
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of Palmarumycin BGs, C1 and Guignardin E†
Xinlei Liu a,
Shuyi Lia,
Xinyu Weia,
Yu Zhaoa,
Daowan Laib,
Ligang Zhoub and
Mingan Wang*a
a,
Shuyi Lia,
Xinyu Weia,
Yu Zhaoa,
Daowan Laib,
Ligang Zhoub and
Mingan Wang*a
aDepartment of Applied Chemistry, College of Sciences, China Agricultural University, Beijing 100193, People's Republic of China. E-mail: wangma@cau.edu.cn
bDepartment of Plant Pathology, College of Plant Protection, China Agricultural University, Beijing 100193, People's Republic of China
First published on 8th January 2020
Abstract
The first total synthesis of Palmarumycin BG1–3, BG5–6, C1 and Guignardin E (1–7) were achieved by the same intermediate Palmarumycin C2 through a N-benzyl cinchoninium chloride-catalyzed epoxidation, an organoselenium-mediated reduction, and a cerium(III) chloride hydrate-promoted regioselective ring-opening and elimination of cyclic α,β-epoxy ketone as the key steps via 6–7 step routes using 1,8-dihydroxynaphthalene (DHN) and 5-methoxytetralone as the starting materials in overall yields of 1.0–17.4%, respectively. Their structures and absolute configurations were characterized and determined by 1H, 13C NMR, IR, HR-ESI-MS and X-ray diffraction data. These compounds displayed significant inhibition activities against HCT116, U87-MG, HepG2, BGC823 and PC9 cell lines.
Introduction
The members of the spirobisnaphthalene family showed broad bioactivities, such as antifungal, antimicrobial, antitumor, anticancer, antiparasitic, anti-inflammatory, and cytotoxic activities.1–4 Palmarumycin BG1–3, and BG5–6 (Fig. 1) were previously isolated from the leaves and stems of the Bruguiera gymnorrhiza plant, which were collected from the Zhanjiang mangrove national nature protection area.5 Guignardins A–F and Palmarumycin C1 were isolated from the endophytic fungus Guignardia sp. KcF8, and the mangrove unidentified fungus BCC 25903, respectively.6,7 Among these compounds, Palmarumycin BG5 showed excellent cytotoxicities against human breast carcinoma MCF-7 with IC50 7.6 μM and promyelocytic leukemia HL60 with IC50 1.9 μM, Guignardins E, F and Palmarumycin C1 exhibited significant cytotoxicities against 10 human tumor cell lines, such as MCF-7 with LD50 8.79, 6.48 and 3.08 μM, HL60 with LD50 2.94, 3.06 and 2.90 μM, and HeLa with LD50 1.32, 0.38 and 1.24 μM et al.6 Many reports related to the total synthesis, structure modification, and biological activity evaluation of the spirobisnaphthalene natural products have appeared in recent years.8–20 The synthesis of Palmarumycin CP17 and its analogues were completed in our laboratory, and the bioassay results showed that they have antifungal activity against several phytopathogens.21 Because of the interesting larvicidal activity of Palmarumycin B6 against Aedes albopictus, its limited access in the fermentation extract of the endophytic fungus Berkleasmium sp., and the importance of the halogen atom in the A-ring.22,23 the total synthesis and structure revision of Palmarumycin B6 were achieved, and found that the location of the chlorine atom plays a crucial role for the larvicidal activity against A. albopictus in the previous report.24 The antitumor activities of Palmarumycin BG5, Guignardin E and Palmarumycin C1 aroused our enthusiasm to gain insights into the structure–activity relationships of these spirobisnaphthalene compounds and explore their mode of action. As such, the first total synthesis of Palmarumycin BGs, Guignardin E and Palmarumycin C1 (1–7) were carried out, and the results would be presented in this paper.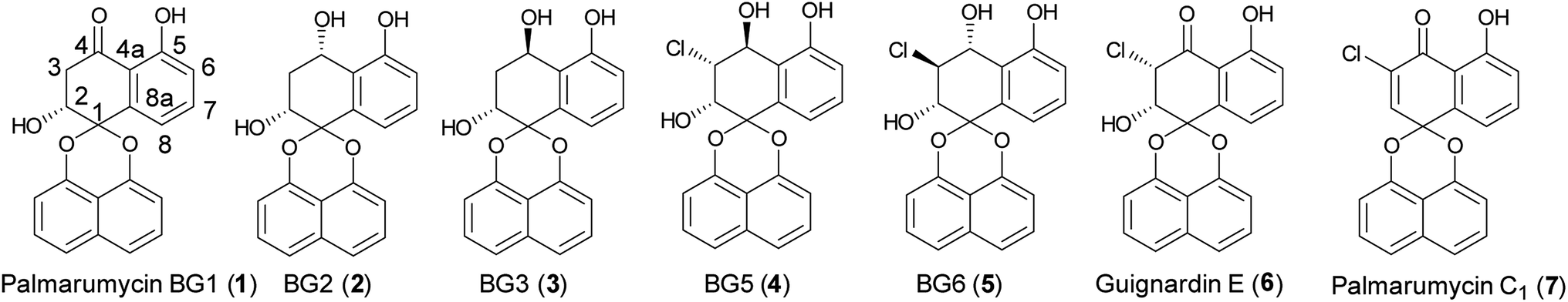 | ||
| Fig. 1 Structures of Palmarumycin BG1–3, BG5–6, Guignardin E, and Palmarumycin C1 (1–7). | ||
Results and discussion
