
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of cytotoxic potential of structurally well-characterized RNA targeted ionic non-steroidal anti-inflammatory (NSAID) Cu(II) & Zn(II) DACH–mefenamato drug conjugates against human cancer cell lines†
Huzaifa Yasir Khan,
Sartaj Tabassum and
Farukh Arjmand*
and
Farukh Arjmand*
Department of Chemistry, Aligarh Muslim University, Aligarh 202002, Uttar Pradesh, India. E-mail: farukh.arjmand18@gmail.com
First published on 24th December 2019
Abstract
New RNA targeted ionic [Cu(DACH)2(H2O)2](mef)2, 1 and [Zn(DACH)2(H2O)2](mef)2, 2 drug conjugates were synthesized and characterized by spectroscopic techniques FT-IR, UV-vis, EPR in case of 1 and 1H and 13C NMR in case of 2, ESI-MS, thermogravimetric analysis and single-crystal X-ray structure determination in case of 1. The interaction studies of 1 & 2 with most likely drug targets like ctDNA and tRNA were performed which demonstrated that the complexes 1 and 2 exhibited strong preferential binding to tRNA as compared to ctDNA, Kb = 2.52(±0.04) × 105 M−1, 7.85(±0.02) × 104 M−1, respectively. Scanning electron microscopy analyses of complex-ctDNA/tRNA condensates suggested the interaction of complexes with ctDNA/tRNA had occurred, followed by lengthening of DNA double helix and bulge region of tRNA. Cytotoxic activity of 1 and 2 against human cancer cell lines namely; MCF-7 (breast), HeLa (cervical), MIA-PA-CA 2 (pancreatic), A-498 (kidney), Hep-G2 (hepatoma) was evaluated by SRB assay. The obtained results showed that copper complex 1 was an outstanding cytotoxic agent with remarkably good GI50 value (<10 μg ml−1) against the tested cancer cell lines except for MIA-PA-CA 2, while zinc complex 2 revealed moderate cytotoxicity against all the tested cancer cell lines.
Introduction
Recently, NSAIDs have emerged as a new prominent class of anticancer pharmaceuticals attenuating their significance as anti-inflammatory drugs.1–3 Most of the NSAIDs such as aspirin, diclofenac, flufenamic acid, indomethacin, naproxen, tolmetin exhibit similar pharmacological (antibacterial, anti-inflammatory, anticancer) behavior,4,5 mechanism of action via COX-independent cell inhibition6 and similar adverse effects specifically risk of gastrointestinal (GI) and cardiovascular complications.7 Typically, NSAIDs contain hydrophilic groups (carboxylic acid or enolic groups) and lipophilic ones, aromatic ring with/without halogen atom which favour amphiphilic properties and on complexation with metal ions prefer to remain in anionic form at physiological pH.8Ionic NSAIDs have attracted considerable research focus owing to their application in medicinal therapy as an estimated 50% of the drugs molecules are administered as salts. Furthermore, ionizable functional groups can overcome the undesirable effect of the parent drug. Metallo-drugs9 can facilitate the muting of toxicity of organic ligands. Additionally, they exhibit synergistic activity when administered with free organic NSAIDs and previous researches demonstrate that on complexation, NSAIDs have proven as more active drug candidates than the parent compounds. Although, there are number of reports describing the interaction of NSAIDs with DNA/serum protein by Psomas et al.10–12 and some earlier reports from our research group.13,14 However, fewer reports are available on RNA targeted metallo-NSAID drug conjugates.
Targeting RNAs is a newer concept which open avenues for pharmaceutical development to treat many chronic diseases.15 Unlike many proteins, biochemical functions or expressions of RNAs has been directly linked to various diseases. RNA is an indispensable drug target, an essential biopolymer of human genome (∼90%) that regulates many vital functions of cell. RNAs play critical roles in transcription or translation, splicing processes, protein synthesis and retroviral replication and RNA structure is accessible to small molecules binding.16
Looking at combinatorial libraries of molecular structures suitable for targeting RNAs, metal complexes are more suitable for the recognition of RNA motifs as they satisfy the criteria; (i) exhibit non-covalent interactions such as vander Waals, electrostatic or hydrogen bonding (ii) are coordinatively saturated (iii) possess rigid planar framework.17 Additionally these complexes have ability to induce RNA scission by different mechanistic pathways and the most plausible mechanism for ionic NSAIDs would be electrostatic interaction. 3D folding of an RNA chain into a scaffold of spatially placed anionic groups creates ‘‘electronegative” pocket that can selectively get bonded by compounds such as metallated drug molecules which exhibit structural electrostatic complementarity.18
Copper – an essential redox metal ion is endowed with unique properties such as Lewis acidity, redox activity, antitumor and apoptosis inducing capabilities. It has been established from previous literature reports that copper plays a significant role in angiogenesis.19 Tumors are dependent on angiogenesis for their growth, invasion and metastasis.20 Copper was found to be a co-factor for several angiogenesis mediators including vascular endothelial growth factor (VEGF),21 basic fibroblast growth factor (bFGF),22 interleukin-1 (IL-1) and IL-8 (ref. 23) essential for tumor angiogenesis. Recent studies have demonstrated that copper(II) phenanthroline complexes containing NSAID, indomethacin were potent against breast cancer stem cells (CSC) which are known to maintain low level of ROS.1 Based on these facts, copper complexes have been listed as new class of anticancer metallodrugs and have appeared in many research and review articles.24,25 Zinc, an endogenously biocompatible metal ion is redox inert exists as a divalent cation in the biological system and is actively involved in DNA synthesis, apoptosis, gene expression, and catalytic functions.26. Zinc-based NSAIDs is capable of showing DNA/RNA recognition that can eventually leads to different cell death mechanism as compared to cisplatin.27
Herein, we describe ionic [Cu(II)/Zn(II)(DACH)2(H2O)2]mefenamic acid conjugates targeted to tRNA. Tethering DACH moiety with NSAID is an innovative strategy for the development of new antitumor therapeutic agents. DACH is a carrier ligand, capable of better cellular uptake and has been known to harness cell inhibition effectively via apoptosis.28 DACH conjugated anticancer drug oxaliplatin, (1,2-DACH) oxaloplatinum(II) was more potent as compared to (S,S)-analog and exhibited less toxic effects.29 A total of ca. 1,2-DACH complexes have entered clinical human trials which were evaluated for their antitumor activity.30 Metallo-drugs derived from NSAIDs have been studied for the treatment of a large number of chronic diseases. A recent review summarizes metallo-drugs of NSAIDs with (a) main group metals, (b) transition metals, (c) metalloids and (d) lanthanides categorized according to their anti-inflammatory, antibacterial and antiproliferative activity and their ability to interact with various intracellular components like DNA, RNA, proteins etc.4 Among the transition metals, copper–NSAIDs tagged with ancillary ligands have gained a prominent therapeutic position as an anti-inflammatory, antiproliferative, antitumor agents owing to the fact that copper is endogenously biocompatible, can act by different mechanism with the biological targets involving ROS species, down regulation of anti-apoptotic proteins (Bcl-2 and Bcl-XL) and anti-angiogenic proteins that trigger cell death even in case of resistant cancer cell lines.31 Previous literature reports reveal that copper-based drug candidates have shown better selectivity, target specificity and improved antitumor efficiency which can be achieved by the selection of appropriate ligand scaffold combinations.1
Results and discussion
Synthesis and characterization
The complexes 1 and 2 [M(DACH)2(H2O)2](mef)2 where M = Cu(II), 1 and Zn(II), 2 were synthesized by reacting methanolic solution of mefenamic acid (deprotonated by KOH), 1,2-diamminocyclohexane and CuCl2·2H2O/ZnCl2·2H2O in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 stoichiometric ratio (Scheme 1).
:2:1 stoichiometric ratio (Scheme 1).
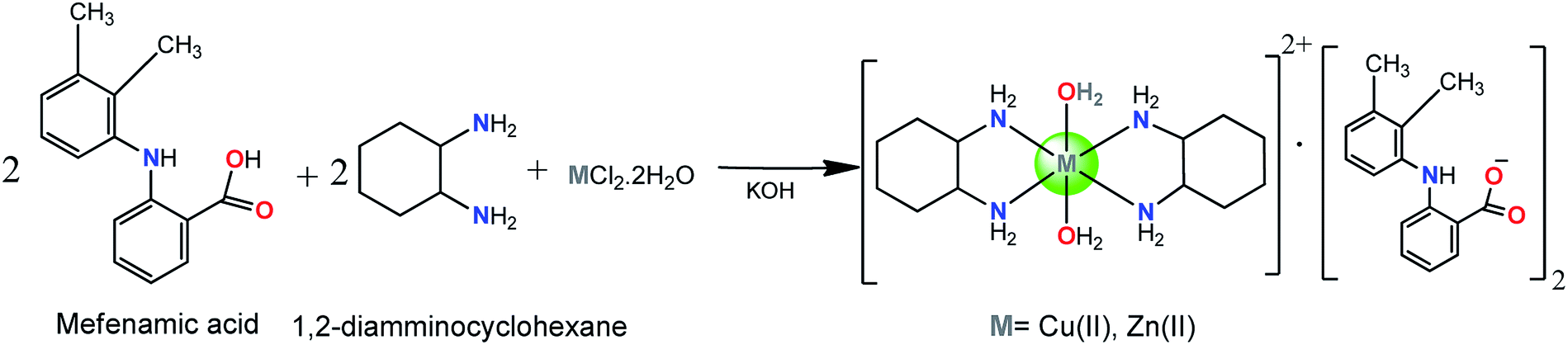 | ||
| Scheme 1 Synthetic route for complexes1 and 2. | ||
The products were found to be air stable, soluble in MeOH and other non-polar solvents like DMF, DMSO. The molecular structure of 1 was established by X-ray diffraction studies. The complexes 1 and 2 were thoroughly characterized by spectroscopic techniques (UV-vis, FT-IR, EPR in case of 1 and 1H NMR, 13CNMR in case of 2), ESI-MS and elemental analysis.
