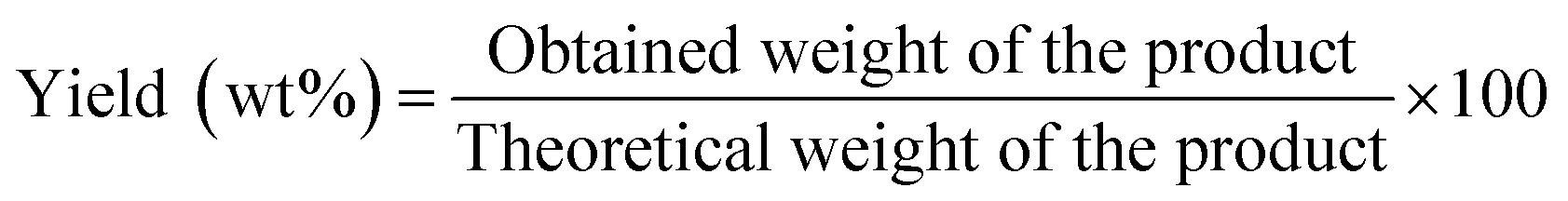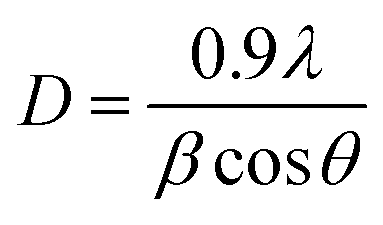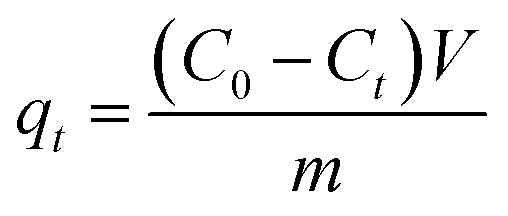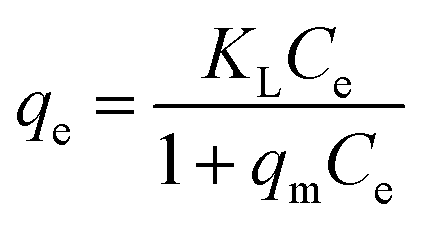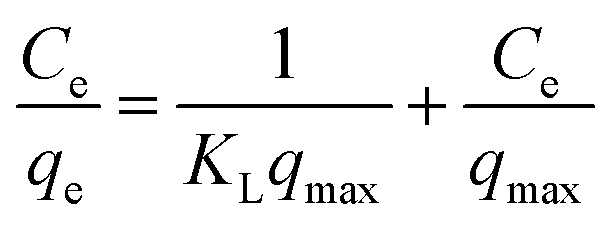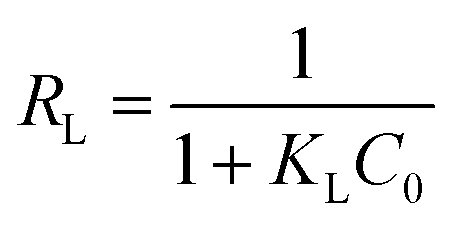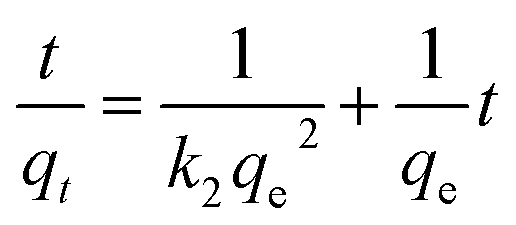DOI:
10.1039/C9RA10300G
(Paper)
RSC Adv., 2020,
10, 9693-9703
Facile fabrication of bimetallic Fe–Mg MOF for the synthesis of xanthenes and removal of heavy metal ions†
Received
8th December 2019
, Accepted 28th February 2020
First published on 6th March 2020
Abstract
This work reported the preparation of Mg-MOF, Fe-MOF and Fe–Mg MOF by a solvothermal technique and their characterization with FT-IR, XRD, SEM, EDS, TEM and SBET analyses. The nanoparticle diameter ranged from 3.1 to 10.9 nm. The acidity of the MOFs was measured by nonaqueous potentiometric titration of n-butylamine. It was observed that the formation of a bimetallic MOF sharply increases the surface acidity and the catalytic activity. The catalytic results of the Fe–Mg MOF catalyzing the synthesis of 14-aryl-14-H-dibenzo[a,j]xanthenes in comparison with those of parent MOFs showed a higher yield of the desired product in a lower time and among various Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mg, the (0.6:1) Fe–Mg MOF showed the highest catalytic activity and acidity. Even after the 4th run, the Fe–Mg MOF catalyst still maintained nearly the initial catalytic activity. The adsorption performance of Mg-MOF, Fe-MOF and Fe–Mg MOF was evaluated by batch experiments. The effect of contact time, the solution pH, the adsorbent dose and the initial concentration of the heavy metal ions was discussed. It was found that the capacity of the bimetallic Fe–Mg MOF for Pb(II), Cu(II) and Cd(II) adsorption was higher than that of the Mg-MOF and Fe-MOF, the kinetic data followed the pseudo-second-order kinetic model and the isothermal data obeyed the Langmuir isotherm model. The mechanism of the removal of the heavy metal ions was discussed.
:Mg, the (0.6:1) Fe–Mg MOF showed the highest catalytic activity and acidity. Even after the 4th run, the Fe–Mg MOF catalyst still maintained nearly the initial catalytic activity. The adsorption performance of Mg-MOF, Fe-MOF and Fe–Mg MOF was evaluated by batch experiments. The effect of contact time, the solution pH, the adsorbent dose and the initial concentration of the heavy metal ions was discussed. It was found that the capacity of the bimetallic Fe–Mg MOF for Pb(II), Cu(II) and Cd(II) adsorption was higher than that of the Mg-MOF and Fe-MOF, the kinetic data followed the pseudo-second-order kinetic model and the isothermal data obeyed the Langmuir isotherm model. The mechanism of the removal of the heavy metal ions was discussed.
1. Introduction
Metal–organic frameworks (MOFs) are a class of compounds constructed by the coordination of clusters or metal ions with organic ligands to compose one, two, or three-dimensional pore structures.1 Recently, MOFs have received great attention owing to their excellent properties, such as versatile porous structures and numerous potential applications.2 MOFs are unique and different to other traditional porous solids owing to their interesting properties. These include a simple composition that is easy to prepare compared with zeolites,3 the presence of coordinative unsaturated active canters, tunable pore structures and ultrahigh surface area.4 Moreover, MOFs have an important advantage that various frameworks can be formed by the participation of several metal cations.5 MOFs can be synthesized with specific properties to improve their performances in reaching the desired targets by deliberately and systematically setting their functionalities and structures; for example, the surface area, pore size and/or shape can be controlled by varying the connectivity of the cations and the type of the organic ligands.6 MOFs have been used in different potential applications as drug delivery,7 catalytic reactions,8 sensing9 and gas adsorption/separation.10
Xanthenes compounds are considered as one of the most important organic compounds due to their importance in the formation of biologically active compounds such as blood palette aggregation inhibitor drugs, calcium channel blocker, antibacterial, antiviral, anti-inflammatory, antihypertensive and antitumor. They are also very useful in laser technologies, fluorescent materials for visualization of biomolecules, dyes and photodynamic therapy.11 Moreover, several polycyclic compounds containing xanthene skeleton have been isolated from natural sources as plant species for their prospective bio-activity. Various methods for the formation of xanthenes derivatives have been reported in the literature.12,13 Numerous catalysts have been used in the preparation of xanthene, for example, Fe3O4-HAD-SO3H,14 sulfated starch,15 ZrO2,16 WOx/ZrO2 (ref. 17) and sulfamic acid/Cr-MIL-101.8 Most of the preparation methods suffer from some drawbacks such as constraints of catalyst reusability, separation of pure products, difficult workup, use of hazardous solvents and prolonged reaction times.
Recently; MOFs have received great attention in adsorption of heavy metal ions owing to unique structural features that are not found in other porous materials.8 In general, the elastic and extremely porous structures of MOFs facilitate the diffusion of the guest ions or molecules easily into the bulk structure. Also; both the size and shape of the formed pores help in increasing the selectivity of adsorbing definite ions or molecules. These distinct advantages make MOFs ideal sorbents in solid phase extraction of heavy metals.18 It was reported by the World Health Organization (WHO) in 2008 that the maximum contaminant level (MCL) of lead in drinking water was 10 μg L−1. Consequently, numerous techniques were applied for the removal of heavy metal ions from environmental samples, including membrane filtration,19 ion exchange20 and/or adsorption.21 Adsorption technology is preferred in comparison to the other traditional techniques due to its superior efficiency and easy application.22 Various adsorbents have been used such as activated carbon,23 zeolite,24 ion imprinted polymer25 and carbon nanotubes.26 However, the applications of these adsorbents have shown some weakness. Therefore some studies have been carried out with the intention of developing new, cheap and sustainable adsorbents for the removal of heavy metal ions from wastewaters. These adsorbents should possess high affinity, selectivity and capacity towards these metals.
Here, bimetallic Fe–Mg MOFs with different Fe:Mg molar ratios have been successfully synthesized via the solvothermal method. Fe–Mg MOFs was utilized for the removal of Pb(II), Cd(II) and Cu(II) from aqueous solution and as a solid Lewis acid catalyst for the synthesis of 14-aryl-14-H-dibenzo[a,j]xanthenes. The structure of the bimetallic Fe–Mg MOF was elucidated by (FT-IR), (XRD), (SEM) and (TEM) analyses.
