the graphitic carbon peak was confirmed by Raman spectroscopy. The XRD results suggest the existence of alkyl amine functionalized GO sheets. The electrochemical studies suggest that the electrochemical stability increases with the increased potential. Furthermore, the corrosion inhibition studies reveal that the pitting corrosion was retarded for the octylamine-functionalized GO-coated Mg alloy surface, as compared with the traditional epoxy coating alloy surface. The AFM studies suggest that the octylamine-functionalized GO-coated Mg alloy has pitting corrosion propagation controlled. Microscopy studies of the Mg alloy surface structure revealed the smooth surface of the octylamine-coated Mg alloy. Computational studies completely support the experimental results, suggesting the high potential reactivity of the octylamine covalently functionalized GO towards oxidizing electrophilic agents.
Conflicts of interest
There is no conflict to declare.
Acknowledgements
N. P. would like to thank the Non-NET fellowship and Central University of Gujarat CIF facilities. The authors are grateful to the UGC-NRC University Hyderabad School of Chemical Science for supporting the research work, and N. P. is thankful to Prof. M. Sathiyendiran.
References
- N. Parhizkar, T. Shahrabi and B. Ramezanzadeh, Corros. Sci., 2017, 123, 55–75 CrossRef CAS.
- N. Palaniappan, I. S. Cole, F. Caballero-Briones, S. Manickam, C. Lal and J. Sathiskumar, RSC Adv., 2019, 9, 8537–8545 RSC.
- N. Palaniappan, I. S. Cole, F. Caballero-Briones, K. Balasubaramanian and C. Lal, RSC Adv., 2018, 8, 34275–34286 RSC.
- B. Jin, D. B. Xiong, Z. Tan, G. Fan, Q. Guo, Y. Su, Z. Li and D. Zhang, Carbon, 2019, 142, 482–490 CrossRef CAS.
- S. Virtanen, Mater. Sci. Eng., B, 2011, 176, 1600–1608 CrossRef CAS.
- F. Cao, G. L. Song and A. Atrens, Corros. Sci., 2016, 111, 835–845 CrossRef CAS.
- W. Shang, C. He, Y. Wen, Y. Wang and Z. Zhang, RSC Adv., 2016, 6, 113967–113980 RSC.
- D. Hoche, C. Blawert, S. V. Lamaka, N. Seharnagl, C. Mendis and M. L. Zheludkevich, Phys. Chem. Chem. Phys., 2016, 18, 1279–1291 RSC.
- M. Razavi and Y. Huang, Biomater. Sci., 2019, 7, 2241–2263 RSC.
- G. Williams, N. Birbilis and H. N. McMurray, Faraday Discuss., 2015, 180, 313–330 RSC.
- D. M. Miskovic, K. Pohl, N. Birbilis, K. J. Laws and M. Ferry, J. Mater. Chem. B, 2016, 4, 2679–2690 RSC.
- Y. J. Wu, X. B. Chen, G. Williams, J. R. Scully, T. Gengenb and N. Birbilis, RSC Adv., 2016, 6, 43408–43417 RSC.
- H. Feng, X. Zhang, G. Wu, W. Jin, Q. Hao, G. Wang, Y. Huang and P. K. Chu, RSC Adv., 2016, 6, 14756–14762 RSC.
- M. Chen, Y. Chen, W. Zhang, S. Zhao, J. Wang, J. Mao, W. Li, N. Zhano, N. Huang and G. Wan, RSC Adv., 2016, 6, 15247–15259 RSC.
- Y. Han and B. Li, RSC Adv., 2015, 5, 46109–46118 RSC.
- A. Li, J. Xie and J. Li, J. Mater. Chem. B., 2019, 7, 509–527 RSC.
- J. liu, P. Wang, C. C. Chu and T. Xi, J. Mater. Chem. B, 2017, 5, 1787–1802 RSC.
- E. G. Yazdi, Z. S. Ghahfarokhia and M. Bagherzadeh, New J. Chem., 2017, 41, 12470–12480 RSC.
- Z. Qiu, R. Wang, J. Wu, Y. Zhang, Y. Qu and X. Wu, RSC Adv., 2015, 5, 44149–44159 RSC.
- A. B. Ikhe, A. B. Kale, J. Jeong, M. J. Reece, S. H. Choi and M. Pyo, Corros. Sci., 2016, 109, 238–245 CrossRef CAS.
