
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDidymellanosine, a new decahydrofluorene analogue, and ascolactone C from Didymella sp. IEA-3B.1, an endophyte of Terminalia catappa†
Ni P. Ariantariab,
Elena Ancheeva a,
Marian Franka,
Fabian Stuhldreierc,
Dieter Meiera,
Yvonne Grönera,
Irene Reimched,
Nicole Teuschd,
Sebastian Wesselborgc,
Werner E. G. Müllere,
Rainer Kalscheuera,
Zhen Liu*a and
Peter Proksch*af
a,
Marian Franka,
Fabian Stuhldreierc,
Dieter Meiera,
Yvonne Grönera,
Irene Reimched,
Nicole Teuschd,
Sebastian Wesselborgc,
Werner E. G. Müllere,
Rainer Kalscheuera,
Zhen Liu*a and
Peter Proksch*af
aInstitute of Pharmaceutical Biology and Biotechnology, Heinrich Heine University Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany. E-mail: zhenfeizi0@sina.com; proksch@uni-duesseldorf.de
bDepartment of Pharmacy, Faculty of Mathematics and Natural Sciences, Udayana University, 80361 Bali, Indonesia
cInstitute of Molecular Medicine I, Medical Faculty, Heinrich Heine University Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany
dDepartment of Biomedical Sciences, Institute of Health Research and Education, University of Osnabrück, Germany
eInstitute for Physiological Chemistry, University Medical Center of the Johannes Gutenberg University Mainz, Duesbergweg 6, 55128 Mainz, Germany
fHubei Key Laboratory of Natural Products Research and Development, College of Biological and Pharmaceutical Sciences, China Three Gorges University, Yichang 443002, People's Republic of China
First published on 18th February 2020
Abstract
Didymellanosine (1), the first analogue of the decahydrofluorene-class of natural products bearing a 13-membered macrocyclic alkaloid conjugated with adenosine, and a new benzolactone derivative, ascolactone C (4) along with eight known compounds (2, 3, 5–10), were isolated from a solid rice fermentation of the endophytic fungus Didymella sp. IEA-3B.1 derived from the host plant Terminalia catappa. In addition, ascochitamine (11) was obtained when (NH4)2SO4 was added to rice medium and is reported here for the first time as a natural product. Didymellanosine (1) displayed strong activity against the murine lymphoma cell line L5178Y, Burkitt's lymphoma B cells (Ramos) and adult lymphoblastic leukemia T cells (Jurkat J16), with IC50 values of 2.0, 3.3 and 4.4 µM, respectively. When subjected to a NFκB inhibition assay, didymellanosine (1) moderately blocked NFκB activation in the triple-negative breast cancer cell line MDA-MB 231. In an antimicrobial assay, ascomylactam C (3) was the most active compound when tested against a panel of Gram-positive bacteria including drug-resistant strains with MICs of 3.1–6.3 µM, while 1 revealed weaker activity. Interestingly, both compounds were also found active against Gram-negative Acinetobacter baumannii with MICs of 3.1 µM, in the presence of a sublethal concentration (0.1 µM) of colistin.
Introduction
Endophytic fungi are microorganisms which reside in inner tissues of host plants, that represent a notable reservoir for a wide array of biologically active molecules.1,2 These metabolites provide natural product pharmacophores which are of importance for the discovery of molecules for pharmaceutical and agricultural purposes.1,3 Examples of bioactive natural products derived from fungal endophytes include the insecticidal compound nodulisporic acid A,4 and a number of potent anti-HIV compounds such as altertoxins I–III and V.5 Moreover, the isolation of remarkable anticancer agents from endophytes, especially those that were originally obtained from host plants, such as paclitaxel,6 podophyllotoxin,7 and camptothecin,8 point to their potential as alternative sources of pharmaceutically valuable metabolites.In our search for bioactive metabolites from endophytes, we investigated Didymella sp. IEA-3B.1, a fungus isolated from leaves of Terminalia catappa (Combretaceae) from Bali, Indonesia. Species of the genus Didymella have been identified as teleomorphs of numerous important plant pathogens formerly only known from their corresponding anamorphs. Examples within the family Didymellaceae include several environmentally relevant species of Ascochyta,9,10 and Phoma.11,12 Literature survey of the genus Didymella revealed the occurrence of the phytotoxin pinolidoxin,10 tricycloalternarene derivatives,13 and desmethyldichlorodiaportintone, a dichloroisocoumarin that showed significant inhibition of NO production.14 Another recent study on this genus afforded five compounds of the decahydrofluorene-class, including cytotoxic ascomylactams A–C, along with phomapyrrolidones A and C.15 In the present study, we describe the isolation and structure elucidation of two newly discovered metabolites (1 and 4) from a solid rice fermentation of Didymella sp. IEA-3B.1, and of ascochitamine (11) obtained from a fungal fermentation on solid rice medium following addition of (NH4)2SO4, as well as the results of cytotoxicity, NFκB inhibition and antimicrobial assays conducted with the isolated compounds.