2. Experimental
2.1. Materials
Mg(NO3)2·6H2O, Fe(NO3)3·9H2O, terephthalic acid and dimethylformamide (DMF) from Sigma Aldrich. Lead nitrate, copper nitrate and cadmium nitrate which provided by BDH. N-Butylamine, acetonitrile, ethyl acetoacetate, benzaldehyde, urea and 2-naphthol were used as received.
2.2. Preparation of MOFs
Fe–Mg bimetallic organic frameworks with different molar ratios of Fe3+:Mg2+, (0.6:1 and 1:1), were synthesized as reported previously.27 1 mmol of Mg(NO3)2·6H2O, Fe(NO3)3·9H2O (0.6 mmol or 1 mmol) and 1 mmol of terephthalic acid (H2BDC) were dissolved in 10 mL of DMF under constant stirring. The mixture was blended completely and heated at 120 °C in a Teflon-lined autoclave for 8 h. Then, the suspension was cooled gradually to room temperature, collected by centrifugation, washed with DMF several times and dried at 120 °C under vacuum. The resulted brown powder were denoted as (0.6:1) Fe–Mg MOF and (1:1) Fe–Mg MOF.
In the same manner, monometallic Fe-MOF and Mg-MOF were synthesized using only Fe(NO3)3·9H2O or Mg(NO3)2·6H2O as the metal precursor.
2.3. Instrumentation
The FTIR spectra were measured using Thermo SCIENTIFIC (NICOLET iS10) FTIR spectrometer from Thermo Electron Corporation in the range 400–4000 cm−1 for a sample of 2 mg diluted with 200 mg KBr. Powder X-ray diffraction was performed using PW150 (Philips) using Cu Kα radiation source and Ni filter. The instrument was operated at a voltage of 40 kV and a current of 45 mA. TEM (transmission electron microscopy) images were obtained using a JEOL-JEM-2100 transition-electron-microscope. SEM (scanning electron microscopy) and EDS (energy dispersive X-ray spectroscopy) images for surface morphological evaluation and composition were obtained using JEOL-JSM-6510LV scanning-electron-microscope, Japan. The porosity of the MOF materials and BET surface area are examined by N2 adsorption/desorption analysis at −196 °C.
2.4. Surface acidity measurements
The surface acidity of MOFs have achieved by nonaqueous titration of 0.03 g of MOF suspended in 10 mL acetonitrile with (0.025 N) n-butylamine at 0.005 mL min−1, which is a basic molecule suitable for the titrating strong acid and medium sites present on the surface of the catalysts.28 The various electrode potential values were measured using a double-junction electrode of an Orion 420 digital A model.
2.5. Catalytic activity
A mixture of (1 mmol) benzaldehyde, (2 mmol) 2-naphthol, and (0.05 g) MOF was placed in a round bottom flask in an oil bath and fitted to a condenser at 120 °C for 2 h for the synthesis of 14-aryl-14-H-dibenzo[a,j]xanthenes. The progress in the reaction was adjusted using TLC. After cooling, the reaction mixture was placed onto crushed ice and mixed for a few minutes. The product was filtrated, washed with cold water and after that recrystallized by using ethanol to yield the pure product which was calculated as follows:| |
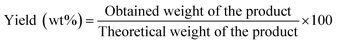 | (1) |
The obtained product (14-aryl-14-H-dibenzo[a,j]xanthenes, pale yellow solid) was characterized by m.p, 184–185 °C, and FT-IR (ν = 742, 805, 960, 1076, 1250, 1401, 1456, 1512, 1594, 1628, 2854, 2924, 3061 cm−1.
2.6. Batch adsorption experiment
A series of 150 mL Erlenmeyer flasks were filled with 100 mL of heavy metal ions. 0.03 g MOF was added to each conical flask and shaken in a thermostated shaker at 150 rpm. Then, 10 mL of the solution was collected through centrifugation at 4000 rpm for 10 min. The filtrate was collected in a polyethylene tube and the concentrations of Pb(II), Cu(II) and Cd(II) were determined by dilution. 1 mL from the filtered solution with 2 mL of 4,2-pyridylazo resorcinol (PAR) and 10 mL buffer (pH = 10) were prepared in measuring flask, then the volume was completed by distilled water to 25 mL, and Unicom 5625 UV/Vis Spectrophotometer, PerkinElmer USA was used for the determination of heavy metal ions concentration. The effect of the initial solution pH on metal ion removal was determined for pH values of 3–7 by the batch removal process described above. The pH was adjusted with 0.1 M HNO3 and 0.1 M NaOH to prepare acidic and neutral solutions, respectively. An adsorption kinetics study of the removal of Pb(II), Cu(II) and Cd(II) by MOF was carried out at the optimum conditions.
3. Results and discussion
3.1. Characterization
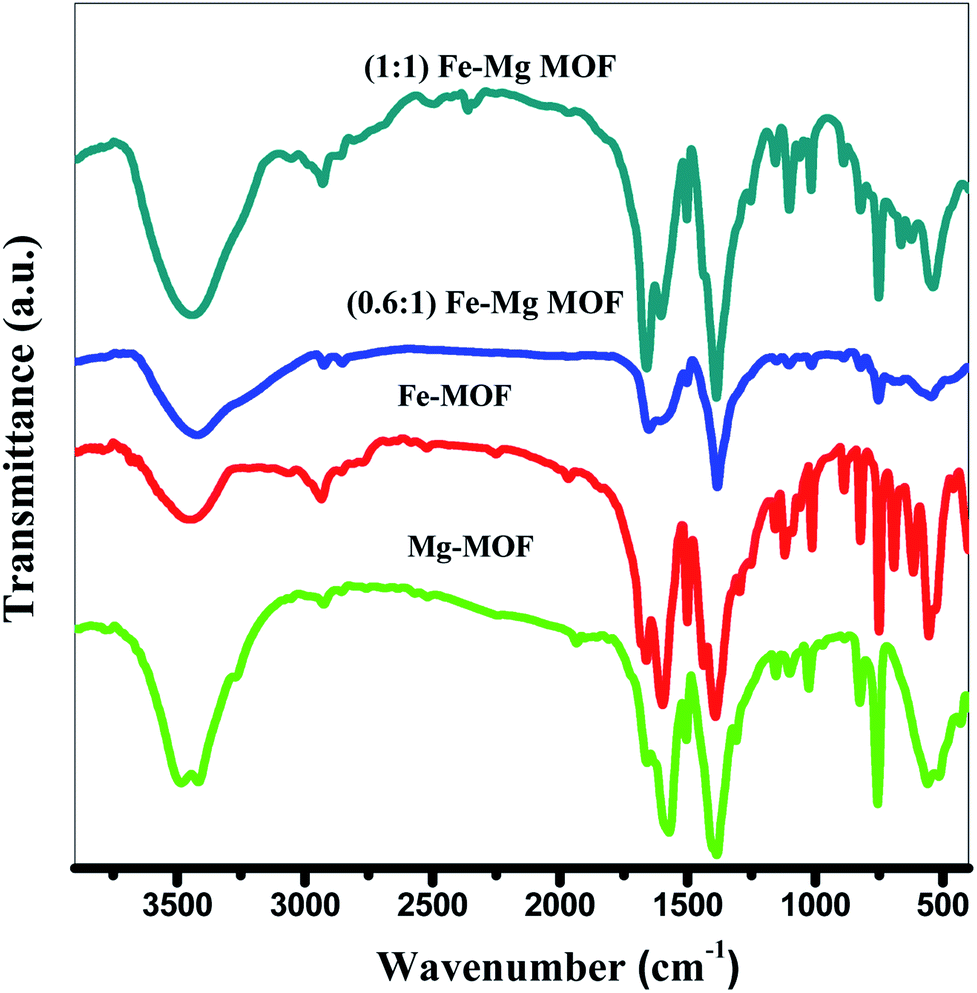
3.1.1. FT-IR analysis. The FTIR spectra of Fig. 1 display a main characteristic peak at ∼3450 cm−1 assigned for the H–O–H stretching vibrations. The peaks which are observed at 1598 cm−1 for Mg-MOF and Fe-MOF, 1646 cm−1 of (0.6:1) Fe–Mg MOF and 1665 cm−1 of (1:1) Fe–Mg MOF have been specified for the strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (–COOH) stretching vibration that may be ascribed to the existence of terephthalic acid in the MOF.29 The appeared peak ∼1390 cm−1 is due to the C–O stretching vibration. The three peaks at 1015, 1100 and 1145 cm−1 were attributed to the bending vibration of the hydroxyl group bound to metal ions (M − OH)29 and the substitution of the aromatic ring is appeared at 825 cm−1. These results suggest that the structure of Fe-MOF, Mg-MOF and Fe–Mg-MOF contains the basic terephthalic acid skeleton and the carboxyl groups of terephthalic acid are deprotonated. In addition, the bands at 758, 760 and 752 cm−1 were ascribed to metal–oxygen vibrations.30