- W. Shang, F. Wu, Y. Wen, C. He, X. Zhang and Y. Li, Ind. Eng. Chem. Res., 2019, 58, 1200–1211 CrossRef CAS.
- S. Dea and J.
L. Lutkenhaus, Green Chem., 2018, 20, 506–514 RSC.
- G. C. Son, D. Kyu Hwang, J. Jang, S. S. Chee, K. Cho, M. M. Myoung and M. H. Ham, Nano. Res., 2019, 12, 19–23 CrossRef CAS.
- L. B. Tong, J. B. Zhang, C. Xu, X. Wang, S. Y. Song, Z. H. Jiang, S. Kamado, L. R. Cheng and H. J. Zhang, Carbon, 2016, 109, 340–351 CrossRef CAS.
- N. Palaniappan, I. S. Cole, A. E. Kuznetsov, K. Balasubramanian and K. R. Justin Thomas, RSC Adv., 2019, 9, 32441–32447 RSC.
- C. Wu, Q. Liu, J. Liu, R. Chen, K. Takahashi, L. Liu, R. Li, P. Liu and J. Wang, New J. Chem., 2017, 41, 12767–12776 RSC.
- B. Dai, L. Fu, L. Liao, N. Liu, K. Yan, Y. Chen and Z. Liu, Nano Res., 2011, 4, 434–439 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Parr and W. G. Yang, Density-functional theory of atoms and molecules, Oxford University Press, Oxford, 1989 Search PubMed.
- A. McLean and D. G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- E. Cances and B. T. Mennucci, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- C. C. Caliman, A. F. Mesquita, D. F. Cipriano, J. C. C. Freitas and A. A. C. Cotta, RSC Adv., 2018, 8, 6136–6145 RSC.
- S. P. Zhang and H. O. Song, New J. Chem., 2012, 36, 1733–1738 RSC.
- T. Ide, J. P. Barham, M. Fujita, Y. Kawato, H. Egami and Y. Hamashima, Chem. Sci., 2018, 9, 8453–8460 RSC.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- M. Majdoub, Y. Essamlali, O. Amadine, I. Ganetri and M. Zahouily, New J. Chem., 2019, 43, 15659–15672 RSC.
- C. Revathi, K. Rajavel, M. Saranya and R. T. Rajendra Kumar, J. Electrochem. Soc., 2016, 163, B627–B632 CrossRef CAS.
- S. K. Krishnan, E. Singh, P. Singh, M. Meyyappan and H. Singh Nalwa, RSC Adv., 2019, 9, 8778–8881 RSC.
- B. Gorska, L. Timperman, M. Anouti, J. Pernaka and F. Beguin, RSC Adv., 2016, 6, 55144–55158 RSC.
- W. Wei, W. Ye, J. Wang, C. Huang, J. B. Xiong, H. Qiao, S. Cui, W. Chen, L. Mi and P. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 32269–32281 CrossRef CAS PubMed.
- W. Wei, J. Wu, S. Cui, Y. Zhao, W. Chen and L. Mi, Nanoscale, 2019, 11, 6243–6253 RSC.
- W. Wei, W. Chen, L. Ding, S. Cui and L. Mi, Nano Res., 2017, 10, 3726–3742 CrossRef CAS.
- M. Fouladi and A. Amadeh, Electrochim. Acta, 2013, 106, 1–12 CrossRef CAS.
- A. D. King, N. Birbilis and J. R. Scully, Electrochim. Acta, 2014, 121, 394–406 CrossRef CAS.
- D. Eaves, G. Williams and H. N. McMurray, Electrochim. Acta, 2012, 79, 1–7 CrossRef CAS.
- G. Williams and R. Grace, Electrochim. Acta, 2011, 56, 1894–1903 CrossRef CAS.
- A. S. Hamdy, I. Doench and H. Mohwal, Electrochim. Acta, 2011, 56, 2493–2502 CrossRef CAS.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC.
- M. W. Akhtar, Y. S. Lee, C. M. Yang and J. S. Kim, RSC Adv., 2016, 6, 100448–100458 RSC.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210–3221 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
I. S. Cole*b and
A. E. Kuznetsov
*a,
I. S. Cole*b and
A. E. Kuznetsov