DOI:
10.1039/C9RA10212D
(Paper)
RSC Adv., 2020,
10, 7509-7522
New, highly versatile bimolecular photoinitiating systems for free-radical, cationic and thiol–ene photopolymerization processes under low light intensity UV and visible LEDs for 3D printing application†
Received
5th December 2019
, Accepted 10th February 2020
First published on 19th February 2020
Abstract
1-Amino-4-methyl-naphthalene-2-carbonitrile derivatives are proposed for the role of photosensitizers of iodonium salt during the photopolymerization processes upon near UV-A and visible ranges. Remarkably, 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives are highly versatile allowing access to photoinitiating systems for (i) the cationic photopolymerization of epoxide monomers with a ring opening mechanism and vinyl ether monomers with chain growth mechanisms (ii) the free-radical photopolymerization of acrylate monomers, (iii) the photopolymerization of interpenetrated polymer networks (IPNs) based on epoxide and acrylate monomers under air and under laminate in an oxygen-free atmosphere (iv) the thiol–ene photopolymerization processes. Excellent polymerization profiles are obtained during all types of photopolymerization processes. The initiation mechanisms are analyzed through steady state photolysis, cyclic voltammetry and fluorescence experiments. Moreover, the newly developed bimolecular photoinitiating systems were investigated by applying an additive manufacturing process under visible light sources. Furthermore, vat photopolymerization processes using IPN compositions, which are polymerizable by using new photoinitiating systems, provide high resolution and speeds. For these reasons, new bimolecular photoinitiating systems are promising initiators for photopolymerization-based 3D printing process to fabricate 3D structures.
Introduction
Currently, the technologies of manufacturing polymer materials are based on processes initiated photochemically.1 The synthesis of polymer materials implemented through photopolymerization is one of the most efficient and widespread methods, and is still developing rapidly.2 Photoinduced polymerization processes find application in many branches of the industry, namely in photolithography to produce printed circuits,3 in the coating industry for photocuring polymer adhesives,4,5 and even in medicine to obtain hydrogel polymer materials.6 Another dynamic direction of application is in the printing industry, where photopolymerization is used in making prints on plastic or metal.7 This process of polymerization also plays a significant role in the design and formation of 3D models8,9 using 3D printing technology.10,11 The growing popularity of the processes of photopolymerization is dictated by its many benefits in comparison to, for instance, conventional methods of polymerization requiring thermal curing.12 The materials obtained by the method of photoinduced polymerization do not contain solvents or other volatile organic compounds (VOCs). Its popularity is also linked to ecological concerns and the restrictive norms relating to the limitation of the use of substances harmful to the working and natural environments. From a technological point of view, the photocuring processes are usually rapid, and it is possible to control them temporally and spatially through switching on or off the light source, or using the appropriate masks to obtain the desired pattern on the cured surface.13 Moreover, demand is decreased for such factors as energy, temperature and spatial requirements for the compact devices for the curing process. The abovementioned set of advantages of photopolymerization encouraged the rapid development of these processes. In industrial practice, the two most commonly used types of polymerization initiated photochemically are radical and cationic polymerization. Each differs in terms of mechanism, as well as type of monomers and initiators.14,15 The basis of current free-radical photoinitating systems is acrylate and methacrylate monomers that are polymerized by the radical mechanism.16 The reason for their popularity is their high reactivity and the option to obtain materials with different properties due to the possibility of modifying the ester chain. The significant disadvantage of these monomers is oxygen inhibition, caused by the presence of atmospheric oxygen during the process of polymerization.17 Oxygen significantly decreases the effectiveness of initiation through quenching the excited initiator states (especially radical initiators of the second type). What is more, oxygen reacts violently with the radicals placed on the carbon atom that constitute the so-called propagating radicals participating in the growth of polymer chains, creating very few reactives in terms of double bond peroxide radicals. As a result, oxygen inhibition creates many technological problems. In addition to the decrease of the speed of polymerization, it generates a long period of induction, and is the cause for the incomplete reaction of double bonds, and the resulting small conversion of monomers and the formation of a non-polarized layer of internal polymer coating.18 In industrial practice, oxygen inhibition can be eliminated by using similar gas shields or light sources with high intensity, as the increased production of radicals can lead to the fast removal of oxygen dissolved in the polymerizing composition.19 In connection with the oxygen inhibition of free radical photopolymerization, much attention is currently paid to the development of technology on cationic photopolymerization,20–22 thiol–ene photopolymerization23,24 and hybrid photopolymerization.25 These methods, initially not very popular, currently attract increasingly more interest due to their resistance to the inhibiting influence of atmospheric oxygen and, in the case of cationic photopolymerization, due to displaying the characteristics of living polymerization. The living character of cationic polymerization processes guarantees that the reaction still takes place effectively, even after switching off the radiation source. The living character of cationic polymerization is the result of the fact that the growth of the polymer chain cannot end. This is the result of the reaction between two active centres, as in the case of free-radical polymerization, whilst the termination of the increase of the polymer chains by cleaving the proton regenerates the protonic acid able to initiate the increase of the next chain. For this reason, cationic polymerization can still take place efficiently, even after turning off the source of radiation. Thanks to this, it is possible to obtain a large degree of conversion, which plays a significant role in industrial practice. As such, photoinduced cationic polymerization is gaining in importance in international markets, as an easy, energy-saving and environmentally friendly method of obtaining cross-linked polymers.26,27 Currently, versatile, high-efficiency photoinitiating systems are sought out, which are useful for obtaining IPN type materials, i.e. those created through hybrid photopolymerization in one step.28–30 The necessity of adding two different types of photoinitiators often creates a problem with the choice of light source for the polymerization process of the IPN type of materials.31 Different types of photoinitiators have different absorption qualities and different efficiency, depending on the wavelength (e.g. some radical photoinitiators can initiate in visible light, while cationic photoinitiators are not adapted for that purpose). IPN photopolymerization carried out in this way causes problems at the stage of choosing the photoinitiating system; however, this type of process allows the obtaining of materials with completely innovative mechanical and resistance user properties, which cannot be achieved during the photopolymerization of one type of monomer.
In connection with the above, growing interest in photopolymerization processes encourages the search for new types of photoinitiators constituting simultaneous initiators of radical and cationic polymerization, ensuring the versatility of the operation, as the efficiency and speed of photopolymerization processes depend on their properties. An additional aspect motivating research is demand in the market of machine park manufacturers for photochemical applications.32 Particularly interesting here is the increase in the production of radiant heaters based on electroluminescent diodes, of type UV-LED or Vis-LED.33 LED technology is increasingly present in daily life, as well as typical industrial applications. Its development in recent years can easily be hailed as a breakthrough since it successfully displaces the current, traditional solutions in the form of medium-pressure mercury lamps used in the photochemical industry.34 The main problem, however, that needs to be solved is the mismatch of the characteristics of absorption of photoinitiators with those of the emission of the sources in the form of medium-pressure mercury lamps, radiators equipped with UV-LEDs with emission lengths above 365 nm and Vis-LEDs. At present, in the case of medium-pressure mercury lamps, only a small part of the energy is emitted in the absorption range of commercial cationic and radical photoinitiators. In industrial practice, this results in commercial photoinitiators (iodonium salts) currently used having an imperfect adjustment of absorption characteristics to the characteristics of the emission of industrial light sources. It is, thus, necessary to solve this problem.
In this work, 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives are proposed for the role of photosensitizers of iodonium salt (initiator) in the photopolymerization processes occurring as a result of exposure to low-power LEDs in both UV-A and visible ranges. Within the framework of implemented works, the usefulness and versatility of two-component initiating systems based on the derivatives of the 1-amino-4-methyl-naphthalene-2-carbonitrile for cationic, free-radical, thiol–ene and hybrid photopolymerization were tested.
As it is widely known, the structure of the chromophore's main core determines spectroscopic properties of the compound. Our experience with 2-amino-4,6-diphenyl-benzene-1,3-dicarbonitrile derivatives35,36 which were demonstrated to show favorable photoinitiating performance encouraged us to design synthetic targets with similar aminonitrile structural motive. We were especially interested how introduction of fused rings system (i.e. replacement of phenyl moiety by naphthalene) will influence spectroscopic characteristics of aromatic conjugated aminonitriles. The synthesis of 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives was based on the synthetic approach analogical to that applied for 2-amino-4,6-diphenyl-benzene-1,3-dicarbonitrile derivatives (one pot, cascade process involving subsequent Knoevenagel condensation, cyclization and aromatization reactions). As a result seven new compounds bearing 1-amino-4-methyl-naphthalene-2-carbonitrile core were obtained.
Experimental part
Research materials
Triethylene glycol divinyl ether (TEGDVE from Sigma Aldrich), (3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (S105 from Lambson) and bis(4-t-butylphenyl)-iodonium hexafluorophosphate (SpeedCure® 938 from Lambson) were applied as a model of vinyl ether monomer, epoxide monomer and a photoinitiator respectively. Trimethylolpropane triacrylate (TMPTA from Sigma Aldrich) was used as an acrylate monomer for the compositions, which are polymerizable by a free-radical mechanism.
Respectively, 1,3,5-triallyl-1,3,5-triazine-2,4,6-trione (TATATO from Sigma Aldrich) and trimethylolpropane tris(3-mercaptopropionate) (MERCAPTO from Sigma Aldrich) were applied as the monomers for the thiol–ene photopolymerization process. 2,4-Diethyl-9H-thioxanthen-9-one (SpeedCure DETX from Lambson) was utilised as a reference sensitizer. All monomers and other chemical compounds were used with the highest available purity and are shown in Scheme 1.
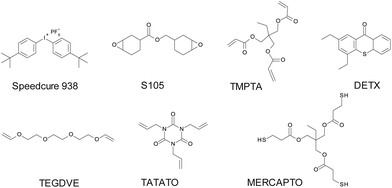 |
| | Scheme 1 Chemical structures of the monomers and iodonium salt. | |
Seven derivatives of 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile were synthesised and used for the role of photosensitizers in a two-component initiating system based on iodonium salt – bis(4-t-butylphenyl)-iodoniumhexa-fluorophosphate. The following derivatives were used for the studies: 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile (J-H), 1-amino-4-methyl-6-(p-tolyl)naphthalene-2-carbonitrile (J-CH3), 1-amino-6-(4-cyanophenyl)-4-methyl-naphthalene-2-carbonitrile (J-CN), 1-amino-6-(4-fluorophenyl)-4-methyl-naphthalene-2-carbonitrile (J-F), 1-amino-6-(4-methoxyphenyl)-4-methyl-naphthalene-2-carbonitrile (J-OCH3), 1-amino-4-methyl-6-(4-methylsulfanylphenyl)-naphthalene-2-carbonitrile (J-SCH3) and 1-amino-4-methyl-6-(4-vinylphenyl)naphthalene-2-carbonitrile (J-vinyl). Details of the synthesis and physicochemical data of the studied derivatives of 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile are given in the ESI.† Structures of the 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile derivatives are shown in Scheme 2.
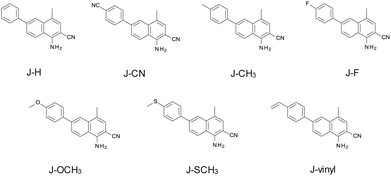 |
| | Scheme 2 Chemical structures of the investigated 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile derivatives. | |
Spectral measurements
Absorbance and fluorescence measurements. For the measurements of the absorption and emission spectra of the 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile derivatives, their solutions were prepared in acetonitrile. The absorption spectra were recorded at 25 °C, using the Silver Nova spectrometer (StellarNet, Inc., USA) in combination with a broadband tungsten-deuterium light source SL5 (from StellarNet, Inc., USA) and a quartz cuvette with 1.0 cm optical path. Next, the absorbance data were converted into extinction coefficients, expressed in classical units [dm3 mol−1 cm−1]. The steady state fluorescence emission and excitation spectra were recorded with a Quanta Master™ 40 spectrofluorometer (from Photon Technology International (PTI), currently a part of Horiba) at varied excitation and observation wavelengths in the range of 200–800 nm. Acetonitrile used for the spectroscopic measurements was of analytical grade from Sigma Aldrich.