O (–COOH) stretching vibration that may be ascribed to the existence of terephthalic acid in the MOF.29 The appeared peak ∼1390 cm−1 is due to the C–O stretching vibration. The three peaks at 1015, 1100 and 1145 cm−1 were attributed to the bending vibration of the hydroxyl group bound to metal ions (M − OH)29 and the substitution of the aromatic ring is appeared at 825 cm−1. These results suggest that the structure of Fe-MOF, Mg-MOF and Fe–Mg-MOF contains the basic terephthalic acid skeleton and the carboxyl groups of terephthalic acid are deprotonated. In addition, the bands at 758, 760 and 752 cm−1 were ascribed to metal–oxygen vibrations.30
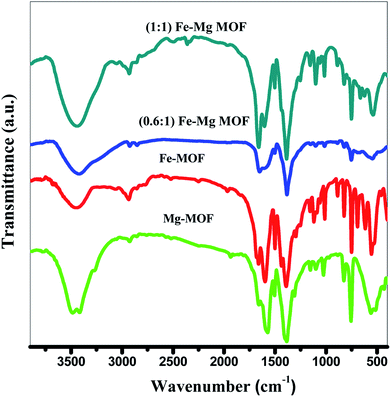 |
| | Fig. 1 FTIR spectra of the prepared MOFs. | |
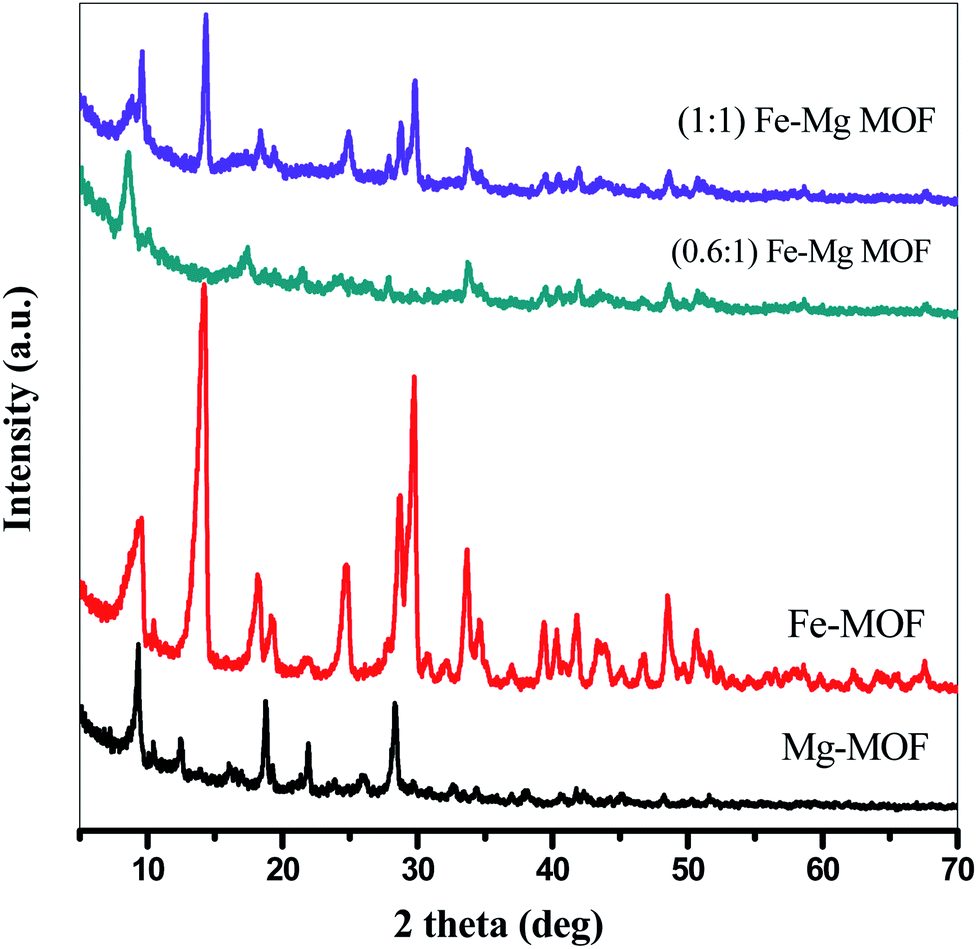
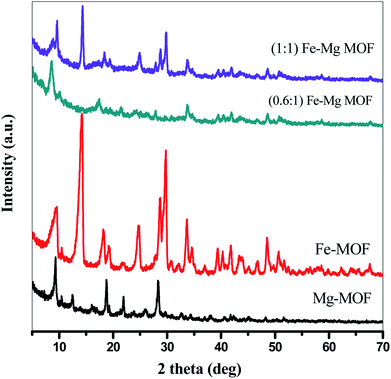
3.1.2. X-ray diffraction analysis. The crystallization behaviors of Fe-MOF, Mg-MOF and ((0.6:1) and (1:1)) Fe–Mg MOF are investigated using XRD analysis and were shown in Fig. 2. XRD patterns characteristic of Fe-MOF are recognized at 2θ = 9.59, 14.31, 18.18, 19.23, 21.85, 24.83, 28.76, 29.70, 33.78, 39.43, 40.34, 41.84, 43.74, 45.25, 46.88, 48.72 and 50.56° (ref. 31) and that of Mg-MOF are recorded at 2θ = 9.33, 12.52, 18.64, 23.68 and 28.24° (ref. 32) confirming the formation of metal–organic framework33,34 with well-developed crystallinity and without any oxide impurities. From the comparison of the low angle diffraction peaks of Fe–Mg MOF (0.6:1) and each of Mg-MOF and Fe-MOF, bimetallic MOF records slight shift toward lower 2θ and the disappearance of the diffraction peaks characteristic for Mg-MOF and Fe-MOF confirming the incorporation of Fe in the as-synthesized architectures. By increasing the Fe:Mg molar ratio to 1:1, the intensity of these XRD peaks increases indicated that the as-prepared Fe–Mg MOF showed the typical diffraction pattern of Fe–Mg MOF, which simultaneously possessed characteristic peaks belonging to Mg-MOF and Fe-MOF. This can be attributed to the high dispersion of Fe crystals on the outer cavities of Mg-MOF surface and inside the pores.35,36 In addition, the observed simultaneous shifts of the XRD patterns to lower and higher angles may be ascribed to the flexibility and/or the breathing effect of Fe-MOF structure.37–39 By increasing the ratio of Fe:Mg to 1:1, the Fe species aggregated on the surface of Mg-MOF which in role decreases the synergistic effect between the two metals and so the intensity of the diffraction peaks characteristic of both Fe and Mg is increased. By applying Scherer eqn (2) for the first peak (111), the crystal size can be easily determined. As seen from the results that tabled in Table 1, the crystal size is decreased by increasing the ratio of Fe:Mg to 0.6:1 then begins to slightly increase by increasing the ratio to 1:1 confirming the effect of Fe species on Mg-MOF.| |
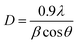 | (2) |
where λ is radiation wavelength (λ = 1.54 Å), D is crystal size, β is the line breadth (radians) and θ = angle of reflection.
 |
| | Fig. 2 XRD of the prepared MOFs. | |
Table 1 Crystallite size, surface area acidity and activity of the prepared MOFs
| Sample name |
Crystallite size (nm) |
SBET (m2 g−1) |
Ei (mV) |
No. of acid sites/g × 10−19 |
Yield (%) of 14-aryl-14-H-dibenzo[a,j]xanthenes |
| From XRD peak (111) |
From TEM |
| Mg-MOF |
10.6 |
11.0–22.3 |
774 |
116.2 |
16.9 |
38.4 |
| Fe-MOF |
12.1 |
8.0–9.6 |
983 |
137.3 |
28.9 |
41.4 |
| (0.6:1) Fe–Mg MOF |
4.5 |
3.1–3.9 |
1320 |
273.1 |
63.7 |
93.5 |
| (1:1) Fe–Mg MOF |
6.1 |
6.5–10.9 |
1162 |
180.5 |
34.9 |
72.8 |
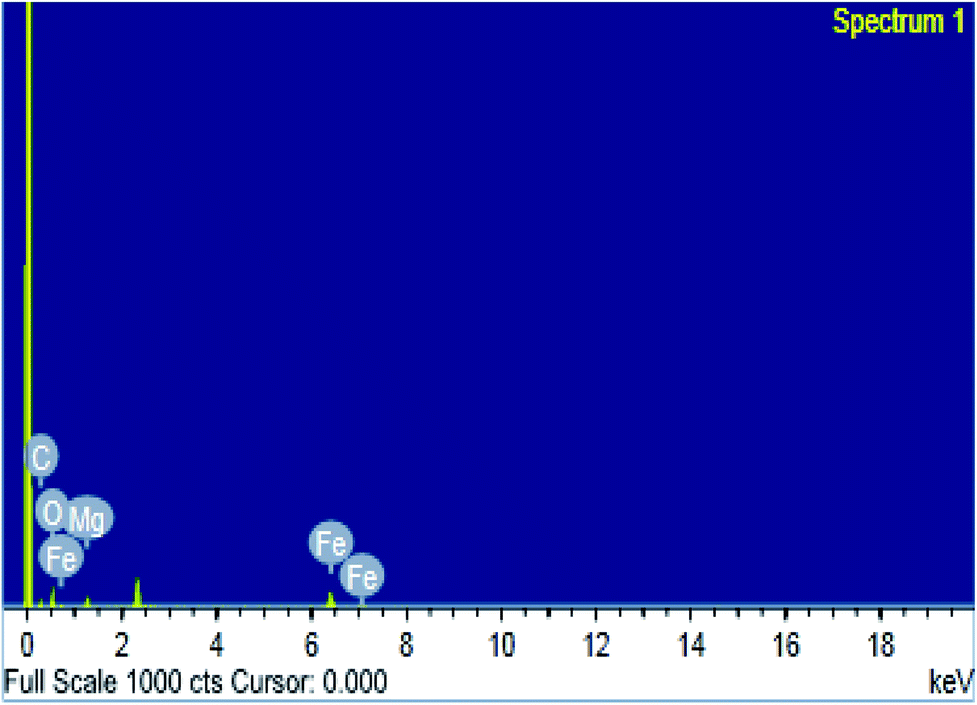
3.1.3. SEM, TEM and EDS studies. The surface morphologies of MOF samples were examined by photographing the surface using scanning and transmission electron microscopes. The images of Mg-MOF and Fe-MOF are shown in Fig. 3A(a, b) and B(a*, b*). As shown from the figures the crystals of Fe-MOF are shaped as a rigid zeotype octahedral structure while that of Mg-MOF appeared as column-like with irregular sizes.40,41 Fig. 3A(c, d) and B(c*, d*) showed the SEM and TEM images of bimetallic Fe–Mg MOF. In the SEM images; the aggregation of Fe–Mg MOF crystals became more visible which shows a clear differentiation between both single and bimetallic MOF crystals. Fe-MOF with a diameter of 8.0–9.6 nm (from TEM measurements, Table 1) is firstly dispersed in the pores of Mg-MOF with diameter 11.0–22.3 nm then on the external surface. However, in TEM images of (1:1) Fe–Mg MOF; the observed larger dark spots on the Mg-MOF surface have resulted from the formation of aggregates of Fe-MOF nanocrystals. Similar agglomerations about bimetallic MOFs were observed in other literature.42,43 The SEM and TEM images are in good agreement with the XRD results in which for (0.6:1) Fe–Mg MOF the peak corresponding to Fe-MOF was found to disappear and on increasing Fe, large aggregates were formed on Mg surface. The EDS spectrum (Fig. 4) of (0.6:1) Fe–Mg MOF confirms the presence of both Fe and Mg in the bimetallic MOF.