The retro-synthetic analysis of Palmarumycin BGs and Guignardin E (1–6) is shown in Scheme 1. Palmarumycin BG2, BG3, BG5 and BG6 can be obtained through reduction of Palmarumycin BG1 and Guignardin E. Palmarumycin BG1 and Guignardin E can be synthesized by the selective reduction and ring-opening of Palmarumycin C2, which was easily prepared by the stereoselectivity epoxidation of Palmarumycin CP1. Palmarumycin CP1 could be obtained from a ketalization of 5-methoxytetralone with 1,8-dihydroxynaphthalene (DHN) in a similar process, such as that used in the synthesis of Palmarumycins CP17 and B6.21,24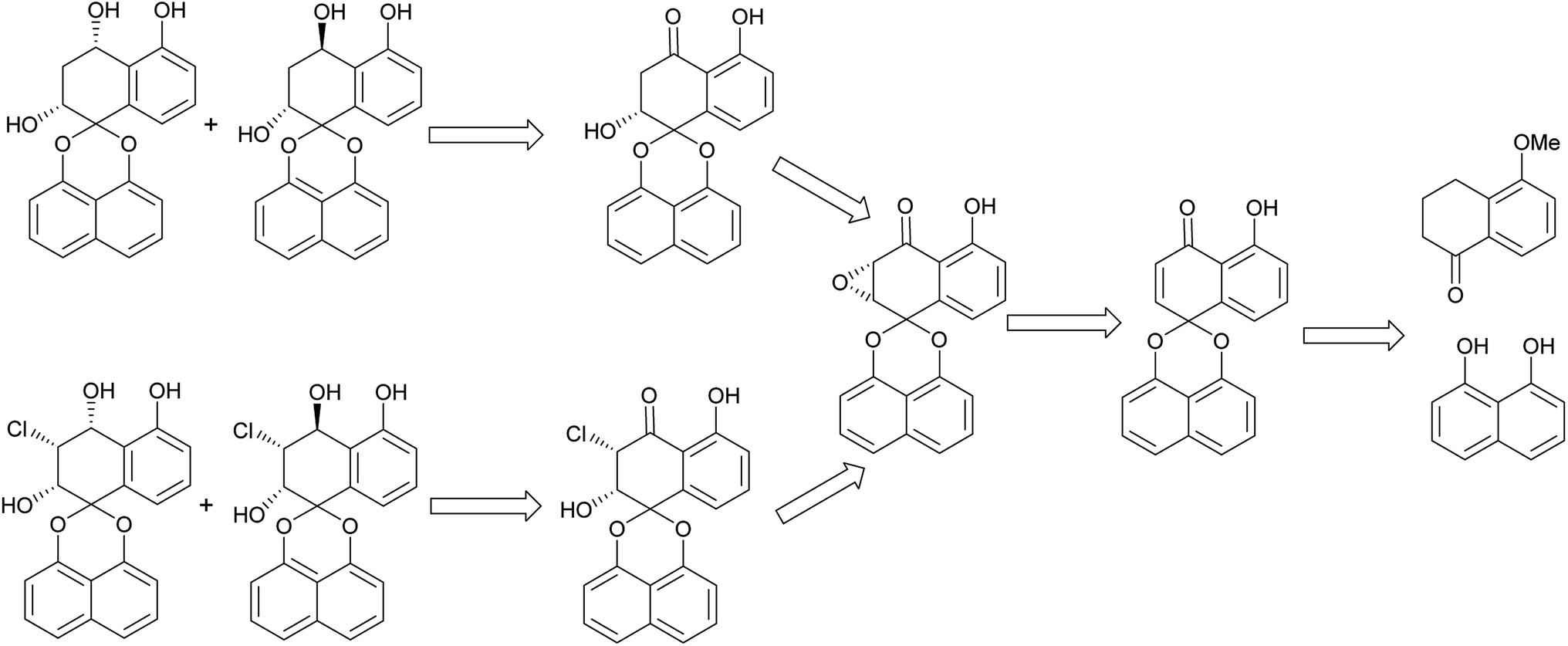 | ||
| Scheme 1 Retro-synthetic analysis of Palmarumycin BG1–6 and Guignardin E. | ||
The synthesis of Palmarumycin BG1–6 and Guignardin E (1–6) started with 5-methoxy-3,4-dihydronaphthalen-1(2H)-one and 1,8-dihydroxynaphthalene (DHN), as shown in Scheme 2. Compound 8 was obtained via the direct ketalization of 5-methoxytetralone with 1,8-dihydroxynaphthalene (DHN). Oxidation of compound 8 with pyridinium dichromate (PDC) and t-BuOOH in benzene gave compound 9, which was further oxidized with DDQ to produce 10. Then, 10 was demethylated with B-bromocatecholborane to afford the target compound Palmarumycin CP1 (11) following the literature procedures.24–27 Next, the stereoselective epoxidation with t-BuOOH of Palmarumycin CP1 catalyzed with N-benzylcinchoninium chloride at 25 °C yielded Palmarumycin C2 (12) in 90.7% ee value. The ee value was improved to be 97.9% at 0 °C.28 The organoselenium-mediated reduction of Palmarumycin C2 afforded Palmarumycin BG1 (1) as the sole product in 92% yield, and the configuration of C-2 did not change.29–31 The NaBH4 reduction of 1 resulted in a mixture of Palmarumycin BG2 (2) and BG3 (3), which were isolated in silica gel to give Palmarumycin BG2 (2) and BG3 (3) in 56% and 30% yields, respectively. Treatment of Palmarumycin C2 (12) with the diluted hydrogen chloride at 25 °C afforded a mixture of Guignardin E (6) and its C-3 epimer (13) in 95% yield (dr value: 5.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).32 When this transformation was performed at 10 °C, the dr value was improved to 25:1. Recrystallization of this mixture yielded pure Guignardin E (6). Guignardin E as the unnatural product was mentioned in mixture form of Guignardin E and its epimer.32,33 but it was first isolated as the natural product from the fungus Guignardia sp. KcF8.6 The NaBH4 reduction of Guignardin E (6) afforded Palmarumycin BG5 (4) and the C-4 epimer (14) of 4 in 22% and 78% yields, respectively. The NaBH4 reduction of the mixture of Guignardin E (6) and its C-3 epimer (13) afforded Palmarumycin BG5 (4), BG6 (5) and the C-4 epimer (14) of 4 in 23%, 9% and 60% yields, respectively, but the C-4 epimer (15) of 5 was not obtained because of the limited quantity of 13 in the mixture and the hindrance effect in the NaBH4 reduction. So we deduced rationally that Guignardin E (6) should be the biosynthetic precursor of Palmarumycin BG5 (4) and BG6 (5). Treatment of Palmarumycin C2 (11) with cerium(III) chloride hydrate at reflux temperature afforded Palmarumycin C1 (7) in 79% yield.34 One possible mechanism is that cerium(III) chloride is hydrolyzed to produce hydrochloride in situ. Then, the regioselective epoxide ring-opening can occur between Palmarumycin C2 and hydrochloride to give the mixture of chlorohydrins Guignardin E (6) and its C-3 epimer (13). A subsequent loss of water could then lead to obtain Palmarumycin C1 at reflux temperature. This approach avoided the use of the strong acid, which removes the possibility of breaking the ketal bond.20 When the mixture of 6 and 13 was treated with diluted hydrochloride in methanol or methanol/water at the reflux temperature, a complex mixture of products were obtained that could not be purified. Palmarumycin C1, which was reported to exhibit antifungal activity against phytopathogen Fusarium oxysporum and cytotoxicities against 10 human tumor cell lines with LD50 values in the range of 1.24–39.2 μM, was isolated from the fungus Guignardia sp. KcF86, and Coniothyrium sp.32