Steady state photolysis experiments
The measurements were carried out similarly to the absorption measurements. During the steady state photolysis measurements, cuvettes with 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile derivatives in acetonitrile were irradiated by the UV-LED-365 M365L2 (from Thorlabs Inc., Tampa, FL, USA) emitting light with wavelength λmax = 365 nm and intensity 13.57 mW cm−2 for 30 min, powered by a DC2200 regulated power supply (from Thorlabs Inc. U.S.)
The photolysis of 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile derivatives in the presence of SpeedCure 938 (1.59 × 10−3 mol dm−3) were determined with the same parameters over 1300 s.
Fluorescence quenching
Fluorescence quenching studies of compounds that act as sensitizers were carried out using the spectrofluorimeter Quanta Master™ 40 (from Photon Technology International (PTI), currently a part of Horiba) at excitation located in the maximum absorption of individual compounds. The appropriate amounts of SpeedCure 938 quenching agent were added to the diluted solutions of the derivatives of 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile so its concentration varied from 0 to 3.0 × 10−2 mol dm−3.
Fluorescence lifetime measurements
Fluorescence decay curves were recorded with spectrophotometer EasyLife™ X (Fluorescence Lifetime Fluorometer from Horiba) with an emission cut-off filter at 320 nm, using a pulsed LED excitation source at 310 nm. In order to use the IRF instrument response function, a solution of colloidal silica Ludox® (from Sigma Aldrich), highly diluted in water, was used as a scatterer. For measurements of fluorescence decay, the diluted solutions of derivatives of 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile in acetonitrile were used. After measurement of fluorescence decay and IRF, the fluorescence lifetime was determined. Fluorescence lifetimes were estimated by fitting the decay curves using a deconvolution procedure implemented with Fluorescence Decay Analysis Software. Fluorescence decay curves were fitted with an exponential function to give lifetime τ.
Determination of electrochemical characteristics of oxidation potentials
The oxidation potentials (Eox vs. Ag/AgCl) of the investigated derivatives of 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile were measured in acetonitrile by cyclic voltammetry with tetrabutylammonium hexafluorophosphate (0.1 M) as a supporting electrolyte (Electrochemical Analyzer M161 and Electrode Stand M164, from MTM-ANKO, Poland). The working electrode was a platinum disk and the reference was a silver chloride electrode, Ag/AgCl; the scan rate of 0.1 V s−1 was used; ferrocene was used as a standard and the potentials were determined from half peak potentials. The Gibbs free energy change ΔGet for electron transfer between photoinitiators (diaryliodonium salt) and co-initiators was calculated from the classical Rehm–Weller37 equation (eqn (1)):| | |
ΔGet = F[Eox (D/D˙+) – Ered (A˙−/A)] – E00 – (Ze2/εa)
| (1) |
where F is the Faraday constant (F = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485.33289(59) C mol−1), and Eox (D/D˙+), Ered (A˙−/A), E00 and (Ze2/εa) are the oxidation potential of the co-initiator, the reduction potential of the diaryliodonium salt, the excited state energy, and the electrostatic interaction energy for the initially formed ion pair respectively. Parameter (Ze2/εa) is generally considered to be negligible in polar solvents.
485.33289(59) C mol−1), and Eox (D/D˙+), Ered (A˙−/A), E00 and (Ze2/εa) are the oxidation potential of the co-initiator, the reduction potential of the diaryliodonium salt, the excited state energy, and the electrostatic interaction energy for the initially formed ion pair respectively. Parameter (Ze2/εa) is generally considered to be negligible in polar solvents.
Molecular orbital calculations
The Gaussian 09 package was used for calculating the energy gap between the first triplet energy (T1) and ground state energy (S0). To begin with, the optimisation of S0 and T1 states was carried out for each of the molecular structures of interest. The optimisation of molecules in the ground state and first triplet excited state were performed using the density functional theory (DFT) method at a B3LYP/6-31G (d,p) level of theory. The energy gap was calculated as the difference of the total energy of the molecule in the first triplet excited state (ET1) and the total energy of the molecule in the ground state (ES0). The frontier molecular orbital properties were analysed and visualised using GaussView 5.0 software. The computational procedures were carried out using the open infrastructure resource PLGrid Infrastructure. PLGrid Infrastructure enables scientific research based on simulations and large-scale calculations using computing clusters, as well as providing convenient access to distributed computing resources.
Preparation of thin-layer samples for monitoring the photopolymerization processes
Thin-layered samples for the study of photopolymerization using the real-time FT-IR method were prepared in vials of dark amber glass in a dark room. Each composition included the appropriate monomer (S105, TMPTA, TEGDVE, mixture S105/TMPTA 1:1 w/w%), the derivative of 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile with a concentration of 7.7 × 10−3 mol dm−3 in relation to the entirety of the composition (which constituted 0.2% mas.) and iodonium salt with a concentration of 1.0% weight in the monomer. The compositions for the study of thiol–ene reaction kinetics included the mixture of monomers of TATATO and MERCAPTO (1:1 w/w%), 1% weight of SpeedCure 938 (18.6 × 10−3 mol dm−3), iodonium salt and the derivative of 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile with a concentration of 3.35 × 10−3 mol dm−3 which constituted approx. 0.1% of the weight in relation to the total composition.
Monitoring the photopolymerization processes in real-time FT-IR
For monitoring the conversion degree of the monomers during the processes of photopolymerization, the real-time FT-IR method was used with an FTIR – i10 NICOLET™ spectrophotometer from Thermo Scientific, equipped with a horizontal attachment. The measurements were carried out for up to 800 s depending on the monomer. The obtained data was recorded in the OMNIC program, dedicated to the processing of the FT-IR spectra. The data was registered in a darkened room, in which lamps emitting red light were used as the only source of illumination. A drop of the prepared composition was placed, using the glass pipette, onto a barium fluoride pellet (for the composition including monomer S105 and TATATO/MERCAPTO) or between two polypropylene films (for compositions containing the TMPTA and TEGDVE monomers). After performing this action, the pellet or foil was positioned on the metal holder and placed in the horizontal attachment of the spectrometer. The measurements were made maintaining the same thickness for the applied layer of composition equaling 25 μm. After 10 s from starting the OMNIC software registering the IR spectrum, the LED diode emitted light of wavelength λmax = 365 nm or λmax = 405 nm. The conversion of functional groups was calculated from the following formula:
where: C – conversion; A – value of the surface area of the band monitored during the photopolymerization process as a function of time; A0 – initial value of the band surface at the monitored wave number.
Cationic photopolymerization (CP) experiments
The evolution of the epoxy group content was continuously followed by real-time FT-IR spectroscopy (Nicolet iS10, from Thermo Scientific U.S.) at about 790 cm−1. For triethylene glycol divinyl ether (TEGDVE), the photopolymerization was followed at about 1630 cm−1.
Free-radical photopolymerization (FRP) experiments
The experiments were carried out in laminated conditions. The polypropylene films (25 μm thick) deposited on a horizontal holder for the FT-IR spectrometer were irradiated. The evolution of the double bond of acrylate TMPTA content was continuously monitored by real-time FT-IR spectroscopy (Nicolet iS10, from Thermo Scientific U.S.) at about 1634 cm−1.
Thiol–ene photopolymerization (T-EP) experiments
The photosensitive formulations which contain 1,3,5-triallyl-1,3,5-triazine-2,4,6-trione (TATATO) and MERCAPTO monomers (50%/50% w/w) were deposited on a BaF2 pellet. The evolution of the thiol (S–H) group content was continuously monitored by real-time FT-IR spectroscopy (Nicolet iS10, from Thermo Scientific U.S.) at approximately 2570 cm−1. FT-IR also followed the double bond conversion of TATATO at about 3083 cm−1. The molar ratio of thiol vs. allyl used in all experiments was 1:1.6.
IPN photopolymerization (IPN) experiments
Photosensitive compositions consisting of an S105 and TMPTA blend (50%:50% w/w) were used. The experiments were carried out in laminate and under air, on a BaF2 pellet. FT-IR spectra were followed at about 790 cm−1 for epoxy groups and at about 1630 cm−1 for acrylate groups by real-time FT-IR spectroscopy (NicoletTM iS10, from Thermo Scientific U.S.)
Source of light for real-time experiments
The light sources for the real-time FT-IR method were 365 nm M365L2 UV-LED diode and 405 nm M405L4 diode (from Thorlabs Inc. U.S.), powered by DC2200 regulated power supply (from Thorlabs Inc. U.S.).
3D printing experiment
For laser writing and 3D printing experiments, a laser diode at 405 nm with an intensity of 100 mW cm−2 (spot size ∼50 μm) was used for the spatially controlled irradiation (NEJE DK-8-KZ 1000 mW Laser Engraver Printer). The photosensitive formulations (2 mm thickness) were deposited onto a microscope slide and were polymerized under air. The generated 3D objects were observed thanks to an optical stereo microscope (Bresser Advance ICD 10–160× Zoom Stereo-Microscope, Bresser GmbH, Germany) and DSX1000 from OLYMPUS.
Results and discussion
Light absorption properties of the 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives
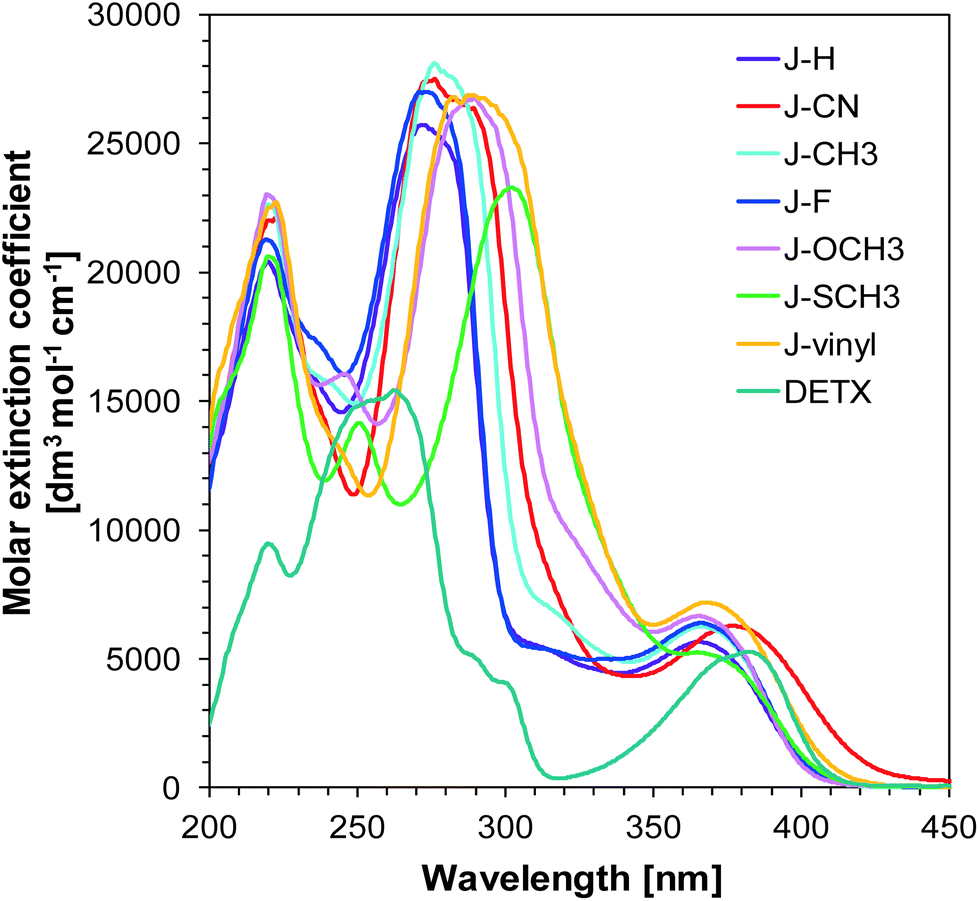
In the presented research, we focused our attention on the application of 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives as components in bimolecular photoinitiating systems for the photopolymerization of different types of monomer. The studied 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives were applied as UV-A and visible light-absorbing molecules in the presence of iodonium salt (SpeedCure 938) and used as efficient photosensitizers. One of the most critical parameters in photopolymerization is the compatibility of the absorption spectrum of photoinitiating systems with the emission spectrum of the light source. Thus, the spectral properties of the photoinitiating system have a significant impact on polymerization rate, which is directly connected with the amount of absorbed light. As shown in Fig. 1, 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives demonstrate broad light absorption for all investigated compounds reaching 420 nm or 440 nm, which exhibits excellent overlap with the emission spectra of the maximum of medium-pressure mercury lamps, and with the emission of UV-A-LED with a maximum at 365 nm. This endows them with the potential to act as UV-A and visible photoinitiators. 1-Amino-4-methyl-naphthalene-2-carbonitrile derivatives possess similar absorption properties. The absorption maxima (λmax) and extinction coefficients (ε) at absorption maximum, as well as the maximum emission wavelengths of the different LEDs for all investigated compounds are listed in Table 1.