 |
| | Fig. 3 (A) SEM images of Mg-MOF (a), Fe-MOF (b), (0.6:1) Fe–Mg MOF (c) and (1:1) Fe–Mg MOF (d). (B) TEM images of Mg-MOF (a*), Fe-MOF (b*), (0.6:1) Fe–Mg MOF (c*) and (1:1) Fe–Mg MOF (d*). | |
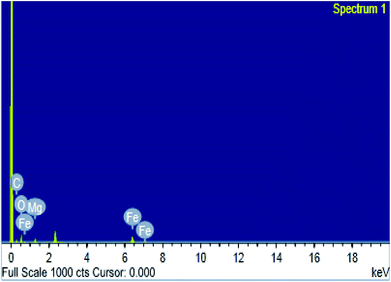 |
| | Fig. 4 EDS image of (0.6:1) Fe–Mg MOF. | |
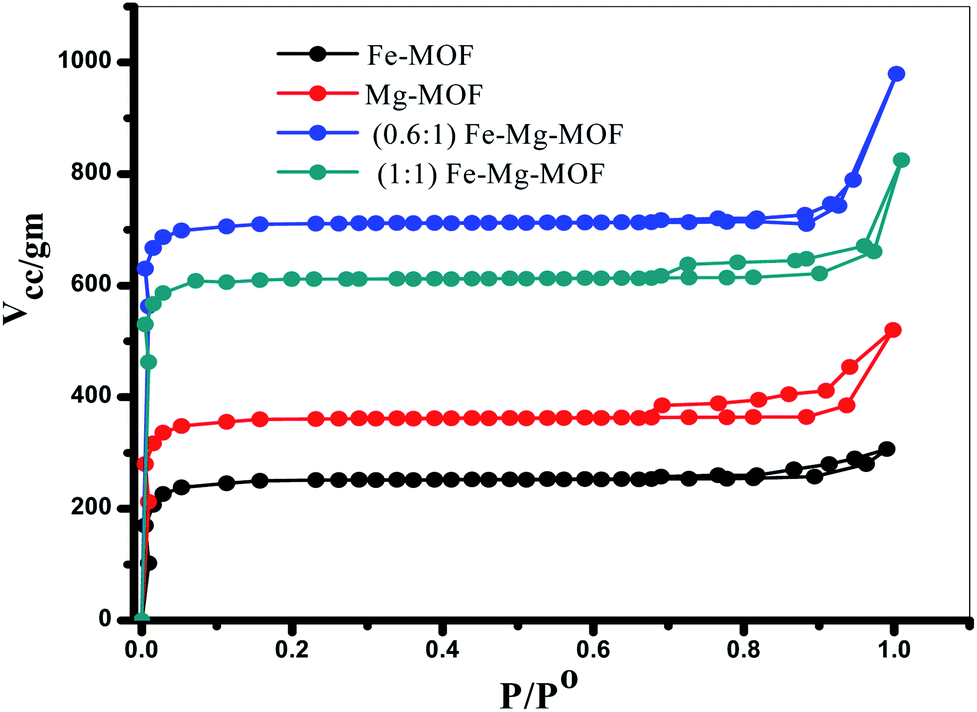
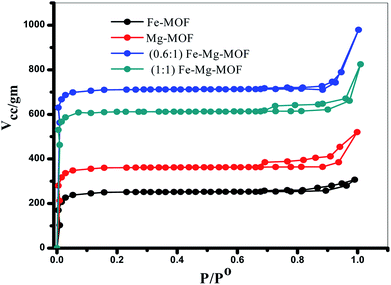
3.1.4. SBET measurements. The porosity of the MOF materials and BET surface area are examined by N2 adsorption/desorption analysis at −196 °C. The isotherms, Fig. 5, reveal that all the prepared MOFs show type I isotherms typical of microporous structure. The SBET of Fe-MOF, Mg-MOF, (0.6:1) Fe–Mg MOF and (1:1) Fe–Mg MOF are 774, 983, 1320 and 1162 m2 g−1, respectively. As illustrated the bimetallic MOFs exhibit higher surface area than the both of monometallic MOFs. This may be ascribed to the creation of new micropores and consequently new active sites by the presence of both two metals Fe and Mg together in the bimetallic MOF.1 The surface area of (0.6:1) Fe–Mg MOF is high compared with that of (1:1) Fe–Mg MOF. This can be explained, as discussed above, due to the synergistic effect between Fe and Mg metals and the high dispersion of Fe crystals inside the pores and on the outer cavities of Mg-MOF surface.35,36 This synergistic effect decreases in (1:1) Fe–Mg MOF material due to the aggregation of Fe species on the surface of Mg-MOF. The BET results confirm the results of XRD, TEM and SEM analysis.
 |
| | Fig. 5 N2 adsorption–desorption isotherms of the prepared MOFs. | |
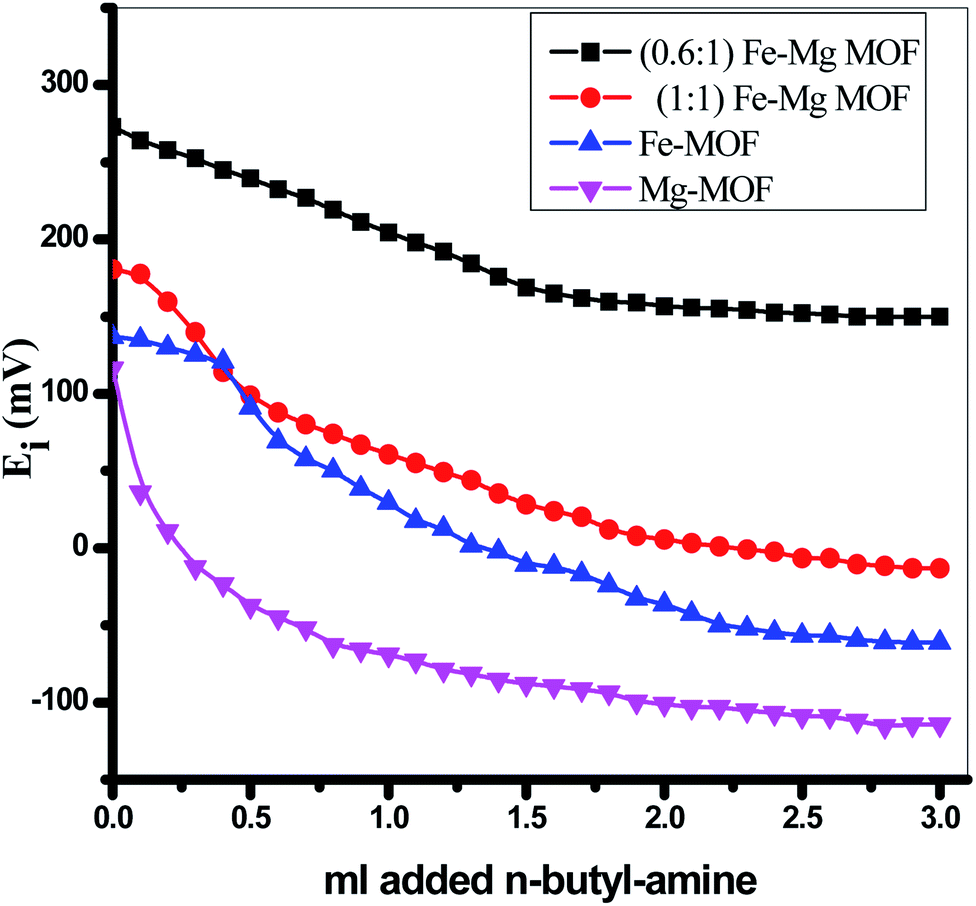
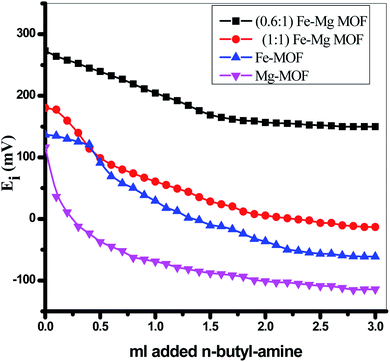
3.2. Surface acidity
The surface acidity of the MOFs was determined using a potentiometric titration method using a basic molecule as n-butylamine. The initial electrode potential (Ei) obtained from this technique displays the maximum acid strength of the catalyst surface while the overall issue of acid sites per gram solid catalyst (meq/g) has been obtained from the domain where a plateau is extended. The acidity measurements resulted from the potentiometric titration curves, Fig. 6 of MOFs catalysts were recorded in Table 1. The following equation successfully used to calculate the overall issue of acid sites/gram catalyst:| |
 | (3) |
where N is Avogadro's number.
 |
| | Fig. 6 Nonaqueous potentiometric titration for the prepared MOFs. | |
It was proposed that the sites with Ei > 100 mV are very strong sites, the sites with 0 < Ei < 100 mV are strong sites, the sites with −100 < Ei < 0 are weak sites and the sites with Ei < −100 mV are very weak sites.28,44,45 From Table 1 and Fig. 6 and according to the above classification, both pure Mg-MOF and pure Fe-MOF samples exhibit strong acid sites, Ei = 116.2 and 137.3 mV, respectively. For bimetallic MOF, the incorporation of Fe-MOF into Mg-MOF increases the surface acidity and the acid strength (Ei = 273.1 mV) for the sample (0.6:1) Fe–Mg MOF as a result of the high surface area and high synergistic effect between Fe and Mg metals due to the dispersion of Fe-MOF particles on the surface and within the pores of Mg-MOF as evident from XRD, TEM and SBET analysis. The decreases in the surface acidity and the acid strength of (1:1) Fe–Mg MOF, (Ei = 180.5 mV) may be related to the decrease in the surface area as a result of the aggregation Fe-MOF species and formation of multilayer on the Mg surface which in role decreases the synergistic effect between the two metals as mentioned above in XRD, SEM, TEM and SBET results.