:1).32 When this transformation was performed at 10 °C, the dr value was improved to 25:1. Recrystallization of this mixture yielded pure Guignardin E (6). Guignardin E as the unnatural product was mentioned in mixture form of Guignardin E and its epimer.32,33 but it was first isolated as the natural product from the fungus Guignardia sp. KcF8.6 The NaBH4 reduction of Guignardin E (6) afforded Palmarumycin BG5 (4) and the C-4 epimer (14) of 4 in 22% and 78% yields, respectively. The NaBH4 reduction of the mixture of Guignardin E (6) and its C-3 epimer (13) afforded Palmarumycin BG5 (4), BG6 (5) and the C-4 epimer (14) of 4 in 23%, 9% and 60% yields, respectively, but the C-4 epimer (15) of 5 was not obtained because of the limited quantity of 13 in the mixture and the hindrance effect in the NaBH4 reduction. So we deduced rationally that Guignardin E (6) should be the biosynthetic precursor of Palmarumycin BG5 (4) and BG6 (5). Treatment of Palmarumycin C2 (11) with cerium(III) chloride hydrate at reflux temperature afforded Palmarumycin C1 (7) in 79% yield.34 One possible mechanism is that cerium(III) chloride is hydrolyzed to produce hydrochloride in situ. Then, the regioselective epoxide ring-opening can occur between Palmarumycin C2 and hydrochloride to give the mixture of chlorohydrins Guignardin E (6) and its C-3 epimer (13). A subsequent loss of water could then lead to obtain Palmarumycin C1 at reflux temperature. This approach avoided the use of the strong acid, which removes the possibility of breaking the ketal bond.20 When the mixture of 6 and 13 was treated with diluted hydrochloride in methanol or methanol/water at the reflux temperature, a complex mixture of products were obtained that could not be purified. Palmarumycin C1, which was reported to exhibit antifungal activity against phytopathogen Fusarium oxysporum and cytotoxicities against 10 human tumor cell lines with LD50 values in the range of 1.24–39.2 μM, was isolated from the fungus Guignardia sp. KcF86, and Coniothyrium sp.32
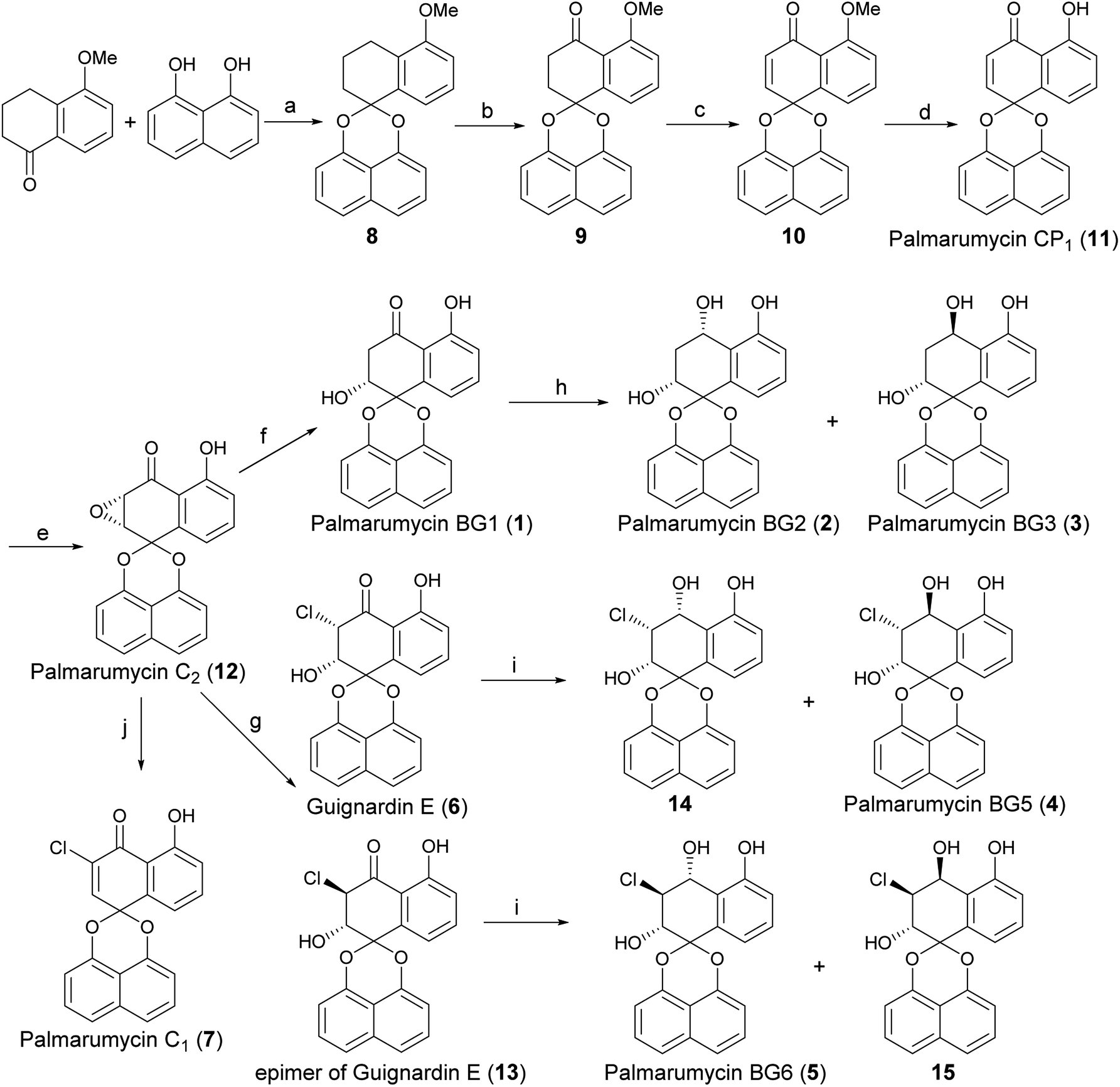 | ||
| Scheme 2 Synthesis of Palmarumycin BG1–6, Guignardin E (1–6) and Palmarumycin C1 (7). Reagents and conditions: (a) TsOH, toluene, reflux, 72 h, 57%; (b) PDC, Celite, t-BuOOH, benzene, rt, 24 h, 74%; (c) DDQ, benzene, reflux, 82%; (d) B-bromocatecholborane, DCM, 0 °C, 75%; (e) N-benzylcinchoninium chloride, t-BuOOH, NaOH (0.1 M), toluene/H2O, 0 °C, 73%; (f) (PhSe)2, NaBH4, AcOH, EtOH, 0 °C, 92%; (g) HCl (1 M), THF/H2O, 10 °C, 3 day, 96% for 6 + 13, dr = 25:1; (h) NaBH4, MeOH, rt, 56% for 2 and 30% for 3, dr = 2:1; (i) NaBH4, MeOH, rt, 23% for 4, 9% for 5, and 60% for 14; (j) CeCl3·7H2O, MeOH/H2O, reflux, 79%. | ||
After finishing the synthesis of the desired compounds, all structures were characterized with the 1H, 13C NMR, IR and HR-ESI-MS data. In order to confirm the absolute configuration, the X-ray diffraction analysis of 14 was performed using Cu Kα radiation and its structure was depicted in Fig. 2, which unambiguously showed that the absolute configuration of C-2, C-3 and C-4 were 2S, 3R and 4R in Flack parameter 0.006(16) (CCDC ID 1947664). Based on this result, the C-2 absolute configuration of Palmarumycin BG1 (1) was 2R, the configuration of C-2 and C-4 of Palmarumycin BG2 (2) were 2R and 4S, those of C-2 and C-4 of Palmarumycin BG3 (3) were 2R and 4R, those of C-2, C-3 and C-4 of Palmarumycin BG5 (4) were 2S, 3R and 4S, those of C-2, C-3 and C-4 of Palmarumycin BG6 (5) were 2S, 3S and 4R, and those of C-2 and C-3 of Guignardin E (6) were 2S and 3S, all of these absolute configurations were consistent with those of the natural products.5,6 Because the organoselenium-mediated reduction of the α,β-epoxy ketone did not change the configuration of the β-carbon,29–31 so the absolute configurations of C-2 and C-3 of 12 would be 2R and 3S, which are in agreement with those of natural Palmarumycin C2,32,33 and Guignardin F,6 implies that Palmarumycin C2 and Guignardin F are the same compound.