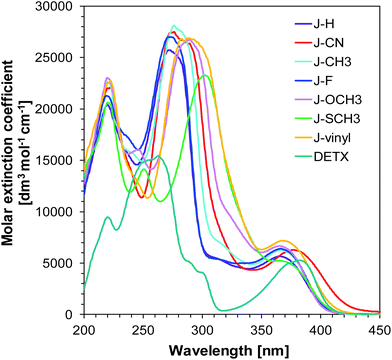 |
| | Fig. 1 UV-visible absorption spectra of the 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives in acetonitrile. | |
Table 1 Light absorption properties of the studied 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives
| Sensitizer |
λmax-ab [nm] |
ελmax [dm3 mol−1 cm−1] |
ε365 nm [dm3 mol−1 cm−1] |
ε405 nm [dm3 mol−1 cm−1] |
| J-H |
365.7 |
5647 |
5649 |
606 |
| J-CN |
377.1 |
6268 |
5672 |
3107 |
| J-CH3 |
366.1 |
6243 |
6244 |
533 |
| J-F |
366.1 |
6383 |
6376 |
599 |
| J-OCH3 |
365.3 |
6651 |
6660 |
464 |
| J-SCH3 |
364.8 |
5243 |
5254 |
781 |
| J-vinyl |
367.9 |
7184 |
7116 |
1514 |
The extinction coefficient at the peak maximum is of the order of 5000–7000 [dm3 mol−1 cm−1], which is relatively low compared to the system composed of conjugated benzene rings, but high enough for the application of 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives as sensitizers. Nevertheless, the magnitude of the extinction coefficient indicates that the long wavelength absorption band originates in the excitation of an electron from a non-bonding (n) orbital of one of the heteroatoms into an antibonding (π*) orbital of one of the benzene rings. The effect of substituents on the absorption of naphthalene is small (i.e. there is up to 20% difference between the highest and lowest value of the extinction coefficient), so the choice of naphthalene for practical applications is sound and can be based on other factors, such as the values of oxidation potential or the electron transfer quantum yield from the excited state, both of which guarantee that the molecule is an efficient photosensitizer.
1-Amino-4-methyl-naphthalene-2-carbonitrile derivatives as a component of bimolecular photoinitiating systems for photopolymerization processes
The kinetics of photopolymerization processes of various monomers in the presence of bimolecular photoinitiation systems were monitored by the real-time FT-IR method. Based on the real-time recorded FT-IR spectra of individual photocurable compositions, under appropriate illumination the sample with an appropriate light source, photopolymerization reactions were observed.
Cationic photopolymerization of monomers
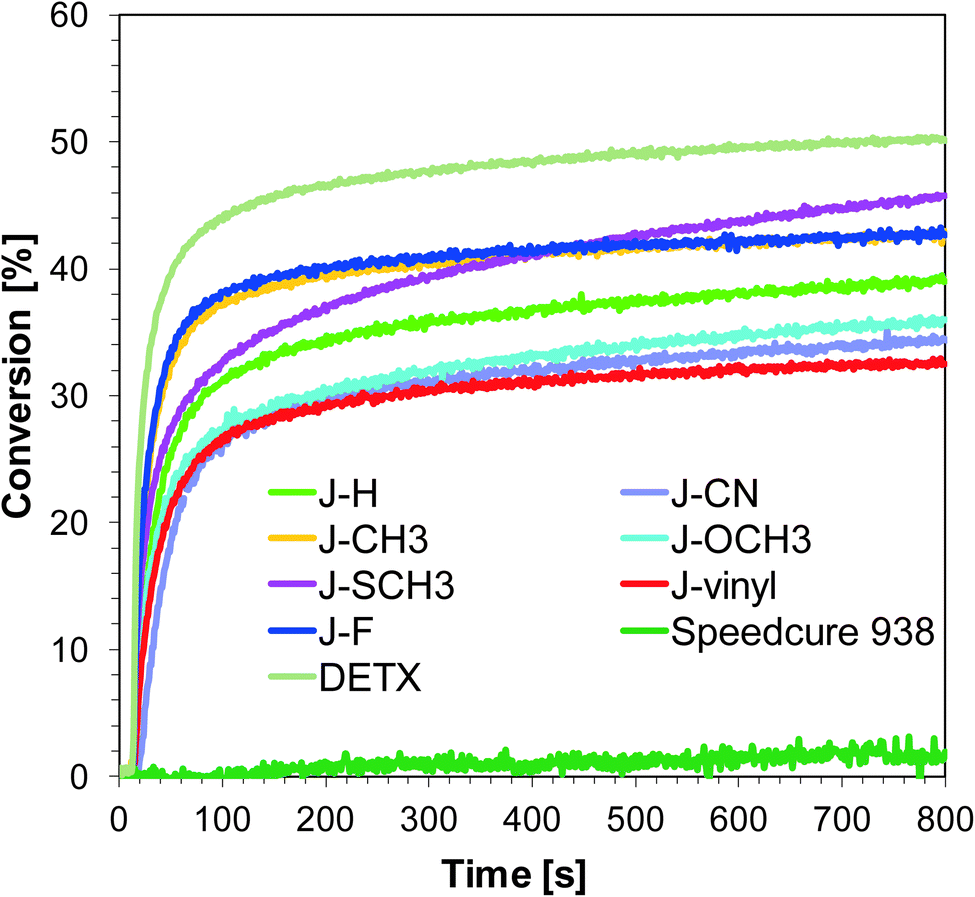
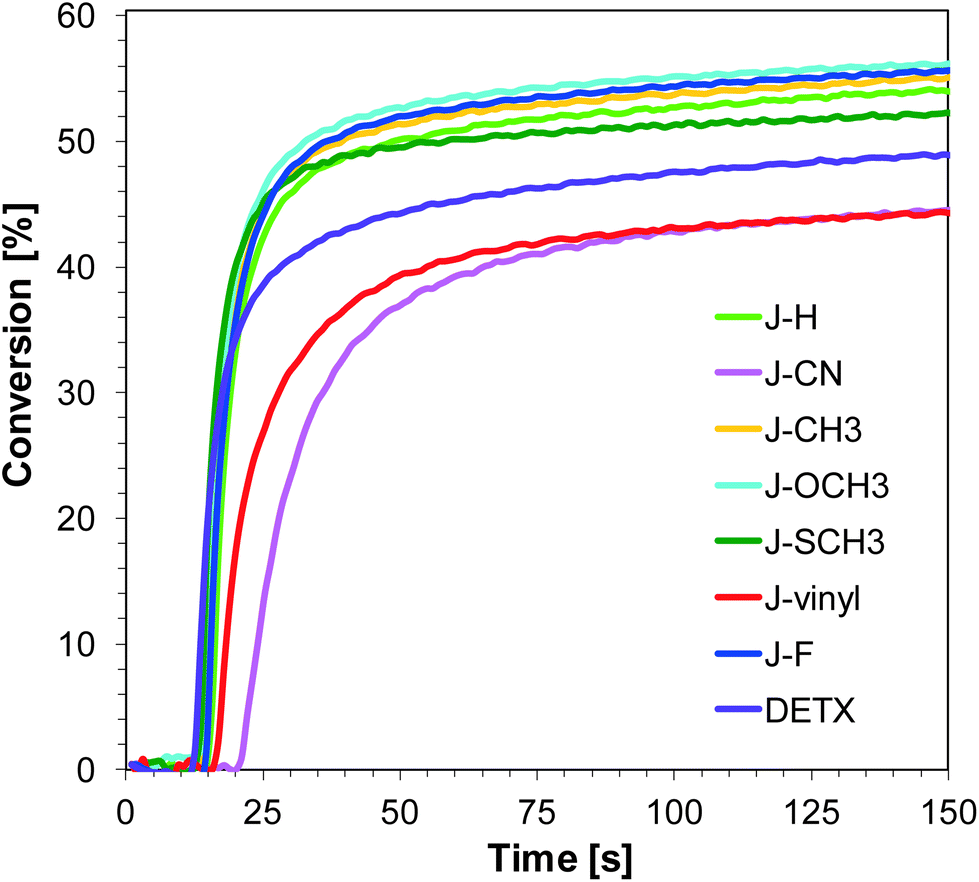
As the first step, the photopolymerization process began with the opening of the oxirane ring of the 3,4-(epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (S105) monomer and was carried out using two different light emitting diodes with maximum emissions at 365 nm and 405 nm. Subsequent experimental studies have shown that the use of initiating systems consisting of derivatives of 1-amino-4-methyl-naphthalene-2-carbonitrile and SpeedCure 938® lead to effective photopolymerization processes under near UV-A and also under visible light sources (Fig. 2 and 3). Conversion values after 800 s of irradiation of the cycloaliphatic epoxide monomer are listed in Table 2. It is worth noting that the photopolymerization of epoxide monomer (S105) did not occur at all under 365 nm, even after long irradiation time, when the composition contained only diphenyliodonium salt (SpeedCure 938®) as a one-component photoinitiator. This is because the maximal absorption of the diphenyliodonium photoinitiator is set at about 230 nm, and the absorption decreases rapidly to near zero at 300 nm.
