
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe acidic hydrolysis of N-acetylneuraminic 4,5-oxazoline allows a direct functionalization of the C5 position of Neu5Ac2en (DANA)†
Paola Rota *a,
Paolo La Roccab,
Federica Cirilloc,
Marco Piccolic,
Pietro Allevia and
Luigi Anastasia*cd
*a,
Paolo La Roccab,
Federica Cirilloc,
Marco Piccolic,
Pietro Allevia and
Luigi Anastasia*cd
aDepartment of Biomedical, Surgical and Dental Sciences, University of Milan, 20133, Milan, Italy. E-mail: paola.rota@unimi.it
bDepartment of Biomedical Sciences for Health, University of Milan, Via Fratelli Cervi 9, Segrate, 20090, Milan, Italy
cLaboratory of Stem Cells for Tissue Engineering, IRCCS Policlinico San Donato, San Donato Milanese, Piazza Malan 2, San Donato Milanese, 20097, Milan, Italy
dUniversity of Vita-Salute San Raffaele, Milan, Italy. E-mail: luigi.anastasia@hsr.it
First published on 24th December 2019
Abstract
The acidic hydrolysis of N-acetylneuraminic 4,5-oxazoline affords the corresponding 2,3-unsaturated amino ester, which was not previously detected (nor isolated) due to the unexpected rearrangement into its corresponding alcohol. In this work, we unveiled the mechanism of these reactions and optimized the conditions to obtain either synthetic intermediate.
Several 2,3-unsaturated derivatives of the N-acetylneuraminic acid (Neu5Ac, 1) have been developed as potent inhibitors of sialidases, the glycolytic enzymes critical for bacteria and virus spread.1–6 This class of inhibitors includes the 5-acetamido-2,6-anhydro-3,5-dideoxy-D-glycero-D-galacto-non-2-enoic acid 2a (Neu5Ac2en, DANA), the N-trifluoroacetylated congener 2b (FANA), the N-isobutyrylated one 2c (BCX 2798), and the 4-guanidino derivative 2d (Zanamivir), which has been widely used in the clinic against the influenza virus (Fig. 1).1–6
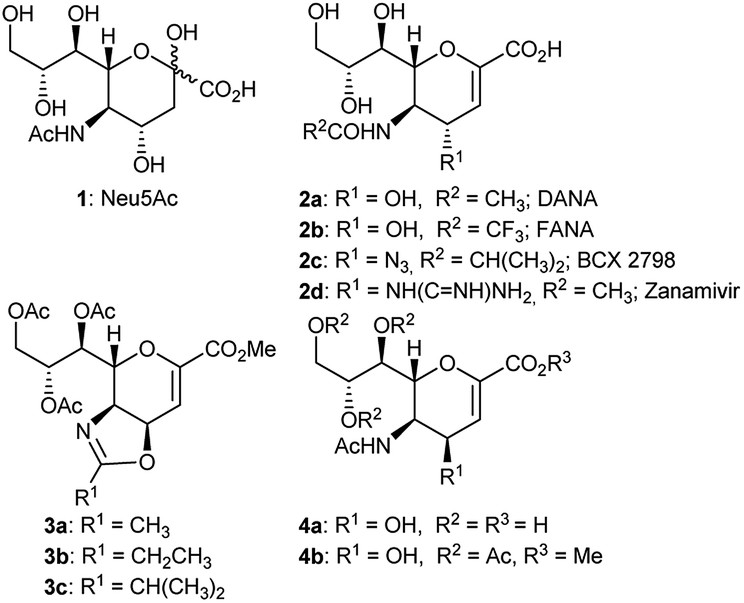 | ||
| Fig. 1 Sialic acid (Neu5Ac), some sialidase inhibitors (DANA, FANA, BCX 2798, Zanamivir) and some 4,5-oxazolines of Neu5Ac. | ||
Interestingly, the 4,5-oxazoline of Neu5Ac 3a has been shown1–12 to be a key intermediate for the synthesis of various C4- or C4- and C5-modified DANA derivatives (e.g. BCX 2798 2c and Zanamivir 2d),1,5,6 of the 4-epi DANA 4a3 (or its protected 4-epi hydroxyl derivative 4b6,9–13), as well as of several 3,4-unsaturated DANA-homologs, which were obtained through a Ferrier reaction.8,14 In this context, our group has revealed the conditions to perform a critical intermolecular nucleophilic attack on 3a at its C4 position, while avoiding its intramolecular formation, and reported the synthesis