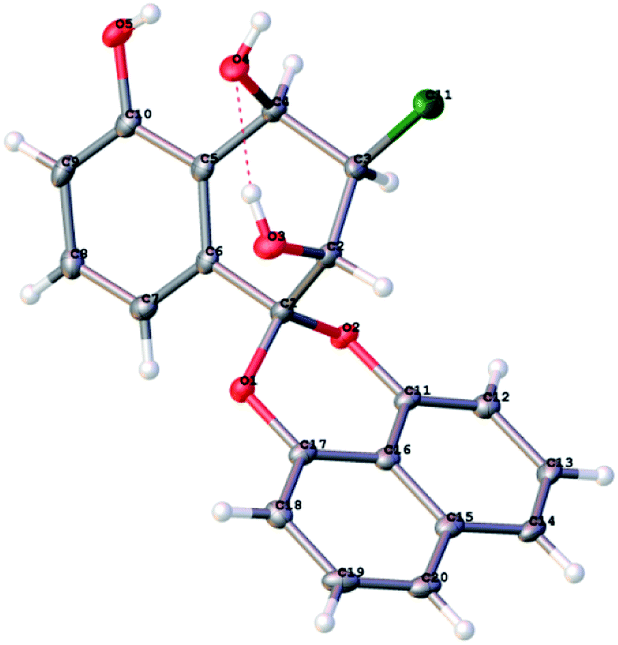 | ||
| Fig. 2 ORTEP drawing of Palmarumycin BG5 C-4 epimer (14). | ||
The cytotoxic activities of 1–7 and 14 against the growth of tumor cell lines (HCT116, U87-MG, HepG2, BGC823 and PC9) were evaluated using a MTT assay22 and the results were shown in Table 1. These results indicated that compounds 1–4, 6–7 and 14 exhibited significant cytotoxicity against several tumor cell lines with an IC50 in the range of 2.45–30.50 μM, while compound 5 was inactive against above mentioned cancer cells (IC50 > 50 μM). When comparison the IC50 data of compounds 1–3 with compounds 4, 6–7 and 14, we found that the compounds bearing a chlorine atom at the C-3 position exhibited stronger cytotoxic activity than those compounds without the chlorine atom at C-3 position. In the other aspect, the reduction products of carbonyl group at C-4 such as 2 and 3, 4, 5 and 14 exhibited weaker cytotoxic activity than those retaining C-4 carbonyl compounds 1 and 6. These results indicated that the chlorine atom at C-3 and the carbonyl at C-4 play a critical role for cytotoxicity.23,24
| Compounds | HCT116 | U87-MG | HepG2 | BGC823 | PC9 |
|---|---|---|---|---|---|
| 1 | 9.14 | 19.40 | 20.36 | 19.15 | 14.87 |
| 2 | 30.08 | >50 | 35.56 | >50 | 30.50 |
| 3 | 29.12 | 26.10 | 28.81 | 24.55 | 22.10 |
| 4 | 6.71 | 9.62 | 4.21 | 6.25 | 5.58 |
| 5 | >50 | >50 | >50 | >50 | >50 |
| 6 | 3.70 | 2.45 | 4.06 | 4.36 | 7.42 |
| 7 | 2.63 | 2.60 | 6.05 | 14.08 | 15.39 |
| 14 | 6.62 | 19.64 | 9.64 | 12.06 | 14.37 |
| Taxol | 0.000616 | 0.000245 | 0.00475 | 0.000642 | 0.000823 |
Conclusions
In summary, Palmarumycin BG1–3, BG5–6, Guignardin E and Palmarumycin C1 (1–7) were first synthesized in overall yields of 1.0–17.4% through a N-benzylcinchoninium chloride-catalyzed epoxidation, an organoselenium-mediated reduction of the α,β-epoxy ketone, and a cerium(III) chloride hydrate-promoted ring-opening and elimination as the key steps via 6–7 step routes using 1,8-dihydroxynaphthalene (DHN) and 5-methoxytetralone as the starting materials. Their structures and absolute configurations were characterized by 1H, 13C NMR, IR, HR-ESI-MS data and X-ray crystallographic data (CCDC 1947664). Compounds 1–4, 6–7 and 14 displayed significant inhibition activity against HCT116, U87-MG, HepG2, BGC823 and PC9 cell lines.Experimental procedures
General experimental procedures
Melting points (uncorrected) were measured on a WRX-4 microscopic melting point apparatus (Shanghai Yi Ce Instrument Factory). All 1H and 13C NMR spectra were obtained on a Bruker DPX 300 spectrometer with CDCl3 and CD3OD as solvents and TMS as an internal standard. HR-ESI-MS spectra were analyzed on a Bruker Apex II mass spectrometer. IR spectra were recorded with a Shimadzu IRTracer 100 FT-IR spectrometer (KBr plate). Optical rotation values were measured with a JASCO P-2000 Polarimeter. The crystal structure was analyzed with a Thermo Fisher ESCALAB 250 four-circle X-ray diffractometer (Xcalibur, Eos, Gemini). Unless otherwise stated, all reagents were purchased from commercial suppliers and used without further purification. Organic solutions were concentrated under reduced pressure using a rotary evaporator or oil pump. Flash column chromatography was performed using Qingdao Haiyang silica gel (200–300 mesh). HPLC analyses were performed on an Agilent 1100 instruments, UV detection was monitored at 254 and 220 nm, an IB N-5 column (5 μm, 4.6 × 250 mm) was used as the chiral stationary phase, and hexane/i-PrOH (90:10) was used as the mobile phase at a flow rate of 1.0 mL min−1.