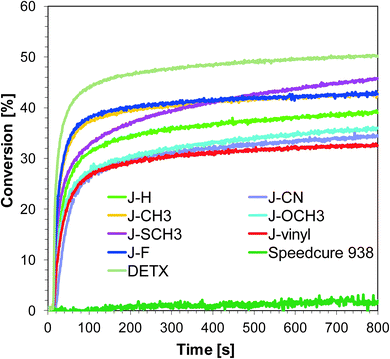 |
| | Fig. 2 Polymerization profiles of S105 (epoxy function conversion vs. irradiation time) upon exposure to the LED@365 nm under air in the presence of different photoinitiating systems based on SpeedCure 938® (1% w/w) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w). The irradiation starts at t = 10 s. | |
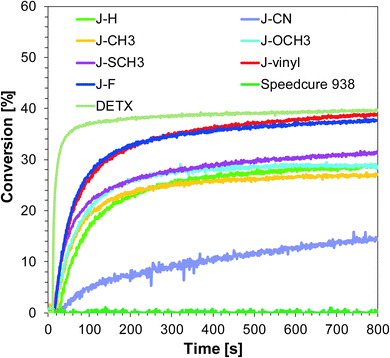 |
| | Fig. 3 Polymerization profiles of S105 (epoxy function conversion vs. irradiation time) upon exposure to the visible LED@405 nm under air in the presence of different photoinitiating systems based on SpeedCure 938® (1% w/w) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w). The irradiation starts at t = 10 s. | |
Table 2 Functional group conversions of epoxy monomer for S105 and vinyl monomer for TEGDVE using photoinitiating system based on bis(4-t-butylphenyl)-iodonium hexafluorophosphate (SpeedCure 938® wt 1%) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w) in the role of co-initiator at 365 nm and 405 nm exposure
| Sensitizer |
Conversion of epoxy monomer S105 [%] |
Conversion of double bonds in TEGDVE [%] |
| @365 nm (3.76 mW cm−2) |
@405 nm (19.82 mW cm−2) |
@365 nm (1.61 mW cm−2) |
@405 nm (3.97 mW cm−2) |
| J-H |
39 |
29 |
86 |
87 |
| J-CN |
34 |
14 |
87 |
92 |
| J-CH3 |
43 |
27 |
86 |
94 |
| J-F |
42 |
37 |
85 |
94 |
| J-OCH3 |
36 |
29 |
85 |
91 |
| J-SCH3 |
46 |
31 |
86 |
89 |
| J-vinyl |
32 |
39 |
85 |
90 |
Although varying the substitution of 1-amino-4-methyl-naphthalene-2-carbonitrile in position six does not significantly influence absorption properties, the studied compounds show slightly different photosensitizing capacity. The highest photoinitiating activity in the cationic photopolymerization of the epoxy monomer upon exposure to LED 365 nm exhibits a two-component system consisting of J-SCH3 and SpeedCure 938. The epoxy group conversion was equal to 46%. This correlates with high molar extinction coefficients at 365 nm and the low value of free energy change (ΔGet = −1.32 eV), which indicates electron transfer between the photosensitizer and iodonium salt. The efficiency trend for the ring-opening cationic photopolymerization of epoxy monomer using LED 365 nm is of the following order:
| J-SCH3 > J-F–J-CH3 > J-H > J-OCH3 > J-CN > J-vinyl. |
Initiating performance of the investigated systems during the cationic photopolymerization process carried out under irradiation LED 405 nm is slightly lower. The highest final conversion was obtained for photoinitiating system J-vinyl/SpeedCure 938, because sensitizer J-vinyl is characterised by the highest molar extinction coefficient at 405 nm. According to Fig. 1, J-vinyl possesses a red-shifted absorption spectrum in comparison to other compounds with electron-donating groups. The trend for the ring-opening cationic photopolymerization of the epoxy monomer using visible LED at 405 nm for the studied sensitizer is of the following order:
| J-vinyl > J-F > J-SCH3 > J-OCH3 > J-H > J-CH3 > J-CN. |
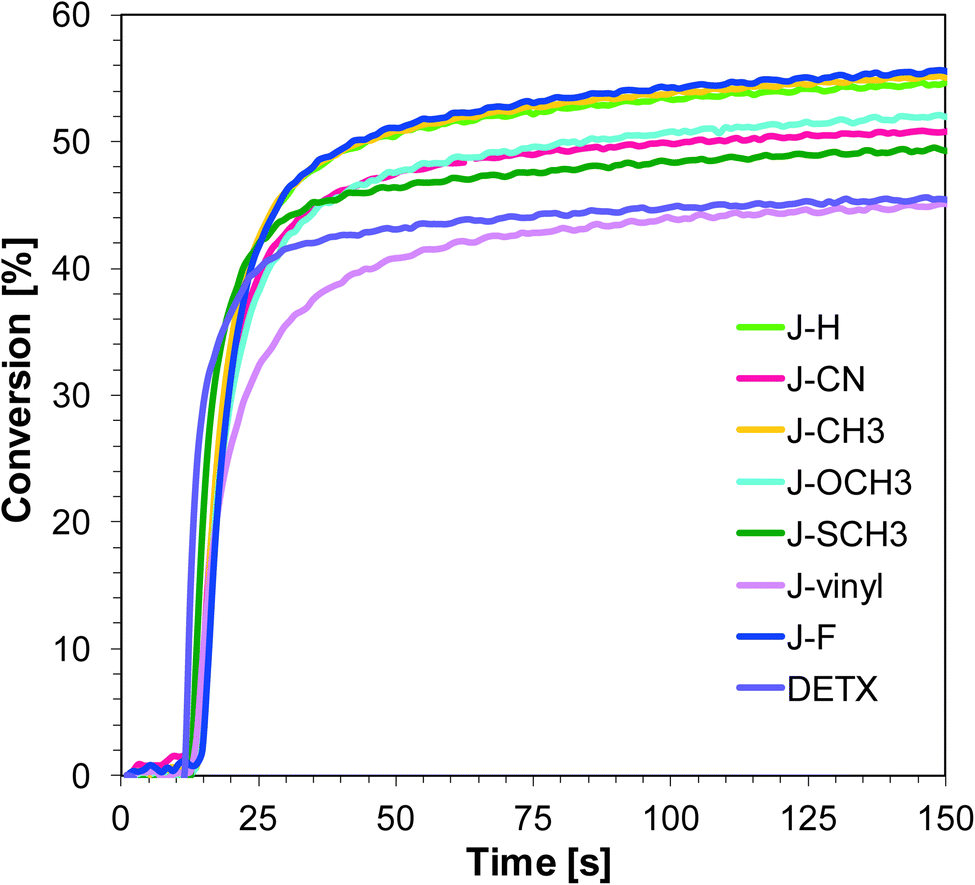
It can be concluded that incorporation of the electron donating group into 1-amino-4-methyl-6-phenyl-naphthalene-2-carbonitrile has led to improvements in photoinitiating efficacy. The best 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives in the role of sensitizers of iodonium salt exhibit similar photosensitizing efficiency to the commercially used sensitizer DETX upon exposure to LED 365 nm and LED 405 nm (final conversions for epoxy groups around 40%).
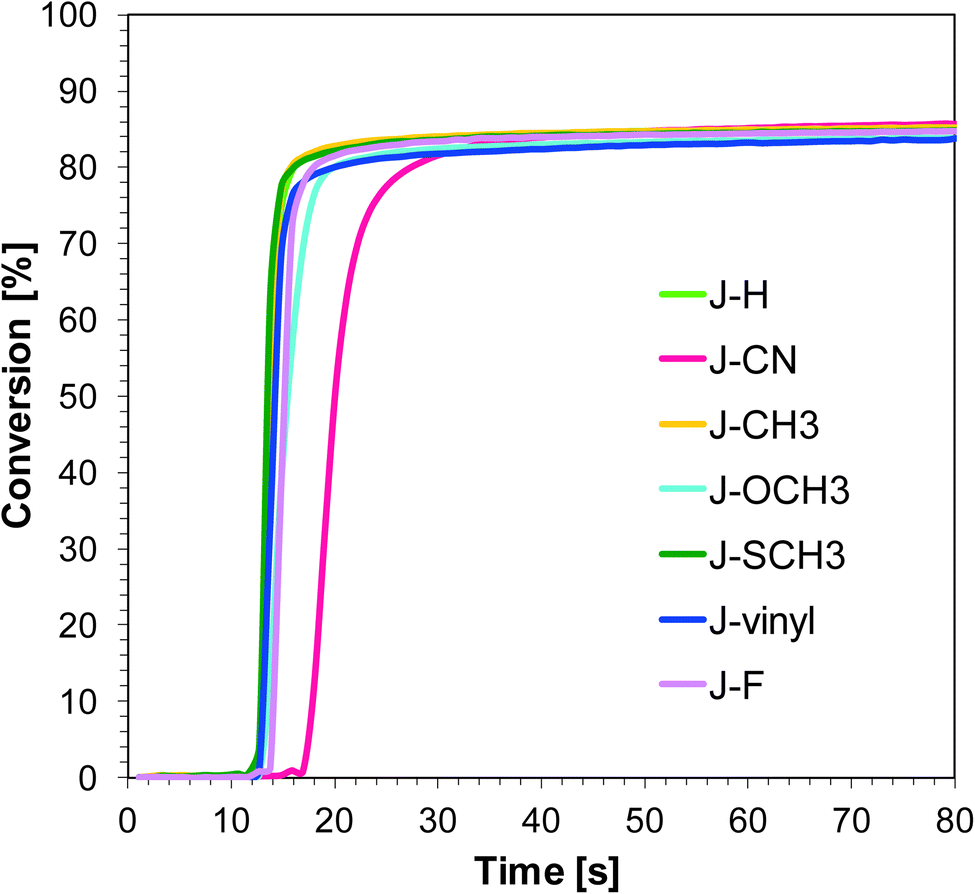
Naphthalene-based photoinitiating systems were also capable of initiating cationic polymerization of vinyl monomer TEGDVE upon exposure to the UV-A and visible LEDs. It was shown that conversion above 80% can be attained for the TEGDVE double bond using the 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives of UV-A LED irradiation (Fig. 4) and above 90% after less than 80 s of Vis-LED irradiation (Fig. 5). Conversion values after 80 s of irradiation are gathered in Table 2. Final conversion values are very similar for all studied sensitizers during polymerization upon exposure to the appropriate wavelength.
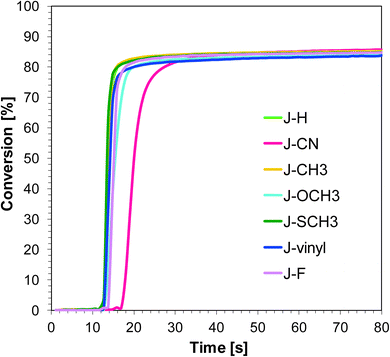 |
| | Fig. 4 Polymerization profiles of TEGDVE (vinyl function conversion vs. irradiation time) upon exposure to the LED@365 nm under laminate in the presence of different photoinitiating systems based on SpeedCure 938® (1% w/w) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w). The irradiation starts at t = 10 s. | |
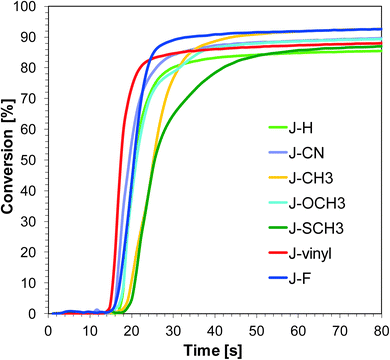 |
| | Fig. 5 Polymerization profiles of TEGDVE (vinyl function conversion vs. irradiation time) upon exposure to the LED@405 nm under laminate in the presence of different photoinitiating systems based on SpeedCure 938® (1% w/w) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w). The irradiation starts at t = 10 s. | |
Nevertheless, the induction time is quite different. Sensitizer J-CN is characterised by the longest induction time under irradiation LED 365 nm. Conversely, during the photopolymerization of vinyl monomers carried out in the presence of LED 405 nm, sensitizers J-CH3 and J-SCH3 have led to a slower polymerization process in comparison to other photoinitiating systems based on naphthalene derivatives.