Results and discussion
Chromatographic workup of the EtOAc extract of the fungal endophyte Didymella sp. cultured on solid rice medium, yielded two new natural products, didymellanosine (1) and ascolactone C (4), together with eight known compounds, phomapyrrolidone A (2),15,16 ascomylactam C (3),15 (9S,11R)-(+)-ascosalitoxin (5),17 ascochitine (6),18,19 fusarimine (7),20 3,6,8-trihydroxy-3-methyl-3,4-dihydroisocoumarin (8),21 3-methoxy-6,8-dihydroxy-3-methyl-3,4-dihydro-isocoumarin (9),21 and 6,8-dihydroxy-3-methyl-isocoumarin (10)22 (Fig. 1). The known compounds were identified by comparison of their NMR and MS data, as well as their specific optical rotations with data reported in the literature. | ||
| Fig. 1 Compounds isolated from Didymella sp. IEA-3B.1. | ||
Didymellanosine (1) was isolated as a white, amorphous solid. The molecular formula of 1 was established as C44H54N6O8 from the HRESIMS data, accounting for twenty-one degrees of unsaturation. The planar structure of 1 was deduced by detailed analysis of 1D and 2D NMR spectra, as well as by comparison of its NMR data to those of reported structurally related alkaloids.15,16,23 The 1H and 13C NMR spectra of 1 (Table 1) aided by HSQC revealed signals of six methyl groups, four methylenes and twenty-one methines, including six aromatic methines at δH 8.47 (H-43), 8.20 (H-39), 7.10 (H-30 and H-34), 6.97 (H-33) and 6.71 (H-31), as well as thirteen quaternary carbons (eleven sp2 and two sp3). The consecutive COSY correlations (Fig. 2) observed from H-7 through H-16, and between H-1/H-20, H-7/H-15, H-8/H-13, H-10/Me-24, and H-14/Me-25, together with the HMBC correlations from Me-20 to C-1, C-2 and C-5, from Me-21 to C-1, C-2 and C-3, from Me-22 to C-3, C-4, C-5, and C-16, and from Me-23 to C-5, C-6 and C-7, indicated the presence of a 5/6/5/6 tetracyclic system with six methyl groups at C-1, C-2, C-4, C-6, C-10 and C-12. The COSY correlations between H-30/H-31, H-33/H-34 along with the HMBC correlations from H-30 and H-34 to C-32, and from H-31 and H-33 to C-29 confirmed the presence of a para-substituted benzene ring in 1. Moreover, the NH signal resonating at δH 7.70 (H-35) showed HMBC correlations to C-18, C-26, C-27 and to a carbonyl C-19, while H-26 showed HMBC correlations to C-18, C-19, and C-27, suggesting the presence of a γ-lactam ring, which was further connected to the benzene ring through a methylene group, as evident from the HMBC correlations from H2-28 to C-26, C-27, C-29 and C-30, and from 27-OH to C-27 and C-28. Additional HMBC correlations from H-15 to C-32, from H-16 and H-26 to C-17, and from 17-OH (δH 11.44) to C-16, C-17 and C-18 indicated an ether bridge between C-15 and C-32 and the linkage of lactam ring and tetracyclic core through C-17. Thus, a macrocyclic decahydrofluorene skeleton similar to 3 was established for 1. The attachment of an additional adenosine moiety at C-26 was deduced by the spin systems from H-46 to H2-50 and between H-26 and NH-36, the HMBC correlations from H-46 to C-41 and C-43, from H-43 to C-41 and C-45, from H-39 to C-37 and C-41, from NH-36 to C-37 and C-45, and from H-26 to C-37, along with the molecular formula of 1. Thus, the planar structure of 1 was elucidated as shown (Fig. 2). Compound 1 shares a similar partial structure with embellicine B,23 and ascomylactam A,15 except for the presence of an adenosine unit in 1 instead of hydroxy or methoxy groups in the former compounds.
| No. | δC, type | δH (J in Hz) | No. | δC, type | δH (J in Hz) |
|---|---|---|---|---|---|
| a Recorded at 600 MHz (1H) and 125 MHz (13C) in DMSO-d6. | |||||
| 1 | 43.5, CH | 2.68, q (7.1) | 27 | 85.7, C | |
| 2 | 138.0, C | 28 | 46.0, CH2 | 2.94, d (12.5); 2.88, d (12.5) | |
| 3 | 131.3, CH | 4.44, s | 29 | 128.9, C | |
| 4 | 52.6, C | 30 | 130.6, CH | 7.10, d (8.4) | |
| 5 | 144.7, C | 31 | 119.4, CH | 6.97, dd (8.4, 2.2) | |
| 6 | 125.5, C | 32 | 157.3, C | ||
| 7 | 46.9, CH | 1.95, t (12.0) | 33 | 122.0, CH | 6.71, dd (8.4, 2.2) |
| 8 | 42.7, CH | 1.54, m | 34 | 129.9, CH | 7.11, d (8.4) |
| 9 | 39.6, CH2 | 2.08, d (12.2); 0.72, m | 35 | 7.70, s | |
| 10 | 31.8, CH | 1.52, m | 36 | 7.46, d (5.1) | |
| 11 | 44.7, CH2 | 1.74, m; 0.68, m | 37 | 152.2, C | |
| 12 | 30.9, CH | 1.79, m | 39 | 150.8, CH | 8.20, s |
| 13 | 56.3, CH | 1.16, m | 41 | 147.8, C | |
| 14 | 87.0, CH | 4.52, dd (8.1, 5.2) | 43 | 140.4, CH | 8.47, s |
| 15 | 52.3, CH | 1.97, m | 45 | 119.8, C | |
| 16 | 46.2, CH | 3.49, d (7.2) | 46 | 87.9, CH | 5.89, d (5.2) |
| 17 | 163.6, C | 47 | 73.7, CH | 4.48, q (5.4) | |
| 18 | 106.6, C | 48 | 70.0, CH | 4.13, q (4.6) | |
| 19 | 169.6, C | 49 | 85.3, CH | 3.94, q (3.9) | |
| 20 | 18.4, CH3 | 0.80, d (7.1) | 50 | 61.1, CH2 | 3.68, ddd (12.0, 4.7, 4.2); 3.52, ddd (12.0, 6.3, 3.8) |
| 21 | 12.3, CH3 | 0.40, s | 17-OH | 11.44, s | |
| 22 | 27.6, CH3 | 0.95, s | 27-OH | 6.62, s | |
| 23 | 15.3, CH3 | 1.73, s | 47-OH | 5.47, d (6.0) | |
| 24 | 22.4, CH3 | 0.92, d (6.5) | 48-OH | 5.18, d (5.1) | |
| 25 | 19.9, CH3 | 1.07, d (6.1) | 50-OH | 5.16, t (5.5) | |
| 26 | 55.5, CH | 4.11, d (5.1) | |||
 | ||
| Fig. 2 COSY and key HMBC correlations of compound 1. | ||
The relative configuration of 1 was deduced through analysis of the NOESY spectrum of 1 and by comparison with those of ascomylactams A and C (3),15 (Fig. 3). The NOE correlations between H-16/H-14, H-14/H-13, H-13/H-7, H-7/Me-22, Me-22/H-16, H-16/H-31, H-31/Me-25, Me-25/H-14, H-14/H-31, Me-25/H-11b, H-11b/H-13 and H-11b/Me-24 suggested these protons to be on the same side of the molecule, whereas NOEs observed for H-1/OH-17, OH-17/H-15, H-15/H-8, H-8/H-12, H-12/H-10, OH-17/H-26, H-26/H-33, and H-33/H-15 indicated them to be on the opposite side. In addition, the NOE correlations between H-26/H-34, H-26/H-33, H-33/H-15, and between NH-35/H-30, H-31/H-14, H-31/Me-25, OH-27/NH-36 indicated restricted rotation of the benzene ring and its parallel orientation to the γ-lactam ring, the same as reported for ascomylactams A and C (3).15 Moreover, the relative configuration of the adenosine moiety was elucidated by the NOE correlations between H-46/H-49, H-46/OH-47, and H-49/OH-48, as well as by comparison of the chemical shifts and coupling constants with literature data.24,25 Thus, the structure of compound 1 was determined as shown, representing the first example within decahydrofluorene-type alkaloids bearing a 13-membered macrocyclic skeleton conjugated with an adenosine moiety.