Spectroscopic measurements
FT-IR spectroscopy of 1 and 2 was used to assign the carboxylate coordination mode of mefenamato anion and the binding mode of DACH ligands. Complexes 1 and 2 exhibited broad absorption bands at 3235 cm−1 and 3545 cm−1, respectively which were assigned to O–H stretching vibration of coordinated water molecule to the metal centre.11 The sharp absorption bands at 3128 cm−1 and 3134 cm−1, respectively attributed to N–H stretching frequency of mefenamato anion or DACH ligand coordinated to M(II) centres.14 Absorption bands due to ν(CH2) vibrations of the cyclohexyl ring of DACH were observed at 2859–2935 cm−1.32 In complexes 1 and 2, two important bands at 1655(s) and 1255(s) cm−1 corresponding to stretching vibrations ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)carboxylic and ν(C–O)carboxylic of carboxylic group (–COOH) were shifted to 1576–1616 cm−1 and 1383–1431 cm−1. In both the complexes, the parameter Δν(CO2) [Δν = νas(CO2) − νs(CO2)] values falls in the range 160–210 cm−1 observed for ionic complexes.11 The absorption peaks observed in the region 1300–1100 cm−1 might be attributed to the symmetric deformation vibrations of the –CH3 group of mefenamato anion in 1 and 2. However, in-plane bending and out-of-plane deformation vibrations of hydrogen atoms were observed in the region 1000–620 cm−1. The absorption bands at 595–427 cm−1 and 563–425 cm−1 in 1 and 2 were found within the range reported for ν(M–N) and ν(M–O) stretching frequencies in the far IR region.33
O)carboxylic and ν(C–O)carboxylic of carboxylic group (–COOH) were shifted to 1576–1616 cm−1 and 1383–1431 cm−1. In both the complexes, the parameter Δν(CO2) [Δν = νas(CO2) − νs(CO2)] values falls in the range 160–210 cm−1 observed for ionic complexes.11 The absorption peaks observed in the region 1300–1100 cm−1 might be attributed to the symmetric deformation vibrations of the –CH3 group of mefenamato anion in 1 and 2. However, in-plane bending and out-of-plane deformation vibrations of hydrogen atoms were observed in the region 1000–620 cm−1. The absorption bands at 595–427 cm−1 and 563–425 cm−1 in 1 and 2 were found within the range reported for ν(M–N) and ν(M–O) stretching frequencies in the far IR region.33
The X-band EPR spectrum of 1 was recorded to assess the oxidation state, geometry, and electronic configuration of the complex. 1 exhibited an isotropic spectra with g‖ = 2.197, g⊥ = 2.049 and gav = 2.83 computed from the expression gav2 = g‖2 + 2g⊥2/3 suggesting an octahedral geometry. The trend g‖ > g⊥ > ge (2.0023) indicated that the unpaired electron was located in the dx2−y2 orbital of the Cu(II) ion which is a characteristic of axial symmetry. (Fig. S1†)34
1H NMR spectrum of 2, recorded in DMSO-d6 did not display O–H proton signal of the mefenamic acid (Hmef) (Fig. 1) expected in the range of 10–13 ppm, confirming its deprotonation. The characteristic aromatic signatures of mefenamato moiety were observed in the region 7.99–6.64 ppm (H2, H5, H7, H3, H9, H8 and H4). Sharp singlets corresponding to methyl protons (H10 and H11) were observed at 2.30 and 2.18 ppm, respectively. Moreover, a complex multiplet in the region 1.37–1.12 ppm and 1.73–1.63 ppm was attributed to DACH ring protons. A singlet at 8.11 ppm may be assigned to amine (–NH) proton sandwiched between two aromatic moieties of mefenamato anion. (Fig. S2†)
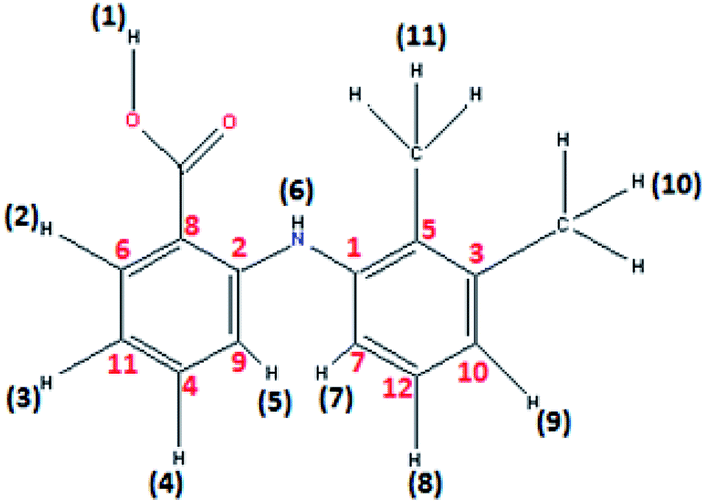 | ||
| Fig. 1 Mefenamic acid (Hmef) with H and C atom numbering in accordance to 1H NMR proton's assignment and 13C NMR carbon's assignment. | ||
1H NMR spectrum of free ligand mefenamic acid display O–H proton signal at 12.56 ppm. The characteristic aromatic signatures were observed in the region 7.31–6.67 ppm. Sharp singlets corresponding to methyl protons (H10 and H11) were observed at 2.34 and 2.18 ppm, respectively. The signal at 8.03 ppm may be assigned to amine (–NH) proton sandwiched between two aromatic moieties of mefenamic acid. (Fig. S4†) 1H NMR spectrum of free ligand DACH display complex multiplet in the region 1.22–0.99 ppm and 2.22–1.61 ppm was attributed to DACH ring protons. (Fig. S5†) A singlet at 5.29 ppm assigned to –NH2 group attached to cyclohexyl ring which causes upfield shift due to Zn(II) and observed at 2.88 ppm in complex 2.
13C NMR spectrum of 2 showed resonances of the cyclohexyl carbons in the range 35.58–25.69 ppm. Two resonance signals corresponding to –CH3 carbons attached to aromatic moiety of mefenamato anion were found at 22.81 and 20.69 ppm, respectively. The aromatic ring carbon atoms displayed their resonance signatures in the range 149.37–113.71 ppm. A significant peak for carboxylate ion was observed at 182.72 ppm with more scans. (Fig. S3†)
13C NMR spectrum of free mefenamic acid exhibited significant peak for carboxylate group at 172.91 ppm. The aromatic ring carbon atoms displayed their resonance signatures in the range 156.02–109.21 ppm. Two resonance signals corresponding to –CH3 carbons attached to aromatic moiety of mefenamato anion were found at 20.61 and 14.05 ppm. (Fig. S5b†) 13C NMR spectrum of free DACH resonances of the cyclohexyl carbons in the range 25.29–57.48 ppm. (Fig. S5c†)
The ESI-MS analyses offer the assignment of characteristic fragmentation peaks. The ESI-MS spectrum of 1 (Fig. S6†) exhibited molecular ion peak at m/z = 792.59 corresponding to [C42H60N6O6Cu + 3H]+ fragment after removal of one water molecule. The base peak observed at m/z = 643.23 attributed to fragment [2{C15H14NO2} + 2H2O + N4H8Cu–2C6H10]. The peak at m/z = 286 was ascribed to cationic moiety having molecular formula [C12H28CuN4 + 5H+] after removal of water molecules. The additional ion peaks at 335 and 599 were observed for the molecular fragments [C42H60N6O6Cu–2C15H14NO2 + 8H+] and [C42H60N6O6Cu–DACH–2H2O–Cu + 5H+], respectively. Similarly, fragmentation scheme of complex 2 revealed peaks at m/z 297.2, 613.4assigned to [C42H60N6O6Zn–2{C15H14NO2}−–2H2O + 5H]+, [C42H60N6O6Zn–DACH–H2O–Zn]+fragments and m/z = 483.3 corresponding to two mefenamato anion with molecular fragments [2C15H14NO2 + 3H]+, respectively. (Fig. S7†)
The UV-vis spectrum of 1 in methanolic solution exhibited a broad and intense band at 630 nm attributed to copper ion in the tetragonally distorted octahedral environment which was followed by strong bands in the UV region at 259 and 305 nm assigned to the π–π* and n–π* ligand centered transitions.35 The electronic spectrum of 2 displayed two well resolved bands centered at 332 nm and 260 nm ascribed to n–π* and intraligand (IL) π–π* transition indicative of the octahedral Zn(II) ion.36
Conductance measurements of 1 and 2 were carried out at 25 °C in DMSO. Molar conductance values were found to be 228 and 186 S cm2 mol−1 for complexes 1 and 2, respectively. These values fall in the range observed for 1:2 electrolytes supporting ionic formulation of 1 and 2.37
Thermogravimetric analyses
Thermogravimetric analyses of 1 and 2 were carried out under nitrogen atmosphere to examine the stability of complexes at elevated temperature. The temperature ranges from 120–240 °C corresponds to the loss of water molecules in 1 accompanied by a weight loss of 65.16% (calculated%: 64.68) while in case of 2, it occured in the range of 60–200 °C with a weight loss of 58.61% (calculated%: 58.56). Furthermore, no significant stabilization in TGA plot has been observed for 1 and 2 corresponding to loss of both the organic ligands even up to 600 °C, thereby leading to the formation of metal oxides as residues (Fig. S8†).Single crystal X-ray description
The molecular structure of 1 was crystallized in the triclinic with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The crystal structure unit of 1 comprises of cationic part having copper(II) bis-DACH moiety at the square base with two H2O molecules at two axial positions, thereby adopting an elongated octahedral geometry while two mefenamato (mef−) anions were located outside the coordination sphere as counter ions (Fig. 2a). The asymmetric unit of 1 revealed copper(II) ion in tetragonally distorted octahedral environment with four nitrogen atoms of DACH at the equatorial positions, having bond distances Cu(1)–N(1)/N(4) = 2.019 Å, Cu(1)–N(3) = 2.018 Å and Cu(1)–N(2) = 2.005 Å. The vacant axial Jahn–Teller positions are occupied by O atom of water with bond distances Cu(1)–O(2w) = 2.619 Å and Cu(1)–O(3w) = 2.522 Å. The Cu–N/and–O bond distances were found to be in agreement with those of similar reported copper complexes in octahedral geometry.38 The crystal architecture of 1 was dominated by inter–/intra molecular hydrogen bonding interactions that were observed between the carboxylato oxygen atoms (O2 and O1) of one mefenamato (mef−) anion and the amino hydrogen atom H(N1) and H(N3) such that O2⋯H1 = 3.065 Å and O1⋯H1 = 2.938 Å (intramolecular). Moreover, the intermolecular H-bonds were observed between the carboxylato oxygen atoms (O2) of one mef− ion and O atom of water molecule such that O(2w)–H⋯O = 2.812 Å (Fig. 2b). The carboxylato oxygen atom (O5) of the other mef− ion makes H-bond with O atom of water molecule such that O(5w)–H⋯O = 3.005 Å. Also, 1 exhibited packing on the basis of π–π stacking interactions between centrosymmetrically orientated phenyl rings in the crystal lattice that reinforces the crystal architecture and plays a significant role in the stabilization of supramolecular framework.
space group. The crystal structure unit of 1 comprises of cationic part having copper(II) bis-DACH moiety at the square base with two H2O molecules at two axial positions, thereby adopting an elongated octahedral geometry while two mefenamato (mef−) anions were located outside the coordination sphere as counter ions (Fig. 2a). The asymmetric unit of 1 revealed copper(II) ion in tetragonally distorted octahedral environment with four nitrogen atoms of DACH at the equatorial positions, having bond distances Cu(1)–N(1)/N(4) = 2.019 Å, Cu(1)–N(3) = 2.018 Å and Cu(1)–N(2) = 2.005 Å. The vacant axial Jahn–Teller positions are occupied by O atom of water with bond distances Cu(1)–O(2w) = 2.619 Å and Cu(1)–O(3w) = 2.522 Å. The Cu–N/and–O bond distances were found to be in agreement with those of similar reported copper complexes in octahedral geometry.38 The crystal architecture of 1 was dominated by inter–/intra molecular hydrogen bonding interactions that were observed between the carboxylato oxygen atoms (O2 and O1) of one mefenamato (mef−) anion and the amino hydrogen atom H(N1) and H(N3) such that O2⋯H1 = 3.065 Å and O1⋯H1 = 2.938 Å (intramolecular). Moreover, the intermolecular H-bonds were observed between the carboxylato oxygen atoms (O2) of one mef− ion and O atom of water molecule such that O(2w)–H⋯O = 2.812 Å (Fig. 2b). The carboxylato oxygen atom (O5) of the other mef− ion makes H-bond with O atom of water molecule such that O(5w)–H⋯O = 3.005 Å. Also, 1 exhibited packing on the basis of π–π stacking interactions between centrosymmetrically orientated phenyl rings in the crystal lattice that reinforces the crystal architecture and plays a significant role in the stabilization of supramolecular framework.