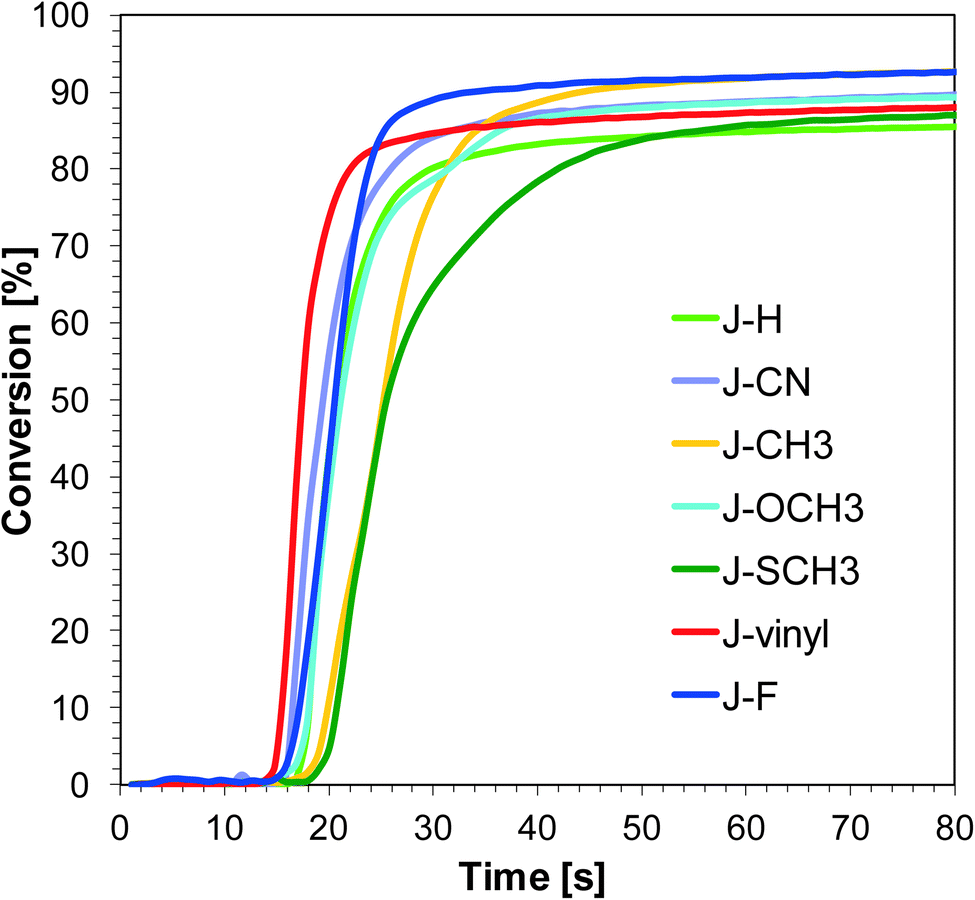
Free-radical photopolymerization of acrylate monomers
The developed photoinitiating systems also fulfill their role in the polymerization reactions of the TMPTA acrylate monomer (Fig. 6 and 7). The calculated conversions of the double bonds of the acrylate monomer are about 50%, both when exposed to UV and visible light (diodes with maximum emission at 365 nm and 405 nm respectively).
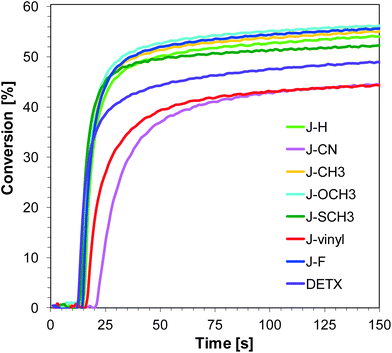 |
| | Fig. 6 Polymerization profiles of TMPTA (acrylate function conversion vs. irradiation time) upon exposure to the LED@365 nm under laminate in the presence of different photoinitiating systems based on SpeedCure 938® (1% w/w) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w). The irradiation starts at t = 10 s. | |
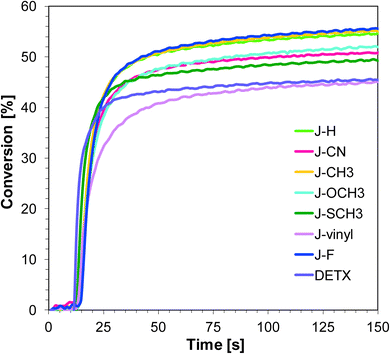 |
| | Fig. 7 Polymerization profiles of TMPTA (acrylate function conversion vs. irradiation time) upon exposure to the LED@405 nm under laminate in the presence of different photoinitiating systems based on SpeedCure 938® (1% w/w) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w). The irradiation starts at t = 10 s. | |
Sensitizers J-OCH3, J-F and J-CH3 are characterised by the best initiation efficiency at 365 nm. This is associated with the high extinction coefficient of these compounds at 365 nm and the fact that electron transfer processes in the sensitizer/iodonium salt system are thermodynamically privileged ΔGet = −1.40 eV for J-OCH3, ΔGet = −1.34 eV for J-F and ΔGet = −1.37 eV for J-CH3.
Even though sensitizers J-vinyl and J-CN are characterised by the highest extinction coefficients at λ = 365 nm and λ = 405 nm, the conversions obtained for the TMPTA monomer are the lowest using the J-vinyl/SpeedCure 938 and J-CN/SpeedCure 938 initiating systems. This is because the ΔGet value for these systems is the highest, so electron transfer processes between sensitizers and iodonium salt are the least favoured.
Thiol–ene photopolymerization of TATATO and MERCAPTO monomers
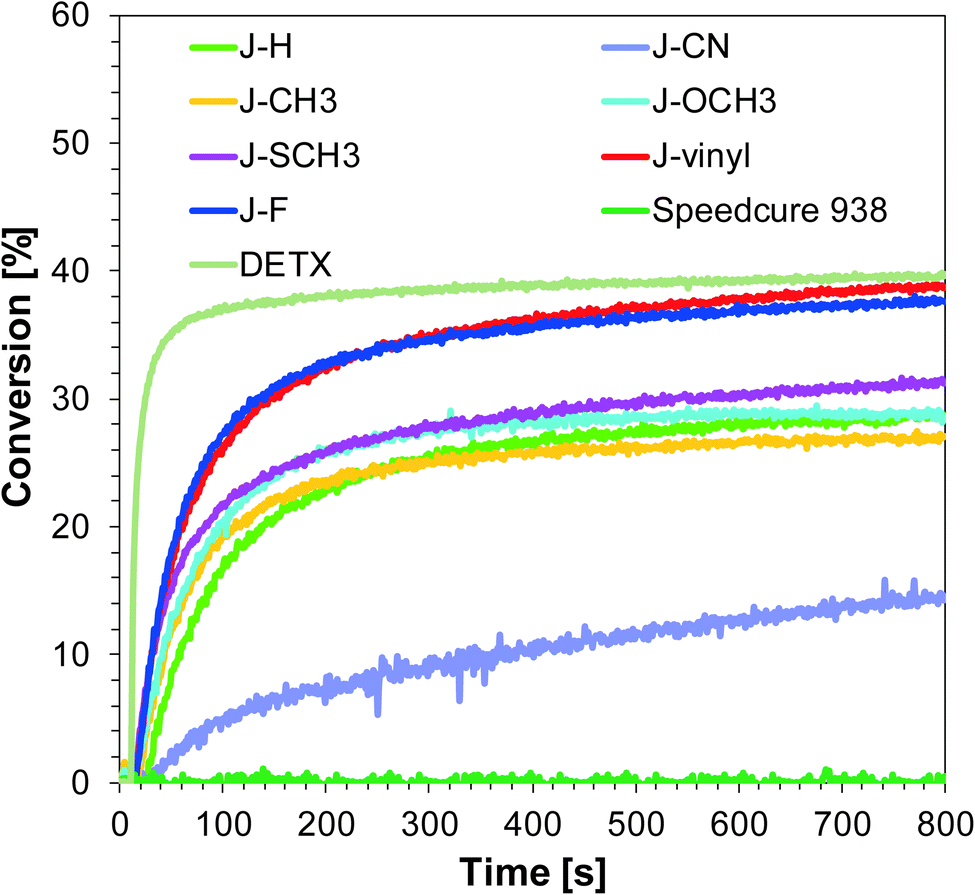
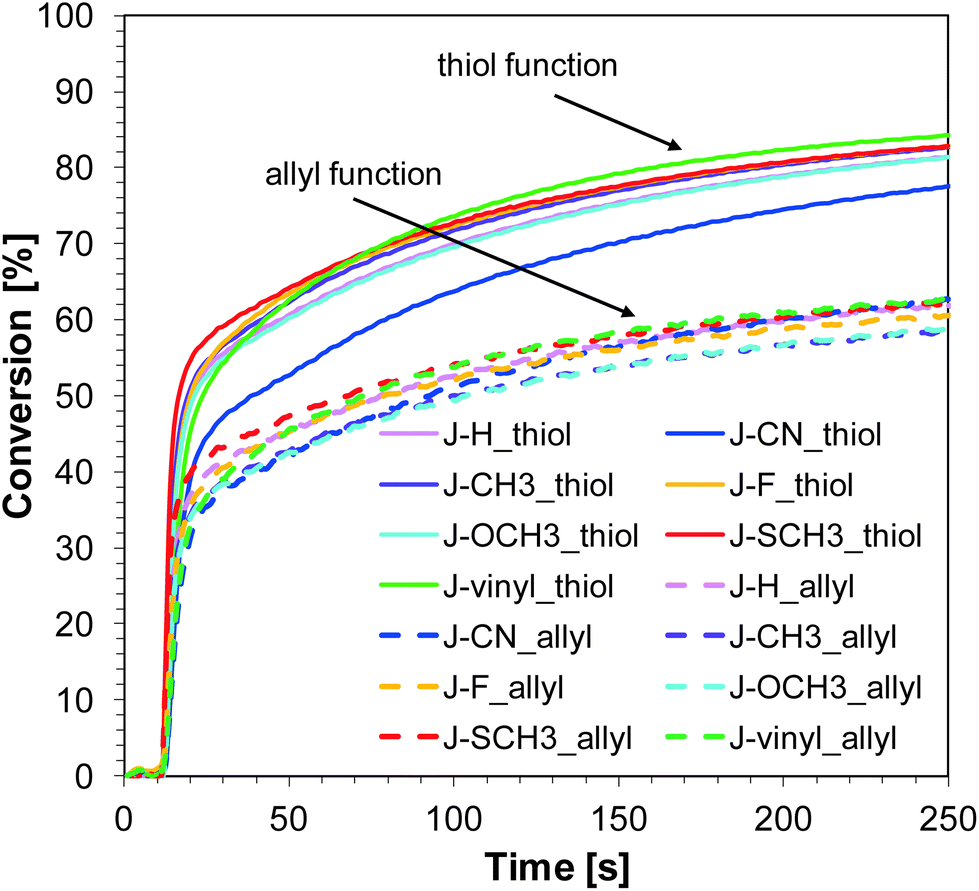
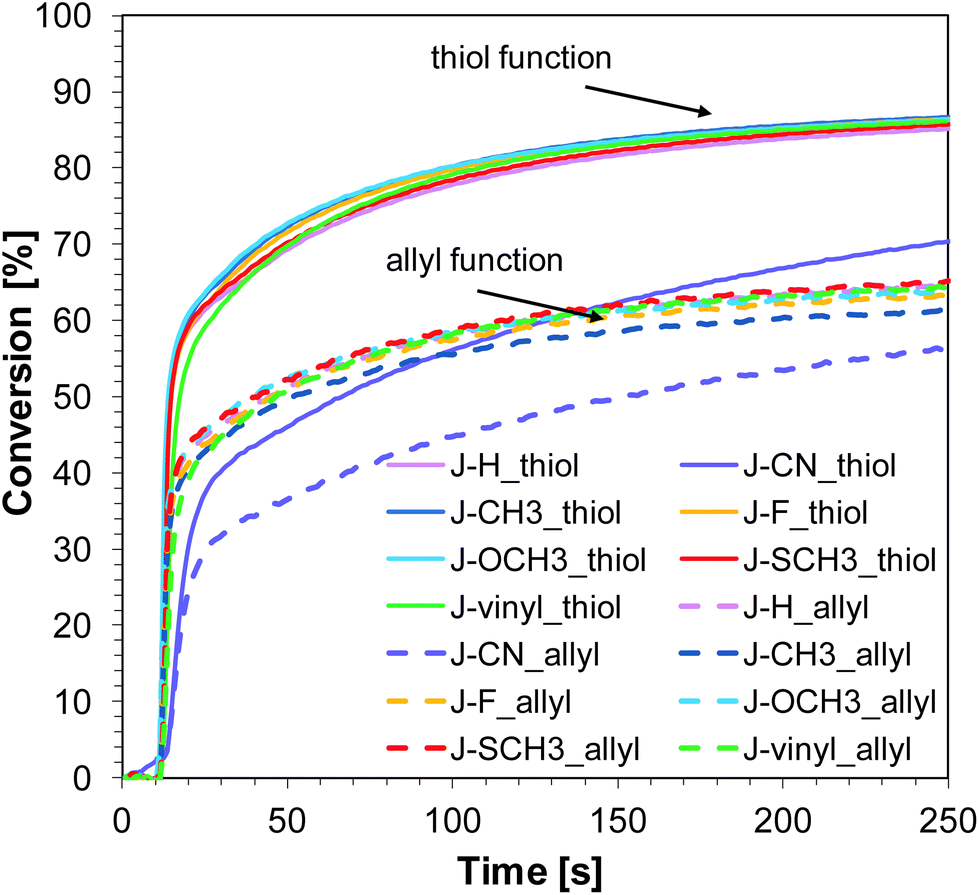
Similar to the free-radical photopolymerization of TMPTA, the studied bimolecular initiating systems also demonstrated the ability to initiate the thiol–ene photopolymerization of allyl monomer 1,3,5-triallyl-1,3,5-triazine-2,4,6-trione (TATATO) and thiol monomer trimethylolpropane tris(3-mercaptopropionate) (MERCAPTO) upon exposure to various irradiation sources. Excellent conversion versus irradiation time profiles for the thiol–ene polymerization in the presence of the investigated bimolecular system are shown in Fig. 8 and 9, while the final conversion values are summarised in Table 3. Conversion for thiol function is higher than for double bonds of allyl groups (∼86% vs. 64% for polymerization upon exposure to LED 365 nm and ∼82% vs. 59% for polymerization under LED 405 nm). TATATO and MERCAPTO monomers were used in the ratio 1:1 by weight.
