
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn exclusive deposition method of silver nanoparticles on TiO2 particles via low-temperature decomposition of silver-alkyldiamine complexes in aqueous media†
Tomohiro Yahagi ab,
Takanari Togashia and
Masato Kurihara*a
ab,
Takanari Togashia and
Masato Kurihara*a
aFaculty of Science, Yamagata University, 1-4-12 Kojirakawa-machi, Yamagata 990-8560, Japan. E-mail: kurihara@sci.kj.yamagata-u.ac.jp
bTechnical Support Center, National Institute of Technology, Tsuruoka College, 104 Sawada, Inooka, Tsuruoka, Yamagata 997-8511, Japan
First published on 28th January 2020
Abstract
An exclusive deposition method of Ag nanoparticles (NPs) on TiO2 particles has been developed. Ag NPs supported on TiO2 particles, Agx/TiO2, with various Ag weight ratios versus total weights of Ag and TiO2 between x = 2 and 16 wt% are prepared via low-temperature thermal decomposition of Ag(I)–alkyldiamine complexes generated by a reaction between AgNO3 and N,N-dimethyl-1,3-propanediamine (dmpda) in an aqueous medium suspending TiO2 particles. The thermal decomposition of the Ag(I)–alkyldiamine complexes is accelerated by TiO2 particles in the dark, indicating that the reaction catalytically occurs on the TiO2 surfaces. Under optimised reaction conditions, the thermal decomposition of the complex precursors is completed within 3 hours at 70 °C, and Ag NPs are almost exclusively deposited on TiO2 particles with high conversion efficiencies (≥95%) of the precursor complexes. The thermal decomposition rates of the precursor complexes are strongly influenced by the chemical structure of a family of water-soluble dmpda analogues, and dmpda with both primary and tertiary amino groups is adopted as a suitable candidate for the exclusive deposition method. The number-averaged particle sizes of the Ag NPs are 6.4, 8.4, 11.8 and 15.2 nm in the cases of Agx/TiO2, x = 2, 4, 8 and 16, respectively. To the best of our knowledge, the as-prepared Agx/TiO2 samples show one of the highest catalytic abilities for the hydrogenation reduction of 4-nitrophenol into 4-aminophenol as a model reaction catalysed by Ag NPs.
1. Introduction
The unique catalytic abilities of Ag metals have been utilised for industrial applications such as the production of ethylene oxides and formaldehydes.1,2 Recently, the focus has been on Ag nanoparticles (NPs) as high performance catalysts for the hydrogenation reduction of aromatic NO2 to NH2 groups,3 the reduction of carbonyl compounds,4 the oxidation of alcohols5 and the oxidation of CO.6 Specifically coloured Ag NPs due to the surface plasmon resonance have elicited interest in discovering new photochemical activities.7 Because Ag is one of the most inexpensive noble metals, the further improved catalytic activities of Ag NPs would strongly contribute to the construction of industrially available reactions for chemical syntheses and environmental purification.8,9Metallic NP catalysts deposited on various supports such as oxide particles of TiO2, Al2O3, and SiO2 have been comprehensively developed.10 These NP catalysts have been advantageously employed because of the ease of separating the support particles from the reaction media and suppressing the agglomeration of the NPs. It is well known that the catalytic activities of metal NPs are greatly affected by their sizes and morphologies, which are derived from the increased surface area by downsizing and the appearance of the specific catalytic crystal faces.11,12 To obtain high performance catalysts, it is ordinarily necessary to increase the surface area of NPs by means of downsizing NPs and densifying NPs on supports, which also saves precious resources.
Various technologies have been developed to deposit Ag NPs on supports.13 Conventional methods such as impregnation,14,15 sol–gel16,17 and colloidal18 techniques have been employed; however, these include complicated and/or time-consuming reaction pathways and/or high-temperature treatments of more than 300 °C for the decomposition reduction of precursor silver salts.13–18 With an eye toward environmentally friendly technologies, the invention of much simpler, lower-temperature and wasteless deposition of downsized and densified Ag NPs on supports has been indispensable.
Photo-deposition19,20 and chemical reduction21–24 are typical low-temperature protocols. In the photo-deposition method, Ag NPs are directly deposited on TiO2 support particles from silver salts dissolved in water by photo-irradiation. The chemical reduction method has been widely utilised for the deposition of Ag NPs on support particles, where AgNO3 is reduced in aqueous media by the addition of water-soluble reducing agents such as NaBH4,21 H2O2,22 formaldehydes23 and trisodium citrate.24 However, a critical problem is the difficulty in the exclusive (wasteless) deposition of Ag NPs on support particles as well as in controlling their size, because the reduced Ag particles also unexpectedly appear and grow throughout the aqueous medium and the reaction vessel's wall. For achieving an exclusive photo-deposition method of Ag NPs on TiO2 support particles, Ma et al. utilised separately prepared Ag NP seeds.25 Dinh et al. succeeded in a size- and population-controllable photo-deposition method of Ag NPs on TiO2 support particles by capping TiO2 surfaces with hydrophobic surfactants.26 Jiang et al. reported on the selective attachment of Ag NPs on the SiO2 support particles via the surface functionalization of SiO2, where the Ag NPs were generated in situ by the chemical reduction of Ag salts or were provided using their separately prepared seeds.27,28 However, the conversion efficiencies of the Ag resources to Ag NPs on the support particles have not definitely been discussed.26–28 Accordingly, the development of easily controllable reaction systems for exclusive deposition methods of Ag NPs on surface-unmodified support particles is still a challenging research subject.