3.3. Catalytic activity
3.3.1. Synthesis of 14-aryl-14-H-dibenzo[a,j]xanthenes. 14-aryl-14-H-dibenzo[a,j]xanthenes can be synthesized using benzaldehyde and 2-naphthol in presence of the prepared MOFs. The yield of xanthenes in the presence of pure Mg-MOF crystals was found to be 38.4% while in presence of Fe-MOF was found to be 41.4%, the yield was significantly increased over bimetallic MOF as shown in Fig. 7. The maximum yield (93.5%) was obtained for (0.6:1) Fe–Mg MOF catalyst which has the maximum number of acid sites. The yield is improved remarkably using bimetallic system than monometallic ones in a good agreement with the surface acidity and the total number of acid sites. This can be ascribed to a synergetic interaction between Fe and Mg in Fe–Mg MOF which plays an important role in catalytic reactions.35,46–48 The decrease in the catalytic activity of the sample (1:1) Fe–Mg MOF was due to the agglomeration of Fe nanoparticles on the surface of Mg which decrease acidity and activity, Fig. 7. The reaction can occur via Fe and Mg metals which considered as a Lewis acid catalyst which activates the carbonyl group of the benzaldehyde. Then, a fast Knoevenagel addition was occurred by condensation of one molecule of β-naphthol to provide an intermediate followed by Michael-type addition of the active methylene of the second molecule of β-naphthol to the intermediate followed by cyclization and hydrolysis afforded the final product 14-aryl-14-H-dibenzo[a,j]xanthenes.49–51
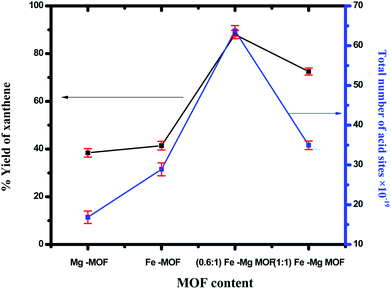 |
| | Fig. 7 Effect of the total number of acid sites of the prepared MOF on synthesis of 14-aryl-14-H-dibenzo[a,j]xanthenes. The error bars represent the standard error. | |
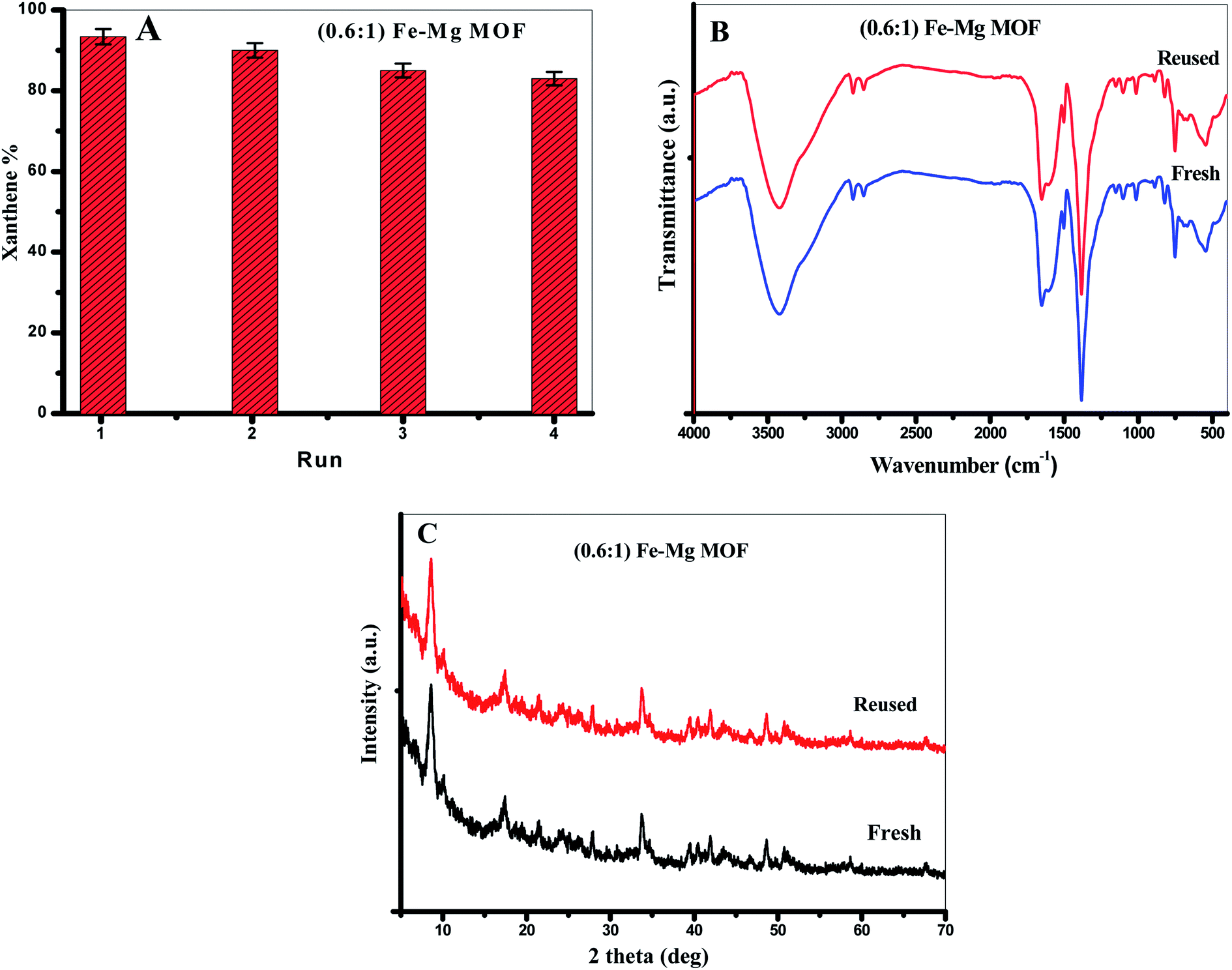
3.3.2. Reusability of the MOF. From the viewpoint of green chemistry, we tried hard to design a green and reused catalyst. We performed the reusing test of the MOF in the model reaction between benzaldehyde (1 mmol) and 2-naphthol (2 mmol) in the presence of 0.03 g of (0.6:1) Fe–Mg MOF at 120 °C. After each run, the MOF was separated, washed with ethanol and dried at 120 °C. From Fig. 8A, (0.6:1) Fe–Mg MOF can be recovered and reused in the xanthenes formation reaction four times with remarkably similar activity proving that Fe has been effectively incorporated in the framework of Mg MOF. The FTIR and XRD analyses were determined for the fresh and reused (0.6:1) Fe–Mg MOF as shown in Fig. 8B and C. To detect any leached amount of Fe or Mg ions during the reaction, the leaching of the catalysts was tested by taking a small amount of the filtrate after centrifugation during recycling and analyzing it by the atomic absorption spectroscopy. No traces are found from both metals in the filtrate which indicate no leaching process is occurred during the reaction.
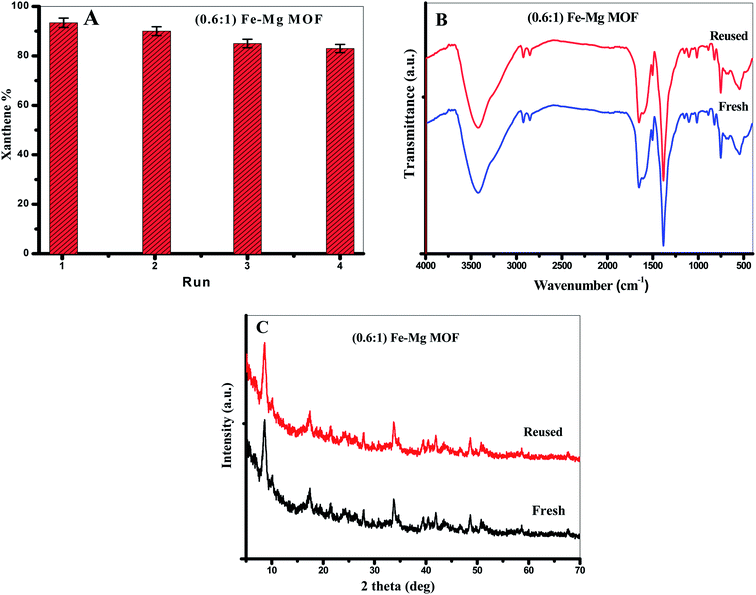 |
| | Fig. 8 (A) The reusability of (0.61) Fe–Mg MOF in the synthesis of xanthenes, (B) XRD and (C) FTIR of the fresh and reused (0.61) Fe–Mg MOF. The error bars represent the standard error. | |
Previously reported synthesis of pharmaceutically 14-aryl-14-H-dibenzo[a,j]xanthenes compared with our catalysts showed that the efficiency of Fe–Mg MOF was so higher than that of several reported catalysts e.g. nano-ZnAl2O4,52 acid activated clay in Kerman Province,53 graphene oxide-copper ferrite nanocomposite,54 palladium nanoparticles supported on amino-functionalized MOFs and zinc oxide nanoparticles.55 The comparison study not only depended on the percentage yield of the desired product but also the condition required optimizing the reaction.