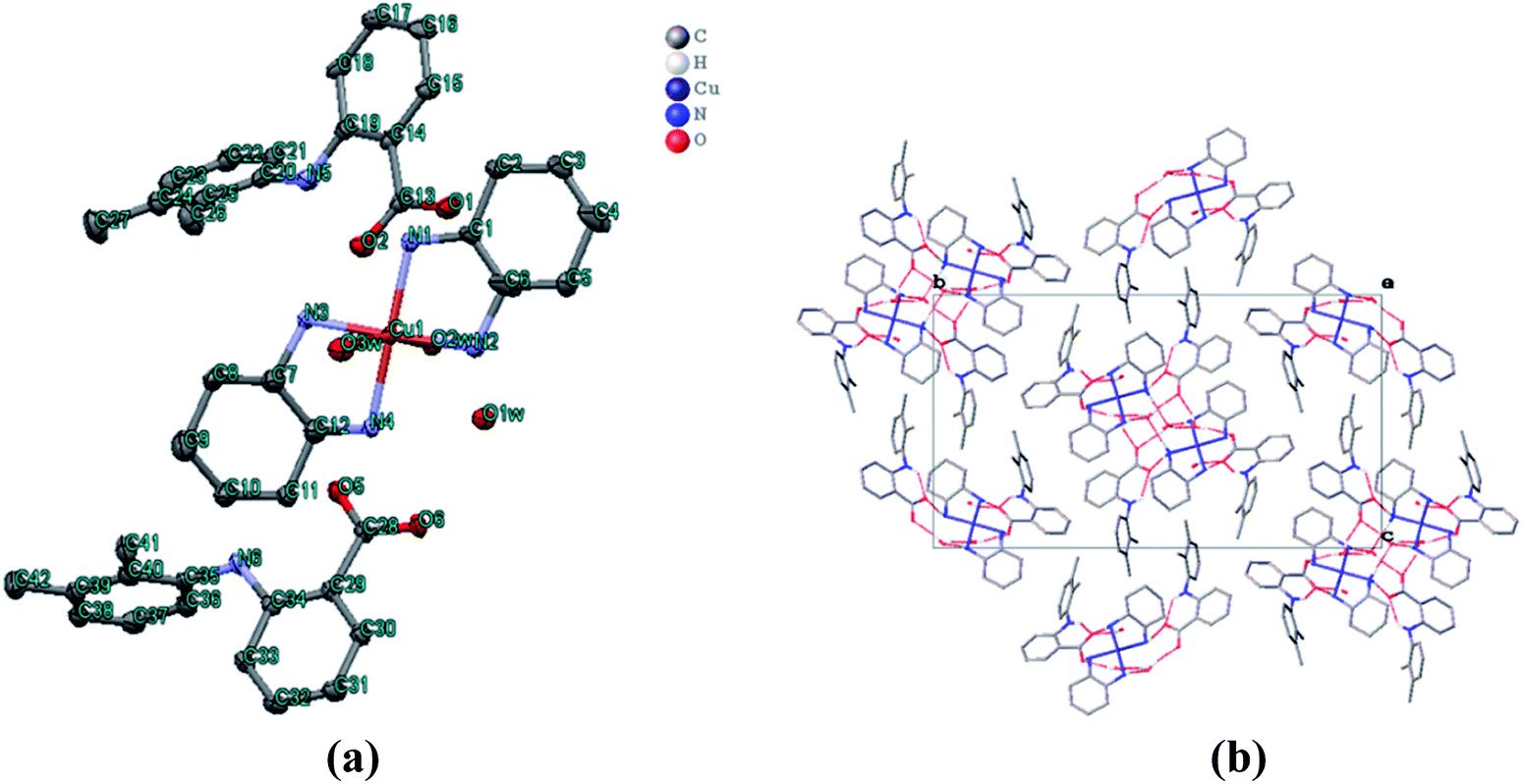 | ||
| Fig. 2 (a) ORTEP diagram of complex 1, [{Cu(DACH)2(H2O)2}(mef)2] along with the atom numbering scheme. The hydrogen atoms have been omitted for clarity. (b) Packing diagram of1showing intra/inter molecular hydrogen bonding. | ||
Hirshfeld surface analysis
The nature of intermolecular interactions in 1 was analyzed through Hirshfeld surfaces and two-dimensional fingerprint plots delineating surfaces that have been mapped over dnorm (−0.5 to 1.5 Å), shape index (−1.0 to 1.0 Å) and curvedness (−4.0 to 0.4 Å) (Fig. 3). The dnorm surface highlighted various close intermolecular interactions in which the red region corresponds to closer intermolecular contacts, whereas the blue region represents larger intermolecular contacts than the van der Waals radii. The deep-red regions observed on the dnorm surfaces of 1 describe close interactions, mainly responsible for H⋯H and C⋯H bonding in the complex. The shape index of 1 depicted red concave regions with ‘bow-tie patterns’ which correspond to C–H⋯π interactions indicating the presence of aromatic stacking interactions whereas the curvedness depicted the electron density present around various intermolecular interactions. | ||
| Fig. 3 Hirshfeld surface map representing (a) dnorm, (b) shape index and (c) curvedness of complex 1. | ||
The 2D-fingerprint plots for 1 and those delineated into C⋯C, C⋯H, N⋯H, H⋯H and O⋯H contacts are illustrated in Fig. 4. The plots were demonstrated by using the expanded 0.6–2.8 Å view with the di and de distance scales displayed on the graph axis. The fingerprint plots described into H⋯H contacts clearly indicate that these interactions made the most significant contribution to the overall Hirshfeld surface, i.e., 67.9%. In the fingerprint plots delineated into C⋯H contacts, the cluster of blue points with (de + di)min ∼ 2.7 Å corresponded to 17.8% contributions to the Hirshfeld surface. The contribution from O⋯H contacts to the Hirshfeld surface i.e., 13.7% reflected the presence of intermolecular C–H⋯O interactions. A pair of sharp spikes having almost equal length with tips at (de + di) ∼ 2.15 Å were characteristic of C–H⋯O bonded chain in 1. The small but significant contributions from C⋯C (0.5%) and N⋯H (0.1%) contact were found due to the short interatomic distances.
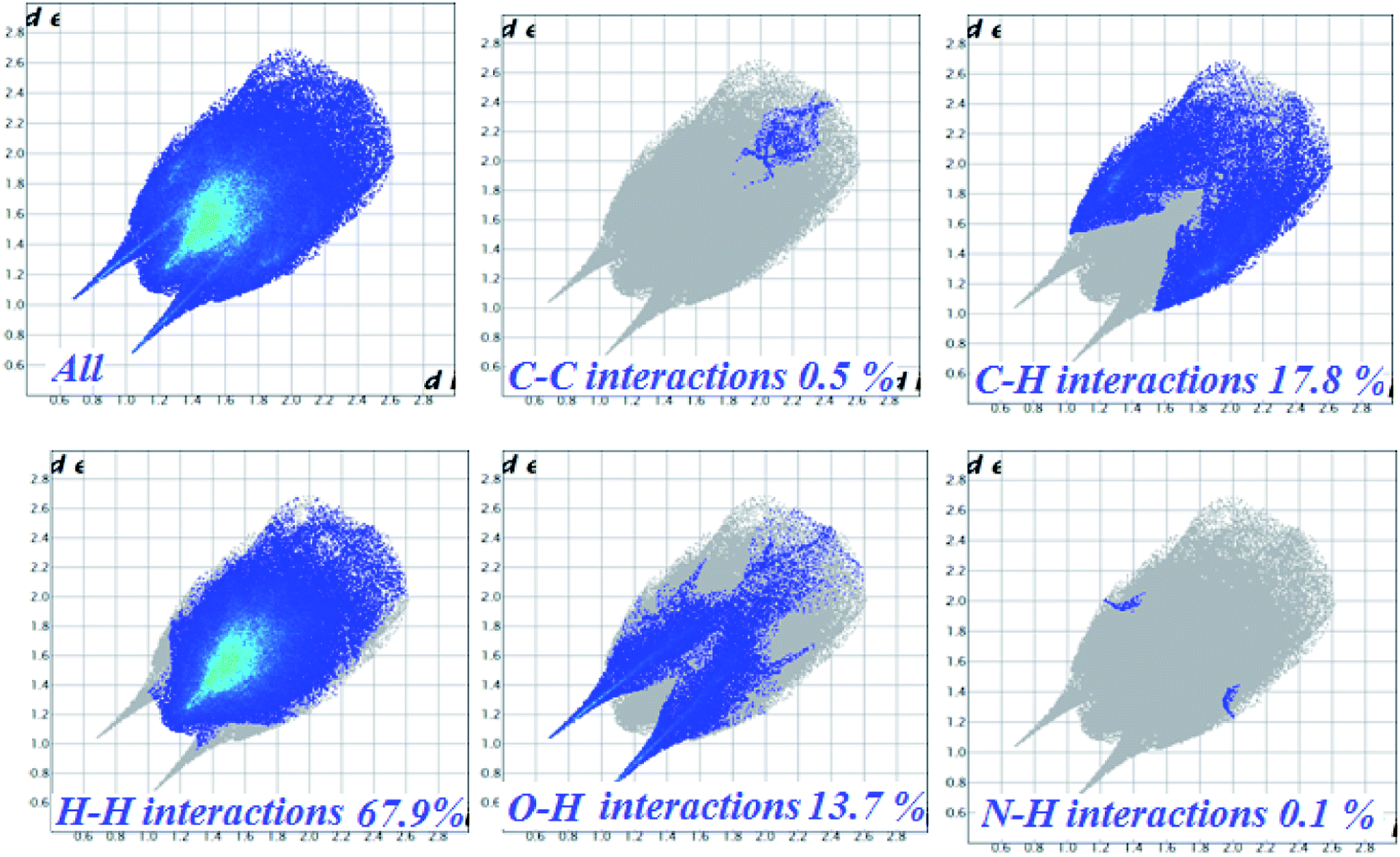 | ||
| Fig. 4 Fingerprint plots of complex 1, full (left) and resolved into C⋯C, C⋯H, H⋯H, O⋯H and N⋯H contacts showing the percentage of contacts contributing to the total Hirshfeld surface area of molecules. | ||
Interaction studies with nucleic acids (DNA and RNA)
Since DNA is the pharmacological target of most of the clinically approved antitumor drugs, and therefore, targeting DNA by these complexes seems intuitively appealing and logical. However RNA's unique structural diversity {hairpin, bulge or stem-loop and pseudoknots} could be utilized to design drug entities to treat various infectious and chronic diseases. Since RNA possess unique binding pockets for the recognition of small molecules, its structural diversity can be exploited to design new class of compounds that can interact with myriad RNA binding sites with high affinity and specificity. Many organic compounds such as aminoglycosides, tetracyclines, macrocycles including NSAIDs are known to modulate RNA function.39 While many metallo-drugs antitumor candidates were found to interact with DNA via electrostatic interactions as evidenced by characteristic ‘hyperchromic effect’ in the UV-vis titrations with DNA, there are fewer reports of interaction studies of these drug candidates with tRNA.Upon progressive addition of ctDNA/tRNA (0–4 × 10−5 M), the UV-vis spectra of 1 and 2 (0.067 × 10−5 M) at absorption band centered at 260 nm exhibited significant “hyperchromism” (48% and 38%, respectively) with tRNA (Fig. 5) and (44% and 31%, respectively) with ct-DNA (Fig. S9†). Similar behaviour of ionic metal–NSAID complexes in presence of ctDNA was previously reported for Cu(II) & Co(II) flufenamate complexes with DACH as co-ligand.13 From the observed “hyperchromic effect”, we can interpret the mechanism involved for the binding of 1 and 2 with ctDNA/tRNA which is presumed to be mainly electrostatic. In our hypothesis, we propose that ionic NSAIDs containing cationic metal bis(diamminocyclohexane) moiety are capable of strong electrostatic interactions with dinegative oxygen atom of phosphate sugar backbone of nucleic acids, causing a contraction and ultimately breakage of DNA/RNA structure. Furthermore, the extent of hyperchromism observed for tRNA is greater in magnitude than with ctDNA implicating more avid high binding affinity for 3D folding region of an RNA chain, perhaps in a very specific manner. Additionally, mefenamato anions could also be engaged in hydrogen bonding with nitrogen atoms of the nucleobases which was validated further by molecular docking experiments.