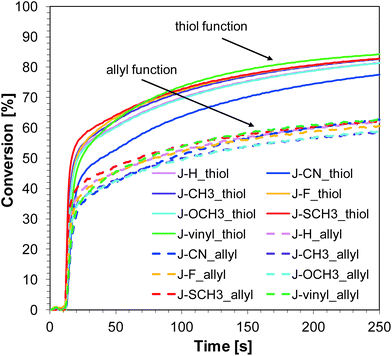 |
| | Fig. 8 Conversion of allyl and thiol functions during thiol–ene polymerization of TATATO and MERCAPTO (50/50% w/w) in the presence of 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.1% w/w) and SpeedCure 938 (1% w/w) in air upon exposure to LED 365 nm. The irradiation starts at t = 10 s. | |
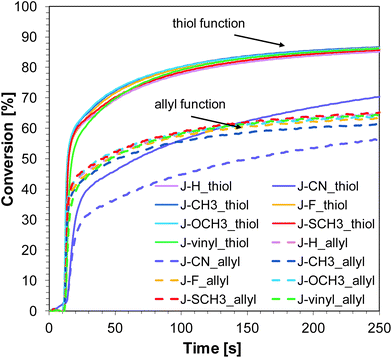 |
| | Fig. 9 Conversion of allyl and thiol functions during thiol–ene polymerization of TATATO and MERCAPTO (50/50% w/w) in the presence of 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.1% w/w) and SpeedCure 938 (1% w/w) in air upon exposure to LED 405 nm. The irradiation starts at t = 10 s. | |
Table 3 Functional group conversions of acrylate monomer TMPTA, allyl monomer TATATO and thiol monomer MERCAPTO using photoinitiating system based on bis(4-t-butylphenyl)-iodonium hexafluorophosphate (SpeedCure 938® wt 1%) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w for FRP and 0.1% w/w for thiol–ene) in the role of co-initiator at 365 nm and 405 nm exposure
| Sensitizer |
Conversion of acrylate monomer TMPTA [%] |
Conversion [%] |
| @365 nm (3.76 mW cm−2) |
@405 nm (19.82 mW cm−2) |
@365 nm (0.11 mW cm−2) |
@405 nm (0.19 mW cm−2) |
| Allyl group |
Thiol group |
Allyl group |
Thiol group |
| J-H |
57 |
57 |
85 |
64 |
81 |
62 |
| J-CN |
47 |
52 |
70 |
56 |
77 |
62 |
| J-CH3 |
58 |
58 |
87 |
61 |
82 |
58 |
| J-F |
58 |
57 |
86 |
63 |
83 |
61 |
| J-OCH3 |
58 |
54 |
86 |
63 |
81 |
59 |
| J-SCH3 |
54 |
51 |
86 |
65 |
83 |
62 |
| J-vinyl |
47 |
47 |
86 |
64 |
84 |
62 |
Therefore, the molar concentration ratio of the functional groups in the photocurable formulation was 1:1,6 (thiol:allyl). Based on the fact that thiol–ene photopolymerization is not sensitive to oxygen, the process was carried out in air (on a BaF2 pellet). Oxygen atmosphere is conducive to additional chain transfer reactions. The peroxide radicals formed by the incorporation of oxygen to the chain are not reactive towards the monomer, but they are able to detach hydrogen from the thiol. As a result of this reaction, alkyl-hydroxy peroxides are formed, and the thiyl radicals are regenerated while the thiol monomer is consumed.
Photopolymerization processes of interpenetrated polymer networks (IPNs)
The developed initiating systems consisting of SpeedCure 938 iodonium salt and derivatives of 1-amino-4-methylnaphthalene-2-carbonitrile have also been used to initiate concomitant cationic and radical polymerization of cycloaliphatic epoxide monomer (S105) and acrylate monomer (TMPTA) blends (50/50% w/w). Hybrid curing was carried out in the presence of air and in laminate when exposed to 365 nm UV diode and 405 nm Vis diode. This led to the acquisition of interpenetrating polymer networks (IPNs).
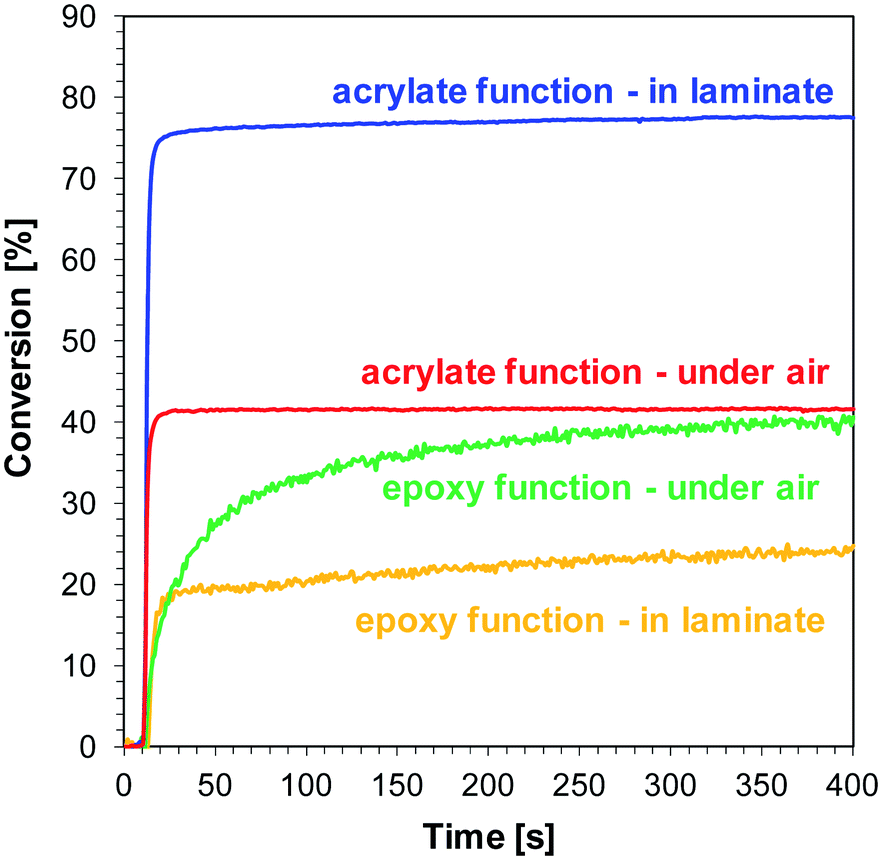
The final conversions of acrylate and epoxy groups are summarised in Table 4. Selected bimolecular photoinitiating systems based on 1-amino-4-methyl-6-(p-tolyl)naphthalene-2-carbonitrile (J-CH3) as photosensitizer of iodonium salt is shown in Fig. 10. Higher conversions were obtained for the acrylate monomer in the laminate, while for the epoxy monomer to the contrary, higher conversions were observed during curing on the pellet in the presence of air. Free radical polymerization of TMPTA occurs faster than cationic polymerization of the epoxy monomer in laminate, because oxygen inhibition is limited and most of the resulting radicals are used to initiate the free radical polymerization reaction of the TMPTA monomer.