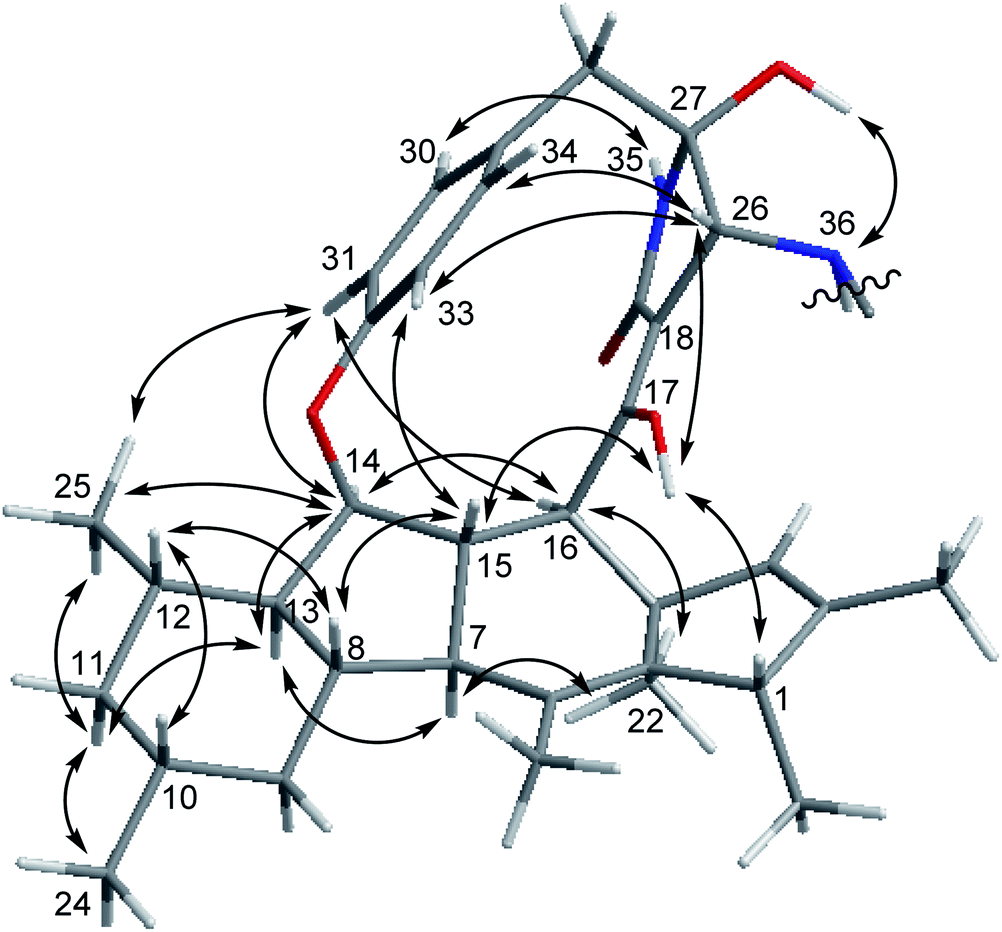 | ||
| Fig. 3 Key NOE correlations of compound 1. | ||
Ascolactone C (4), was obtained as a white, amorphous solid. Its HRESIMS spectrum exhibited a prominent pseudomolecular ion peak at m/z 279.1226 [M + H]+, which was attributed to the molecular formula C15H18O5, corresponding to 7 degrees of unsaturation. Inspection of the 1H NMR data of 4 (Table 2) revealed resonances of one aromatic singlet, four methyl groups, one set of methylene protons and one methine. The aromatic proton which appeared at δH 6.58 (H-7), exhibited HMBC correlations to C-3a, C-5 and C-6, while the aromatic methyl singlet at δH 2.09 (Me-14) showed HMBC correlations to C-4, C-5 and C-6, suggesting the presence of a penta-substituted benzene ring bearing a methyl substituent at the meta-position (Fig. 4). The COSY correlations between Me-11/H2-10/H-9/Me-12, along with the HMBC correlations from H-9, H2-10 and Me-12 to a ketone carbonyl at δC 210.8 (C-8), allowed the establishment of 2-methyl-1-oxobutyl moiety. The HMBC correlations from Me-13 (δH 1.67) to C-1, C-7a and C-8, and from H-7 to C-1, confirmed the connection of 2-methyl-1-oxobutyl moiety to the aromatic ring through C-1. Moreover, the weak HMBC correlation detected from H-7 and to the carbonyl C-3, together with the chemical shift of C-1 (δC 90.7) and the remaining one degree of unsaturation, indicated the presence of a γ-lactone fused to the benzene ring, thus forming the benzolactone skeleton. In addition, two hydroxy groups were deduced at C-4 and C-6 of the benzene ring based on the chemical shifts of C-4 and C-6 and the molecular formula of 4. Accordingly, the planar structure of 4 was established as shown (Fig. 4). Compound 4 is structurally related to ascolactones A and B, except for the replacement of the carboxy group at C-5 by a methyl group in 4.19 The absolute configuration of the latter two compounds was determined by TDDFT-ECD calculations and chemical reactions. The absolute configuration of 4 was concluded to be the same as that of ascolactone B (1S,9R) on the basis of a good agreement of the 1H NMR data with regard to resonances of the side chain (δH 0.49, 1.06 for Me-11,12 in ascolactone A compared to δH 0.85, 0.78 for Me-11,12 in ascolactone B), as well as based on similar negative values of their specific optical rotations.19
| Position | δC, typeb | δH (J in Hz) |
|---|---|---|
| a Recorded at 600 MHz (1H) and 150 MHz (13C).b Data were extracted from HSQC and HMBC. | ||
| 1 | 90.7, C | |
| 3 | 160.7, C | |
| 3a | 102.8, C | |
| 4 | 156.2, C | |
| 5 | 112.7, C | |
| 6 | 163.7, C | |
| 7 | 101.3, CH | 6.58, s |
| 7a | 149.0, C | |
| 8 | 210.8, C | |
| 9 | 42.2, CH | 2.83, m |
| 10 | 26.6, CH2 | 1.71, ddd (14.0, 7.4, 6.7) |
| 1.36, ddd (14.0, 7.4, 6.7) | ||
| 11 | 11.7, CH3 | 0.87, t (7.4) |
| 12 | 16.7, CH3 | 0.79, d (6.8) |
| 13 | 23.4, CH3 | 1.67, s |
| 14 | 7.7, CH3 | 2.09, s |
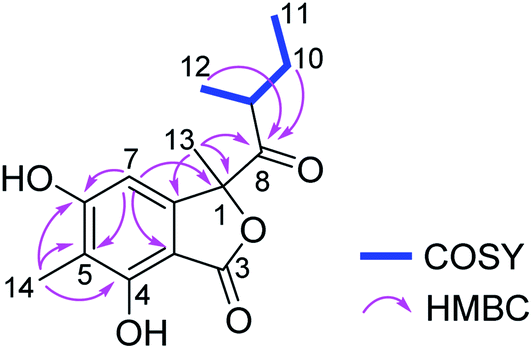 | ||
| Fig. 4 COSY and key HMBC correlations of compound 4. | ||
In an attempt to influence the metabolite pattern of the fungus Didymella sp. IEA-3B.1, the strain was subjected to further fermentation in presence of 3.5 g (NH4)2SO4 that had been added to solid rice medium. HPLC analysis of the extracts resulting from fungal fermentation on rice with and without addition of (NH4)2SO4, revealed distinct differences of the metabolite profiles between these two cultures (Fig. S1†). The production of the main fungal metabolite during fermentation on solid rice medium, ascochitine (6), an azaphilone contributing to the green color of this culture, dramatically decreased when (NH4)2SO4 was added to rice medium, thus resulting in a white coloration of the fungal culture in the presence of (NH4)2SO4 (Fig. S2†). Similarly, isocoumarins (8–10) and decahydrofluorene analogues (1–3) were significantly down-regulated in the salt containing culture. In contrast, compound 11 was only detected in the presence of (NH4)2SO4. The 1H NMR data of 11 were similar to those of ascochitine (6) and fusarimine (7), two azaphilone derivatives co-isolated in this study. Detailed investigation of HRESIMS and 2D NMR data of 11 identified it as the previously reported synthetic compound, ascochitamine, which was prepared by adding NH3 or NH4OH to ascochitine (6).26 The formation of nitrogen containing azaphilones through substitution of the pyrane oxygen by nitrogen derived from endogenous ammonia or exogenous amino acids during fungal fermentation is well known.27,28 The different pH values and