of several C5-perfluorinated 4-epi DANA derivatives.2,3,7,15,16 Overall, the synthetic strategies involving the synthon 3a could be summarized with three distinctive reactions (Scheme 1): (a) a nucleophilic attack – via SN2 – on the carbon at C4, which eventually opens up the oxazoline ring, thus restoring the acetyl group at C5 (analogously to the reaction used for the insertion of the azido group at C4),17 (b) a hydrolytic ring-opening caused by the direct attack of a water molecule on the sp2-carbon of the oxazoline ring, which affords the 4β-hydroxy derivative 4b,6,9–13 and (c) a nucleophilic attack on the anomeric carbon (C2), followed by a shift of the double bond and the restoration of the N-acetyl group through a Ferrier reaction.8,14
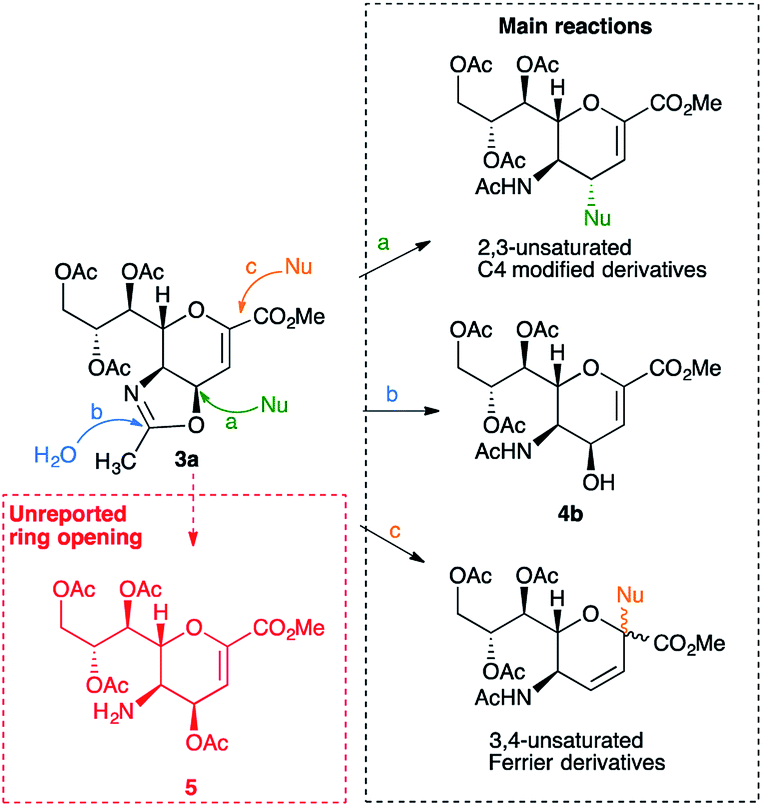 | ||
| Scheme 1 Main reported reactions on the 4,5-oxazoline 3a: (a) nucleophilic attack via SN2 at the C4; (b) hydrolytic ring-opening with the formation of 4b; (c) nucleophilic attack at the C2 and formation of 3,4-unsaturated Ferrier products. | ||
To date, the formation of the amino ester 5, an isomer of 4b and derived from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond cleavage of the oxazoline ring, has never been described. Indeed, differently to the well-established oxazoline chemistry,18 it was reported that the acidic treatment of the oxazoline 3a afforded the 4-epi hydroxyl derivative 4b, as a single product, in variable yields (42–88%), calling for further studies.6,9–13
N bond cleavage of the oxazoline ring, has never been described. Indeed, differently to the well-established oxazoline chemistry,18 it was reported that the acidic treatment of the oxazoline 3a afforded the 4-epi hydroxyl derivative 4b, as a single product, in variable yields (42–88%), calling for further studies.6,9–13
As a result of our continuing interest in the sialic acid chemistry/biochemistry,2,3,7,14–16,19–23 in this work we solved this mechanistic puzzle, as we unveil that, the acidic hydrolysis of the 4,5-oxazoline 3a affords the amino ester 5, which could be isolated in good yields and fully characterized under appropriate work-up conditions. Furthermore, we detailed the shortened synthesis of the precursor of BCX 2798 (2c) and other sialidase inhibitors (such as BCX 2755 and various C4 triazole derivatives) further highlighting the synthetic utility of 5.1,24,25 Finally, we report an improved procedure for the quantitative preparation of the isomeric alcohol 4b and clarified the chemical structure of several by-products observed during the acid hydrolysis12,13 of the oxazoline 3a.