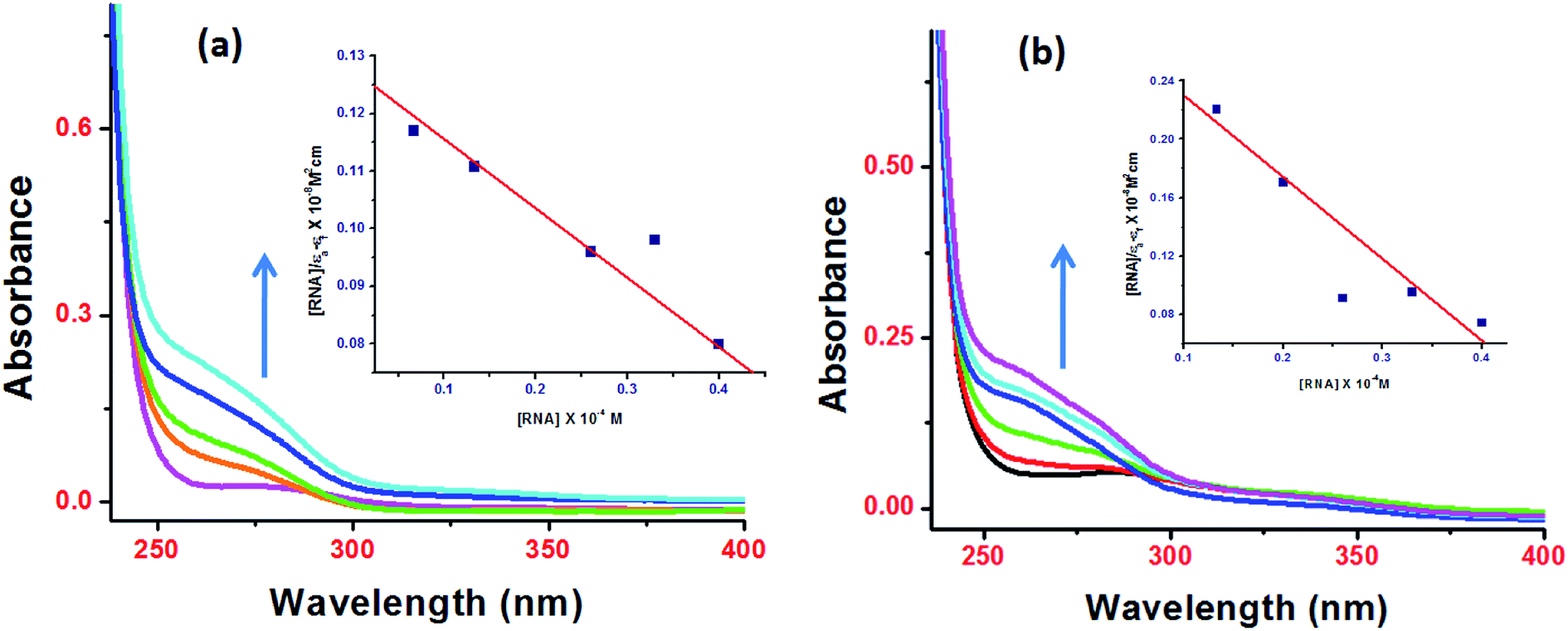 | ||
| Fig. 5 Absorption spectra of complexes 1 (a) and 2 (b) in presence of increasing tRNA concentration. | ||
The binding strength of 1 and 2 toward ctDNA/tRNA was quantified in terms of intrinsic binding constant, Kb values as shown in Table 1. The Kb values validate that 1 and 2 were preferentially bound to tRNA as compared to ctDNA, with copper–mefenamato 1 exhibiting the higher binding constant with tRNA [Kb = 2.52(±0.04) × 105 M−1] than their Zn(II) analogue 2 attenuating the effect of different transition metal ions which can also be correlated to mutual synergetic effect between copper and mefenamic acid.40 The results of binding experiments were in good agreement with the cytotoxicity studies. Furthermore, 1 exhibited significantly pronounced cytotoxicity with tested human cancer cell lines as compared to 2.
| Complexes | Kb values (M−1) | K values (M−1) | Ksv values | % hyperchromism | ||||
|---|---|---|---|---|---|---|---|---|
| ctDNA | tRNA | ctDNA | tRNA | ctDNA | tRNA | ctDNA | tRNA | |
| 1 | 1.16(±0.05) × 105 | 2.52(±0.04) × 105 | 1.71(±0.06) × 105 | 2.30(±0.03) × 105 | 1.05 | 3.00 | 44 | 48 |
| 2 | 3.12(±0.08) × 104 | 7.85(±0.02) × 104 | 1.65(±0.05) × 104 | 5.27(±0.04) × 104 | 0.68 | 0.77 | 31 | 38 |
Fluorescence spectra of 1 and 2, at their fixed concentration (0.067 × 10−4 M) were recorded without and after concomitant addition of ctDNA/tRNA under similar physiological conditions as in UV experiments which demonstrated an enhancement in the fluorescence intensity (Fig. S10 and S11†). The enhancement in the emission intensity arises due to the penetration of complexes into the hydrophobic environment of interior tRNA/ctDNA helixes as has been reported in many research articles describing the interaction studies of small molecules with nucleic acids.41–43 As studies revealed greater binding propensity for tRNA as compared to ctDNA which could be attributed to the conformation of RNA having an L-shaped tertiary structure, where nucleobases are pushed outwards from the helix axis in the minor groove direction and tilted substantially with respect to the helix axis, funneling the complexes to the interior of the narrow grooves of RNA. Furthermore binding strength of 1 and 2 was quantified by employing the Scatchard equation44 and the evaluated data is shown in Table 1.
Steady state competitive binding studies using ethidium bromide (EB) as a fluorescent probe were also performed for both the complexes. The decrease in the peak intensities of 1 and 2 was observed in presence of ctDNA/tRNA with progressive addition of 1 and 2 in the ratio of 1:1 to 6:1 (Fig. S12 and S13†) which showed significant quenching of fluorescence probe ethidium bromide. However, the observed decrease in emission intensity was found to be lower as compared to classical intercalators,45 ruling out the possibility of an intercalative mode and confirmed the external groove binding nature of 1 and 2.
Quenching efficiency is related to the Stern–Volmer constant which could be determined by equation;
| I0/I = 1 + Ksvr |
The calculated values of Ksv for both 1 and 2 are summarized in Table 1. The resulting Kb, K and Ksv values validated that complex 1 exhibited the higher binding constant values with tRNA as compared to ctDNA.
Circular dichroism (CD), a sensitive optical technique was further used to analyze the conformational changes in ctDNA/tRNA and the binding modes of metal-based mefenamate drug candidates 1 and 2.
On incubation of 1 and 2 to ctDNA (r = [complex 1/complex 2]/[ctDNA] = 1), a decrease in intensity in characteristic stacking and helicity bands at 245 and 275 nm, respectively was observed (Fig. S14†) without any apparent shift in band positions. These negligible perturbations in both base signatures revealed that the complexes possibly unwind the secondary structure of DNA helix leading to the loss of helicity of DNA.
On the contrary, the CD spectrum of tRNA depicted characteristic peaks (two positive peaks at 227 nm and 270 nm & two negative peaks at 210 nm and 240 nm). Upon addition of 1 and 2 to tRNA solution, a significant decrease in the intensity of a negative band at 240 nm was observed with a blue shift while positive bands at 227 nm and 270 nm showed an amplification in the respective bands. However, the band at 270 nm exhibited no band shifting (Fig. 6). The major intensity changes of the bands at 210 nm, 227 nm and 240 nm could be due to tRNA aggregation upon addition of complexes.46
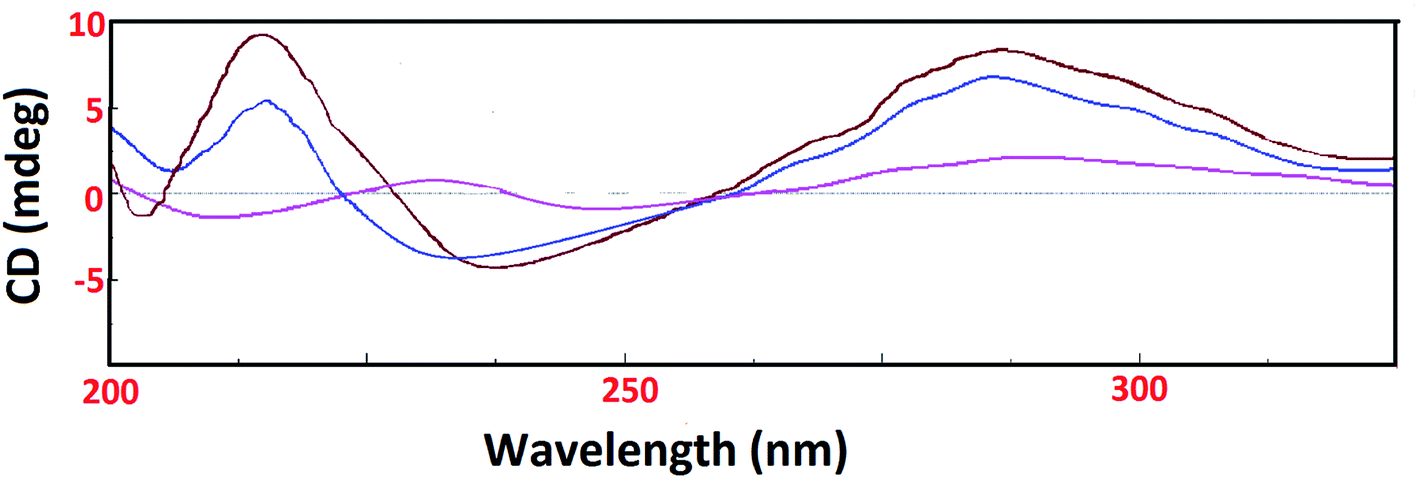 | ||
| Fig. 6 CD spectra of tRNA alone (pink) and in presence of complexes 1 (blue) and 2 (brown) in Tris–HCl buffer at 25 °C. [Complex 1/2] = 1 × 10−4 M, [tRNA] = 1 × 10−4 M. | ||
Metallo-drug complexes are known to possess unique electrochemical behavior due to different redox potential and were investigated for their anticancer potential via cyclic voltammetric binding studies.47 The variations in the votammetric responses of redox active metal complex either in terms of changes in current or potential or both can be further utilized to determine their interaction with ctDNA/tRNA.