nitrogen sources [(NH4)2SO4, NaNO3 and peptone], in the culture medium of Monascus anka have been shown to affect the composition and color of Monascus pigments as well.29 Therefore, the accumulation of ascochitamine (11) in this study is presumably due to the fungal response to the presence of ammonium present in the culture medium containing (NH4)2SO4. This result also provided further evidence of the effects of media composition (e.g. addition of salts) on the profile of azaphilone pigments, as we reported recently for bulgarialactone D isolated from a mistletoe-associated fungus, Bulgaria inquinans, cultured on solid Czapek medium containing different salt mixtures (MgSO4, NaNO3 and NaCl).30
All isolated compounds were investigated for their cytotoxicity towards the murine lymphoma cell line L5178Y. Compound 1 showed pronounced activity with an IC50 value of 2.0 µM, even more active than that of the positive control, kahalalide F (IC50 4.3 µM), while the remaining compounds were inactive. Furthermore, 1 was also active when assayed against two human cancer cell lines, Burkitt's lymphoma B cells (Ramos) and the adult lymphoblastic leukemia T cells (Jurkat J16) with IC50 values of 3.3 and 4.4 µM, respectively (Table 3). As a counter screen for general toxicity, 1 was assayed against non-malignant human fetal lung fibroblast MRC5 cells which resulted in an IC50 value of merely 21.2 µM. Thus, 1 is approximately 5–6 times more active against the tested cancer cell lines than against MRC5 cells, indicating moderate selectivity of 1 for cancer cells.
| L5178Ya | Ramosb | Jurkat J16b | MRC5c | |
|---|---|---|---|---|
| a Kahalalide F (IC50 4.3 µM) as positive control.b Staurosporine (IC50 2.5 µM) as positive control.c Kahalalide F (IC50 10.3 µM) as positive control.d Selectivity index (SI): IC50 value against MRC5 cells divided by IC50 values against cancer cells. | ||||
| 1 | 2.0 | 3.3 (6.4)d | 4.4 (4.8)d | 21.2 |
Furthermore, we evaluated the impact of compound 1 on NFκB activity. For NFκB inhibition study, 1 was tested in the triple-negative breast cancer (TNBC) cell line NFκB-MDA-MB-231 which is stably transfected with a NFκB-dependent luciferase reporter gene. The IC50 value of 1 amounted to 15.5 µM in this assay. To exclude that inhibition of NFκB activation is caused by cytotoxicity, cell viability of TNBC cells was determined in parallel. Compound 1 was about 3 times more potent in the NFκB inhibition assay compared to its cytotoxicity (IC50 45.4 µM), thereby suggesting that the antitumor activity of compound 1 (Table 3) may be due to blockade of NFκB activation.
Moreover, all isolated compounds were subjected to an antibacterial screening. Compound 3 was the most active substance against drug-susceptible and drug-resistant strains of the Gram-positive bacteria Staphylococcus aureus, Enterococcus faecalis and Enterococcus faecium, with MIC values ranging from 3.1 to 6.3 µM (Table 4). Compound 1 likewise inhibited the growth of the latter bacteria with MICs ranging from 6.3 to 12.5 µM, whereas much weaker activity was observed for 2 compared to the two aforementioned analogues. Intriguingly, 1 and 3 were also found to be active against the Gram-negative bacterium Acinetobacter baumannii, with comparable MIC values of 3.1 µM, both tested in the presence of a sublethal concentration of colistin (0.1 µM). The activity of 1 against Gram-negative A. baumannii is not due to general toxicity of the compound as shown by comparison of the MIC of 1 with the IC50 against MRC5 cells (Tables 3 and 4). Compounds with activity against A. baumannii are of substantial interest, as the increasing emergence of multidrug-resistant strains has led to serious clinical challenges due to the limited number of effective antimicrobial drugs.31,32 Combination therapy of colistin with other antibacterial agents is currently considered as a promising alternative for the treatment of A. baumannii infections.33,34 All compounds were inactive against the Gram-positive bacterium Mycobacterium tuberculosis when tested up to 100 µM. On the basis of the antimicrobial activity of these alkaloids, the presence of a γ-lactam substructure as present in 1 and 3 seems to be preferable for the activity rather than a succinimide moiety as in 2. As a lower potency towards Gram-positive bacteria was observed for 1 in comparison to 3, it is suggested that the adenosine conjugate in 1 might attenuate its antimicrobial activity with regard to particular strains, while it had no effect on the tested Gram-negative bacterium.