Via thermal decomposition, Ag(I)–alkylamine complexes can be directly transformed into alkylamine-passivated Ag NPs whose temperatures are lower than those of the original Ag(I) inorganic salts.29–33 In a typical case, Ag(I) oxalate, Ag2(C2O4), is reacted with various alkylamines to form dinuclear Ag(I)–alkylamine complexes bridged by an oxalato ligand. The decomposition temperature ranges from 100 to 150 °C, while that of the original Ag2(C2O4) is more than 200 °C.29,30 The counter anion, C2O42−, functions as a reducing agent itself and is decomposed with the evolution of CO2. If the counter anion shows a poor reducing ability in the case of AgNO3, the thermal decomposition of the Ag(I)–alkylamine complexes into Ag NPs has been realised by adopting suitable structures of alkylamines as reducing agents. Kim et al. reported that AgNO3 was decomposed between 45 and 50 °C in absolute ethanol containing butylamine for the selective deposition of Ag NPs on SiO2 support particles by using a reaction vessel made of polyethylene to avoid the co-generation of Ag metals on glass (SiO2) wares; nevertheless, the conversion efficiency of AgNO3 to Ag NPs on SiO2 support particles was not disclosed.34–36 In our previous study, we reported on a low-temperature decomposition reaction of a water-soluble Ag2(C2O4) complex coordinated by a specific alkylamine, N,N-dimethyl-1,3-propanediamine (dmpda), catalysed by TiO2 support particles.37 However, the role of the alkylamine leading to the thermal decomposition has not been elucidated.
In this study, we have explored suitable reaction conditions leading to the exclusive deposition of Ag NPs on TiO2 support particles via the thermal decomposition of Ag(I)–alkyldiamine complexes derived from a general Ag(I) resource, AgNO3. It is revealed that Ag(I)–alkyldiamine complexes are catalytically decomposed and deposited on the TiO2 particle surfaces in an environmentally friendly solvent, water, at moderate temperatures, and the conversion efficiencies of the precursor complexes into Ag NPs exceed 95% by adjusting a crucial reaction factors of the chemical structures of alkyldiamines, the decomposition temperatures and the decomposition times. The number-averaged particle sizes of the Ag NPs are controlled by the concentrations of Ag(I)–alkyldiamine complexes. To the best of our knowledge, the as-prepared Ag NPs on TiO2 support particles show one of the highest catalytic abilities via hydrogenation reduction from 4-nitrophenol to 4-aminophenol using NaBH4 as a model reaction catalysed by Ag NPs.
2. Experimental
2.1 Materials
AgNO3 (99.7%) and anatase-type TiO2 (>99.0%) particles (50–300 nm) were purchased from Kanto Chemicals and Merck, respectively. N,N′-Dimethyl-1,3-propanediamine (>97%) was purchased from Aldrich. Propylamine (>98%), 1,3-propanediamine (>98%), N-methyl-1,3-propanediamine (>98%), N,N-dimethyl-1,3-propanediamine (dmpda) (>99%) and N,N,N′,N′-tetramethyl-1,3-propanediamine (>98%) were obtained from Tokyo Chemical Industry. Nitric acid (60–61%), deuterium oxide (>99.9%), NaBH4 (>95%) and 4-nitrophenol (>99%) were supplied by Wako Pure Chemical Industries Ltd. All chemicals were used without further purification.2.2 Methods
In a typical method for preparing the Ag nanoparticles (NPs) supported on TiO2 particles (Agx/TiO2) with various Ag weight ratios versus total weights of Ag and TiO2 between x = 2 and 16 wt%, aqueous solutions of Ag(I)–alkyldiamine (–dmpda) complexes were prepared by adding various volumes (378 μL (x = 2), 772 μL (4), 1.61 mL (8) and 3.53 mL (16)) of an AgNO3 solution (1.00 mol L−1) into an ice-cooled mixture of water (10 mL) and dmpda with vigorous stirring, where the molar ratio of dmpda to AgNO3 was fixed at 4. An aqueous suspension (90 mL) of TiO2 particles (2.00 g) was controlled at various constant temperatures under a nitrogen atmosphere. The aqueous solution of the Ag(I)–dmpda complexes was added into the aqueous suspension of TiO2 particles with vigorous stirring. The thermal decomposition reaction of the Ag(I)–dmpda complexes was carried out at a constant temperature in the dark. After the reaction was completed, Agx/TiO2 samples were obtained by centrifugal separation and washing with water four times and then vacuum drying at room temperature.2.3 Evaluation of the catalytic abilities of Agx/TiO2
The catalytic activities of Agx/TiO2 were evaluated by a typical model reaction catalysed by Ag NPs, i.e. a reduction reaction of 4-nitrophenol to 4-aminophenol using NaBH4. A mixture of aqueous suspension (3.2 mL) of Agx/TiO2 (0.020 mg) and an aqueous solution of 4-nitrophenol (0.40 mL, 6.0 × 10−4 mol L−1) was placed in an optical quartz cell. A freshly prepared aqueous solution of NaBH4 (0.40 mL, 0.30 mol L−1) was mixed quickly to initiate the catalytic reaction. Time-course changes in the UV-Vis absorption spectra were measured at 5 second intervals at a constant temperature of 25 °C using a photodiode array spectrometer (Agilent 8453) to pursue the decreasing absorption band of 4-nitrophenol.2.4 Characterisation