Synthesis of intermediates 8–12
The intermediates 8–11 were synthesized following the literature procedures, and their analytical data were consistent with the reported data.25–27Palmarumycin CP1 (11, 120 mg, 0.38 mmol) was added into a toluene (10 mL) solution of N-benzylcinchoninium chloride (48 mg, 0.114 mmol, 0.3 eq.) in a 50 mL round-bottom flask. NaOH solution (0.1 M; 5.7 mL, 1.5 eq.) was added dropwise in the mixture, followed by the addition of t-BuOOH (0.524 mL, 7.2 M, 3.77 mmol) at 0 °C (the ice-water bath) and stirred for 6 h. After completion of the reaction, the solution was diluted with EtOAc (50 mL), washed with 0.2 M HCl solution (2 × 20 mL), brine, and the organic phase was dried over anhydrous Na2SO4. The solvent was removed and the crude product was purified by flash column chromatography on silica gel (petroleum ether/EtOAc, 20:1) to give Palmarumycin C2 (12, 100 mg, 79%) as a yellow solid.28 The ee value of the key intermediate 12 was analyzed by HPLC to be 97.9%. Compound 12, a yellow solid, mp 218–220 °C (lit. 11 mp 219–221 °C; lit. 28 mp 225 °C; lit. 32 mp 228 °C); [α]20D = −335 (c = 0.66, CHCl3), (lit. 11 [α]20D = −340, c = 1.00, CHCl3; lit. 28 [α]20D = −300, c = 1.00, CH2Cl2; lit. 32 [α]20D = −341, c = 1.00, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 11.37 (1H, s), 7.64 (1H, dd, J = 8.4, 8.0 Hz), 7.62–7.50 (3H, m), 7.45 (2H, dd, J = 8.0, 7.5 Hz), 7.19 (1H, d, J = 7.5 Hz), 7.14 (1H, d, J = 8.4 Hz), 6.92 (1H, d, J = 7.5 Hz), 4.09 (1H, d, J = 4.0 Hz), 3.68 (1H, d, J = 4.0 Hz); 13C NMR (75 MHz, CDCl3) δ: 196.65, 162.00, 147.07, 146.81, 137.77, 137.02, 134.31, 127.90, 127.76, 121.56, 121.46, 120.17, 119.18, 112.92, 112.41, 110.28, 109.45, 96.12, 53.40, 53.36; IR νmax 3426, 3051, 1651, 1612, 1454, 1412, 1381, 1269, 1238, 1177, 1115, 1065, 968, 876, 806, 752 cm−1; ESI-MS, m/z: 333 [M + H]+. These data were identical with the published data.11,28,32
Synthesis of (2R)-2,5-dihydroxy-2,3-dihydro-4H-spiro[naphthalene-1,2′-naphtho[1,8-de] [1,3]dioxin]-4-one (Palmarumycin BG1, 1)
The reduction reagent (Na[PhSeB(OEt)3]) was prepared following the protocol in the literature.29,30 (PhSe)2 (245 mg, 0.79 mmol) and EtOH (15 mL) were added into a 100 mL round-bottom flask at 0 °C, then NaBH4 (60 mg, 1.58 mmol) was gradually added into the mixture. After vigorous evolution of hydrogen ceased and NaBH4 was thoroughly consumed, AcOH (0.108 mL, 1.89 mmol) was added and the mixture was stirred for 10 min to obtain the reduction reagent solution. Then a solution of Palmarumycin C2 (12, 131 mg, 0.39 mmol) in EtOH/AcOH (1:1, 10 mL) was added into the reduction reagent solution, stirred for 10 min and removed from the ice-water bath. After completion of the reaction, the solution was diluted with EtOAc (100 mL), washed with brine, and the organic phase was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography on silica gel (petroleum ether/EtOAc, 5:1) to give Palmarumycin BG1 (1, 121 mg, 92%) as a white solid. mp 73–75 °C; [α]20D = −152 (c = 0.60, CHCl3); (lit. 5 [α]17D = −151, c = 0.50, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 12.35 (1H, s), 7.57 (1H, d, J = 8.0 Hz), 7.54 (2H, d, J = 8.4 Hz), 7.49 (1H, dd, J = 8.0, 7.6 Hz), 7.44 (1H, dd, J = 8.4, 7.6 Hz), 7.34 (1H, dd, J = 8.0, 1.1 Hz), 7.08 (2H, d, J = 8.4 Hz), 6.91 (1H, d, J = 7.6 Hz), 4.59 (1H, s), 3.24 (1H, dd, J = 17.8, 4.0 Hz), 2.94 (1H, dd, J = 17.8, 4.0 Hz), 2.38 (1H, s); 13C NMR (75 MHz, CDCl3) δ: 201.12, 162.21, 147.24, 146.42, 138.03, 137.16, 134.30, 127.82, 127.78, 121.52, 121.22, 119.94, 118.10, 115.46, 113.26, 109.66, 108.97, 98.83, 67.36, 41.40; IR νmax 3433, 3059, 2924, 1643, 1608, 1585, 1454, 1412, 1381, 1346, 1269, 1234, 1165, 1119, 1069, 976, 891, 822, 756 cm−1; HR-ESI-MS, m/z: C20H14O5 [M–H]−, calcd. 333.0768, found: 333.0791. These data were consistent with the published data.5,6
Synthesis of (2R,4S)-3,4-dihydro-2H-spiro[naphthalene-1,2′-naphtho[1,8-de][1,3]dioxine]-2,4,5-triol and (2R,4R)-2,3-dihydro-2H-spiro[naphthalene-1,2′-naphtho[1,8-de][1,3]dioxine]-2,4,5-triol (Palmarumycin BG2 and BG3, 2 and 3)
NaBH4 (38 mg, 1.00 mmol) was added into a mixture of palmarumycin BG1 (1, 107 mg, 0.32 mmol) and MeOH (10 mL) in a 25 mL round-bottom flask at 0 °C. The mixture was stirred at room temperature for 2 h. The solution was extracted with EtOAc (2 × 30 mL). The organic phase was washed with brine, and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, the residue was subjected to flash column chromatography on silica gel and eluted with petroleum ether/EtOAc (3:1) to afford palmarumycin BG2 (2, 61 mg, 56%) as a white solid and palmarumycin BG3 (3, 31 mg, 30%) as a white solid. Palmarumycin BG2 (2), mp 189–191 °C; [α]20D = −108 (c = 0.64, CHCl3), (lit. 5 [α]16D = −40, c = 0.055, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 7.55 (1H, d, J = 8.4 Hz), 7.52 (1H, d, J = 8.4 Hz), 7.48 (1H, d, J = 7.4 Hz), 7.45 (1H, dd, J = 7.6, 1.5 Hz), 7.42 (1H, dd, J = 8.4, 7.5 Hz), 7.40 (1H, dd, J = 8.0, 7.6 Hz), 7.09 (1H, dd, J = 7.6, 1.5 Hz), 7.04 (1H, d, J = 7.0 Hz), 6.90 (1H, s), 6.87 (1H, dd, J = 7.5, 0.3 Hz), 5.0 (1H, dd, J = 10.7, 5.3 Hz), 4.43 (1H, d, J = 3.7 Hz), 3.40 (1H, d, J = 11.2 Hz), 2.55 (1H, dd, J = 15.3, 5.6 Hz), 2.51 (1H, s), 2.40 (1H, ddd, J = 15.3, 4.5, 1.7 Hz); 13C NMR (75 MHz, CDCl3) δ: 155.65, 147.33, 146.99, 134.34, 132.08, 130.30, 127.83, 127.67, 124.88, 121.40, 120.95, 119.65, 118.24, 113.47, 109.43, 109.10, 99.68, 65.86, 63.41, 31.89; IR νmax 3210, 3059, 2928, 1636, 1609, 1474, 1412, 1377, 1339, 1269, 1119, 1042, 972, 891, 822, 790, 756 cm−1; HR-ESI-MS, m/z: C20H16O5 [M–H]−, calcd. 