The electrochemical investigations of complex–ctDNA/tRNA interaction were carried out in DMSO solution at room temperature at a scan rate of 0.2 V s−1 by cyclic voltammetry in the potential range of −1 to 1 V. The CV of 1 demonstrated a quasi–reversible one electron redox process involving a shuttle between Cu(II)/Cu(I) redox pairs with two anodic waves at 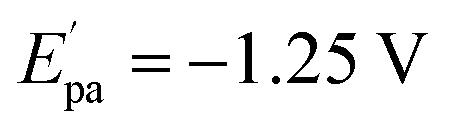 and Epa = −0.75 V followed by a cathodic peak at Epc = −0.50 V. The current intensity
and Epa = −0.75 V followed by a cathodic peak at Epc = −0.50 V. The current intensity 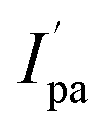 and Ipa were observed at −0.00045 A and −0.0005 A. Two anodic waves at Epa and
and Ipa were observed at −0.00045 A and −0.0005 A. Two anodic waves at Epa and 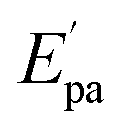 can be attributed to the oxidation of metallic copper and Cu(I), respectively. The CV of 2 featured reduction of Zn(II)/Zn(I) form at a cathodic peak potential of Epc = −0.50 V and an oxidation peak appeared at Epa = −0.10 V (Fig. 7)
can be attributed to the oxidation of metallic copper and Cu(I), respectively. The CV of 2 featured reduction of Zn(II)/Zn(I) form at a cathodic peak potential of Epc = −0.50 V and an oxidation peak appeared at Epa = −0.10 V (Fig. 7)
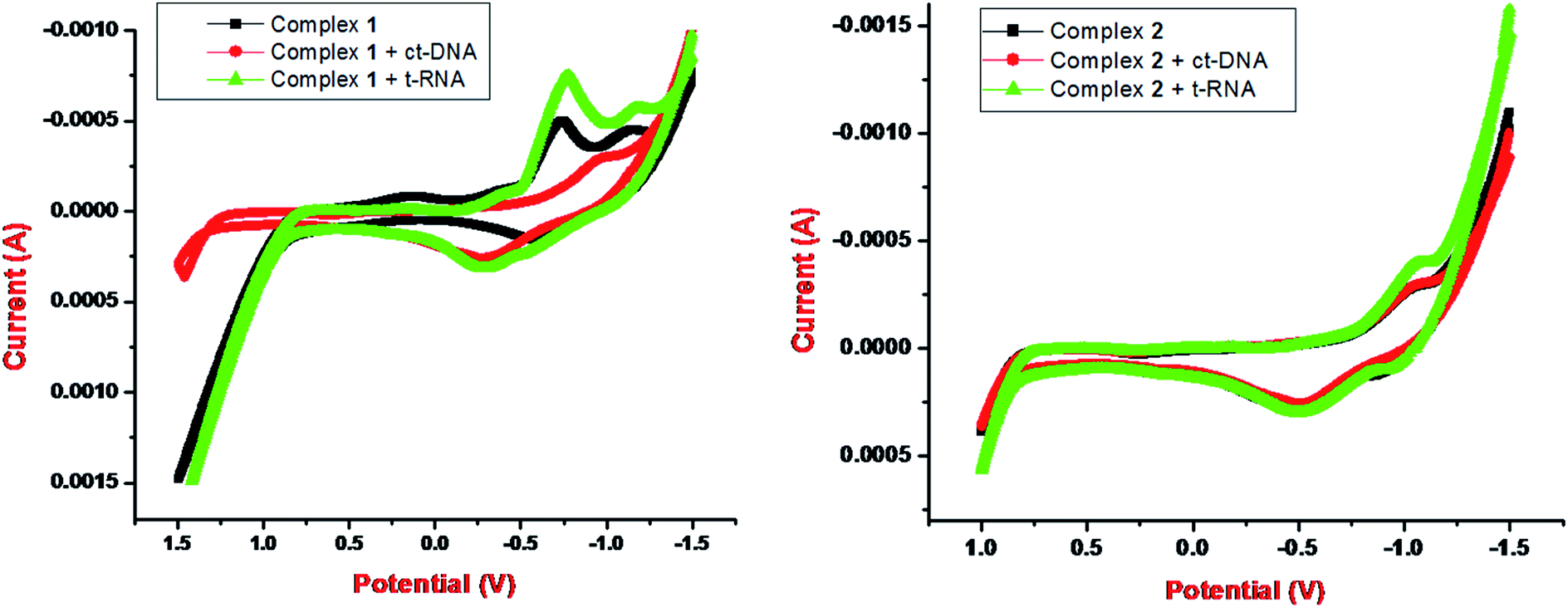 | ||
| Fig. 7 Cyclic voltammogram (1:2 DMSO: buffer solution, 25 °C) of 1 and 2 in the presence of tRNA. | ||
On incubation with ctDNA, anodic peak potential, Epa and the current intensity Ipa of complex 1 was observed at −1.0 V and −0.00025 A, respectively. The shift in peak potential and the current intensity was observed as ΔEpa = −0.25 V and ΔIpa = 0.00025 with ct-DNA. However, with tRNA, anodic peak potential, Epa and the current intensity Ipa were observed at −0.8 V and −0.00075 A, respectively. The shift in peak potential and the current intensity was observed as ΔEpa = −0.05 V and ΔIpa = −0.00025 A with t-RNA. Our results implicated that greater shifts in potential parameters and decrease in current ratios of complex 1 with tRNA was observed as compared with DNA. This indicated an interaction between each complex and ctDNA/tRNA explained in terms of an equilibrium mixture of free and DNA/RNA bound complexes to the electrode surface.42,48
Morphological studies
Recently, the structural studies of complex-DNA/RNA condensates were utilized to identify the specific interactions of drug candidates to ctDNA/tRNA. Controllable drug DNA/RNA condensation is a newer approach for transportation of drug candidates across the biological membranes to specific target at the molecular level.49 RNA possesses highly structured active sites that can dictate the formation of different shape particles due to its thermodynamic stability, base stacking capabilities and tertiary interactions.50 The molecular interactions between DNA/RNA and the cationic component determine many vital biological processes such as recombination, replication, transcription and repair.51 Literature revealed that morphology of condensates vary with the change in solution properties (ionic strength and solvent polarity), nature of condensing agent (charge density) and the surface of substrate.52 In the present study, DNA/RNA condensates of 1 and 2 were prepared by evaporating an equimolar mixture of 1, 2 and ctDNA/tRNA in Tris–HCl buffer. The scanning electron microscopy (SEM) analyses revealed that 1 displayed hollow rod like structures while 2 showed some concrete rock like structures(Fig. 8a and d). The DNA/RNA molecules upon condensation with 1 and 2 represented different structural features i.e., mixing of complexes and DNA/RNA particles. This clearly indicated the condensation of ctDNA/tRNA molecules into a compact, massive structures. Such SEM micrograph patterns reveal that hydrophobic interaction of 1 and 2 has taken place via oxygen atoms of mefenamato carboxylate moiety with nucleobases of ctDNA/tRNA which resulted in lengthening of ctDNA/tRNA helix due to insertion of 1 and 2 in the grooves of DNA helix/bulge region of tRNA, which could be also be envisaged as ‘threading partial intercalation’.13 | ||
| Fig. 8 SEM images showing surface morphology of complex 1 (a) & 2 (d) alone and b (1) & (e) 2 upon condensation with ctDNA. (c) 1 & (f) 2 upon condensation with tRNA. | ||
Molecular modelling studies
Docking Simulation is a useful tool to understand the complex–nucleic acid interactions and also gaining a better understanding of the experimental results. Docked complexes conformation was analyzed in terms of energy and hydrophobic interaction with drug candidates and B-DNA (PDB ID = 1BNA)/tRNA (PDB ID = 6TNA). From the docking scores, the free energy of binding of complexes 1 and 2 was calculated and results are shown in Table 2.| Complexes | Binding energy (kJ mol−1) | |
|---|---|---|
| ctDNA | tRNA | |
| 1 | −260 | −306.45 |
| 2 | −178.72 | −283.45 |
The docking results revealed that 1 interacts with the DNA helix through the G–C region present inside the minor groove region. The complex 1 was found in the close proximity of C21, G22, C23, G24, G2, C3 and G4 DNA base pairs (Fig. 9b). On the other hand, complex 2 revealed preferential binding within the wide A–T region of major groove of the DNA helix (Fig. S15b†). The minor groove is particularly an attractive target for such molecules as the closer proximity of the strands allows more intimate contact in surface area and tightly binds with best fitting.53
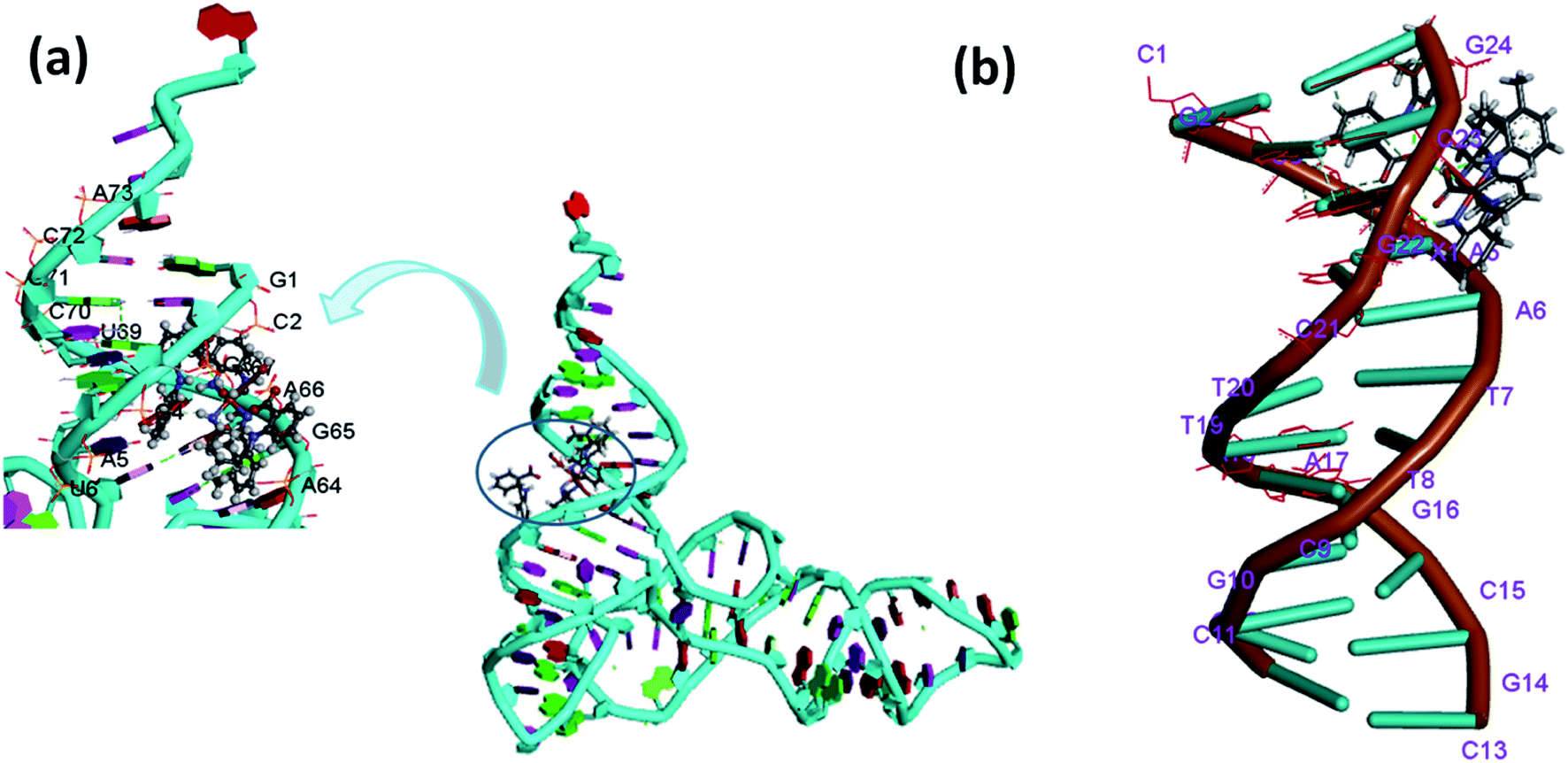 | ||
| Fig. 9 Molecular docked model of complex 1 with (a) tRNA (b) ctDNA. | ||
It is well known that tRNA is characterized by well defined 3D structure regions like D arm, acceptor stem, T arm, ψ loop and anticodon arm. The docking results with tRNA implicated that both the complexes 1 and 2 were inserted into the active pocket located between upper and lower stem in close proximity to C-2, A-64, A-66, A-38, G-65, C-40, U-69 and G-4.54 (Fig. 9a and S15a†). The results obtained in terms of free energy of binding and hydrophobic interaction correlate well with other experimental spectroscopic techniques suggesting 1 possess a higher binding ability with tRNA as compared to ctDNA.