The morphologies and sizes of Ag NPs of Agx/TiO2 were characterised via transmission electron microscope (TEM) images (JEOL JEM 2100) operating at 200 kV. All specimens for TEM were prepared by dropping a diluted suspension of Agx/TiO2 on a collodion membrane-coated Cu grid. The number-averaged particle sizes, dav, were calculated by measuring the long-axis lengths of more than 100 particles. The powder X-ray diffraction (XRD) patterns were obtained on an X-ray diffractometer (Rigaku MiniFlex II) equipped with a Cu Kα X-ray source (30 kV, 15 mA). 109Ag nuclear magnetic resonance (NMR) spectra of the Ag(I)–alkyldiamine complexes in aqueous solutions were measured with an NMR spectrometer (JEOL ECX-400), where an aqueous solution of AgNO3 was used as a reference for calibrating chemical shifts (δ). The concentrations of Ag ions in aqueous media and the Ag contents of Agx/TiO2 were determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES, SII Nanotechnology, SPS 3500-DD). X-ray photoelectron spectroscopy (XPS) was carried out using a Shimadzu ESCA-3200 photoelectron spectrometer equipped with a Mg Kα X-ray source (hν = 1253.6 eV, 10 kV, 30 mA). The binding energy of the Ag NPs of Agx/TiO2 was calibrated by setting the C 1s peak to 284.8 eV.3. Results and discussion
3.1 Exclusive deposition of Ag NPs on TiO2 particles
Ag NPs supported on TiO2 particles, Agx/TiO2, with various Ag weight ratios versus total weights of Ag and TiO2 between x = 2, 4, 8 and 16 wt% were prepared via low-temperature thermal decomposition of Ag(I)–alkyldiamine (–dmpda) complexes. The concentration of Ag components dissolving in aqueous media decreased and almost disappeared within 3 hours by low-temperature heating at 70 °C (Fig. 1). In the absence of TiO2 particles, the rate of decrease of Ag components was significantly suppressed, vide infra (Fig. 6a, red line), and the TiO2 particles initially exhibited purple in colour along with decreasing concentrations of Ag components. The colour originated from the surface plasmon resonance of metallic Ag NPs. These results suggest that the Ag(I)–dmpda complexes are catalytically decomposed on the TiO2 particles in aqueous media to immobilise on their surfaces as Ag NPs. | ||
| Fig. 1 Time-course changes in concentrations of Ag components dissolving in the reaction aqueous media at 70 °C in the presence of suspended TiO2 particles. Added volumes of an AgNO3 solution (1.00 mol L−1) were 378 μL (○), 772 μL (◇), 1.61 mL (□) and 3.53 mL (△). The decomposition reactions were investigated in the dark. | ||
In the XRD pattern of the pristine TiO2 particles, the signals appearing at 25.3, 36.9, 37.8, 38.5, 48.0, 53.9, 55.1, 62.1, 62.7, 68.8, 70.3, 75.0 and 76.1° were derived from the anatase type (PDF: 01-070-7348); these signals were consistent with the (101), (103), (004), (112), (200), (105), (211), (213), (204), (116), (220), (215) and (301) faces, respectively (Fig. 2). Although the XRD-signal profile was not changed after the low-temperature thermal decomposition of Ag(I)–dmpda complexes, new weak XRD signals appeared at 38.1, 44.1, 64.5 and 77.3° in the cases of Ag8/TiO2 and Ag16/TiO2. These signals are assigned to the (111), (200), (220) and (311) faces of the face-centred cubic phase of metallic Ag (PDF: 01-071-3762). The signals are significantly broadened, indicating that metallic Ag is deposited as NPs on TiO2 surfaces. Similar XRD signals due to metallic Ag cannot be clearly observed in the cases of Ag2/TiO2 and Ag4/TiO2, due to the signal broadening and low concentration of the deposited metallic Ag on TiO2 surfaces. These results suggest that Ag(I) ions are successfully reduced to Ag(0) via the low-temperature thermal decomposition of Ag(I)–dmpda complexes occurring catalytically on the TiO2 surfaces in the dark.
 | ||
| Fig. 2 XRD patterns of the pristine TiO2 particles (a) and the Ag NP-deposited Agx/TiO2, x = 2 (b), 4 (c), 8 (d) and 16 (e). The XRD signal positions due to metallic Ag are marked by ▼. | ||
The morphology and sizes of Ag NPs of Agx/TiO2 were investigated via TEM images (Fig. 3). Innumerable NPs (Fig. 3b–e) were observed as dark contrasts on the surfaces of TiO2 particles (Fig. 3a) between 50 and 300 nm in dimension. From high-resolution TEM images of the deposited NPs (Fig. 3f), lattice fringes can be clearly observed with lattice spacing of 0.24 nm which is consistent with (111) faces of the face-centred cubic metallic Ag.
 | ||
| Fig. 3 TEM images of the pristine TiO2 particles (a) and the Ag NP-deposited Agx/TiO2, x = 2 (b), 4 (c), 8 (d) and 16 (e), and a magnified high-resolution lattice image of Ag NPs in the case of Ag2/TiO2 (f). | ||
The deposited amounts of Ag on TiO2 particles of Agx/TiO2 were determined via an ICP-AES technique using aqueous solutions of Ag NPs oxidatively dissolved by HNO3. When the initial Ag contents were adopted as Ag(I)–dmpda complexes as a precursor for preparing Agx/TiO2, x = 2, 4, 8 and 16 wt%, the actual amounts of the deposited Ag NPs on TiO2 particles were 1.9, 3.9, 7.9 and 15.5 wt%, respectively. It is noted that the Ag(I)–dmpda complex precursors are almost exclusively deposited on TiO2 particles through the catalytic low-temperature decomposition of the precursors on the TiO2 surfaces with high conversion efficiencies of 95, 98, 99 and 97% in the cases of x = 2, 4, 8 and 16, respectively. The number-averaged particle sizes, dav, of Ag NPs on the TiO2 surfaces were calculated based on more than 100 particles from each prepared sample of Agx/TiO2 (Fig. 3). The total number-averaged particle size, dav,total, and the standard deviation, σ, were estimated from several dav with sample dependency. Fig. 4 shows the plot of dav,total of Agx/TiO2 versus the initial Ag contents of the Ag(I)–dmpda complex precursors, x = 2, 4, 8 and 16 wt%. The dav,total were 6.4, 8.4, 11.8 and 15.2 nm in the cases of x = 2, 4, 8 and 16, respectively. The particle size distributions of the deposited Ag NPs from the representative samples of Agx/TiO2 are shown as histograms in Fig. 5. The size distribution of the Ag NPs becomes wider with increasing x. At this stage, the dimension of the Ag NPs is still uncontrollable in the case of x = 16, because the distribution is largely deviated from dav.