| M. tuberculosisa | S. aureusb | E. faecalisb | E. faeciumb | A. baumanniib | ||||
|---|---|---|---|---|---|---|---|---|
| H37Rv | ATCC 29213c | ATCC 700699d | ATCC 29212c | ATCC 51299e | ATCC 35667c | ATCC 700221f | BAA 1605g | |
| a Rifampicin as positive control.b Moxifloxacin as positive control.c Drug-susceptible strain.d Methicillin-resistant strain of S. aureus (MRSA).e Vancomycin-resistant strain of E. faecalis.f Vancomycin-resistant strain of E. faecium.g Tested in the presence of a sublethal concentration (0.1 µM) of colistin. | ||||||||
| 1 | >100 | 6.3 | 6.3 | 6.3 | 12.5 | 12.5 | 6.3 | 3.1 |
| 2 | >100 | 25 | 12.5 | 100 | 50 | 25 | 12.5 | 12.5 |
| 3 | >100 | 3.1 | 3.1 | 6.3 | 3.1 | 6.3 | 3.1 | 3.1 |
| 6 | >100 | 50 | 50 | >100 | >100 | 50 | >100 | >100 |
Of note, biosynthetically related fungal alkaloids belonging to the decahydrofluorene-class, such as GKK1032s,35,36 pyrrocidines,37–40 hirsutellones,41,42 trichobamide A,43 and penicipyrrodiether A,44 were found in previous studies to exhibit antibacterial activity against Gram-positive bacteria including drug-resistant strains,36–38,44 and against Mycobacterium tuberculosis.41,42 These compounds furthermore exhibited antifungal activity,37 were active as inhibitors of prolyl oligopeptidase,39 and were shown to be cytotoxic against numerous cancer cell lines.35,40,43 The complex molecular architecture, consisting of a tricyclic polyketide fused to a 12 or 13-membered macroether ring which contains a γ-lactam or a succinimide moiety, combined with their intriguing bioactivities, have triggered tremendous efforts in synthetic and biosynthetic studies as well.45–47 However, decahydrofluorenes featuring a tetracyclic core as encountered in compound 1 are rarely reported. To date, only embellicines A and B, possessing cytostatic, cytotoxic and NFκB inhibitory activities, from a fungal endophyte Embellisia eureka,23 antitubercular phomapyrrolidones A–C from an endophytic Phoma sp. NRRL 46751,16 and cytotoxic ascomylactams A–C from a mangrove associated fungus Didymella sp. CYSK-4,15 stand as examples. Thus, in light of the bioactivity results reported for didymellanosine (1) in this study and its new chemical feature bearing an adenosine unit attached to a pyrrolidinone, further studies on the pharmacological properties of this metabolite seem promising.
Experimental section
General procedures
HPLC analysis was carried out with a Dionex UltiMate 3000 system coupled with an UltiMate 3000 pump linked to a photodiode array detector (DAD 3000 RS). Detection wavelengths were set at 235, 254, 280, and 340 nm. The column was prefilled with Eurospher 100-10 C18, 125 × 4 mm (Knauer, Germany). The routine HPLC analysis was performed with the following gradient (MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1% HCOOH in H2O): 0 min (10% MeOH); 5 min (10% MeOH); 35 min (100% MeOH); 45 min (100% MeOH). Semipreparative HPLC was conducted with a Merck Hitachi Chromaster HPLC system (UV detector 5410; pump 5110; column Eurospher 100-10 C18, 300 × 8 mm, Knauer; flow rate at 5 mL min−1). Silica gel 60 M (Macherey-Nagel) was used for vacuum liquid chromatography (VLC) and Sephadex LH-20 for column chromatography. TLC plates pre-coated with silica gel 60 F254 (Macherey-Nagel) were used for routine analysis. One- and two-dimensional NMR spectra were recorded on Bruker AVANCE DMX 600 or 500 NMR spectrometers. ESIMS and HRESIMS data were acquired by a Finnigan LCQ Deca mass spectrometer and an UHR-QTOF maXis 4G (Bruker Daltonics) mass spectrometer, respectively. FT-IR spectra were recorded on a Bruker Tensor 37 IR spectrometer in a range of 4000–400 cm−1 with the resolution of 4 cm−1 as a KBr pellet. Optical rotations were measured on a Jasco P-2000 polarimeter.
:0.1% HCOOH in H2O): 0 min (10% MeOH); 5 min (10% MeOH); 35 min (100% MeOH); 45 min (100% MeOH). Semipreparative HPLC was conducted with a Merck Hitachi Chromaster HPLC system (UV detector 5410; pump 5110; column Eurospher 100-10 C18, 300 × 8 mm, Knauer; flow rate at 5 mL min−1). Silica gel 60 M (Macherey-Nagel) was used for vacuum liquid chromatography (VLC) and Sephadex LH-20 for column chromatography. TLC plates pre-coated with silica gel 60 F254 (Macherey-Nagel) were used for routine analysis. One- and two-dimensional NMR spectra were recorded on Bruker AVANCE DMX 600 or 500 NMR spectrometers. ESIMS and HRESIMS data were acquired by a Finnigan LCQ Deca mass spectrometer and an UHR-QTOF maXis 4G (Bruker Daltonics) mass spectrometer, respectively. FT-IR spectra were recorded on a Bruker Tensor 37 IR spectrometer in a range of 4000–400 cm−1 with the resolution of 4 cm−1 as a KBr pellet. Optical rotations were measured on a Jasco P-2000 polarimeter.