To this purpose, we dissolved 1 mmol of the oxazoline 3a in a solution of THF–H2O (15 mL, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) containing TFA (0.2 mL), at 23 °C for 5–15 minutes (alternatively CH3CN–H2O, 15 mL, 1:1 v/v as a solvent system), under the same experimental conditions previously described for the preparation of 4b.9 The reaction was monitored by TLC on silica (AcOEt eluent). After a few minutes, the starting oxazoline 3a (Rf = 0.34) disappeared and was completely converted into a single more polar compound (Rf = 0.22). After a careful isolation using weak basic resin (IRA-67), we could assign to this molecule the structure of the 2,3-unsaturated amino ester 5, on the bases of its physicochemical properties (correct mass, 1H and 13C NMR spectra), and reactivity (such as a brownish-yellow spot when sprayed with an alcoholic solution of ninhydrin).‡ The obtained compound 5 was sufficiently stable under anhydrous conditions, but, as expected, in the presence of water, it could undergo a slow rearrangement into the alcohol 4b, possibly explaining previous inconsistent results reported in the literature.
:1 v/v) containing TFA (0.2 mL), at 23 °C for 5–15 minutes (alternatively CH3CN–H2O, 15 mL, 1:1 v/v as a solvent system), under the same experimental conditions previously described for the preparation of 4b.9 The reaction was monitored by TLC on silica (AcOEt eluent). After a few minutes, the starting oxazoline 3a (Rf = 0.34) disappeared and was completely converted into a single more polar compound (Rf = 0.22). After a careful isolation using weak basic resin (IRA-67), we could assign to this molecule the structure of the 2,3-unsaturated amino ester 5, on the bases of its physicochemical properties (correct mass, 1H and 13C NMR spectra), and reactivity (such as a brownish-yellow spot when sprayed with an alcoholic solution of ninhydrin).‡ The obtained compound 5 was sufficiently stable under anhydrous conditions, but, as expected, in the presence of water, it could undergo a slow rearrangement into the alcohol 4b, possibly explaining previous inconsistent results reported in the literature.
Moreover, we observed that the amino ester 5 was partially transformed into the isomeric amido alcohol 4b (Rf = 0.11) when the reaction mixture was neutralized12,13 with a saturated sodium hydrogen carbonate solution or when the crude residue of the reaction was chromatographed9–11 on silica gel column (ethyl acetate–methanol 100:1 v/v).§
Time-laps NMR experiments further clarified the reaction mechanism by revealing the initial formation of compound 5 as an ammonium salt, its successive transformation into the free amine form and its slow rearrangement into the alcohol 4b. Remarkably, by performing the reaction directly in an NMR tube, by dissolving oxazoline 3a in CD3CN–D2O (1:1 v/v) and successively adding TFA, we could observe the fast and complete transformation of the oxazoline 3a into the ammonium form of 5 (see ESI, S6†). The successive elution of the NMR solution through a small column containing a weak basic resin (IRA-67) resulted in the formation of the free amine 5, as revealed by the shift of the H-4, H-5 and H-6 proton signals (δH4 5.50, δH5 3.82 and δH6 4.59 ppm for ammonium salt of 5 and δH4 5.25, δH5 3.04 and δH6 4.12 ppm for 5), together with the formation of some traces of the isomeric alcohol 4b. The reaction was further monitored by 1H-NMR, revealing the complete transformation of amine 5 into the 4-hydroxyl derivative 4b as the only product after six days.