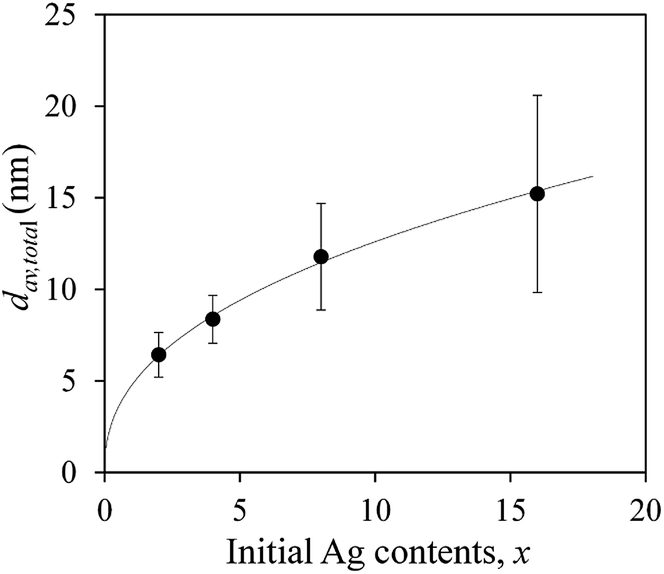 | ||
| Fig. 4 Plots of total number-averaged particle sizes, dav,total, of Agx/TiO2 versus the initial Ag contents of the Ag(I)–dmpda complex precursors, x = 2, 4, 8 and 16 wt%. The error bars show the standard deviation, ±σ. | ||
 | ||
| Fig. 5 The particle size distributions of the deposited Ag NPs from the representative samples of Agx/TiO2, x = 2 (a), 4 (b), 8 (c) and 16 (d). | ||
3.2 Investigation of the optimised reaction conditions for the exclusive deposition
As the decreasing rate of the Ag components was already shown in Fig. 1, the decreasing rates, i.e. the thermal decomposition rates of the Ag(I)–dmpda complexes dissolving in an aqueous medium, were investigated depending on the reaction temperatures between 0 and 100 °C in the dark (Fig. 6). In the absence of TiO2 particles (Fig. 6a), almost no consumption (no thermal decomposition) of the Ag(I)–dmpda complexes was observed at less than 60 °C within 3 hours, while the Ag(I)–dmpda complexes were suddenly consumed at elevated temperatures of ≥70 °C and disappeared from the aqueous medium within 1 hour at 100 °C. It is obvious that the presence of TiO2 particles accelerates the thermal decomposition rate of the Ag(I)–dmpda complexes, as compared with the elevated temperatures of ≥60 °C (Fig. 6b). Consequently, we adopted 70 °C as one reaction temperature suitable for completing the exclusive deposition of Ag NPs on TiO2 particles within a few hours. | ||
| Fig. 6 Time-course changes in consumption rates of Ag components dissolving in the reaction aqueous media at various temperatures of 0, 60, 70 and 100 °C in the absence (a) or in the presence (b) of suspended TiO2 particles. The added volume of an AgNO3 solution (1.00 mol L−1) was 772 μL. The consumption rates were expressed as [Ag]t/[Ag]0, where [Ag]0 and [Ag]t are the initial concentration of the Ag components in the reaction media at time = 0 and the concentrations of the Ag components at various reaction times = t, respectively. The decomposition reactions were investigated in the dark. | ||
Intriguingly, the realisation of the exclusive deposition reaction of Ag NPs on TiO2 particles was strongly influenced by both the addition amounts and the chemical structures of water-soluble alkyldiamines (Table 1). The photographs of aqueous solutions of AgNO3 in the presence of different molar ratios of dmpda are shown in Fig. S1.† The aqueous solutions were immediately clouded by black particles when an aqueous solution of AgNO3 was added to an aqueous solution of dmpda with molar ratios of dmpda to AgNO3 of 1.0 and 1.5 mol mol−1 (Fig. S1(i) and (ii)†). The XRD pattern of the black precipitates isolated from the aqueous solutions was derived from Ag2O crystals. In the case of molar ratios of ≥2.0, colourless transparent solutions were obtained (Fig. S1(iii)–(v)†). In a 109Ag-NMR spectrum of the transparent and colourless aqueous mixture of AgNO3 and dmpda with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mol mol−1, one single signal was observed at 362 ppm (Fig. S2a†), when one signal of an aqueous solution of AgNO3 without alkyldiamines was employed as a standard one of 0 ppm for evaluating chemical shifts. In case of Ag:dmpda = 1:2 mol mol−1, the 109Ag-NMR signal appeared at 348 ppm. This result suggests that dmpda ligands are dynamically exchanged on Ag+ via the weak coordination of H2O as a solvent molecule, depending on the concentrations of dmpda. The 109Ag-NMR chemical-shift value was consistent with 363 ppm of Ag+ coordinated by propylamine with a primary amino group in the case of Ag:propylamine = 1:4 (Fig. S2b†). As a result, it is generally understood that (i) Ag+ ions can be coordinated by dmpda as a monodentate ligand to form [Ag(dmpda)2]+ via dynamical ligand-exchange with H2O for dissolving in water, (ii) Ag+ ions are coordinated by dmpda via the primary amino group, but not the bulky tertiary amino group, and (iii) an insoluble Ag2O precipitate is generated by the basic aqueous conditions without stoichiometrically sufficient dmpda ligands of Ag:dmpda = 1:2 mol mol−1.