Table 4 Functional group conversions of epoxy monomer S105 and acrylate monomer TMPTA during INPs synthesis in laminate and under air, using photoinitiating system based on bis(4-t-butylphenyl)iodonium hexafluorophosphate (SpeedCure 938 wt 1%) and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives (0.2% w/w) in the role of photosensitizers upon exposure to LED 365 nm (3.76 mW cm−2)
| Sensitizer |
Conversion [%] |
| @365 nm (3.76 mW cm−2) |
@405 nm (19.82 mW cm−2) |
| Acrylate function |
Epoxy function |
Acrylate function |
Epoxy function |
| Under air |
In laminate |
Under air |
In laminate |
Under air |
In laminate |
Under air |
In laminate |
| J-H |
43 |
79 |
43 |
26 |
36 |
76 |
35 |
15 |
| J-CN |
32 |
77 |
48 |
23 |
40 |
78 |
42 |
20 |
| J-CH3 |
28 |
76 |
38 |
15 |
41 |
77 |
40 |
19 |
| J-F |
44 |
75 |
44 |
39 |
35 |
76 |
28 |
17 |
| J-OCH3 |
49 |
78 |
41 |
20 |
37 |
77 |
29 |
17 |
| J-SCH3 |
45 |
77 |
44 |
16 |
38 |
79 |
28 |
19 |
| J-vinyl |
39 |
79 |
49 |
25 |
38 |
78 |
27 |
18 |
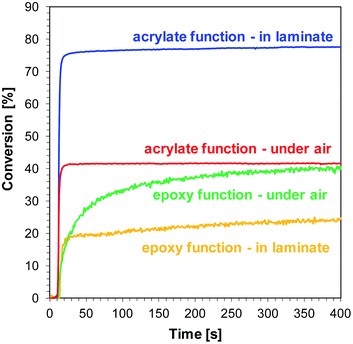 |
| | Fig. 10 Photopolymerization profiles of TMPTA (acrylate function conversion vs. irradiation time) and S105 (epoxy function vs. time) during polymerization of a S105/TMPTA blend (1:1, w/w), upon exposure to the LED@405 nm under air and in laminate, in the presence of SpeedCure 938 (1% w/w) and J-CH3 (0.2% w/w). The irradiation starts at t = 10 s. | |
Photochemical mechanism of bimolecular photoinitiating systems based on 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives
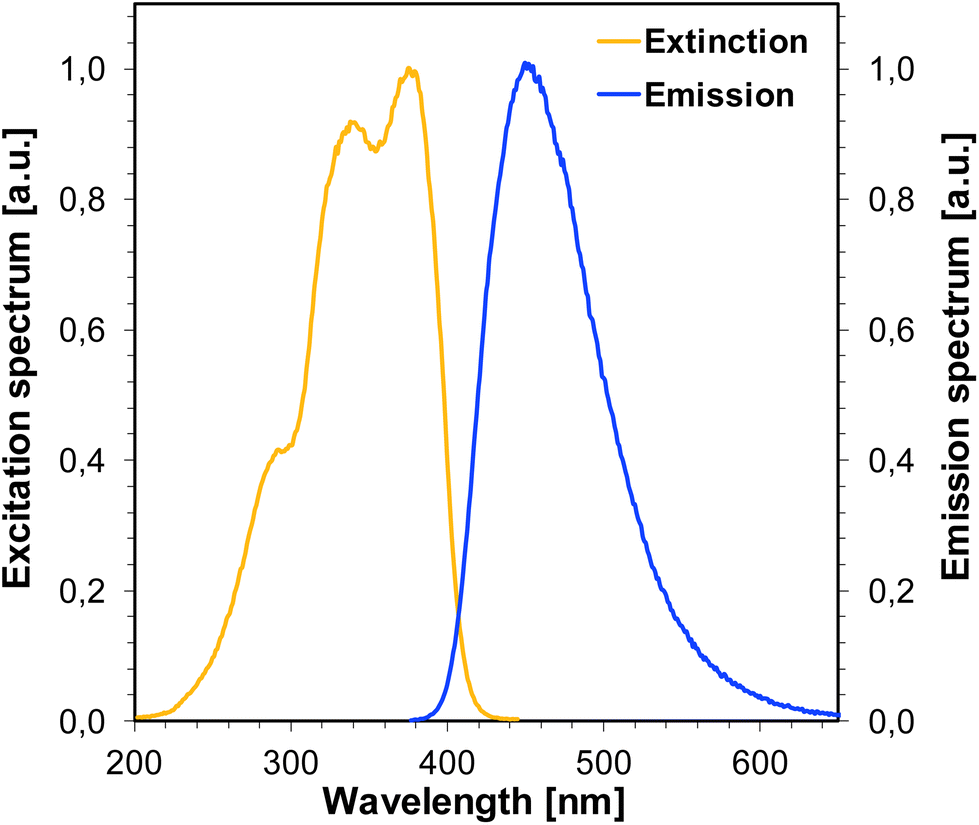
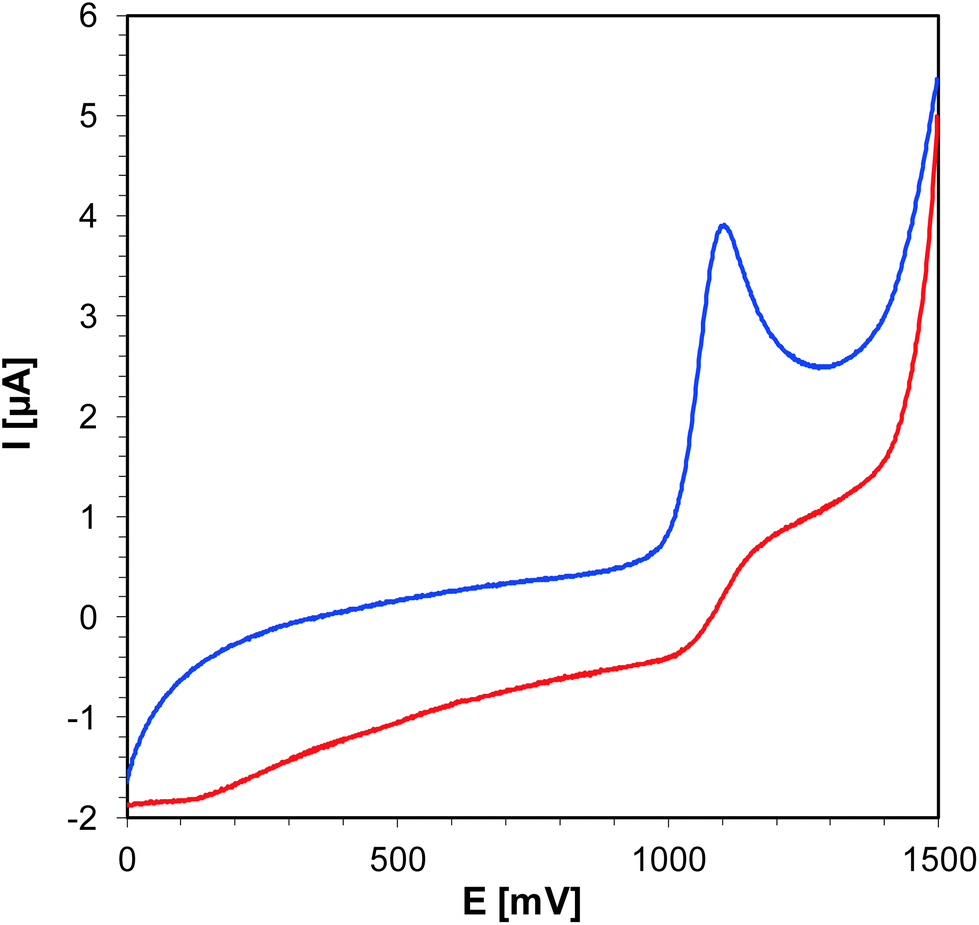
For the photosensitization of iodonium salt to be successful, both a photosensitizer and light are required. As a first step, photoexcitation takes place, the photosensitizer absorbs light and is excited to the first singlet state. The excited state lifetime is highly important for the efficient electron transfer to another molecule, namely iodonium salt. For the studied bimolecular photoinitiator systems based on iodonium salt and 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives, it is assumed that the initiation of photopolymerization proceeds by an electron transfer mechanism. The electron transfer from the 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives to the iodonium salt is feasible if the change of the free energy for the photoinduced polymerization process (ΔGet) is negative. The free energy (ΔGet) values for all studied bimolecular photoinitiating systems were estimated using the Rehm–Weller equation. In order to determine ΔGet, which accompanies the electron transfer in the studied two component initiation systems, the energy of singlet state (E00) was determined based on measurements of excitation and emission spectra (Fig. 11). The oxidation potential (Eox) of the 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives were measured by cyclic voltammetry (for example, Fig. 12). All the obtained data are collected in Table 5. The calculated values of (ΔGet) for the studied photoinitiating systems oscillate in the range between −1.40 and −1.12 eV for the singlet excited state. These calculations show that the electron transfer process of the investigated systems is thermodynamically allowed and suggest that the photoredox pairs should effectively generate radicals and radical cations able to initiate the photopolymerization processes of monomers. Structural modifications of the 1-amino-4-methyl-6-phenylnaphthalene-2-carbonitrile core have led to changes in its electrochemical properties. Based on the research and calculations carried out, it was found that the electron transfer process in the J-OCH3/iodonium salt system is the most efficient (ΔGet = −1.40 eV), because 1-amino-6-(4-methoxyphenyl)-4-methyl-naphthalene-2-carbonitrile (J-OCH3) exhibits the lowest oxidation potential. Conversely, sensitizer 1-amino-6-(4-cyanophenyl)-4-methyl-naphthalene-2-carbonitrile (J-CN) possesses the highest value of oxidation potential, so this system is the least effective of all presented.
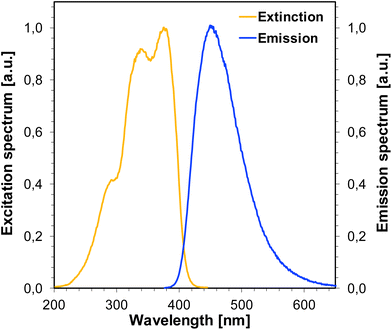 |
| | Fig. 11 Normalized extinction and emission spectra for J-SCH3. | |
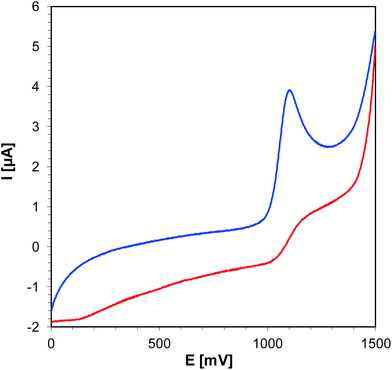 |
| | Fig. 12 Cyclic voltammogram for J-SCH3 in acetonitrile. | |
Table 5 Electrochemical and thermodynamical properties of sensitizers
| Sensitizer |
Eox vs. SCE [mV] |
ES1 [eV] |
ΔGet(S1)a [eV] |
ET1 [eV] |
ΔGet(T1)a [eV] |
KSV [M−1] |
τ [ns] |
kq [M−1s−1] |
Φet(S1) |
| Calculated from the classical Rehm–Weller equation: ΔGet = F[Eox (D/D˙+) – Ered (A˙−/A)] – ES1/T1 – Ze2/εa. Eox (D/D˙+) – the electrochemically determined oxidation potential of the sensitizer (vs. SCE). Ered (A˙−/A) – the electrochemically determined reduction potential of the electron acceptor, −0.68 V for the diaryliodonium salt (vs. SCE).38,39 ES1 – singlet state energy of the sensitizer determined based on excitation and emission spectra. ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G (d,p) level of theory). Ze2/εa – interaction energy for the initially formed ion pair, negligible in polar solvents. Φet – the electron transfer quantum yields from the excited singlet state from the equation: Φet = KSV[IOD]/(1 + KSV[IOD]), [IOD] = 0.0186 M. |
| J-H |
1029 |
3.08 |
−1.36 |
2.31 |
−0.60 |
103.57 |
6.05 |
1.71 × 1010 |
0.66 |
| J-CN |
1059 |
2.86 |
−1.12 |
2.22 |
−0.48 |
173.75 |
7.57 |
2.30 × 1010 |
0.76 |
| J-CH3 |
1023 |
3.08 |
−1.37 |
2.32 |
−0.61 |
148.63 |
6.83 |
2.18 × 1010 |
0.73 |
| J-F |
1045 |
3.07 |
−1.34 |
2.30 |
−0.57 |
149.54 |
6.77 |
2.21 × 1010 |
0.74 |
| J-OCH3 |
1011 |
3.09 |
−1.40 |
2.32 |
−0.63 |
80.56 |
5.69 |
1.42 × 1010 |
0.60 |
| J-SCH3 |
1039 |
3.05 |
−1.32 |
2.30 |
−0.58 |
103.18 |
5.82 |
1.77 × 1010 |
0.66 |
| J-vinyl |
1121 |
2.99 |
−1.19 |
2.28 |
−0.48 |
115.98 |
7.24 |
1.60 × 1010 |
0.68 |
As a next step, the fluorescence quenching of the bimolecular system based on naphthalene derivatives with iodonium salt was carried to study the interaction between the photosensitizers in question and SpeedCure 938.