Overall, our results support a reaction mechanism which is in line with other oxazoline chemistry and in disagreement with the state of art of the oxazoline.18 In particular, the opening of the oxazolinic ring and its transformation into the ammonium salt of 5 could involve both the nucleophilic attack of a water molecule on the sp2 carbon atom of the oxazoline ring and, at the same time, the protonation of the nitrogen group (Scheme 2). Then, the subsequent formation of the alcohol 4b could be explained by the relative instability of 5 due to the presence of a free amino group in a cis configuration, supporting the migration of the acetyl group between the C-4 and C-5 position.
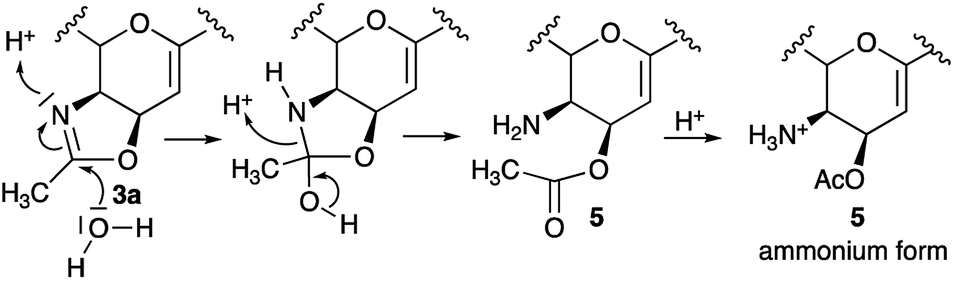 | ||
| Scheme 2 Oxazoline ring opening with the formation of the ammonium salt of 5. | ||
Finally, to further confirm the structure of compound 5, a classical reactivity-based approach was performed. In particular, the amino ester 5, reacted with propionyl chloride in the presence of a basic resin (IRA-67) affording the desired propionylamido derivative 6, as expected (Scheme 3). Moreover, we tested whether the exchange reaction of the acyl group between the C5 and C4 was general in scope. To this purpose, we transformed compound 6 into its oxazoline derivative 3b, using the standard reaction conditions.7 Compound 3b was achieved in good yield (70%) and high purity (>98% by 1H NMR). Then, the propionyl-oxazoline 3b was treated with moist TFA and, after its disappearance, directly reacted with an excess of acetyl chloride in the presence of a basic resin (IRA-67), affording compound 7 in high yield (80%).
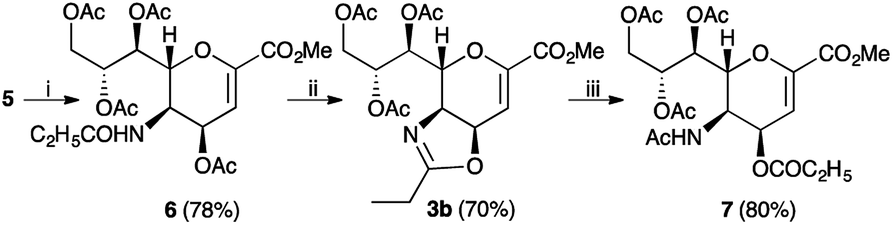 | ||
| Scheme 3 Exchange of an acyl group between the C5 and C4 positions. Reaction conditions: (i) propionyl chloride, weak basic resin IRA 67, CH2Cl2, 0 °C to 23 °C, 0.5 h; (ii) BF3 Et2O, CH2Cl2, 23 °C to 80 °C, 20 min; (iii) TFA, CH3CN–H2O, 1:1 v/v, 23 °C, 5–15 min, then acetyl chloride, weak basic resin IRA 67, CH2Cl2, 0 °C to 25 °C, 0.5 h. | ||
The discovery that 3a could be easily converted into the amino ester 5 is of high synthetic value, as the direct acylation of its C5 amino group opens up a novel and easy way to replace the acetamido function present in DANA derivatives with different suitable acyl amido substituents. Indeed, as a proof of concept, we were able to directly functionalize the C5 position, ultimately achieving the desired isobutyl amide derivative 8 with this new one-pot reaction from the starting oxazoline 3a in good overall yield (78%, Scheme 4), which could be easily converted into 9, the key precursor of several sialidase inhibitors such as BCX 2798 (2c) and BCX 2755.1,24,25
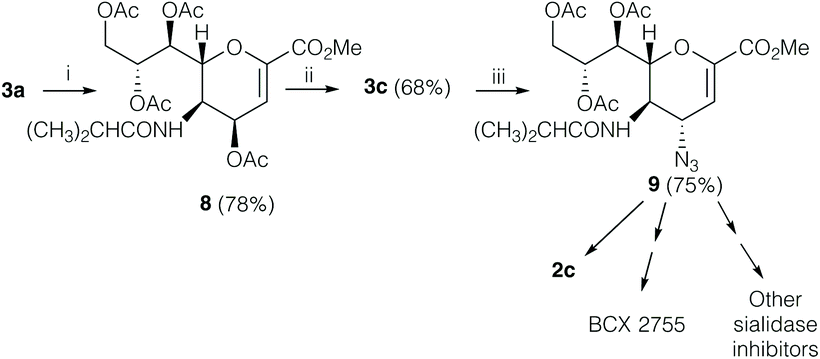 | ||
| Scheme 4 Synthesis of precursor 9 of BCX 2798, BCX2755 and other sialidase inhibitors. Reaction conditions: (i) TFA, CH3CN–H2O, 1:1 v/v, 23 °C, 5–15 min, then isobutyric anhydride, weak basic resin IRA 67, CH2Cl2, 0 °C to 25 °C, 1 h; (ii) BF3 Et2O, CH2Cl2, 23 °C to 80 °C, 20 min; (iii) TMSN3, tBuOH, 80 °C, overnight. | ||