:4 mol mol−1, one single signal was observed at 362 ppm (Fig. S2a†), when one signal of an aqueous solution of AgNO3 without alkyldiamines was employed as a standard one of 0 ppm for evaluating chemical shifts. In case of Ag:dmpda = 1:2 mol mol−1, the 109Ag-NMR signal appeared at 348 ppm. This result suggests that dmpda ligands are dynamically exchanged on Ag+ via the weak coordination of H2O as a solvent molecule, depending on the concentrations of dmpda. The 109Ag-NMR chemical-shift value was consistent with 363 ppm of Ag+ coordinated by propylamine with a primary amino group in the case of Ag:propylamine = 1:4 (Fig. S2b†). As a result, it is generally understood that (i) Ag+ ions can be coordinated by dmpda as a monodentate ligand to form [Ag(dmpda)2]+ via dynamical ligand-exchange with H2O for dissolving in water, (ii) Ag+ ions are coordinated by dmpda via the primary amino group, but not the bulky tertiary amino group, and (iii) an insoluble Ag2O precipitate is generated by the basic aqueous conditions without stoichiometrically sufficient dmpda ligands of Ag:dmpda = 1:2 mol mol−1.
| Alkyldiamines | Formation of Ag(I)–alkyldiamine complexesa | Thermal decomposition ratesb (%) |
|---|---|---|
| a The alkyldiamine (3.09 mmol) was dissolved in water (10 mL), and an AgNO3 aqueous solution (1.00 mol L−1, 772 μL) was dropwise added to the alkyldiamine solution with vigorous stirring under ice cooling, where the molar ratio is Ag:alkyldiamine = 1:4 mol mol−1.b The thermal decomposition rates (%) of Ag(I)–alkyldiamine complexes for 3 h at 70 °C under the same reaction conditions and analyses as Fig. 6b. |
||
 |
○ | 7% |
 |
○ | 31% |
 |
○ | 100% |
 |
× | N.A. |
 |
× | N.A. |
A family of dmpda analogues was investigated to choose water-soluble alkyldiamines suitable for low-temperature thermal decomposition on TiO2 particles, as listed in Table 1. Transparent and colourless solutions were similarly obtained by the reaction of AgNO3 with 1,3-propanediamine or N-methyl-1,3-propanediamine, indicating that water-soluble Ag(I)–alkyldiamine complexes were stably formed by the coordination bonding of the primary amino group, whereas N,N′-dimethyl-1,3-propanediamine and N,N,N′,N′-tetramethyl-1,3-propanediamine without primary amino groups produced black particles of Ag2O. The thermal decomposition behaviour of Ag(I)–alkyldiamine complexes composed of 1,3-propanediamine and N-methyl-1,3-propanediamine was compared to that of the Ag(I)–dmpda complexes. The Ag(I)–(1,3-propanediamine) complexes were scarcely decomposed at 70 °C even after 3 hours, and a moderate decomposition reaction was observed in the case of the Ag(I)–(N-methyl-1,3-propanediamine) complexes which reached 31% decomposition after 3 hours (Fig. 7, Table 1). It is evident that stable coordination to Ag+ ions occurs via the primary amino groups, and low-temperature decomposition is promoted in the order of tertiary > secondary > primary amino groups of the distal position. This order is consistent with the reduction abilities of alkylamino groups due to increased electron density on the nitrogen atom. On the basis of these results, we adopted dmpda as a suitable water-soluble alkyldiamine to realise the exclusive deposition of Ag NPs on TiO2 particles from aqueous solutions of Ag(I)–alkyldiamine complexes.
 | ||
| Fig. 7 Time-course changes in consumption rates of Ag components dissolving in the reaction aqueous media at 70 °C in the presence of TiO2 using 1,3-propanediamine (×), N-methyl-1,3-propanediamine (△) and dmpda (◇). The aqueous solutions of Ag(I)–alkyldiamine complexes were prepared by the addition of an aqueous solution (772 μL) of AgNO3 (1.00 mol L−1) into to a mixture of the alkyldiamines and water (10 mL), where the molar ratio of the AgNO3 and alkyldiamines is 1:4 mol mol−1. The time-course changes in consumption rates of the Ag components were monitored after the addition of the aqueous solutions of Ag(I)–alkyldiamine complexes into an aqueous suspension (90 mL) of TiO2 particles (2.00 g) at a constant temperature of 70 °C. The decomposition reactions were investigated in the dark. | ||
3.3 Evaluation of the catalytic abilities of Agx/TiO2