In vitro cytotoxic evaluation
The ionic mefenamato complexes 1 and 2 were tested for their antiproliferative activity in vitro against five human cancer cell lines namely; MCF-7(breast), HeLa (cervix), A-498 (Kidney), MIA-PA-CA-2 (Pancreas), Hep-G2 (Hepatoma) by SRB assay. The values have been evaluated in terms of GI50, TGI and LC50 for both the complexes and are summarized in Table 3. Complex 1 showed significantly low GI50 value (<1 μM) against all the tested cancer cell lines except MIA-PA-CA 2 cell line. However, Complex 2 revealed moderate cytotoxicity against the mentioned cell lines. (Fig. S16†) Since, Copper accumulates in tumor due to selective permeability of the cancer cell membranes to copper compounds; thus, a number of copper complexes have been screened for anticancer activity and some of them were found to be active in vivo and in vitro. It was earlier reported in the literature that metallotherapeutics of NSAIDs containing Cu(II) ion bound more strongly to nucleotides55 thus, inhibiting the proliferation of cancer cells. In this way, we observed a positive correlation between the cytotoxic activity and binding affinity of complexes 1 and 2 which implicated that NSAIDs Cu(II) complex 1 with strong binding propensity results in inhibition of cancer cells at very low micromolar concentration causing cell death. The morphological changes observed in the cell lines exposed to complex 1 and 2 for a period of 48 h are shown in Fig. 10 which implicated that on treating these cell lines with complex 1 and 2, a reduction in morphology as well as cell adhesion capacity was observed as compared to the control drug.| Cell line | MCF-7 | HeLa | A-498 | MIA-PA-CA-2 | Hep-G2 | |
|---|---|---|---|---|---|---|
| a GI50 = growth inhibition of 50% (GI50) calculated from [(Ti − Tz)/(C − Tz)] × 100 = 50, drug concentration that results in a 50% reduction in the net protein increase. ADR = adriamycin (positive control). GI50 value < 10 μg ml−1 is considered to demonstrate activity. TGI = tumor growth inhibition. LC50 = lethal concentration of 50% (LC50). | ||||||
| GI50 | Complex 1 | <1 | <1 | <1 | 1.85 | <1 |
| Complex 2 | 1.826 | 3.204 | 3.21 | 3.172 | 4.406 | |
| ADR | <1 | <1 | <1 | <1 | <1 | |
| TGI | Complex 1 | 5.16 | 1.50 | 1.67 | 6.93 | 2.92 |
| Complex 2 | 4.80 | 5.35 | 5.46 | 5.57 | 6.26 | |
| ADR | 3.93 | <1 | <1 | <1 | <1 | |
| LC50 | Complex 1 | NE | 5.29 | 5.71 | NE | 5.94 |
| Complex 2 | 7.77 | 7.49 | 7.71 | 7.97 | >8 | |
| ADR | NE | NE | NE | NE | 4.19 | |
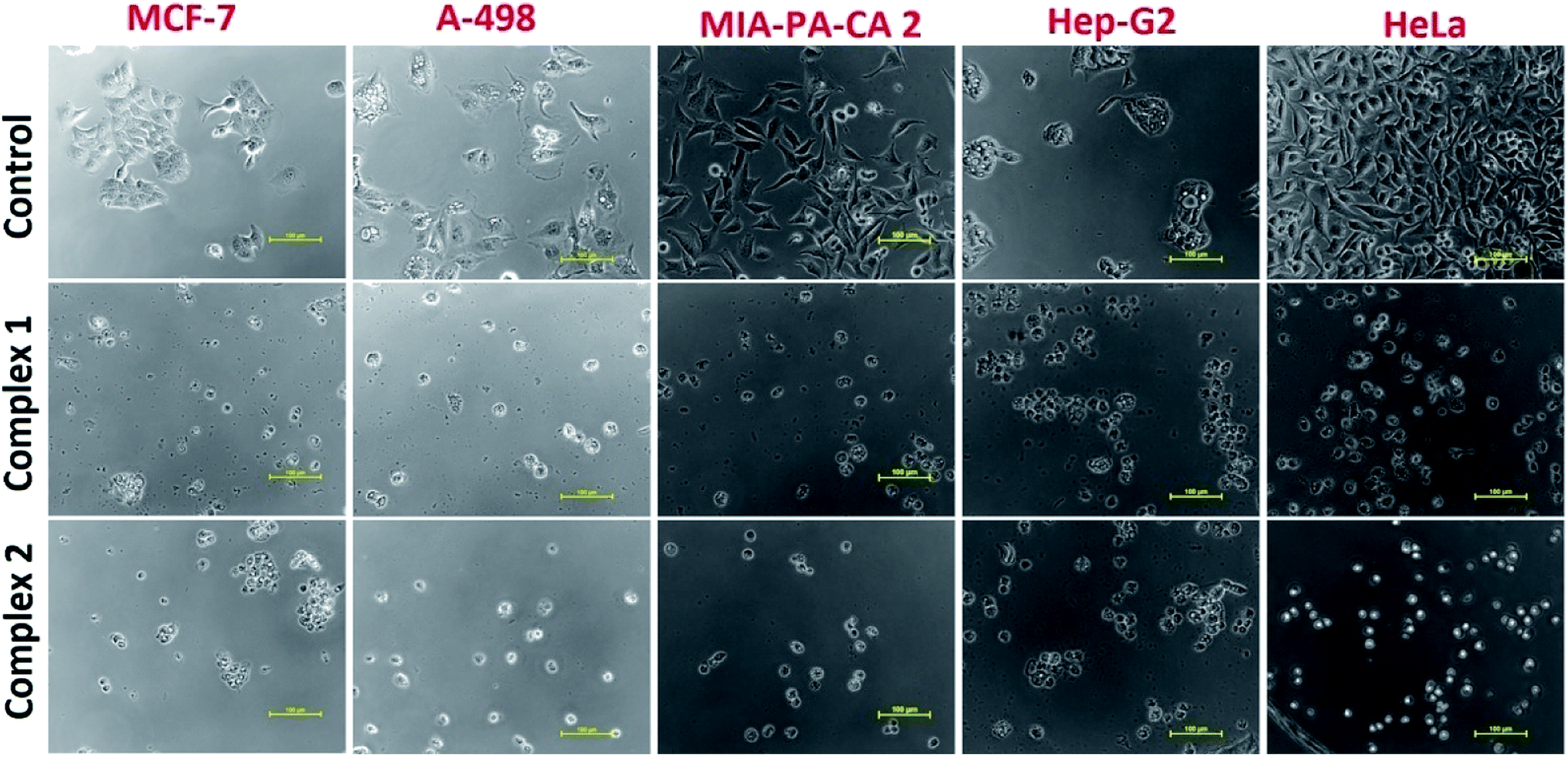 | ||
| Fig. 10 Morphological changes observed in cancer cell lines on treating with complexes 1 and 2. | ||
Conclusions
Two novel ionic [Cu(DACH)2(H2O)2](mef)2, 1 and [Zn(DACH)2(H2O)2](mef)2, 2 antitumor drug conjugates were synthesized and thoroughly characterized. Single-crystal X-ray structure determination of 1 confirmed that each DACH ligand coordinates to copper(II) ion through two nitrogen atoms at the equatorial positions in tetragonally distorted octahedral environment. The vacant axial Jahn–Teller positions are occupied by O atom of water molecules while two mefenamato (mef−) anions acted as counter ions. The interaction studies of these complexes with nucleic acids (ctDNA/tRNA) were carried out by UV-vis, fluorescence, circular dichroic and cyclic voltammetric studies which demonstrated their preferential binding with tRNA as compared to ctDNA through electrostatic interaction. Ionic NSAID complexes containing cationic metal bis(diamminocyclohexane) moiety and mefenamato anions could interact with dinegative oxygen atom of phosphate sugar backbone of nucleic acid helix by electrostatic mode of binding. Furthermore, mefenamato anions could also be engaged in hydrogen bonding with nitrogen atoms of RNA nucleobases. These experiments have demonstrated higher binding propensity of copper complex 1 as compared to its zinc analogue 2. Complexes 1 and 2 were screened for cytotoxic activity against MCF-7 (breast), HeLa (cervical), MIA-PA-CA 2 (pancreatic), A-498 (kidney), Hep-G2 (hepatoma) cell lines. Complex 1 was found to be an outstanding cytotoxic agent with remarkably good GI50 value (<10 μg ml−1) against the tested cancer cell lines except for MIA-PA-CA 2, while complex 2 revealed moderate cytotoxicity against all the tested cancer cell lines. This study leads to the development of new copper-based NSAID promising antitumor drug candidate. Further, it gives impetus to the understanding of the mechanisms of action involved in the process of cancer cell inhibition which can be instrumental for rational development of more efficacious RNA targeted NSAID therapeutics.Experimental
Materials and instrumentation
Mefenamic acid, 1,2–diaminocyclohexane(Sigma Aldrich), KOH, CuCl2·2H2O and ZnCl2·2H2O (Merck) were purchased and used as received. All the solvents used were of purity grade. Elemental analysis was carried out on a Carlo Erba Analyser Model 1106. Infrared spectra were obtained (KBr disk, 400–4000 cm−1) on a Perkin–Elmer Model 1320 spectrometer. The EPR spectrum of the 1 was acquired at X-band frequency (9.167 GHz) at room temperature on JEOL FA-200 continuous wave spectrophotometer (g = 2.02055). NMR spectra of 2 were obtained on a Bruker DRX-400 spectrometer with Me2SO-d6 as solvent. Electro spray ionization mass (ESI-MS) spectra were recorded on a Waters (Micromass MS technologies) Q-Tof MS analyzer spectrometer. Molar conductance was measured on Eutech con 510 electronic conductivity bridge. The TGA analysis of 1 and 2 was performed on Schimadzu DTG-60H analyzer under nitrogen atmosphere from 20 to 850 °C at a heating rate of 20 °C min−1. Electronic spectra were recorded on a PerkinElmer Lambda 25 using cuvettes of 1 cm path length, and the data were reported in λmax/nm.The experiments involving the interaction of the complexes with ctDNA/tRNA were carried out in Tris–HCl buffer (pH = 7.3). The concentration per base pairs for both DNA and RNA was determined spectrophotometrically at 260 nm assuming ε = 6600 and 7700 M−1 cm−1.56 Emission spectra were acquired on a Shimadzu RF-5301 PC spectrofluorophotometer. CD spectra were measured on a Jasco J-815-CD spectropolarimeter at room temperature. Cyclic voltammetry was carried out at CH instrument electrochemical analyzer. High purity H2O and DMSO (95:5) was employed for the cyclic voltammetry studies with 0.4 M KNO3 as a supporting electrolyte. A three electrode configuration was used comprising of a Pt disk working electrode, Pt wire counter electrode and Ag/AgCl as reference electrode. Electrochemical measurements were made under N2 atmosphere.