335.0925, found: 335.0954. Palmarumycin BG3 (3), mp 145–147 °C; [α]20D = −262 (c = 0.56, MeOH); (lit. 5 [α]17D = −261, c = 0.14, acetone); 1H NMR (300 MHz, CD3OD) δ: 7.51 (1H, dd, J = 8.4, 1.2 Hz), 7.49 (1H, dd, J = 8.4, 1.2 Hz), 7.45 (1H, dd, J = 8.4, 7.3 Hz), 7.42 (1H, dd, J = 8.4, 7.3 Hz), 7.20 (2H, d, J = 4.2 Hz), 7.01 (1H, dd, J = 7.2, 1.3 Hz), 6.92–6.85 (2H, m), 5.30 (1H, dd, J = 8.8, 6.2 Hz), 4.31 (1H, dd, J = 5.8, 2.2 Hz), 2.43 (1H, ddd, J = 13.4, 6.1, 6.1 Hz), 2.28 (1H, ddd, J = 13.5, 8.8, 2.3 Hz); 13C NMR (75 MHz, CD3OD) δ: 157.16, 149.40, 148.74, 135.89, 135.54, 129.94, 128.68, 128.42, 126.38, 121.38, 121.30, 119.77, 117.52, 114.69, 110.47, 109.22, 100.75, 67.62, 65.21, 36.18; IR νmax 3152, 2924, 2851, 1636, 1605, 1474, 1412, 1377, 1315, 1269, 1115, 1072, 972, 883, 822, 802, 756 cm−1; HR-ESI-MS, m/z: C20H16O5 [M–H]−, calcd. 335.0925, found: 335.0939. These data were identical with the published data.5
Synthesis of (2S,3S)-3-chloro-2,5-dihydroxy-2,3-dihydro-4H-spiro [naphthalene-1,2′-naphtho[1,8-de] [1,3]dioxin]-4-one (Guignaridin E, 6)
A solution of HCl (1 M, 10 mL) was added into a mixture of Palmarumycin C2 (12, 32 mg 0.10 mmol) and THF (12 mL) in a 50 mL round-bottom flask at 25 °C, and the mixture was stirred at room temperature for 24 h. The solution was diluted with EtOAc (50 mL), washed with brine, and the organic phase was dried over anhydrous Na2SO4. The solvent was removed, and the crude product was purified by flash column chromatography on silica gel (petroleum ether/EtOAc, 2:1) to give the mixture of Guignardin E and its C-3 epimer (6 and 13, 34 mg, 95%) as a yellow solid (dr = 5.7:1).
A solution of HCl (1 M, 10 mL) was added into a palmarumycin C2 (12, 91 mg, 0.27 mmol) and THF (20 mL) in a 100 mL round-bottom flask at 0 °C, and the mixture was stirred at 10 °C for 72 h. Workup as routine, and the crude product was purified by flash column chromatography on silica gel (petroleum ether/EtOAc, 2:1) to afford the mixture of Guignardin E and its C-3 epimer (98 mg, 96%) as a yellow solid (dr = 25:1). The mixture (dr = 25:1) was recrystallized repeatedly with methanol to provide Guignardin E (6, 61 mg, 60%) as a yellow solid, mp 178–180 °C; [α]20D = −191 (c = 0.53, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 11.84 (1H, s), 7.66 (1H, t, J = 8.1 Hz), 7.61 (1H, d, J = 8.1 Hz), 7.58 (1H, d, J = 8.1 Hz), 7.52 (1H, dd, J = 8.5, 7.4 Hz), 7.48 (1H, d, J = 8.5 Hz), 7.46 (1H, dd, J = 8.5, 8.4 Hz), 7.16 (1H, dd, J = 7.4, 1.0 Hz), 7.15 (1H, dd, J = 7.4, 1.0 Hz), 6.93 (1H, dd, J = 7.5, 0.8 Hz), 5.31 (1H, d, J = 2.2 Hz), 4.70 (1H, dd, J = 2.8, 2.7 Hz), 2.68 (1H, d, J = 2.7 Hz); 13C NMR (75 MHz, CDCl3) δ: 194.14, 162.17, 146.74, 146.10, 138.03, 137.48, 134.34, 127.93, 127.84, 121.85, 121.70, 120.46, 118.83, 114.28, 113.06, 110.13, 109.24, 98.69, 72.34, 62.55; IR νmax 3471, 1663, 1609, 1585, 1458, 1412, 1377, 1265, 1196, 1172, 1115, 1065, 987, 894, 818, 760, 694 cm−1; HR-ESI-MS, m/z: C20H13ClO5 [M–H]−, calcd. 367.0379, found: 367.0397. These data were consistent with the published data.6
Synthesis of (2S,3R,4S)-3-chloro-3,4-dihydro-2H-spiro [naphthalene-1,2′-naphtho[1,8-de] [1,3]dioxine]-2,4,5-triol, (2S,3R,4R)-3-chloro-3,4-dihydro-2H-spiro[naphthalene-1,2′-naphtho [1,8-de][1,3]dioxine]-2,4,5-triol and (2S,3S,4R)-3-chloro-3,4-dihydro-2H-spiro [naphthalene-1,2′-naphtho[1,8-de][1,3]dioxine]-2,4,5-triol (Palmarumycin BG5, its C-4 epimer and BG6, 4, 14 and 5)
NaBH4 (7 mg, 0.18 mmol) was added into a solution of Guignardin E (6, 23 mg, 0.06 mmol) in MeOH (1.5 mL) in a 10 mL round-bottom flask. The mixture was stirred at room temperature for 2 h. The solution was extracted with EtOAc (2 × 10 mL). The organic phase was washed with brine, and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, the residue was subjected to flash column chromatography on silica gel and eluted with petroleum ether/EtOAc (2:1) to afford Palmarumycin BG5 (4, 5 mg, 22%) as a white solid and its C-4 epimer (14, 18 mg, 78%) as a white solid. Palmarumycin BG5 (4), mp 108–110 °C; [α]20D = −316 (c = 0.96, CHCl3), (lit. 5 [α]16D = −314.7, c = 0.75, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 8.38 (1H, s), 7.59 (1H, d, J = 8.5 Hz), 7.55 (1H, dd, J = 8.5, 0.9 Hz), 7.49 (1H, dd, J = 8.3, 7.5 Hz), 7.45 (1H, dd, J = 7.6, 7.6 Hz), 7.43–7.37 (2H, m), 7.09 (1H, dd, J = 7.4, 0.9 Hz), 7.07 (1H, dd, J = 7.0, 1.5 Hz), 6.92 (1H, dd, J = 7.5, 1.0 Hz), 5.47 (1H, d, J = 9.1 Hz), 4.68 (1H, dd, J = 9.1, 1.9 Hz), 4.47 (1H, d, J = 1.9 Hz), 3.23 (1H, s), 2.40 (1H, s); 13C NMR (75 MHz, CDCl3) δ: 155.95, 147.08, 146.35, 134.36, 132.68, 130.90, 127.85, 127.80, 121.73, 121.25, 120.03, 119.88, 119.19, 113.19, 109.75, 109.33, 99.82, 71.77, 70.86, 63.95; IR νmax 3325, 2963, 2920, 2851, 1609, 1462, 1412, 1381, 1342, 1265, 1107, 1022, 891, 799, 756 cm−1; HR-ESI-MS, m/z: C20H15ClO5 [M–H]−, calcd. 369.0535, found: 369.0548. These data were consistent with the published data.5 The C-4 epimer of Palmarumycin BG5 (14), mp 200–202 °C; [α]20D = −96 (c = 0.58, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 7.58 (1H, d, J = 7.6 Hz), 7.56 (1H, d, J = 8.5 Hz), 7.51–7.42 (4H, m), 7.12 (1H, dd, J = 7.3, 2.0 Hz), 7.07 (1H, dd, J = 7.6, 0.9 Hz), 6.91 (1H, dd, J = 7.5, 1.0 Hz), 6.38 (1H, s), 5.08 (1H, s), 4.77 (1H, dd, J = 4.8, 1.7 Hz), 4.48 (1H, s), 3.43 (1H, d, J = 9.8 Hz), 2.86 (1H, s); 13C NMR (75 MHz, CDCl3) δ: 155.22, 146.90, 146.61, 134.39, 131.59, 131.15, 127.90, 127.79, 123.53, 121.73, 121.37, 120.29, 118.79, 109.72, 109.32, 100.26, 100.17, 71.59, 66.25, 57.59; IR νmax 3464, 2928, 1609, 1585, 1466, 1416, 1377, 1323, 1180, 1115, 1057, 1038, 984, 887, 822, 791, 764, 691 cm−1; HR-ESI-MS, m/z: C20H15ClO5 [M–H]−, calcd. 369.0535, found: 369.0547.