3.4. Adsorption of Pb(II), Cd(II) and Cu(II) ions by Mg, Fe and Fe–Mg MOFs
Pb(II), Cd(II) and Cu(II) ions present in positive forms; therefore, can easily undergo favorable electrostatic interactions with the MOF adsorbents having negatively charged framework (surface). Mg, Fe and Fe–Mg MOF were successfully used to adsorb Pb(II), Cd(II) and Cu(II) ions which are different in ionic radius. The individual influences of solution pH, adsorption time, initial cations concentration and adsorbent dose on the uptake efficiency of Pb(II), Cd(II) and Cu(II) ions by MOF have been studied to detect the optimum range of every single operating factor. In order to achieve this, we noticed the influence resulted by changes in a single parameter while the other operating parameters were maintained constant.
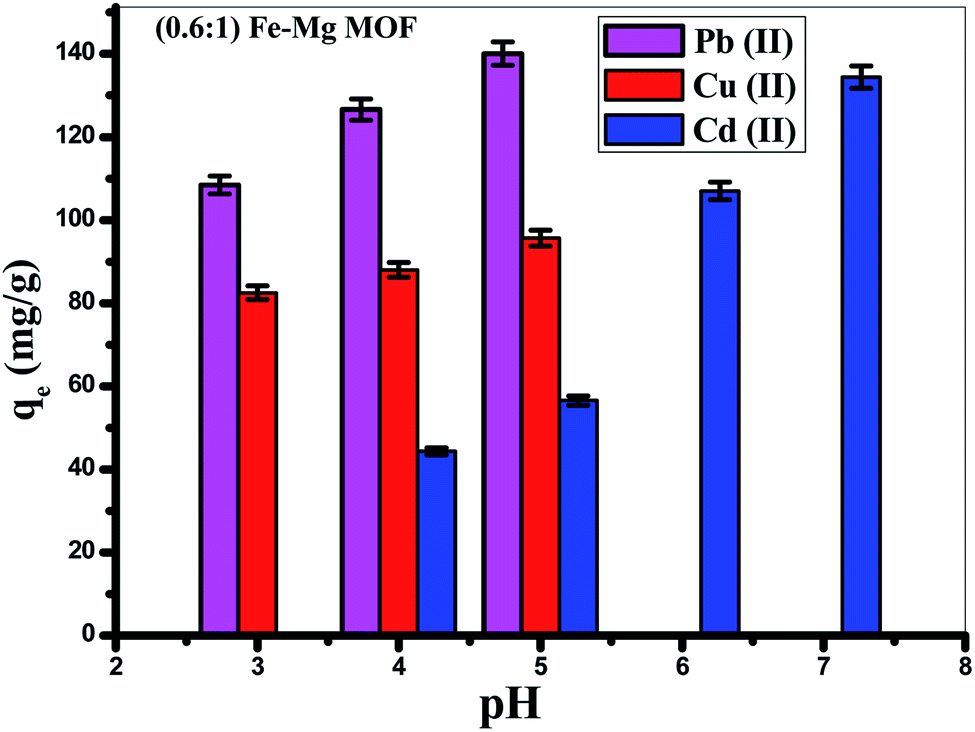
3.4.2. Effect of pH. The solution pH is an important variable influencing the acid–base property of the adsorbent surface. Fig. 9 showed that the adsorption of Pb(II) is increased from (108.4 mg g−1) at pH = 3 to reach maximum (140.6 mg g−1) at pH 5, but the removal of Cd(II) is increased from (44.3 mg g−1) at pH 4 to reach maximum around pH 7 (134.4 mg g−1), while, the removal of Cu(II) is increased from 82.5 mg g−1 at pH = 3 to reach 95.6 mg g−1 at pH = 5. Thus, the optimum pH is achieved at pH = 5 for Pb(II), Cu(II) and 7 for the removal of Cd(II).59 At low pH (pH less than 3), MOF offered a positive surface charge, since functional groups of the surface were protonated,60 which gives rise to electrostatic repulsion among MOF and Pb(II), Cu(II) and Cd(II) resulting in low cationic metal ions removal. When the pH of the solution is increased, a negative surface charge appeared on the MOF surface because of the deprotonating of function groups which was favorable for the surface electrostatic attraction of positively charged Pb(II), Cu(II) and Cd(II) found in solution which make the ion exchange process occurred. In acidic solution, hydrated hydrogen ions (H3O+) competed with Pb(II), Cd(II) and Cu(II) on the available adsorption sites. As the pH increases, the concentration of H3O+ decreases resulting in increasing the adsorption rate of Pb(II), Cd(II) and Cu(II). Moreover, increasing the adsorption capacity with increasing the pH is due to the greater stability of MOF structure in addition to the increase of the negatively charged active centers. Furthermore, considering precipitation of both (Pb(OH)+ and Pb(OH)2), (Cu(OH)+ and Cu(OH)2) that appear at pH > 5 and precipitation of both (Cd(OH)+ and Cd(OH)2) that seem at pH > 7.
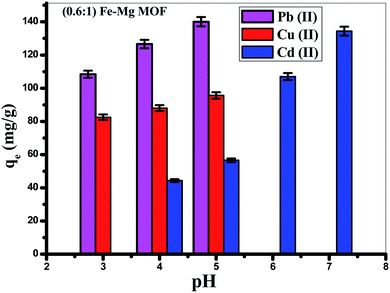 |
| | Fig. 9 Effect of pH on the removal of Pb(II), Cd(II) and Cu(II) ions by (0.6:1) Fe–Mg MOF sample. The error bars represent the standard error. | |
3.4.3. Adsorbent dose. The effect of adsorbents weight is one of the most significant factors that affect the adsorption. In this test different amounts of MOFs (10–70 mg) were added to 50 mL solution with concentration of (100 mg L−1) and appropriate pH, then shacked for 2 h at 25 °C and by UV/Vis spectrophotometer, the adsorption of Pb(II), Cd(II) and Cu(II) were evaluated. Fig. S2,† shows the adsorption amount as a function of the mass of MOFs. The experimental results demonstrated that the adsorption is increased with the increase in the weight of MOFs catalyst from (10–50 mg) and then almost constant. This is may be due to the increase of the number of the deprotonating function groups on the solid MOF by increasing its weight up to 50 mg which was favorable for the surface electrostatic attraction of positively charged cations.61–63 The almost constant adsorption after 50 mg of MOF may be attributed to the saturation of the surface active sites available for the adsorbed cations.
3.4.4. Effect of the initial concentration of the heavy metal ions. The adsorption isotherms of the Pb(II), Cd(II) and Cu(II) by MOFs were studied in a series of batches using various initial concentrations ranging from (50–200 mg L−1) at 25 °C. From Fig. S3,† It is clear that the adsorption increases with the increase in the initial concentrations of the heavy metal ions up to 150 mg L−1 then almost steady constant. This can be ascribed to the saturation of most available active sites of MOFs with raising the initial concentration of Pb(II), Cd(II) and Cu(II).59 Cations adsorption efficiency were significantly higher for initial concentration up to 150 mg L−1 due to plenty of freely active sites on the adsorbent surface which has been saturated at extremely higher cation concentrations and as a result, the adsorption slowed increases due to the competition between ions for the available binding sites.8,57
3.4.5. Adsorption isotherms. For a solid–liquid system, one of the most important physico-chemical studies in the characterization of sorption behavior is the equilibrium isotherm of sorption which used to characterize the experimental adsorption data. The Langmuir isotherm is the most applied adsorption model and aspects monolayer sorption behavior on the adsorbent surface which contains a homogeneous and definite number of active sites.64 The model generally expressed in the well-known form represented in the following equation as:| |
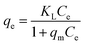 | (5) |
A linear form of this model expression is:
| |
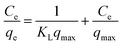 | (6) |
where
Ce (mg L
−1) is the equilibrium concentration of the adsorbed molecule;
qe (mg g
−1) is the adsorption capacity of the adsorbent at equilibrium per unit mass of adsorbent;
qmax (mg g
−1) indicates the theoretical sorption capacity and
KL in L mg
−1 is the Langmuir sorption isotherm constant. One of the most important descriptions of the Langmuir isotherm equation is the dimensionless separation factor (
RL), represented in the equation as:
65| |
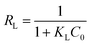 | (7) |
where
KL in L mg
−1 is the Langmuir sorption constant and
C0 in mg L
−1 is the initial concentration of the adsorbed molecules. The
RL values describe the isotherm type to be either irreversible (
RL = 0), linear (
RL = 1), unfavorable (
RL > 1) or favorable (0 <
RL < 1). On the other hand, Freundlich isotherm model is represented by the well-known logarithmic form which expressed in the following equation:
66,67
A linear form of this expression can be represented as follows:
| | |
lnqe = (1/n)lnCe + lnKF
| (9) |
where
n and
KF are the Freundlich isotherm exponents.
KF describes the adsorption or distribution coefficient giving an indication of the definite quantity of the adsorbed molecule on an adsorbent surface at equilibrium while
n indicates the favorability of the adsorption isotherm and measure the surface heterogeneity or adsorption intensity. The process is more heterogeneous when the value of 1/
n gets nearer to zero. A value of 1/
n lower than unity is indicative of Langmuir isotherm while 1/
n above unity indicating of co-operative sorption.