The catalytic abilities of Agx/TiO2 were evaluated via a typical model reaction catalysed by Ag NPs, i.e. hydrogenation reduction of 4-nitrophenol to 4-aminophenol using NaBH4.38 In a typical time-course change in the UV-Vis absorption spectra in the case of Ag2/TiO2 (Fig. 8), the absorption band of 4-nitrophenol at 400 nm gradually decreased in intensity after the addition of NaBH4, and completely disappeared within 6 minutes. A new absorption band originating from 4-aminophenol appeared at 300 nm with isosbestic points at 282 and 314 nm. The hydrogenation reduction of 4-nitrophenol to 4-aminophenol was catalysed by the Ag NPs deposited on TiO2 particles because a similar spectral change was not observed in the case of pristine TiO2 particles. Similar absorption spectral changes were also observed in the cases of x = 4, 8 and 16 of Agx/TiO2, and the catalytic reaction of 4-nitrophenol was accelerated by increasing amounts, x of the Ag NPs, as shown in the time-course changes in the absorbance at 400 nm (Fig. 9a). In the initial stage, induction periods were observed where the absorbance did not change. The induction periods became longer as the deposited amounts of the Ag NPs were decreased. It has been explained that the induction period is caused by the reconstitution of catalyst surfaces, i.e. removal of oxygenated components on the surfaces of Ag NPs and/or the consumption of dissolved oxygen.39,40 In order to evaluate the catalytic activities dependent on the deposited amounts of Ag NPs, the apparent rate constants, k, were calculated. When the largely excess amount of NaBH4 to 4-nitrophenol was used in concentration, the concentration of NaBH4 was assumed to be constant during the catalytic reaction. Thus, the catalytic reaction can be treated as pseudo-first-order kinetic behaviour,38 and the apparent rate constants, k, can be expressed by the absorbance (A) at a certain reaction time (t) using the following eqn (1):
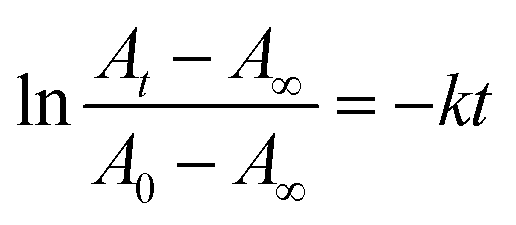 | (1) |
 | ||
| Fig. 8 UV-Vis absorption spectral change of an aqueous solution of 4-nitrophenol in 30 second intervals after the addition of NaBH4 in the presence of Ag2/TiO2 at a constant temperature of 25 °C, where initial concentrations of 4-nitrophenol, NaBH4, Ag2/TiO2 are 6.0 × 10−5, 3.0 × 10−2 and 5.0 mg L−1, respectively. | ||
 | ||
| Fig. 9 (a) Time-course changes in absorbance at 400 nm and (b) their transformed plots by eqn (1) in the cases of Agx/TiO2, x = 2 (○), 4 (□), 8 (△) and 16 (◇). The concentrations of Agx/TiO2 per volumes (L) of the reaction media were prepared as 5.0 mg L−1 (see Table 2). | ||
The time-course changes in absorbance (Fig. 9a) were transformed by eqn (1) (Fig. 9b). The reaction rate constants of k of Agx/TiO2 were calculated by the linear approximation of plots excluding the induction periods, and were 2.3 × 10−2, 4.0 × 10−2, 4.7 × 10−2 and 5.8 × 10−2 s−1 in the cases of x = 2, 4, 8 and 16, respectively. In order to compare the catalytic abilities of Agx/TiO2 to those of the previously reported Ag catalysts, we used the normalised values, k/[Ag].41,42 As summarised in Table 2, the concentration of catalysts, [cat.], was expressed by the weight (mg) of the catalyst per volume (L) of the reaction medium, and the concentration, [Ag], was of the actual Ag weight (g) contained in each catalyst per volume (L) of the reaction medium. The k/[Ag] values of Agx/TiO2 were 240, 210, 120 and 75 s−1 g−1 L in the cases of x = 2, 4, 8 and 16, respectively. The catalytic abilities are improved as dav,total are decreased, i.e. as the surface areas of the Ag NPs are increased. The size-dependent nature of the catalytic abilities of the Ag NPs is also understandable via the turnover numbers (TONs). The catalytic abilities of Agx/TiO2 are much higher than those of the previously reported Ag catalysts, on the basis of the k/[Ag]. To the best of our knowledge, Agx/TiO2 samples show one of the highest catalytic abilities for a model reaction, hydrogenation reduction of 4-nitrophenol to 4-aminophenol. There is a tendency for Ag NPs on metal-oxide substrates, especially on TiO2, to present relatively higher catalytic activities. As shown in Fig. 3f, the Ag NPs connect with the TiO2 surfaces in hemisphere shapes, suggesting formation of strong junctions between Ag NPs and TiO2 particles. In fact, XPS analysis showed that the binding energy (367.6 eV) of Ag NPs on TiO2 particles was shifted to a lower energy than that of the intrinsic Ag metal (368.2 eV) (Fig. S3†). The specific electronic interaction between Ag NPs and TiO2 particles would be responsible for the enhanced catalytic abilities of Agx/TiO2. In addition, the particle sizes, morphologies, and distribution of the Ag NPs on TiO2 particles were not so significantly changed after the catalytic reactions from the TEM images of the Agx/TiO2 (Fig. S4†).