Hirshfeld surface analysis
Hirshfeld analysis along with 2D fingerprint plot for complex 1 was generated using crystal Explorer 3.1 (ref. 57) software based on results of single crystal X-ray diffraction studies.Synthesis of [Cu(DACH)2(H2O)2](mef)2, 1
Complex 1 was synthesized by reacting methanolic solution of mefenamic acid (0.482 g, 2 mmol, deprotonated with KOH), 1,2-diaminocyclohexane (0.228 g, 2 mmol and CuCl2·2H2O (0.170 g, 1 mmol)) in a molar ratio of 2:2:1.The reaction mixture was stirred for ca. 3 h till the completion of reaction and then kept aside at room temperature. Violet crystals appeared after a few days, which were separated from the mother liquor and dried in air. Complex 1 was found to be air stable and soluble in organic solvents like MeOH, DMF and DMSO. The molecular structure of 1 was established by single crystal X-ray diffraction studies and other spectroscopic techniques.
:2 electrolyte). Anal. calc. for [C42H60N6O6Cu] (%): C, 62.39; H, 7.48; N, 10.39; found: C, 62.28; H, 7.13; N, 10.72. UV-vis(1× 10−4 M, DMSO, λmax nm): 259 (π–π*), 305 (n–π*), 630 (d–d). FT–IR (KBr pallet, υmax/cm−1):3235 ν(O–H), 3128 ν(N–H), 2929 ν(CH2), 1576ν(CO), 563 ν(Cu–N), 427 ν(Cu–O). g‖ = 2.197, g⊥ = 2.049. ESI-MS (m/z): 792.59 [C42H60N6O6Cu + 3H+–H2O]+, 643.23 [2{C15H14NO2} + 2H2O + N4H8Cu–2C6H10], 599 [C42H60N6O6Cu–DACH–2H2O–Cu + 5H+], 335 [C42H60N6O6Cu–2C15H14NO2 + 8H+], 286 [C12H28CuN4 + 5H+].Synthesis of [Zn(DACH)2(H2O)2](mef)2, 2
Complex 2 was synthesized in a similar manner as complex 1 with the use of ZnCl2·2H2O instead of CuCl2·2H2O till the completion of reaction (which was monitored on TLC). The resulting solution was kept aside for slow evaporation at room temperature. Brown precipitate was collected after few days which was soluble in organic solvents like MeOH, DMF and DMSO.:2 electrolyte). Anal. calc. for [C42H60N6O6Zn] (%): C, 62.25; H, 7.46; N, 10.37; found: C, 62.72; H, 7.55; N, 10.23. UV-vis(1 × 10−4 M, DMSO, λmax nm): 260 (π–π*), 332 (n–π*). FT-IR (KBr pallet, υmax/cm−1):3545ν(O–H), 3135ν(N–H), 2932 ν(CH2), 1579 ν(CO), 596 ν(Zn–N), 424 ν(Zn–O).1H NMR (400 MHz, CDCl3-d6, δ, ppm): 1.37–1.12 ppm and 1.73–1.63 ppm (complex m, 8H, –CH2– of cyclohexyl), 2.18 (6H, s, H11–mef), 2.30 (6H, s, H10–mef), 2.88 (s, 4H, –NH2), 8.11 (s, H6–mef), 7.97 (d, H2–mef), 7.48 (m, H4–mef), 7.20 (d, H7–mef), 7.15 (m, H8-mef), 7.04 (m, H9–mef), 6.89 (d, H5–mef), 6.65 (m, H3–mef). 13C NMR (100 MHz, CDCl3–d6, δ, ppm): 182.72 ppm (COO−), 149.37 (C-2), 147.74 (C-1), 137.87 (C-3), 132.14 (C-4), 131.49 (C-5), 130.38 (C-6), 125.69 (C-7), 124.91 (C-8, 12), 120.51 (C-9, 10), 113.71 (C-11), 35.58 (–CH–cyclohexyl), 31.62–22.81 (–CH2 cyclohexyl ring carbons), 20.69, 14.08 (–CH3 group attached at β, α position to –NH group in aromatic moiety of mefenamic acid, respectively).ESI-MS (m/z): 613.4[C42H60N6O6Zn–DACH–H2O–Zn]+, 483.3 [2C15H14NO2 + 3H]+, 297.2 [C42H60N6O6Zn–2{C15H14NO2}−–2H2O + 5H]+.
X-ray diffraction studies
Single crystal X-ray data for 1 was collected on a Bruker SMART APEX CCD diffractometer (100 K) using graphite monochromatic Mo–Kα radiation (k = 0.71073 Å). The linear absorption coefficients, scattering factors for the atoms and the anomalous dispersion corrections were referred from the International Tables for X-ray crystallography. The data integration and reduction were carried out with SAINT software. Empirical absorption correction was applied to the collected reflections with SADABS program58 and the space group was determined using XPREP. The structure was solved by direct methods using SHELXTL-97 and refined on F2 by full-matrix least-squares using the SHELXTL-97 programme package.59 A summary of selected crystallographic information is given in table.| Crystal data and structure refinement for complex 1 | |
|---|---|
| CCDC | 1916426 |
| Empirical formula | C42H60CuN6O7 |
| Formula weight | 822.51 |
| Temperature/K | 100(2) |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a/Å | 7.6603(4) |
| b/Å | 30.6640(17) |
| c/Å | 17.5010(11) |
| α/° | 90 |
| β/° | 90.996(2) |
| γ/° | 90 |
| Volume/Å3 | 4110.3(4) |
| Z | 4 |
| ρcalc g cm−3 | 1.3291 |
| μ/mm−1 | 0.588 |
| F(000) | 1750.1 |
| Crystal size/mm3 | 0.33 × 0.21 × 0.16 |
| Radiation | Mo Kα (λ = 0.71073) |
| 2θ range for data collection/° | 4.66 to 50 |
| Index ranges | −10 ≤ h ≤ 10, −40 ≤ k ≤ 40, −23 ≤ l ≤ 23 |
| Reflections collected | 65619 |
| Independent reflections | 7222 [Rint = 0.0594, Rsigma = 0.0403] |
| Data/restraints/parameters | 7222/0/518 |
| Goodness-of-fit on F2 | 1.059 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0653, wR2 = 0.1411 |
| Final R indexes [all data] | R1 = 0.0747, wR2 = 0.1493 |
| Largest diff. peak/hole/e Å−3 | 1.86/−0.93 |
Morphological studies
The condensates for SEM analysis were prepared from an equimolar mixture of methanolic solution of complexes 1, 2 and ct-DNA/tRNA (Tris–HCl buffer, pH = 7.2). The samples were air dried at room temperature for 12 h. Before taking the SEM micrographs, the samples were coated with platinum using JEOL JFC-1600 auto fine coater at 20 mA. SEM was recorded on Zeiss supra 55VP scanning electron microscope. The observed images were captured and saved digitally.Cytotoxicity studies
The cytotoxic activity of 1 and 2 were evaluated against a panel of five human cancer cell lines viz., MIA-PACA-2 (Pancreatic), MCF-7 (breast), A-498 (Renal cell), HeLa (cervical) and Hep-G2 (Hepatoma) by SRB assay. Adriamycin, standard anticancer drug was taken as positive control. These human malignant cell lines were procured and grown in RPMI-1640 medium supplemented with 10% Fetal Bovine Serum (FBS) and antibiotics to study growth pattern of these cells. The proliferation of the cells upon treatment with chemotherapy was determined by SRB assay. The dose response parameters such as growth inhibition 50% (GI50), total growth inhibition (TGI) and lethal concentration 50% (LC50) were calculated. Cells were seeded in 96 well plates at an appropriate cell density to give optical density in the linear range (from 0.5 to 1.8) and were incubated at 37 °C in CO2 incubator for 24 h. Stock solutions of the complexes were prepared as 100 mg ml−1 in DMSO and four dilutions i.e. 10 μL, 20 μL, 40 μL, 80 μL, in triplicates were tested, each well receiving 90 μL of cell suspension and 10 μL of the drug solution. The plates were incubated further for 48 h. Termination of experiment was done by gently layering the cells with50 μL of chilled 30% TCA (in case of adherent cells) and 50% TCA (incase of suspension cell lines) for cell fixation and kept at 4 °C for 1 h. All experiments were repeated three times.Molecular docking
Molecular docking studies were performed using HEX 8.0 software,60 which is an interactive molecular graphics program for calculating and displaying feasible docking modes of enzymes and DNA molecules. Visualization of the docked pose was performed using the Discovery Studio molecular graphics program.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The author (Huzaifa Yasir Khan) gratefully acknowledges financial support from Department of Science and Technology, New Delhi for providing DSTINSPIRE Senior Research Fellowship (DST/AORC–IF/UPGRD/IF160130). The authors extend thanks to Dr Musheer Ahmad, Department of Applied Chemistry, Aligarh Muslim University, Aligarh for refining single X-ray crystal data of complex 1 and IIT Kanpur for solving the structure. The authors also extend their appreciation to the University Sophisticated Instruments Facility (USIF) and Instrumentation Centre, Department of Chemistry, Aligarh Muslim University, Aligarh for spectroscopic analysis. The authors are highly thankful to anticancer drugs screening facility (ACDSF) of ACTREC, Tata Memorial Centre, Navi Mumbai for carrying out in vitro anti-cancer activity of complexes by SRB assay.References
- J. N. Boodram, I. J. Mcgregor, P. M. Bruno, P. B. Cressey, M. T. Hemann and K. Suntharalingam, Angew. Chem., 2016, 128, 2895–2900 CrossRef.