68 Langmuir and Freundlich sorption isotherms for Pb(
II), Cd(
II) and Cu(
II) using (0.6
:
1) Fe–Mg MOF as adsorbent are seen in Fig. S4.
† The correlation factor coefficients,
R2 and isotherm parameters are calculated and recorded at
Table 2. The Langmuir model with
R2 factor ranging from 0.9980 to 9982 for Pb(
II), 0.981 to 0.998 for Cd(
II) and 0.985 to 0.999 for Cu(
II), respectively, represents the accurate fit of the experimental data and more acceptable than that of Freundlich isotherm. It gives an indication of the monolayer adsorption of Pb(
II), Cd(
II) and Cu(
II) ions on the homogeneous surface of Fe–Mg MOF adsorbent. The maximum monolayer capacity obtained from Langmuir model for the uptake of Pb(
II), Cd(
II) and Cu(
II) is found to be 196, 191 and 175 mg g
−1, respectively. Moreover, the dimensionless separation constant
RL values between 0.033–0.213, 0.117–0.293 and 0.020–0.227 for of Pb(
II), Cd(
II) and Cu(
II) respectively indicate the favorability of the sorption process for the three cations, also the value of 1/
n lower than unity is indicative of Langmuir isotherm as shown in
Table 2. Moreover, it is obviously from the adsorption figures and
Table 2 that the (0.6
:
1) Fe–Mg MOF material show the better adsorption efficiency among the other prepared MOFs. This can be explained on the basis of increasing surface area due to the synergistic effect between the two metals and creation of new active sites that facilitate and improve the adsorption efficiency in this sample.
Table 2 Langmuir and Freundlich parameter for adsorption of heavy metals on the prepared MOFs
| Sample |
Heavy metal ions |
Langmuir isotherm |
Freundlich isotherm |
| qmax (mg g−1) |
KL (L mg−1) |
R2 |
RL |
KF (mg g−1) |
n |
R2 |
| Mg-MOF |
Pb(II) |
158 |
0.237 |
0.97243 |
0.078 |
1.61 |
2.32 |
0.96677 |
| Fe-MOF |
179 |
0.074 |
0.95238 |
0.213 |
2.88 |
2.27 |
0.90465 |
| 0.6:1 Fe–Mg MOF |
196 |
0.582 |
0.99980 |
0.033 |
4.28 |
5.37 |
0.98474 |
| 1:1 Fe–Mg MOF |
186 |
0.282 |
0.99741 |
0.066 |
3.98 |
3.70 |
0.97055 |
| Mg-MOF |
Cd(II) |
130 |
0.048 |
0.98339 |
0.293 |
1.36 |
2.09 |
0.96358 |
| Fe-MOF |
163 |
0.103 |
0.96483 |
0.163 |
3.78 |
2.17 |
0.96402 |
| 0.6:1 Fe–Mg MOF |
191 |
0.151 |
0.97059 |
0.117 |
3.85 |
4.32 |
0.91991 |
| 1:1 Fe–Mg MOF |
185 |
0.145 |
0.94089 |
0.121 |
3.73 |
2.15 |
0.93427 |
| Mg-MOF |
Cu(II) |
119 |
0.078 |
0.96236 |
0.204 |
3.34 |
1.02 |
0.92998 |
| Fe-MOF |
161 |
0.290 |
0.97734 |
0.065 |
3.43 |
1.03 |
0.97205 |
| 0.6:1 Fe–Mg MOF |
175 |
0.079 |
0.97512 |
0.020 |
4.47 |
2.57 |
0.96631 |
| 1:1 Fe–Mg MOF |
172 |
0.068 |
0.95824 |
0.227 |
3.89 |
2.04 |
0.94908 |
3.4.7. Mechanism of adsorption. Adsorption mechanism is generally described through three stages are as follows:(i) Movement of adsorbate molecules towards the external surface of the adsorbent from the bulk of the solution (film diffusion).
(ii) Particle diffusion, which involves the transfer of ions to the intra particular active sites from the surface.
(iii) Finally adsorption of ions by the contact with active sites in the adsorbent.
The third step does not consider the rate which controls steps because it is a very rapid, consequently the rate which controls steps mainly either particle diffusion or film diffusion.
Weber and Morris who first suggested the model of intraparticle diffusion, which summarized that the adsorption was proportional to the square root of the contact time69 as demonstrated from the following equation:
3.4.8. Boyd model. To differentiate between particle diffusion and film diffusion in order to identify the slowest step in the process of adsorption kinetic equation Boyd was utilized,70 which is represented as:| |
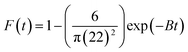 | (13) |
Both intraparticle diffusion and Boyd plots are applied in order to predict the rate limiting step. The lines don't pass through the origin in intraparticle diffusion plot Fig. S5A,† subsequently; the rate limiting step is not only intraparticle diffusion and indicates the effect of film diffusion (boundary layer diffusion). It is a controlled adsorption process for all samples by film diffusion, due to the plots of Boyd are linear but do not pass through the origin the parameters of intraparticle diffusion and Boyd plot for adsorption heavy metals Pb(II), Cd(II) and Cu(II) ions onto investigated catalysts are displayed in Fig. S5B.†
4. Conclusion
This paper offers an Fe–Mg MOF with various advantages such as being easily prepared in larger amounts, high catalytic activity, straight forward synthetic route under mild conditions, simple handling and an easy recovery without losing its activity which makes it an eco-friendly, green and efficient catalyst. Mg-MOF, Fe-MOF and Fe–Mg-MOF were prepared successfully, characterized using sets of different techniques e.g. XRD, FT-IR, SEM, EDS analysis and TEM. The formation of a bimetallic MOF sharply increased the surface acidity and the catalytic activity. The catalytic activity of the formation of 14-aryl-14-H-dibenzo[a,j]xanthenes was related to the acidity of the catalysts. The prepared MOFs were used for removal of Pb(II), Cd(II) and Cu(II) ions from their aqueous solutions. The effect of the experimental parameters such as pH, initial concentration, contact time and MOF weight on the adsorption of Pb(II), Cd(II) and Cu(II) were studied. Both Langmuir and Freundlich isotherm models were used to analyze the equilibrium.
Conflicts of interest
There are no conflicts to declare.
References
- W. S. Abo El-Yazeed and A. I. Ahmed, RSC Adv., 2019, 9, 18803–18813 RSC.
- J. Zhang, Z. Xiong, C. Li and C. Wu, J. Mol. Liq., 2016, 221, 43–50 CrossRef CAS.
- I. Ahmed and S. H. Jhung, Mater. Today, 2014, 17, 136–146 CrossRef CAS.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- J. J-Alcaniz, R. Gielisse, A. B. Lago, E. V. R-Fernandez, P. S-Crespo, T. Devic, N. Guillou, C. Serre, F. Kapteijn and J. Gascon, Catal. Sci. Technol., 2013, 3, 2311–2318 RSC.
- S. H. Jhung, N. A. Khan and Z. Hasan, CrystEngComm, 2012, 14, 7099–7109 RSC.
- M. O. Rodrigues, M. V. De Paula, K. A. Wanderley, I. B. Vasconcelos, S. Alves and T. A. Soares, Int. J. Quantum Chem., 2012, 112, 3346–3355 CrossRef CAS.
- S. A. El-Hakam, S. E. Samra, S. M. El-Dafrawy, A. A. Ibrahim, R. S. Salama and A. I. Ahmed, RSC Adv., 2018, 8, 20517–20533 RSC.
- X.-F. Wang, X.-Z. Song, K.-M. Sun, L. Cheng and W. Ma, Polyhedron, 2018, 152, 155–163 CrossRef CAS.
- J. A. Villajos, G. Orcajo, C. Martos, J. A. Botas, J. Villacanas and G. Calleja, Int. J. Hydrogen Energy, 2015, 40, 5346–5352 CrossRef CAS.
- P. Iniyavan, S. Sarveswari and V. Vijayakumar, Tetrahedron Lett., 2015, 56, 1401–1406 CrossRef CAS.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng and M. Grätzel, Energy Environ. Sci., 2016, 9, 873–877 RSC.
- G. Shen, L. Zhao, Y. Wang, W. Xia, M. Yang and T. Zhang, RSC Adv., 2016, 6, 84748–84751 RSC.
- H. Naeimi and Z. Ansarian, J. Taiwan Inst. Chem. Eng., 2018, 1–8 Search PubMed.
- J. Safari, P. Aftabi, M. Ahmadzadeh, M. Sadeghi and Z. Zarnegar, J. Mol. Struct., 2017, 114215, 33–39 CrossRef.
- P. Bansal, N. Kaur, C. Prakash and G. R. Chaudhary, Vacuum, 2018, 157, 9–16 CrossRef CAS.
- V. S. R. Ganga, M. K. Choudhary, R. Tak, P. Kumari, S. H. R. Abdi, R. I. Kureshy and N. H. Khan, Catal. Commun., 2017, 945, 5–8 CrossRef.
- S. H. Huo and X. P. Yan, Analyst, 2012, 137(15), 3445–3451 RSC.
- Y. G. Abou El-Reash, A. M. Abdelghany and A. Abd Elrazak, Int. J. Biol. Macromol., 2016, 86, 789–798 CrossRef CAS PubMed.
- Y. Ibrahim, E. Abdulkarem, V. Naddeo, F. Banat and S. W. Hasan, Sci. Total Environ., 2019, 69010, 167–180 CrossRef PubMed.
- A. M. Youssef, A. I. Ahmed, M. I. Amin and U. A. El-Banna, Desalin. Water Treat., 2015, 54, 1694–1707 CAS.