Briefly, by simple treatment of 8 with BF3 Et2O in dichloromethane, we obtained the unreported isobutyl-oxazoline 3c (68% yield). Remarkably, we performed the C4α-insertion of an azido group using azidotrimethylsilane (TMSN3) in t-butanol, under the same reaction conditions reported17 to obtain the azido derivative on the oxazoline 3a. Thus, we were able to isolate the key intermediate 9 in high yield (75%). The hydrolysis1 of the azide 9 afforded the final BCX 2798 (2c).
Overall, this novel procedure avoids several steps of protection and deprotection at C5 that were needed in previous synthetic routes for the synthesis of BCX 2798 (2c), as well as of several sialidase inhibitors.1,24,25
Finally, while studying and optimizing the conditions for 3a ring-opening we were able to develop a more efficient synthetic route to the 4-epi hydroxyl alcohol 4b, which was previously obtained with variable yields (42–88%).6,9–13 Remarkably, we were able to obtain 4b in high yields by a “one-pot” reaction from oxazoline 3a. To this purpose, 3a (1 mmol) was treated with TFA in THF–H2O as previously described and, after its disappearance (monitored by TLC), triethylamine (5 mmol) was added. After 40 min at r.t. the intermediate 5 was completely transformed into a single spot (by TLC) corresponding to the desired alcohol 4b. The 1H NMR analysis of crude product of reaction confirmed the quantitative formation of this compound, which, after chromatographic purification from the salts, was obtained in 89% isolated yield.
Intrigued by these results, we carefully scrutinized the more widely used hydrolytic method (AcOH in of AcOEt–H2O mixture) to obtain 4b, in order to assess the possible formation of other byproducts. Interestingly, after 1 h, we observed by TLC the partial reduction of the starting oxazoline 3a, to afford a spot (Rf = 0.22) corresponding to the expected compound 5, and two additional spots (Rf = 0.16 and Rf = 0.11), the most polar one corresponded to 4b. As the reaction proceeded overnight, both 3a and 5 disappeared and were transformed in the two most polar spots in TLC, which remain unchanged even after the basic (NaHCO3) work-up extraction. After two flash chromatographies (see ESI†), three different products were isolated, corresponding to the expected alcohol 4b (51%), and to two less polar products 10a (7%) and 10b (18%) that previously unreported (Fig. 2).
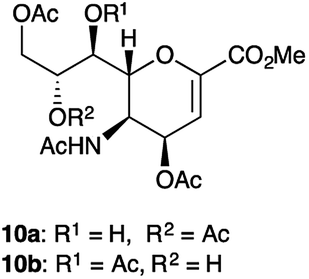 | ||
| Fig. 2 Unreported byproducts of acetic acid hydrolysis of oxazoline 3a. | ||
Indeed, under acidic conditions, the acetyl scrambling between C4 and C5 positions could compete with that among either the C7 or the C8 and the C5, differently from what we observed under basic conditions. However, it cannot be excluded that one of the two products 10a or 10b may derive from the simple migration of the acetyl group between the hydroxyl functions present in the glycerol side chain.