Fungal isolation, identification and cultivation
The fungus, Didymella sp. IEA-3B.1, was isolated from healthy leaves of Terminalia catappa (Combretaceae), collected in April 2018, in Jimbaran in the south of Bali, Indonesia. The fungal culture was identified by a standard molecular biology protocol, through DNA amplification and sequencing of the ITS region as described before.48 The sequence data have been submitted to the GenBank with accession No. MN227696.1. The voucher strain was deposited in the Institute of Pharmaceutical Biology and Biotechnology, Düsseldorf, Germany. The fungus was cultured on twenty 1 L Erlenmeyer flasks, each flask containing 100 g rice in 100 mL distilled water followed by autoclaving. Another set of fungal fermentation was carried out on five 1 L Erlenmeyer flasks by adding 3.5 g (NH4)2SO4 to 100 g rice in 100 mL distilled water to each flask, followed by autoclaving. The fungal culture was maintained under static conditions at room temperature until the rice medium was completely covered by the fungus (30 days).Extraction and isolation
The fungal culture grown on solid rice medium was extracted twice, each with 500 mL EtOAc added to each flask. The EtOAc extract was concentrated in vacuo and the obtained crude extract (32.9 g) was subjected to liquid–liquid partitioning between n-hexane and 90% aqueous MeOH. The 90% aqueous MeOH extract (5.0 g) was chromatographed on silica gel 60 (VLC) by a step gradient elution with n-hexane–EtOAc followed by CH2Cl2–MeOH to afford 13 fractions (V1–V13). Based on HPLC chromatograms, fractions V3, V4, V5, V9, and V10 eluted with n-hexane–EtOAc (6:4), (4:6), (2:8), and CH2Cl2–MeOH (9:1), (7:3), respectively, were subjected to further separation. Fraction V3 (315.7 mg) was applied to a Sephadex LH-20 column and eluted with CH2Cl2–MeOH (1:1) to obtain 4 subfractions. Purification of subfraction V3.2 (164.6 mg) was carried out with semipreparative HPLC employing MeOH–H2O as mobile phase (from 85% to 100% MeOH) to afford 2 (45.6 mg). Meanwhile, compounds 4 (1.8 mg), 5 (5.8 mg) and 10 (2.1 mg) were obtained after purification of subfraction V3.3 (34.3 mg) by semipreparative HPLC with gradient elution of MeOH–H2O (from 50% to 100% MeOH). Separation of fraction V4 (200.3 mg) on Sephadex LH-20 using CH2Cl2–MeOH (1:1) as eluent, followed by purification using semipreparative HPLC eluted with MeOH–0.1% HCOOH in H2O (from 85% to 100% MeOH), yielded 3 (3.4 mg). In a similar manner, fraction V5 (625.5 mg) was submitted to a Sephadex LH-20 column, to give 6 (263.0 mg) along with 5 subfractions. Subfraction V5.6 (37.8 mg) was purified by semipreparative HPLC, employing MeOH–0.1% HCOOH in H2O (from 30% to 70% MeOH) as mobile phase, to yield 8 (11.6 mg) and 9 (2.2 mg). Furthermore, compound 7 (30.0 mg) was obtained by separation of fraction V9 (272.8 mg) on a Sephadex LH-20 column employing CH2Cl2–MeOH (1:1) as eluent. Compound 1 (20.8 mg) was afforded through separation of fraction V10 (308.6 mg) over a Sephadex LH-20 column, eluted with CH2Cl2–MeOH (1:1), followed by purification using semipreparative HPLC with MeOH–H2O (from 85% to 100% MeOH). Following the same procedure, the fungal culture by supplementing rice medium with (NH4)2SO4 was extracted with EtOAc, taken to dryness, and followed by liquid–liquid partition between n-hexane and 90% aqueous MeOH. The resulting MeOH fraction (340.2 mg) was rinsed with various solvents which yielded the pure compound 11 (3.0 mg).
Cytotoxicity assay
Cytotoxicity was tested against the murine lymphoma cell line L5178Y using the MTT method. Kahalalide F (IC50 4.3 µM) and culture media containing 0.1% DMSO were included as positive and negative controls, respectively.30 Briefly, cells were seeded in 96-well plates (270 cells per well) and treated with tested compounds in the range of concentrations from 0.13 to 13 µM in five points for 72 h. Next, 20 µL of MTT (5 mg mL−1) was added to each well and incubated for 3 h. After cell lysis, the absorbance of reduced MTT was measured by a Varioskan Flash (Thermo Scientific) at 595 nm.Meanwhile, cytotoxicity of 1 against the human cell lines Ramos (Burkitt's lymphoma B cells), Jurkat J16 (adult lymphoblastic leukemia T cells) and non-malignant cells MRC5 (human fetal lung fibroblast cells) was determined by the resazurin reduction assay. In brief, cells were plated in 96-well plates (5 × 104 cells per well) and incubated with the indicated compound in concentrations ranging from 0.01 to 30 µM for 72 h, respectively. Subsequently, resazurin was added to a final concentration of 40 µM. After 3 h of incubation fluorescence of resorufin (excitation: 535 nm, emission: 590 nm) was measured via a microplate spectrophotometer. The reduction of resazurin to resorufin is proportional to aerobic respiration and therefore can be used as an indicator for cell viability.49 Staurosporine (IC50 2.5 µM) was employed as a positive control for the cytotoxicity assay against Ramos and Jurkat J16 cells, while kahalalide F (IC50 10.3 µM) was used as a positive control for the assay against MRC5 cells. Culture medium containing 1% DMSO (concentration equal to the highest concentration of DMSO used in the dilutions of the tested compound) served as a negative control for the assay.
Cell culture for MDA-MB-231 cells and materials
Culture medium and supplements were purchased from Gibco (Fisher Scientific, Schwerte, Germany). Cell plates were purchased by Greiner bio-one (Frickenhausen, Germany). Cells were grown and incubated in a humidified 5% CO2 atmosphere at constant 37 °C. The metastatic breast cancer cell line, MDA-MB-231, was obtained from the European Collection of Authenticated Cell Cultures (ECACC, Salisbury, UK). Subculture was done in RPMI 1640 medium (Cat# 21875-034) supplemented with 15% (v/v) fetal calf serum (FCS) and 1% (v/v) penicillin–streptomycin (pen–strep) (10000 U mL−1). The monoclonal NFκB-MDA-MB-231 cell line contains a NFκB response element to control the luciferase reporter gene. For the procedure of cell line generation see the publication of Sperlich et al. (2017).50 Subculture was performed in high glucose DMEM (Cat# 41966-029) supplemented with 10% (v/v) FCS, 1% (v/v) pen–strep (10000 U mL−1) and 400 µg mL−1 hygromycin B (Life Technologies, Darmstadt, Germany; Cat# 10687010) for selection. Starvation medium for the NFκB inhibition assay was composed of high glucose DMEM medium supplemented with 1% (v/v) FCS, 1% (v/v) pen–strep (10000 U mL−1). Cell detachment occurred by trypsinization in 0.25% trypsin–EDTA and cell counting was performed at 1:1 (v/v) dilution in erythrosin B (BioCat, Heidelberg, Germany; #L13002) using the LUNA II automated cell counter (BioCat). Compound 1 used for NFκB inhibition and cell viability assay, was dissolved in DMSO to a final concentration of 10 mM and diluted in cell culture medium with final assay conditions of 1% DMSO.