- M. I. Nandasiri, S. R. Jambovane, B. P. McGrail, H. T. Schaef and S. K. Nune, Chem. Rev., 2016, 311, 38–52 CAS.
- C. R. Aguiar, É. Fontana, J. A. Valle, A. A. Souza, A. F. Morgado and S. Souza, Can. J. Chem. Eng., 2016, 94, 947–955 CrossRef CAS.
- A. M. Anielak and R. Schmidt, Pol. J. Environ. Stud., 2011, 20, 15–19 CAS.
- M. Monier, D. A. Abdel-Latif and Y. G. Abou El-Reash, J. Colloid Interface Sci., 2016, 469, 344–354 CrossRef CAS PubMed.
- N. V. P. Aguilar, E. M. Sandoval and J. R. R. Mendez, J. Nanopart. Res., 2010, 12, 467–480 CrossRef.
- Y. Gu, D. Xie, Y. Wang, W. Qin, H. Zhang, G. Wang, Y. Zhang and H. Zhao, Chem. Eng. J., 2019, 357, 579–588 CrossRef CAS.
- A. I. Ahmed, S. A. El-Hakam, A. S. Khder and W. S. Abo El-Yazeed, J. Mol. Catal. A: Chem., 2013, 366, 99–108 CrossRef CAS.
- Z. Zhang, Z. Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 RSC.
- Z. Li, S. Deng, G. Yu, J. Huang and V. C. Lim, Chem. Eng. J., 2010, 161, 106–113 CrossRef CAS.
- Z. Zhang, X. Li, B. Liu, Q. Zhao and G. Chen, RSC Adv., 2016, 6, 4289–4295 RSC.
- Q. G. Zhai, X. Bu, C. Mao, X. Zhao and P. Feng, J. Am. Chem. Soc., 2016, 138, 2524–2527 CrossRef CAS PubMed.
- M. N. Cele, H. B. Friedrich and M. D. Bala, Catal. Commun., 2014, 57, 99–102 CrossRef CAS.
- X. Yi, W. Dong, X. Zhang, J. Xie and Y. Huangm, Anal. Bioanal. Chem., 2016, 408, 8805–8812 CrossRef CAS PubMed.
- W. Yu, Y. P. Hsu and C. S. Tan, Appl. Catal., B, 2016, 196, 185–192 CrossRef CAS.
- Y. Miao, S. Li, L. Gao and G. Xiao, Mol. Catal., 2017, 436, 128–137 CrossRef.
- M. H. Pham, G. T. Vuong, A. T. Vu and T. O. Do, Langmuir, 2011, 27, 15261–15267 CrossRef CAS PubMed.
- M. Ma, A. Betard, I. Weber, N. S. Al-Hokbany, R. A. Fischer and N. N. Metzler, Cryst. Growth Des., 2013, 13, 2286–2291 CrossRef CAS.
- A. K. Singh and A. K. Singh, Spectrochim. Acta, Part A, 2012, 96, 986–991 CrossRef CAS PubMed.
- J. Tang, M. Yang, M. Yang, J. Wang, W. Dong and G. Wang, New J. Chem., 2015, 39, 4919–4923 RSC.
- L. Yang, Y. Bai, H. Zhang, J. Geng, Z. Shao and B. Yia, RSC Adv., 2017, 7, 22610–22618 RSC.
- A. M. Ebrahim and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2013, 5, 10565–10573 CrossRef CAS PubMed.
- J. Yang, C. Zheng, P. Xiong, Y. Li and M. Wei, J. Mater. Chem. A, 2014, 2, 19005–19010 RSC.
- A. I. Ahmed, S. E. Samra, S. A. El-Hakam, A. S. Khder, H. Z. El-Shenawy and W. S. Abo El-Yazeed, Appl. Surf. Sci., 2013, 282, 217–225 CrossRef CAS.
- A. I. Ahmed, S. A. El-Hakam, M. A. Abd Elghany and W. S. Abo El-Yazeed, Appl. Catal., A, 2011, 407, 40–48 CrossRef CAS.
- T. A. Maark and A. A. Peterson, J. Phys. Chem. C, 2014, 118, 4275–4281 CrossRef CAS.
- L. Ai, X. Liu and J. Jiang, J. Alloys Compd., 2014, 625, 164–170 CrossRef.
- R. Mohammadi, E. Eidi, M. Ghavami and M. Z. Kassaee, J. Mol. Catal. A: Chem., 2014, 393, 309–316 CrossRef CAS.
- H. Naeimi and Z. S. Nazif, Appl. Catal., A, 2014, 477, 132–140 CrossRef CAS.
- J. Mondal, M. Nandi, A. Modak and A. Bhaumik, J. Mol. Catal. A: Chem., 2012, 363–364, 254–264 CrossRef CAS.
- J. Safari, P. Aftabi, M. Ahmadzadeh, M. Sadeghi and Z. Zarnegar, J. Mol. Struct., 2017, 1142, 33–39 CrossRef CAS.
- T. R. Mandlimath, B. Umamahesh and K. I. Sathiyanarayanan, J. Mol. Catal. A: Chem., 2014, 391, 198–207 CrossRef CAS.
- H. Rouhani, A. Sarrafi and M. Tahmooresi, Chem. Eng. Commun., 2016, 203, 289–299 CrossRef CAS.
- A. Kumar, L. Rout, L. S. K. Achary, R. S. Dhaka and P. Dash, Sci. Rep., 2017, 7, 42975–42993 CrossRef PubMed.
- R. Kardanpour, R. S. Tangestaninejad, V. Mirkhani, M. Moghadam, I. Mohammadpoor-Baltork, A. R. Khosropour and F. Zadehahmadi, J. Organomet. Chem., 2014, 761, 127–133 CrossRef CAS.
- S. Wang, C. W. Ng, W. Wang, Q. Li and Z. Hao, Chem. Eng. J., 2012, 197, 34–40 CrossRef CAS.
- M. K. Uddin, Chem. Eng. J., 2017, 308, 438–462 CrossRef CAS.
- B. Das, N. K. Mondal, R. Bhaumik and P. Roy, Int. J. Environ. Sci. Technol., 2014, 11, 1101–1114 CrossRef CAS.
- J. H. Deng, X. R. Zhang, G. M. Zeng, J. L. Gong, Q. Y. Niu and J. Liang, J. Chem. Eng., 2013, 226, 189–200 CrossRef CAS.
- P. Shao, D. Liang, L. Yang, H. Shi, Z. Xiong, L. Ding, X. Yin, K. Zhang and X. Luo, J. Hazard. Mater., 2020, 387, 121676 CrossRef PubMed.
- W. I. Mortada, I. M. M. Kenawy, Y. G. Abou El-Reash and A. A. Mousa, Int. J. Biol. Macromol., 2017, 101, 490–501 CrossRef CAS PubMed.
- I. M. M. Kenawy, W. I. Mortada, Y. G. Abou El-Reash and A. H. Hawwas, Can. J. Chem., 2016, 94, 221–228 CrossRef CAS.
- S. Hei, Y. Jin and F. Zhang, J. Chem., 2014, 546956 Search PubMed.
- N. Murakami, N. Takebe, T. Tsubota and T. Ohno, J. Hazard. Mater., 2012, 212, 83–87 CrossRef PubMed.
- H. Li, N. An, G. Liu, J. Li, N. Liu, M. Jia, W. Zhang and X. Yuan, J. Colloid Interface Sci., 2016, 466, 343–351 CrossRef CAS PubMed.
- M. R. Sohrabi, Z. Matbouie, A. A. Asgharinezhad and A. Dehghani, Microchim. Acta, 2013, 180, 589–597 CrossRef CAS.
- M. R. Sohrabi, Microchim. Acta, 2014, 181, 435–444 CrossRef CAS.
- P. Shao, L. Ding, J. Luo, Y. Luo, D. You, Q. Zhang and X. Luo, ACS Appl. Mater. Interfaces, 2019, 11(33), 29736–29745 CrossRef CAS PubMed.
- X. Yin, P. Shao, L. Ding, Y. Xi, K. Zhang, L. Yang, H. Shi and X. Luo, Environ. Sci.: Nano, 2019, 3307–3315 RSC.
- H. Yu, P. Shao, L. Fang, J. Pei, L. Ding, S. G. Pavlostathis and X. Luo, Chem. Eng. J., 2019, 359, 176–185 CrossRef CAS.
- N. N. Nassar, J. Hazard. Mater., 2010, 184, 538–546 CrossRef CAS PubMed.
- R. Hu, X. K. Wang, S. Y. Dai, D. D. Shao, T. Hayat and A. Alsaedi, Chem. Eng. J., 2015, 260, 469–477 CrossRef CAS.
- N. Zhang, G. L. Zang, C. Shi, H. Q. Yu and G. P. Sheng, J. Hazard. Mater., 2016, 316, 11–18 CrossRef CAS PubMed.
- X. Weng, X. Jin, J. Lin, R. Naidu and Z. Chen, Ecol. Eng., 2016, 97, 32–39 CrossRef.
- W. K. Kim, T. Shim, Y. S. Kim, S. Hyun, C. Ryu, Y. K. Park and J. Jung, Bioresour. Technol., 2013, 138, 266–270 CrossRef CAS PubMed.
- Y. Shen and B. L. Chen, Environ. Sci. Technol., 2015, 49, 7364–7372 CrossRef CAS PubMed.
- Z. N. Garba, N. I. Ugbaga and A. K. Abdullahi, J. Basic Appl. Sci., 2016, 5, 170–179 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10300g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Y. G. Abou El-Reasha,
L. A. Elatwya and
Awad I. Ahmed
*ab,
Y. G. Abou El-Reasha,
L. A. Elatwya and
Awad I. Ahmed