NaBH4 (50 mg, 1.32 mmol) was added into a mixture of Guignardin E and its C-3 epimer (6 and 13, 161 mg, 0.44 mmol, dr = 5.7:1), and MeOH (10 mL) in a 25 mL round-bottom flask. The mixture was stirred at room temperature for 2 h. The solution was extracted with EtOAc (2 × 30 mL). The organic phase was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, the residue was subjected to flash column chromatography on silica gel and eluted with petroleum ether/EtOAc (2:1) to afford Palmarumycin BG5 (4, 37 mg, 23%), the C-4 epimer of Palmarumycin BG5 (14, 96 mg, 60%), and Palmarumycin BG6 (5, 15 mg, 9%) as a yellow solid. Palmarumycin BG6 (5), mp 164–166 °C; [α]20D = +56 (c = 0.50, CHCl3), (lit. 5 [α]20D = +60, c = 0.04, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 8.13 (1H, s), 7.51 (1H, d, J = 8.1 Hz), 7.50 (1H, d, J = 8.3 Hz), 7.45 (1H, dd, J = 8.2, 7.5 Hz), 7.41 (1H, dd, J = 8.1, 7.7 Hz), 7.18 (1H, dd, J = 8.0, 8.0 Hz), 7.03 (1H, d, J = 7.9 Hz), 6.96 (2H, d, J = 8.0 Hz), 6.90 (1H, d, J = 7.4 Hz), 5.32 (1H, d, J = 7.7 Hz), 4.63 (1H, dd, J = 8.7, 7.8 Hz), 4.38 (1H, d, J = 8.7 Hz), 3.51 (1H, brs), 2.64 (1H, brs); 13C NMR (75 MHz, CDCl3) δ: 156.04, 148.06, 147.12, 134.08, 134.04, 130.56, 127.68, 127.66, 120.93, 120.76, 119.21, 118.87, 118.53, 112.85, 108.72, 108.49, 99.32, 74.58, 73.73, 64.35; IR νmax 3319, 2933, 1609, 1585, 1465, 1415, 1381, 1344, 1266, 1181, 1115, 1062, 986, 893, 796, 756 cm−1; HR-ESI-MS, m/z: C20H15ClO5 [M–H]−, calcd. 369.0535, found: 369.0547. These data were in agreement with the reported data.5
Synthesis of 3-chloro-5-hydroxy-4H-spiro[naphthalene-1,2′-naphtho[1,8-de][1,3]dioxin]-4-one (Palmarumycin C1, 7)
CeCl3·7H2O (64 mg, 0.172 mmol), Palmarumycin C2 (12, 52 mg, 0.157 mmol), MeOH (9 mL), and H2O (3 mL) were added in a 25 mL round-bottom flask. The mixture was stirred at reflux temperature for 16 h, cooled to room temperature and the mixture was diluted with EtOAc (50 mL). The organic phase was washed with brine, and dried over anhydrous Na2SO4. The solution was concentrated under vacuum. The crude product was purified by flash column chromatography on silica gel (petroleum ether/EtOAc, 20:1) to give palmarumycin C1 (7, 41 mg, 79%) as a yellow solid. mp 272–273 °C (lit. 32 mp > 280 °C); 1H NMR (300 MHz, CDCl3) δ: 11.87 (1H, s), 7.69 (1H, dd, J = 8.4, 7.5 Hz), 7.61 (2H, dd, J = 8.4, 1.0 Hz), 7.49 (2H, dd, J = 8.4, 7.5 Hz), 7.47 (1H, dd, J = 7.5, 1.0 Hz), 7.18 (1H, dd, J = 8.4, 1.0 Hz), 7.17 (1H, s), 7.00 (2H, dd, J = 7.5, 1.0 Hz); 13C NMR (75 MHz, CDCl3) δ: 182.09, 162.32, 146.93, 138.63, 137.40, 135.84, 135.18, 134.34, 127.84, 121.81, 120.28, 119.85, 113.30, 112.95, 110.24, 93.70; IR νmax 3063, 2924, 1651, 1628, 1612, 1497, 1458, 1412, 1381, 1269, 1231, 1169, 1119, 1069, 941, 903, 845, 826, 802, 756 cm−1; HR-ESI-MS, m/z: C20H10ClO4 [M–H]−, cacld. 349.0273, found: 349.0276. These data were identical with the published data.6,32,33
X-ray diffraction analysis of Palmarumycin BG5 C-4 epimer (14)
Colorless plate-like crystals of compound 14 were obtained from a slowly evaporating methanol solution. A 0.35 × 0.24 × 0.03 mm3 crystal was selected for analysis. The parameters and structure information for compound 14 have been deposited at the Cambridge Crystallographic Data Centre. CCDC ID 1947664 contains the supplementary crystallographic data for this paper.Cytotoxic activity evaluation of title compounds
The cytotoxicity of the title compounds was evaluated against HCT116, U87-MG, HepG2, BGC823 and PC9 cell lines using a microculture tetrazolium (MTT) assay as in the previous report.22 Taxol was used as the positive control. All of human carcinoma cell lines (colon cancer cell HCT116, glioblastoma cell U87-MG, hepatocellular carcinoma cell HepG2, gastric cancer cell BGC823 and lung carcinoma cell PC9) were obtained from the American Type Culture Collection (ATCC) and purchased from the Cell Culture Center of Institute of Basic Medical Sciences, Peking Union Medical College, Chinese Academy of Medical Sciences. These cell lines were cultured in Dulbecco's minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin (100 U mL−1)–streptomycin (100 μg mL−1). The cell culture medium, serum and antibiotics were purchased from Invitrogen. All the cells were maintained at 37 °C in 5% CO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (No. 21772229) and National Key R & D Program of China (2017YFD0201105) are appreciated for the financial supports. Ms Stephanie A. Blaszczyk of the University of Wisconsin-Madison and Dr John Anderson are thanked for their helping to polish the language of this manuscript.Notes and references
- K. Miyashita and T. Imanishi, Chem. Rev., 2005, 105, 4515–4536 CrossRef CAS PubMed.