| Catalysts | da (nm) | Ag contents in catalysts (wt%) | Concentrations of catalysts, [cat.]b (mg L−1) | Rate constants (reaction temperature), k (10−3 s−1) | Catalytic abilities based on Ag, k/[Ag]c (s−1 g−1 L) | TONsd | Ref. |
|---|---|---|---|---|---|---|---|
| a Averaged particle sizes or size ranges of Ag NPs.b The catalyst weights (mg) per volume (L) of a reaction medium.c The rate constants, k, are normalized by the concentration of the actual Ag weights (g) contained in each catalyst per volume (L) of the reaction medium, [Ag].d TONs are expressed by the number ratios of the initial substrate molecule (4-nitrophenol) versus the Ag atoms, when 4-nitrophenol is completely converted into 4-aminophenol for 360, 200, 125 and 100 seconds in the cases of Agx/TiO2, x = 2, 4, 8 and 16, respectively (see Fig. 9). | |||||||
| Ag2/TiO2 | 6.4 | 1.9 | 5.0 | 23 (25 °C) | 2.4 × 102 | 68 | This work |
| Ag4/TiO2 | 8.4 | 3.9 | 5.0 | 40 (25 °C) | 2.1 × 102 | 33 | This work |
| Ag8/TiO2 | 11.8 | 7.9 | 5.0 | 47 (25 °C) | 1.2 × 102 | 16 | This work |
| Ag16/TiO2 | 15.2 | 15.5 | 5.0 | 58 (25 °C) | 75 | 8 | This work |
| Ag NPs (no support) | 14.3 | 100 | 482 | 0.47 (R.T.) | 9.8 × 10−4 | 43 | |
| Ag/porous carbon | 10 | 55.2 | 333 | 1.7 (R.T.) | 9.2 × 10−3 | 44 | |
| Graphene oxide/Ag NPs-Fe3O4 | 9–20 | 24.3 | 33.3 | 27 (R.T.) | 3.3 | 45 | |
| Ag/micron-SiO2 sphere | 10–60 | 43.0 | 250 | 3.6 (22 °C) | 3.3 × 10−2 | 46 | |
| Fe3O4@SiO2–Ag | 3.7 | 8.89 | 16.7 | 7.7 (25 °C) | 5.2 | 47 | |
| Ag/fibrous nano-silica | 4 | 8.97 | 66.2 | 10 (20 °C) | 1.7 | 48 | |
| TiO2@Ag | 10 | 9.28 | 13.0 | 7.5 (25 °C) | 6.2 | 25 | |
| Ag/TiO2 | <1 | 0.75 | 16.0 | 20 (25 °C) | 1.7 × 102 | 49 | |
4. Conclusion
In this study, we focus on developing an exclusive deposition method of Ag NPs on metal oxide particles through a low-temperature decomposition reaction of water-soluble Ag(I)–alkyldiamine complexes catalysed by TiO2 in aqueous media. It is revealed that stable coordination to Ag+ ions occurs via the primary amino group, and low-temperature decomposition is promoted in the order of tertiary > secondary > primary amino groups of the distal position in a water-soluble family of alkyldiamines. We adopt N,N-dimethyl-1,3-propanediamine (dmpda) as one of the most promising candidates for realising an exclusive deposition method where water-soluble dmpda is a commercially available and inexpensive reagent. At the optimised reaction temperature of 70 °C, Ag NPs are completely deposited on TiO2 particles within a few hours and the conversion efficiencies from Ag(I)–dmpda complexes into Ag NPs exceed 95%. The number-averaged particle sizes are increased from 6 to 15 nm, depending on the increased concentrations of Ag(I)–dmpda complexes with Ag weight ratios versus total weights of Ag and TiO2 from 2 to 16 wt%. At this stage, the particle sizes are never controlled in the case of an Ag ratio of 16 wt% because the size distribution is becoming seriously wider as the concentrations of Ag(I)–dmpda complexes are increased. Nevertheless, the as-developed exclusive deposition method is of interest for advantageously preparing high-density Ag catalysts bearing immobilised nanosized Ag particles on the limited surface areas of TiO2 particles. Our research is progressing toward our next reports that will clarify the crystal-growth mechanism of Ag NPs on TiO2 particles and find the crucial factors for size-controllable and high-density deposition of Ag NPs on TiO2 particles. It is noted that the water-based exclusive deposition method will be environmentally friendly, simple, low cost, and capable of the one-step and large-scale production of high-performance Ag NP catalysts on TiO2 particles (Table 2), as well as saving expensive metal resources.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. E. van den Reijen, S. Kanungo, T. A. J. Welling, M. Versluijs-Helder, T. A. Nijhuis, K. P. de Jong and P. E. de Jongh, J. Catal., 2017, 356, 65–74 CrossRef CAS.
- A. Nagy and G. Mestl, Appl. Catal., A, 1999, 188, 337–353 CrossRef CAS.
- K. Shimizu, Y. Miyamoto and A. Satsuma, J. Catal., 2010, 270, 86–94 CrossRef CAS.
- T. Mitsudome, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem.–Eur. J., 2013, 19, 5255–5258 CrossRef CAS PubMed.
- T. Mitsudome, Y. Mikami, H. Funai, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 138–141 CrossRef CAS PubMed.
- H. Liu, D. Ma, R. A. Blackley, W. Zhou and X. Bao, Chem. Commun., 2008, 2677–2679 RSC.
- Z. Zhao, Y. Wang, J. Xu and Y. Wang, RSC Adv., 2015, 5, 59297–59305 RSC.
- X.-Y. Dong, Z.-W. Gao, K.-F. Yang, W.-Q. Zhang and L.-W. Xu, Catal. Sci. Technol., 2015, 5, 2554–2574 RSC.
- C. Wen, A. Yin and W.-L. Dai, Appl. Catal., B, 2014, 160–161, 730–741 CrossRef CAS.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed.
- M. Okumura, T. Fujitani, J. Huang and T. Ishida, ACS Catal., 2015, 5, 4699–4707 CrossRef CAS.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- P. Claus and H. Hofmeister, J. Phys. Chem. B, 1999, 103, 2766–2775 CrossRef CAS.
- N. Bogdanchikova, F. C. Meunier, M. Avalos-Borja, J. P. Breen and A. Pestryakov, Appl. Catal., B, 2002, 36, 287–297 CrossRef CAS.
- J. Zhang, Y. Li, Y. Zhang, M. Chen, L. Wang, C. Zhang and H. He, Sci. Rep., 2015, 5, 12950 CrossRef CAS PubMed.
- A. Hernández-Gordillo, M. Arroyo, R. Zanella and V. Rodríguez-González, J. Hazard. Mater., 2014, 268, 84–91 CrossRef PubMed.
- B. Xin, L. Jing, Z. Ren, B. Wang and H. Fu, J. Phys. Chem. B, 2005, 109, 2805–2809 CrossRef CAS PubMed.