Conclusions
In this work, we report the direct conversion of 3a in the corresponding amine 5, a useful synthon for the preparation of several sialidase inhibitors with anti-viral activity. The understanding of this reaction mechanism clarified several inconsistent and unexplained results in the literature, and it allowed the revision and improvement of the synthesis of alcohol 4b, another key intermediate for the preparation of sialic acid derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Prof. Mario Anastasia for the valuable suggestions and his unrelenting encouragement, and Ms Irene Delcarro for her skilled technical assistance.Notes and references
- I. M. El-Deeb, P. Guillon, M. Winger, T. Eveno, T. Haselhorst, J. C. Dyason and M. von Itzstein, J. Med. Chem., 2014, 57, 7613 CrossRef CAS PubMed.
- P. Rota, P. La Rocca, M. Piccoli, M. Montefiori, F. Cirillo, L. Olsen, M. Orioli, P. Allevi and L. Anastasia, ChemMedChem, 2018, 13, 236 CrossRef CAS PubMed.
- P. Rota, N. Papini, P. La Rocca, M. Montefiori, F. Cirillo, M. Piccoli, R. Scurati, L. Olsen, P. Allevi and L. Anastasia, MedChemComm, 2017, 8, 1505 RSC.
- P. Guillon, L. Dirr, I. M. El-Deeb, M. Winger, B. Bailly, T. Haselhorst, J. C. Dyason and M. von Itzstein, Nat. Commun., 2014, 5, 5268 CrossRef CAS PubMed.
- L. Dirr, I. M. El-Deeb, P. Guillon, C. J. Carroux, L. M. Chavas and M. von Itzstein, Angew. Chem., Int. Ed., 2015, 54, 2936 CrossRef CAS PubMed.
- J. Li, M. Zheng, W. Tang, P.-L. He, W. Zhu, T. Li, J.-P. Zuo, H. Liu and H. Jiang, Bioorg. Med. Chem. Lett., 2006, 16, 5009 CrossRef CAS PubMed.
- I. S. Agnolin, P. Rota, P. Allevi, A. Gregorio and M. Anastasia, Eur. J. Org. Chem., 2012, 6537 CAS.
- K. Ikeda, Y. Ueno, S. Kitani, R. Nishino and M. Sato, Synlett, 2008, 1027 CrossRef CAS.
- E. Schreiner, E. Zbiral, R. G. Kleineidam and R. Schauer, Liebigs Ann. Chem., 1991, 129 CrossRef CAS.
- G. B. Kok, D. R. Groves and M. von Itzstein, Chem. Commun., 1996, 2017 RSC.
- G. B. Kok, D. Groves and M. von Itzstein, J. Chem. Soc., Perkin Trans. 1, 1999, 2109 RSC.
- J. Scheigetz, R. Zamboni, M. A. Bernstein and B. Roy, Org. Prep. Proced. Int., 1995, 27, 637 CrossRef CAS.
- V. Kumar, S. W. Tanenbaum and M. Flashner, Carbohydr. Res., 1982, 101, 155 CrossRef CAS.
- P. La Rocca, P. Rota, M. Piccoli, F. Cirillo, M. Orioli, A. Ravelli, P. Allevi and L. Anastasia, J. Org. Chem., 2019, 84, 5460 CrossRef CAS PubMed.
- P. Rota, I. S. Agnolin, P. Allevi and M. Anastasia, Eur. J. Org. Chem., 2012, 2508 CrossRef CAS.
- P. Rota, P. Allevi, I. S. Agnolin, R. Mattina, N. Papini and M. Anastasia, Org. Biomol. Chem., 2012, 10, 2885 RSC.
- M. Chandler, M. J. Bamford, R. Conroy, B. Lamont, B. Patel, V. K. Patel, I. P. Steeples, R. Storer, N. G. Weir, M. Wright and C. Williamson, J. Chem. Soc., Perkin Trans. 1, 1995, 1173 RSC.