- L. Zhou, J. Zhao, T. Shao, X. Cai and Y. Peng, Mini-Rev. Med. Chem., 2010, 10, 977–989 CrossRef CAS PubMed.
- Y. S. Cai, Y. W. Guo and K. Krohn, Nat. Prod. Rep., 2010, 27, 1840–1870 RSC.
- X. Liu, Y. Zhao, W. Wang, M. Wang and L. Zhou, Chin. J. Org. Chem., 2017, 37, 2883–2894 CrossRef CAS.
- Y. Cai, T. Kurtan, Z. Miao, A. Mandi, I. Komaromi, H. Liu, J. Ding and Y. Guo, J. Org. Chem., 2011, 76, 1821–1830 CrossRef CAS PubMed.
- W. Ai, X. Wei, X. Lin, L. Sheng, Z. Wang, Z. Tu, X. Yang, X. Zhou, J. Li and Y. Liu, Tetrahedron, 2014, 70, 5806–5814 CrossRef CAS.
- T. Bunyapaiboonsri, S. Yoiprommarat, R. Nopgason, K. Intereya, R. Suvannakad and J. Sakayaroj, Tetrahedron, 2015, 71, 5572–5578 CrossRef CAS.
- E. E. Erika, Synthetic progress toward parvistemonine, spiroxins A and B, and generation of palmarumycin analogues, PhD. dissertation, University of Pittsburgh, USA, 2008.
- A. C. Murphy, S. R. A. Devenish, A. C. Muscroft-Taylor, J. W. Blunt and M. H. G. Munro, Org. Biomol. Chem., 2008, 6, 3854–3862 RSC.
- K. Krohn, S. Wang, I. Ahmed, S. Altun, A. Aslan, U. Florke, I. Kock and S. Schlummer, Eur. J. Org. Chem., 2010, 4476–4481 CAS.
- A. Aslan, S. Altun, I. Ahmed, U. Florke, B. Schulz and K. Krohn, Eur. J. Org. Chem., 2011, 1176–1188 CrossRef CAS.
- H. Tsukamoto, S. Hanada, K. Kumasaka, N. Kagaya, M. Izumikawa, K. Shinya and T. Doi, Org. Lett., 2016, 18, 4848–4851 CrossRef CAS PubMed.
- H. Tsukamoto, Y. Nomura, K. Fujiwara, S. Hanada and T. Doi, Org. Lett., 2018, 20, 3140–3143 CrossRef CAS PubMed.
- H. Tsukamoto, S. Hanada, Y. Nomura and T. Doi, J. Org. Chem., 2018, 83, 9430–9441 CrossRef CAS PubMed.
- T. Doi, H. Tsukamoto and Y. Nomura, Heterocycles, 2019, 99, 549–565 CrossRef.
- Y. Ando, A. Hanaki, R. Sasaki, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2017, 56, 11460–11465 CrossRef CAS PubMed.
- B. S. Chinta and B. Baire, Eur. J. Org. Chem., 2017, 3457–3460 CrossRef CAS.
- S. Martinez-Luis, G. Della-Togna, P. Coley, T. Kursar, W. H. Gerwick and L. Cubilla-Rios, J. Nat. Prod., 2008, 71, 2011–2014 CrossRef CAS PubMed.
- J. Dong, H. Song, J. Li, Y. Tang, R. Sun and L. Wang, J. Nat. Prod., 2008, 71, 952–956 CrossRef CAS.
- M. L. Macías-Rubalcava, B. E. Hernández-Bautista, M. Jemenez-Estrada, M. C. Gonzalez, A. E. Glenn, R. T. Hanlin, S. Hernández-Ortega, A. Saucedo-Garcia, J. M. Muria-Ganzalez and A. L. Anaya, Phytochemistry, 2008, 69, 1185–1196 CrossRef PubMed.
- R. Wang, G. Liu, M. Yang, M. Wang and L. Zhou, Molecules, 2016, 21, 600 CrossRef PubMed.
- T. Shan, J. Tian, X. Wang, Y. Mou, Z. Mao, D. Lai, J. Dai, Y. Peng, L. Zhou and M. Wang, J. Nat. Prod., 2014, 77, 2151–2160 CrossRef CAS PubMed.
- J. Tian, X. Liu, Z. Liu, D. Lai and L. Zhou, Pest Manag. Sci., 2016, 72, 961–965 CrossRef CAS.
- X. Liu, W. Wang, Y. Zhao, D. Lai, L. Zhou, Z. Liu and M. Wang, J. Nat. Prod., 2018, 81, 1803–1809 CrossRef CAS.
- J. Ragot, M. Alcaraz and R. Taylor, Tetrahedron Lett., 1998, 39, 4921–4924 CrossRef CAS.
- J. Ragot, C. Steeneck, M. Alcaraz and R. Taylor, J. Chem. Soc., Perkin Trans. 1, 1999, 8, 1073–1082 RSC.
- A. G. M. Barrett, D. Hamprecht and T. Meyer, Chem. Commun., 1998, 8, 809–810 RSC.
- A. Barrett, F. Blaney, A. Campbell, D. Hamprecht, T. Meyer, A. White, D. Witty and D. Williams, J. Org. Chem., 2002, 67, 2735–2750 CrossRef CAS PubMed.
- M. Miyashita, T. Suzuki, M. Hoshino and A. Yoshikoshi, Tetrahedron, 1997, 53, 12469–12486 CrossRef CAS.
- T. Suzuki and M. Miyashita, Yuki Gosei Kagaku Kyokaishi, 1998, 56, 736–744 CrossRef CAS.
- Y. Yamashita, Y. Hirano, A. Takada, H. Takikawa and K. Suzuki, Angew. Chem., Int. Ed., 2013, 52, 6658–6661 CrossRef CAS PubMed.
- K. Krohn, A. Michel, U. Flouke, H.-J. Aust, S. Draeger and B. Schulz, Liebigs Ann. Chem., 1994, 11, 1099–1108 CrossRef.
- G. Bringmann, S. Busemann, K. Krohn and K. Beckman, Tetrahedron, 1997, 53, 1655–1664 CrossRef CAS.
- A. G. Montalban, L.-O. Wittenberg and A. Mckillop, Tetrahedron Lett., 1999, 40, 5893–5896 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1947664. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10316c |
| This journal is © The Royal Society of Chemistry 2020 |