- S. Cai, H. Rong, X. Yu, X. Liu, D. Wang, W. He and Y. Li, ACS Catal., 2013, 3, 478–486 CrossRef CAS.
- S. C. Chen and M. A. Barteau, Langmuir, 2005, 21, 5588–5595 CrossRef PubMed.
- H. Zhang, G. Wang, D. Chen, X. Lv and J. Li, Chem. Mater., 2008, 20, 6543–6549 CrossRef CAS.
- N. Niño-Martínez, G. A. Martínez-Castañón, A. Aragón-Piña, F. Martínez-Gutierrez, J. R. Martínez-Mendoza and F. Ruiz, Nanotechnology, 2008, 19, 065711 CrossRef PubMed.
- X. You, F. Chen, J. Zhang and M. Anpo, Catal. Lett., 2005, 102, 247–250 CrossRef CAS.
- Q. Liu, Y. Cao, W.-L. Dai and J.-F. Deng, Catal. Lett., 1998, 55, 87–91 CrossRef CAS.
- B. Naik, V. S. Prasad and N. N. Ghosh, Powder Technol., 2012, 232, 1–6 CrossRef CAS.
- J. Ma, X. Guo, Y. Zhang and H. Ge, Chem. Eng. J., 2014, 258, 247–253 CrossRef CAS.
- C.-T. Dinh, T.-D. Nguyen, F. Kleitz and T.-O. Do, ACS Appl. Mater. Interfaces, 2011, 3, 2228–2234 CrossRef CAS PubMed.
- Z.-J. Jiang, C.-Y. Liu and L.-W. Sun, J. Phys. Chem. B, 2005, 109, 1730–1735 CrossRef CAS PubMed.
- Z. Jiang and C. Liu, J. Phys. Chem. B, 2003, 107, 12411–12415 CrossRef CAS.
- M. Itoh, T. Kakuta, M. Nagaoka, Y. Koyama, M. Sakamoto, S. Kawasaki, N. Umeda and M. Kurihara, J. Nanosci. Nanotechnol., 2009, 9, 6655–6660 CrossRef CAS PubMed.
- T. Yamada, K. Fukuhara, K. Matsuoka, H. Minemawari, J. Tsutsumi, N. Fukuda, K. Aoshima, S. Arai, Y. Makita, H. Kubo, T. Enomoto, T. Togashi, M. Kurihara and T. Hasegawa, Nat. Commun., 2016, 7, 11402 CrossRef CAS PubMed.
- D. Wang, T. Xie, Q. Peng and Y. Li, J. Am. Chem. Soc., 2008, 130, 4016–4022 CrossRef CAS PubMed.
- M. Yamamoto, Y. Kashiwagi and M. Nakamoto, Langmuir, 2006, 22, 8581–8586 CrossRef CAS PubMed.
- T. Togashi, K. Saito, Y. Matsuda, I. Sato, H. Kon, K. Uruma, M. Ishizaki, K. Kanaizuka, M. Sakamoto, N. Ohya and M. Kurihara, J. Nanosci. Nanotechnol., 2014, 14, 6022–6027 CrossRef CAS PubMed.
- H. K. Park, J. K. Yoon and K. Kim, Langmuir, 2006, 22, 1626–1629 CrossRef CAS PubMed.
- K. Kim, H. S. Kim and H. K. Park, Langmuir, 2006, 22, 8083–8088 CrossRef CAS PubMed.
- K. S. Shin, Y. K. Cho, J.-Y. Choi and K. Kim, Appl. Catal., A, 2012, 413–414, 170–175 CrossRef CAS.
- T. Yahagi, T. Togashi, K. Kanaizuka and M. Kurihara, Chem. Lett., 2016, 45, 1195–1197 CrossRef CAS.
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- N. Pradhan, A. Pal and T. Pal, Colloids Surf., A, 2002, 196, 247–257 CrossRef CAS.
- E. Menumerov, R. A. Hughes and S. Neretina, Nano Lett., 2016, 16, 7791–7797 CrossRef CAS PubMed.
- C. Kästner and A. F. Thünemann, Langmuir, 2016, 32, 7383–7391 CrossRef PubMed.
- L. R. S. Lara, A. D. Zottis, W. C. Elias, D. Faggion Jr, C. E. M. de Campos, J. J. S. Acuña and J. B. Domingos, RSC Adv., 2015, 5, 8289–8296 RSC.
- W. Wu, M. Lei, S. Yang, L. Zhou, L. Liu, X. Xiao, C. Jiang and V. A. L. Roy, J. Mater. Chem. A, 2015, 3, 3450–3455 RSC.
- S. Tang, S. Vongehr and X. Meng, J. Phys. Chem. C, 2010, 114, 977–982 CrossRef CAS.
- J. Qu, C. Ren, Y. Dong, Y. Chang, M. Zhou and X. Chen, Chem. Eng. J., 2012, 211–212, 412–420 CrossRef CAS.
- M. Wang, D. Tian, P. Tian and L. Yuan, Appl. Surf. Sci., 2013, 283, 389–395 CrossRef CAS.
- Y. Chi, Q. Yuan, Y. Li, J. Tu, L. Zhao, N. Li and X. Li, J. Colloid Interface Sci., 2012, 383, 96–102 CrossRef CAS PubMed.
- Z. Dong, X. Le, X. Li, W. Zhang, C. Dong and J. Ma, Appl. Catal., B, 2014, 158–159, 129–135 CrossRef CAS.
- X. Wang, Z. Zhao, D. Ou, B. Tu, D. Cui, X. Wei and M. Cheng, Appl. Surf. Sci., 2016, 385, 445–452 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10307d |
| This journal is © The Royal Society of Chemistry 2020 |