Cell viability of MDA-MB-231 cells in presence of 1
Cytotoxicity of 1 against triple-negative breast cancer was determined using the cell line MDA-MB-231. Using the CyBio® Well vario pipetting robot (Analytik Jena, Jena, Germany; #OL3381-24-730), 18 µL of the cell density of 2.8 × 105 cells per mL were seeded per well on a 384-well plate (Greiner; #781074) and incubated for 24 h. For treatment in quadruples, 2 µL of RPMI + 1% DMSO (negative control) or the ten-fold concentrated substance was applied to reach a final volume of 20 µL per well. The final concentration for compound ranged in two-fold serial dilution steps from 100 µM to 0.78 µM in eight points. As a positive control, doxorubicin (Sigma, CAS# 25316-40-9) was applied in a final concentration of 30 µM. Compound stimulation endured for 2 h and final cell lysis and measurement was done as prescribed in the manufacturer's instruction of the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Mannheim, Germany; #G7570). In short, it was applied the equal volume of CellTiter-Glo® and luminescence was measured using the Spark® microplate reader (TECAN, Männedorf, Switzerland).NFκB inhibition in NFκB-MDA-MB-231 cells in presence of 1
For testing compound 1 concerning NFκB inhibitory activity, 4 × 104 NFκB-MDA-MB-231 cells were seeded in total 100 µL medium per well on a 96-well plate (Greiner; Cat# 655098). On the next day, medium was exchanged and cells pre-incubated for 20 min without (negative control) or with diluted substance in total 100 µL starvation medium. The final concentration of the compounds ranged a two-fold serial dilution from 100 µM downwards to 0.78 µM in 8 points. To activate NFκB signaling, untreated cells (positive control) or compound treated cells were subsequently stimulated for 2 h with 1 µg mL−1 lipopolysaccharide (Sigma-Aldrich, Taufkirchen, Germany; #L2630). Last, cell lysis and measurement was done according to the manufacturer's instruction of the Nano-Glo Luciferase Assay System (Promega; #N1110). In short, it was applied the equal volume of 1:50 (v/v) diluted Nano-Glo reagent and luminescence was measured using the Spark® microplate reader (TECAN).
Antimicrobial activity
The tested bacteria included Staphylococcus aureus (ATCC 29213 and 700699), Enterococcus faecium (ATCC 35667 and 700221), Enterococcus faecalis (ATCC 29212 and 51299), Acinetobacter baumannii (ATCC BAA1605) and Mycobacterium tuberculosis (H37Rv). Except for M. tuberculosis, antimicrobial activity was evaluated utilizing the microdilution method in accordance with the CLSI guidelines.51 Activity against M. tuberculosis was assessed employing the resazurin dye reduction method as described previously.52 Moxifloxacin was used as positive control for all tested Gram-positive and Gram-negative bacteria except for Mycobacterium tuberculosis, where rifampicin was used as a positive control. The tested compounds were pre-dissolved in DMSO and a final amount of 1% DMSO was included as a negative control.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. P. A. wishes to thank DAAD Program Biodiversity and Health for the doctoral scholarship. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 270650915/GRK 2158 (to P. P., S. W. and R. K.). P. P. also wants to thank the Manchot Foundation for support. Furthermore, we wish to thank Mrs Birgit Tommes for technical assistance in FT-IR measurements.Notes and references
- A. H. Aly, A. Debbab, J. Kjer and P. Proksch, Fungal Divers., 2010, 41, 1–16 CrossRef.
- G. Strobel, J. Fungi, 2018, 4, 57 CrossRef PubMed.
- H. Gao, G. Li and H. X. Lou, Molecules, 2018, 23, 646 CrossRef.
- J. G. Ondeyka, G. L. Helms, O. D. Hensens, M. A. Goetz, D. L. Zink, A. Tsipouras, W. L. Shoop, L. Slayton, A. W. Dombrowski, J. D. Polishook, D. A. Ostlind, N. N. Tsou, R. G. Ball and S. B. Singh, J. Am. Chem. Soc., 1997, 119, 8809–8816 CrossRef CAS.
- B. P. Bashyal, B. P. Wellensiek, R. Ramakrishnan, S. H. Faeth, N. Ahmad and A. A. L. Gunatilaka, Bioorg. Med. Chem., 2014, 22, 6112–6116 CrossRef CAS.
- A. Stierle, G. Strobel and D. Stierle, Science, 1993, 260, 214–216 CrossRef CAS PubMed.
- S. C. Puri, A. Nazir, R. Chawla, R. Arora, S. Riyazul-Hasan, T. Amna, B. Ahmed, V. Verma, S. Singh, R. Sagar, A. Sharma, R. Kumar, R. K. Sharma and G. N. Qazi, J. Biotechnol., 2006, 122, 494–510 CrossRef CAS.
- S. Kusari, S. Zühlke and M. Spiteller, J. Nat. Prod., 2009, 72, 2–7 CrossRef CAS PubMed.
- M. I. Chilvers, J. D. Rogers, F. M. Dugan, J. E. Stewart, W. Chen and T. L. Peever, Mycol. Res., 2009, 113, 391–400 CrossRef CAS PubMed.
- A. Cimmino, A. Andolfi, S. Fondevilla, M. A. Abouzeid, D. Rubiales and A. Evidente, J. Agric. Food Chem., 2012, 60, 5273–5278 CrossRef CAS PubMed.
- M. M. Aveskamp, J. de Gruyter, J. H. C. Woudenberg, G. J. M. Verkley and P. W. Crous, Stud. Mycol., 2010, 65, 1–60 CrossRef CAS PubMed.
- Q. Chen, J. R. Jiang, G. Z. Zhang, L. Cai and P. W. Crous, Stud. Mycol., 2015, 82, 137–217 CrossRef CAS PubMed.
- C. Li, Z. Hu, Q. Liu, X. Wu and S. Cao, Tetrahedron Lett., 2018, 59, 3381–3383 CrossRef CAS.
- Y. Chen, Z. Liu, H. Liu, Y. Pan, J. Li, L. Liu and Z. She, Mar. Drugs, 2018, 16, 54 CrossRef PubMed.
- Y. Chen, Z. Liu, Y. Huang, L. Liu, J. He, L. Wang, J. Yuan and Z. She, J. Nat. Prod., 2019, 82, 1752–1758 CrossRef CAS PubMed.
- E. M. K. Wijeratne, H. He, S. G. Franzblau, A. M. Hoffman and A. A. L. Gunatilaka, J. Nat. Prod., 2013, 76, 1860–1865 CrossRef CAS PubMed.
- M. Leyte-Lugo, M. González-Andrade, M. d. C. González, A. E. Glenn, C. M. Cerda-Garcia-Rojas and R. Mata, J. Nat. Prod., 2012, 75, 1571–1577 CrossRef CAS PubMed.
- L. Colombo, C. Gennari, G. S. Ricca, C. Scolastico and F. Aragozzini, J. Chem. Soc., Perkin Trans. 1, 1980, 675–676 RSC.
- S. F. Seibert, E. Eguereva, A. Krick, S. Kehraus, E. Voloshina, G. Raabe, J. Fleischhauer, E. Leistner, M. Wiese, H. Prinz, K. Alexandrov, P. Janning, H. Waldmann and G. M. König, Org. Biomol. Chem., 2006, 4, 2233–2240 RSC.