- L. V. Pavlova and F. Y. Rachinskii, Russ. Chem. Rev., 1968, 37, 587 CrossRef.
- P. Rota, P. Allevi, M. L. Costa and M. Anastasia, Tetrahedron: Asymmetry, 2010, 21, 2681 CrossRef CAS.
- P. Allevi, P. Rota, I. S. Agnolin, A. Gregorio and M. Anastasia, Eur. J. Org. Chem., 2013, 4065 CrossRef CAS.
- P. Rota, F. Cirillo, M. Piccoli, A. Gregorio, G. Tettamanti, P. Allevi and L. Anastasia, Chem.–Eur. J., 2015, 21, 14614 CrossRef CAS PubMed.
- P. Rota, L. Anastasia and P. Allevi, Org. Biomol. Chem., 2015, 13, 4931 RSC.
- P. Rota, P. Allevi and L. Anastasia, Asian J. Org. Chem., 2015, 4, 1315 CrossRef CAS.
- M. Von Itzstein, I. El-Deeb, L. Dirr, P. Guillon and M. Winger, WO Pat., 2016/033660, 2016.
- I. V. Alymova, G. Taylor, T. Takimoto, T. H. Lin, P. Chand, Y. S. Babu, C. Li, X. Xiong and A. Portner, Antimicrob. Agents Chemother., 2004, 48, 1495 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10215a |
| ‡ Preparation of compound 5: to a solution of oxazoline 3a1 (413 mg, 1.0 mmol) in THF–H2O, 2:1 v/v (15 mL) or in CH3CN–H2O, 1:1 v/v (15 mL), TFA (0.2 mL) was added and the solution was stirred at 23 °C until the complete disappearance of the starting material (5–15 minutes), by monitoring with TLC (AcOEt). At this time, the reaction was quenched by the addition of a weak basic resin (IRA-67) until neutral pH, filtered and evaporated to give the desired amine 5 (401 mg, 93%). Compound 5 appeared as white foam: [α]D −130.3 (c 1.0 in CH2Cl2); δH (500 MHz, CDCl3) 6.15 (1H, d, J3,4 = 5.7 Hz, 3-H), 5.63 (1H, dd, J7,6 = 1.4, J7,8 = 6.1 Hz, 7-H), 5.44 (1H, ddd, J8,9a = 2.3, J8,7 = J8,9b = 6.1 Hz, 8-H), 5.28 (1H, dd, J4,5 = 4.2, J4,3 = 5.7 Hz, 4-H), 4.69 (1H, dd, J9a,8 = 2.3, J9a,9b = 12.6 Hz, 9a-H), 4.27 (1H, dd, J9b,8 = 6.1, J9b,9a = 12.6 Hz, 9b-H), 4.01 (1H, dd, J6,7 = 1.4, J6,5 = 10.8 Hz, 6-H), 3.79 (3H, s, COOCH3), 2.93 (1H, dd, J5,4 = 4.2, J5,6 = 10.8 Hz, 5-H), 2.15 (3H, s, OCOCH3), 2.10 (6H, overlapping, 2 × OCOCH3), 2.06 (3H, s, OCOCH3) and 1.65–1.47 (2H, overlapping, NH2); δC (125 MHz, CDCl3) 170.6 (2C, OCOCH3 at C-7 and OCOCH3 at C-9), 170.2 (OCOCH3 at C-4), 169.9 (1C, OCOCH3 at C-8), 162.0 (C-1), 146.2 (C-2), 105.9 (C-3), 75.5 (C-6), 70.8 (C-8), 68.6 (C-7), 65.7 (C-4), 62.0 (C-9), 52.5 (COOCH3), 47.3 (C-5), 20.9 (2C, 2 × OCOCH3), 20.7 (OCOCH3) and 20.6 (OCOCH3); MS (ESI positive): m/z 432.1 [M + H]+, 454.1 [M + Na]+; elemental analysis (found: C, 50.24; H, 5.79; N, 3.10. C18H25NO11 requires C, 50.12; H, 5.84; N, 3.25%). |
| § One or more of these occurrences, could explain the transformation of the compound 5 into the isomer 4b and the discrepancy with literature data. |
| This journal is © The Royal Society of Chemistry 2020 |