- M. J. Thun, S. J. Henley and C. Patrono, J. Natl. Cancer Inst., 2002, 94, 252–266 CrossRef CAS PubMed.
- J. Cuzick, F. Otto, J. A. Baron, P. H. Brown, J. Burn, P. Greenwald, J. Jankowski, C. La Vecchia, F. Meyskens, H. J. Senn and M. Thun, Lancet Oncol., 2009, 10, 501–507 CrossRef CAS PubMed.
- C. N. Banti and S. K. Hadjikakou, Eur. J. Inorg. Chem., 2016, 3048–3071 CrossRef CAS.
- F. D. Hart and E. C. Huskisson, Drugs, 1984, 27, 232–255 CrossRef CAS PubMed.
- M. -L. Smith, G. Hawcroft and M. A. Hull, Eur. J. Cancer, 2000, 36, 664–674 CrossRef CAS PubMed.
- C. Sostres, C. J. Gargallo, M. T. Arroyo and A. Lanas, Best Pract. Res., Clin. Gastroenterol., 2010, 24, 121–132 CrossRef CAS PubMed.
- A. A. Gouda, M. I. Kotb EI-Sayed, A. S. Amin and R. EI Sheikh, Arabian J. Chem., 2013, 6, 145–163 CrossRef CAS.
- I. Yousuf, M. Usman, M. Ahmad, S. Tabassum and F. Arjmand, New J. Chem., 2018, 42, 506–519 RSC.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissoglou and G. Psomas, Dalton Trans., 2010, 39, 4517–4528 RSC.
- R. P. Sharma, S. Kumar, P. Venugopalan, V. Ferretti, A. Tarushi, G. Psomas and M. Witwicki, RSC Adv., 2016, 6, 88546–88558 RSC.
- A. Tarushi, S. Perontsis, A. G. Hatzidimitriou, A. N. Papadopoulos, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2015, 149, 68–79 CrossRef CAS PubMed.
- H. Y. Khan, S. Zehra, S. Parveen, I. Yousuf, S. Tabassum and F. Arjmand, ChemistrySelect, 2018, 3, 12764–12772 CrossRef CAS.
- F. Arjmand, I. Yousuf, T. ben Hadda and L. Toupet, Eur. J. Med. Chem., 2014, 81, 76–88 CrossRef CAS PubMed.
- (a) E. S. DeJong, B. Luy and J. P. Marino, Curr. Top. Med. Chem., 2002, 2, 289–302 CrossRef CAS PubMed; (b) J. Gallego and G. Varani, Acc. Chem. Res., 2001, 34, 836–843 CrossRef CAS PubMed.
- F. Arjmand, Z. Afsan, S. Sharma, S. Parveen, I. Yousuf, S. Sartaj, H. R. Siddique and S. Tabassum, Coord. Chem. Rev., 2019, 387, 47–59 CrossRef CAS.
- K. B. Turner, A. S. Kohlway, N. A. Hagan and D. Fabris, Biopolymers, 2008, 91, 283–296 CrossRef PubMed.
- (a) T. Hermann and E. Westhof, Structure, 1998, 6, 1303–1314 CrossRef CAS; (b) T. Hermann and E. Westhof, J. Mol. Biol., 1998, 276, 903–912 CrossRef CAS PubMed.
- F. Tisato, C. Marzano, M. Porchia, M. Pellei and C. Santini, Med. Res. Rev., 2010, 30, 708–749 CAS.
- J. Folkman, N. Engl. J. Med., 1971, 285, 1182–1186 CrossRef CAS PubMed.
- C. K. Sen, S. Khanna, M. Venojarvi, P. Trikha, E. C. Ellison, T. K. Hunt and S. Roy, Am. J. Physiol.: Heart Circ. Physiol., 2002, 282, H1821–H1827 CrossRef CAS PubMed.
- A. Nasulewicz, A. Mazur and A. Opolski, J. Trace Elem. Med. Biol., 2004, 18, 1–8 CrossRef CAS PubMed.
- M. Moriguchi, T. Nakajima, H. Kimura, T. Watanabe, H. Takashima, Y. Mitsumoto, T. Katagishi, T. Okanoue and K. Kagawa, Int. J. Cancer, 2002, 102, 445–452 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- L. Ruiz-Azuara and M. E. Bravo-Gomez, Curr. Med. Chem., 2010, 17, 3606–3615 CrossRef CAS PubMed.
- S. Liu, W. Cao, L. Yu, W. Zheng, L. Li, C. Fana and T. Chen, Dalton Trans., 2013, 42, 5932–5940 RSC.
- G. Psomas and D. P. Kessissoglou, Dalton Trans., 2013, 42, 6252–6276 RSC.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- J. D. Hoeschele, H. D. H. Showalter, A. J. Kraker, W. L. Elliott, B. J. Roberts and J. W. Kamp, J. Med. Chem., 1994, 37, 2630–2636 CrossRef CAS PubMed.
- F. Dufrasne and M. Galanski, Curr. Pharm. Des., 2007, 13, 2781–2794 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- G. Du, A. Ellern and L. K. Woo, Inorg. Chem., 2003, 42, 873–877 CrossRef CAS PubMed.
- S. Yurdakul and S. Badoglu, Struct. Chem., 2009, 20, 423–434 CrossRef CAS.
- B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- M. N. Patel, P. A. Dosi and B. S. Bhatt, Polyhedron, 2010, 29, 3238–3245 CrossRef CAS.
- A. Tarushi, G. Psomas, C. P. Raptopoulou and D. P. Kessissoglou, J. Inorg. Biochem., 2009, 101, 898–905 CrossRef PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- S. Tsiliou, L.-A. Kefala, A. G. Hatzidimitriou, D. P. Kessissoglou, F. Perdih, A. N. Papadopoulos, I. Turel and G. Psomas, J. Inorg. Biochem., 2016, 160, 125–139 CrossRef CAS PubMed.
- (a) T. Hermann, Top. Med. Chem., 2017, 7, 111 Search PubMed; (b) B. Kong, T. Joshi, M. J. Belousoff, Y. Tor, B. Graham and L. Spicciaa, J. Inorg. Biochem., 2016, 162, 334–342 CrossRef CAS PubMed; (c) E. C. Sherer, Annu. Rep. Comput. Chem., 2010, 6, 139–166 CAS.
- F. Dimiza, S. Fountoulaki, A. N. Papadopoulos, C. A. Kontogiorgis, V. Tangoulis, C. P. Raptopoulou, V. Psycharis, A. Terzis, D. P. Kessissoglou and G. Psomas, Dalton Trans., 2011, 40, 8555–8568 RSC.
- F. Arjmand, Z. Afsan and T. Roisnel, RSC Adv., 2018, 8, 37375–37390 RSC.
- S. Sharma, L. Toupet, M. Ahmad and F. Arjmand, RSC Adv., 2016, 6, 79372–79382 RSC.
- Z. Afsan, T. Roisnel, S. Tabassumand and F. Arjmand, Appl. Organomet. Chem., 2019, 33, 4958 CrossRef.
- E. F. Hearly, J. Chem. Educ., 2007, 84, 1304–1307 CrossRef.
- N. W. Luedtke, J. S. Hwang, E. Nava, D. Gut, M. Kol and Y. Tor, Nucleic Acids Res., 2003, 31, 5732–5740 CrossRef CAS PubMed.
- E. Froehlich, J. S. Mandeville, D. Arnold, L. Kreplak and H. A. Tajmir-Riahi, Biomacromolecules, 2012, 13, 282–287 CrossRef CAS PubMed.
- N. Arshad and S. I. Farooqi, Appl. Biochem. Biotechnol., 2018, 186, 1090–1110 CrossRef CAS PubMed.
- H. Y. Khan, M. O. Ansari, G. G. H. A. Shadab, S. Tabassum and F. Arjmand, Bioorg. Chem., 2019, 88, 102963 CrossRef CAS PubMed.
- K. V. H. Prashanth, S. M. Dharmesh, K. S. J. Rao and R. N. Tharanathan, Carbohydr. Res., 2007, 342, 190–195 CrossRef CAS PubMed.
- L. A. Gugliotti, D. L. Feldheim and B. E. Eaton, J. Am. Chem. Soc., 2005, 127, 17814–17818 CrossRef CAS PubMed.
- S. Tabassum, G. C. Sharma, F. Arjmand and A. Azam, Nanotechnology, 2010, 21, 195102 CrossRef PubMed.
- X. D. Liu, H. Y. Diao and N. Nishi, Chem. Soc. Rev., 2008, 37, 2745–2757 RSC.
- S. Neidle, Nat. Prod. Rep., 2001, 18, 291–309 RSC.
- D. Agudelo, P. Bourassa, M. Beauregared, G. Berube and H.-A. Tajmir-Riahi, PLoS One, 2013, 8, e69248 CrossRef CAS PubMed.
- D. K. -Demertzi, D. H. -Litina, M. Staninska, A. Primikiri, C. Kotoglou and M. A. Demertzis, J. Enzyme Inhib. Med. Chem., 2009, 24, 742–752 CrossRef PubMed.
- I. Yousuf, F. Arjmand, S. Tabassum, L. Toupet, R. A. Khan and M. A. Siddiqui, Dalton Trans., 2015, 44, 10330–10342 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- (a) Software Users Guide, v. 6.0, Bruker Analytical X-ray Systems, Madison, WI, 1999 Search PubMed; (b) G. M. Sheldrick, SADABAS; Area-Detector Absorption Correction, v. 2.03, University of Gottingen, Germany, 1999, SADABAS, v. 2008 Search PubMed.
- G. M. Sheldrick, SHELXT-97, Prog.Crystal-Structure Refinement, 1997 Search PubMed.
- G. Macindoe, L. Mavridis, V. Venkatraman, M.-D. Devignes and D. W. Ritchie, Nucleic Acids Res., 2010, 38, W445–W449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1916426. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07464c |
| This journal is © The Royal Society of Chemistry 2020 |