- S.-X. Yang, J. Xiao, H. Laatsch, J. J. Holstein, B. Dittrich, Q. Zhang and J.-M. Gao, Tetrahedron Lett., 2012, 53, 6372–6375 CrossRef CAS.
- K. Kameda, H. Aoki, H. Tanaka and M. Namiki, Agric. Biol. Chem., 1973, 37, 2137–2146 CrossRef CAS.
- D. S. Zinad, K. A. Shaaban, M. A. Abdalla, M. T. Islam, A. Schüffler and H. Laatsch, Nat. Prod. Commun., 2011, 6, 45–48 CrossRef CAS PubMed.
- W. Ebrahim, A. H. Aly, V. Wray, A. Mándi, M.-H. Teiten, F. Gaascht, B. Orlikova, M. U. Kassack, W. Lin, M. Diederich, T. Kurtán, A. Debbab and P. Proksch, J. Med. Chem., 2013, 56, 2991–2999 CrossRef CAS PubMed.
- H. Rosemeyer, G. Toth and F. Seela, Nucleosides Nucleotides, 1989, 8, 587–597 CrossRef CAS.
- P. Ciuffreda, S. Casati and A. Manzocchi, Magn. Reson. Chem., 2007, 45, 781–784 CrossRef CAS PubMed.
- H. Mishima, M. Kurabayashhi, H. Oku and I. Iwai, Annu. Rep. Sankyo Res. Lab., 1970, 22, 67–79 CAS.
- T. F. Lin, K. Yakushijin, G. H. Büchi and A. L. Demain, J. Ind. Microbiol., 1992, 9, 173–179 CrossRef CAS.
- X. Wang, J. G. S. Filho, A. R. Hoover, J. B. King, T. K. Ellis, D. R. Powell and R. H. Cichewicz, J. Nat. Prod., 2010, 73, 942–948 CrossRef CAS PubMed.
- K. Shi, D. Song, G. Chen, M. Pistolozzi, Z. Wu and L. Quan, J. Biosci. Bioeng., 2015, 120, 145–154 CrossRef CAS PubMed.
- N. P. Ariantari, G. Daletos, A. Mándi, T. Kurtán, W. E. G. Müller, W. Lin, E. Ancheeva and P. Proksch, RSC Adv., 2019, 9, 25119–25132 RSC.
- J. Li, C. R. Rayner, R. L. Nation, R. J. Owen, D. Spelman, K. E. Tan and L. Liolios, Antimicrob. Agents Chemother., 2006, 50, 2946–2950 CrossRef CAS PubMed.
- L.-C. Kuo, C.-C. Lai, C.-H. Liao, C.-K. Hsu, Y.-L. Chang, C.-Y. Chang and P.-R. Hsueh, Clin. Microbiol. Infect., 2007, 13, 196–198 CrossRef CAS PubMed.
- S. Biswas, J.-M. Brunel, J.-C. Dubus, M. Reynaud-Gaubert and J.-M. Rolain, Expert Rev. Anti-Infect. Ther., 2012, 10, 917–934 CrossRef CAS PubMed.
- A. M. Peri, Y. Doi, B. A. Potoski, P. N. A. Harris, D. L. Paterson and E. Righi, Diagn. Microbiol. Infect. Dis., 2019, 94, 413–425 CrossRef CAS PubMed.
- F. Koizumi, Jpn. Kokat Tokyo Koho, Japanese Patent P2001-247574A, 2001.
- X. Qi, X. Li, J. Zhao, N. He, Y. Li, T. Zhang, S. Wang, L. Yu and Y. Xie, J. Antibiot., 2019, 72, 237–240 CrossRef CAS PubMed.
- H. He, H. Y. Yang, R. Bigelis, E. H. Solum, M. Greenstein and G. T. Carter, Tetrahedron Lett., 2002, 43, 1633–1636 CrossRef CAS.
- T. M. Casella, V. Eparvier, H. Mandavid, A. Bendelac, G. Odonne, L. Dayan, C. Duplais, L. S. Espindola and D. Stien, Phytochemistry, 2013, 96, 370–377 CrossRef CAS PubMed.
- Y. Shiono, A. Kosukegawa, T. Koseki, T. Murayama, E. Kwon, S. Uesugi and K.-i. Kimura, Phytochem. Lett., 2012, 5, 91–95 CrossRef CAS.
- S. Uesugi, N. Fujisawa, J. Yoshida, M. Watanabe, S. Dan, T. Yamori, Y. Shiono and K.-i. Kimura, J. Antibiot., 2016, 69, 133–140 CrossRef CAS.
- M. Isaka, N. Rugseree, P. Maithip, P. Kongsaeree, S. Prabpai and Y. Thebtaranonth, Tetrahedron, 2005, 61, 5577–5583 CrossRef CAS.
- M. Isaka, W. Prathumpai, P. Wongsa and M. Tanticharoen, Org. Lett., 2006, 8, 2815–2817 CrossRef CAS.
- S. Chen, H. Shen, P. Zhang, H. Cheng, X. Dai and L. Liu, Chem. Commun., 2019, 55, 1438–1441 RSC.
- T. Song, M. Chen, Z.-W. Ge, W. Chai, X.-C. Li, Z. Zhang and X.-Y. Lian, J. Org. Chem., 2018, 83, 13395–13401 CrossRef CAS PubMed.
- H. Oikawa, J. Org. Chem., 2003, 68, 3552–3557 CrossRef CAS.
- H. Uchiro, R. Kato, Y. Arai, M. Hasegawa and Y. Kobayakawa, Org. Lett., 2011, 13, 6268–6271 CrossRef CAS PubMed.
- A. Ear, S. Amand, F. Blanchard, A. Blond, L. Dubost, D. Buisson and B. Nay, Org. Biomol. Chem., 2015, 13, 3662–3666 RSC.
- J. Kjer, A. Debbab, A. H. Aly and P. Proksch, Nat. Protoc., 2010, 5, 479–490 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef PubMed.
- J. Sperlich, R. Kerr and N. Teusch, Mar. Drugs, 2017, 15, 262 CrossRef PubMed.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard Ninth ed. CLSI document M07-A9, Clinical and Laboratory Standards Institute, Wayne, PA, 2012 Search PubMed.
- N. Rehberg, E. Omeje, S. S. Ebada, L. van Geelen, Z. Liu, P. Sureechatchayan, M. U. Kassack, T. R. Ioerger, P. Proksch and R. Kalscheuer, Antimicrob. Agents Chemother., 2019, 63, e00136-19 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MS, 1D and 2D NMR spectra of compounds 1, 4 and 11. See DOI: 10.1039/c9ra10685e |
| This journal is © The Royal Society of Chemistry 2020 |