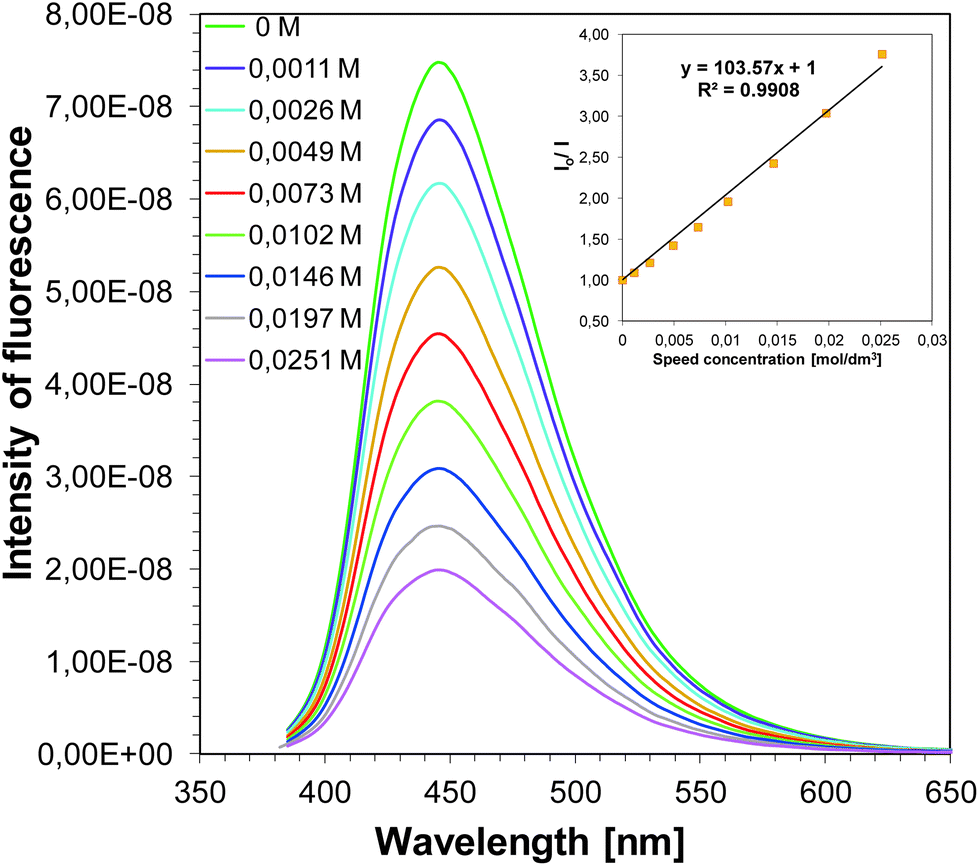
Strong fluorescence quenching of singlet excited states of sensitizers by iodonium salt (SpeedCure 938) was observed (example in Fig. 13, data for all compounds in SI). Stern–Volmer coefficients for all sensitizers Ksv are in the range 70–170 M−1. According to the classical Stern–Volmer equation:
where
I0 and
I stand for the fluorescence intensity of the sensitizer in the absence and presence of the additive (iodonium salt) respectively,
τ0 stands for the lifetime of the excited state of the sensitizer in the absence of the additive (the fluorescence lifetimes of sensitizers are listed in
Table 5).
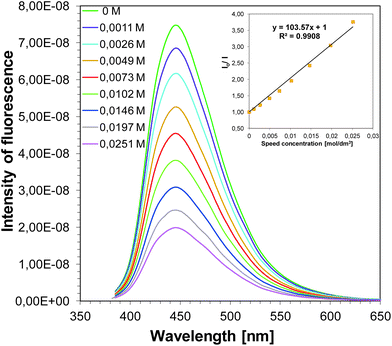 |
| | Fig. 13 Fluorescence quenching of J-H by iodonium salt (SpeedCure 938) in acetonitrile and Stern–Volmer treatment for the J-H/SpeedCure 938 quenching. | |
The interaction rate constants kq between the co-initiator and SpeedCure 938 were calculated. The interaction rate constants kq are almost diffusion controlled ∼1010 M−1 s−1 for 1-amino-4-methyl-naphthalene-2-carbonitrile derivatives as photosensitizers of SpeedCure 938 (Table 5).
Further, the electron transfer quantum yields from the excited singlet state (Φet) were also calculated based on equation:
| Φet(S1) = KSV[IOD]/(1 + KSV[IOD]) |
All results are listed in Table 5. High values (∼0.70) of electron transfer quantum yields in the excited singlet state Φet were calculated for all naphthalene derivatives. The high values of electron transfer quantum yields from S1 indicate that naphthalene derivatives mainly react through a singlet state pathway.
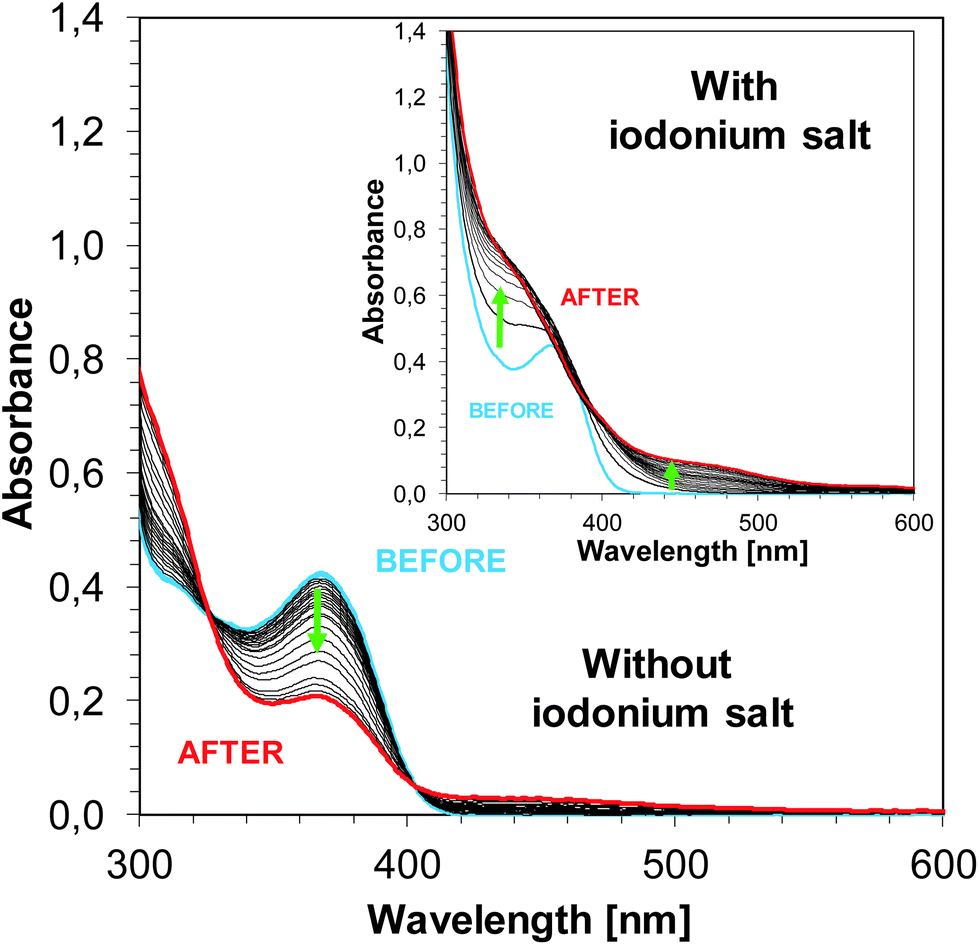
The steady state photolysis for sensitizers alone (without the addition of SpeedCure 938) was observed. The absorption spectrum of photosensitizer J-H changes upon irradiation of UV LED 365 nm over 30 min. In the presence of iodonium salt, the steady state photolysis of J-H occurs very fast. After 600 s, a new photo-product is formed (this is characterised by a new absorption band ∼500 nm – Fig. 14). This also indicates the interaction of compound J-H with iodonium salt SpeedCure 938.
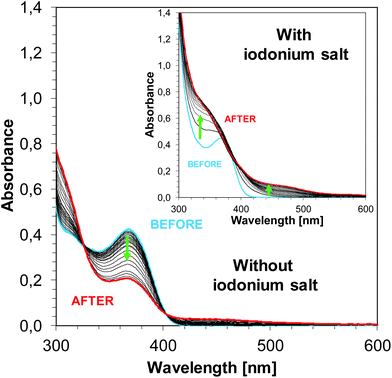 |
| | Fig. 14 Photolysis of J-H in acetonitrile without and with SpeedCure 938 upon exposure to LED 365 nm. | |
3D printing experiment (laser writing)
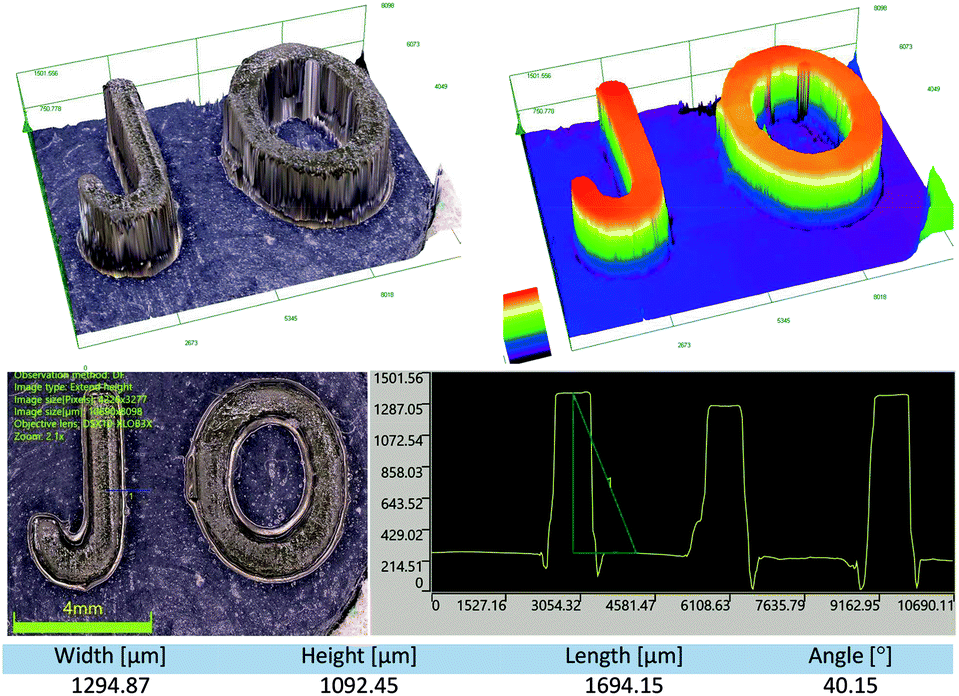
3D printing experiments with laser diode irradiation 405 nm were carried out in air using the investigated bimolecular photoinitiating systems of the TMPTA/S105 blend. The high photosensitivity of the obtained formulations allows efficient photopolymerization in the irradiated area. Patterns were obtained with high spatial resolution in very short time (less than 1 min). Two component photoinitiating systems based on naphthalene derivatives and iodonium salt are excellent candidates for application in 3D print resins. The obtained samples were analysed microscopically, and the patterns are shown in Fig. 15 and 16.
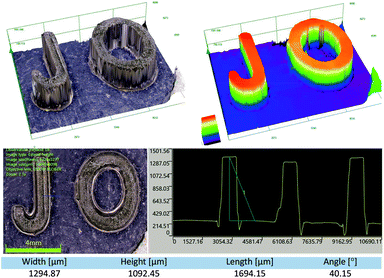 |
| | Fig. 15 The patterns obtained after the 3D printing experiment based on formulations J-OCH3 (0.05% w/w)/SpeedCure 938 (1.0% w/w)/TMPTA/S105 (1:1 w/w). | |
 |
| | Fig. 16 The patterns obtained after the 3D printing experiment based on formulations J-OCH3 (0.05% w/w)/SpeedCure 938 (1.0% w/w)/TMPTA/S105 (1:1 w/w), (A) logo of the Cracow University of Technology, photo from optical stereo-microscope, (B) part of the logo from picture (A), pattern from OLYMPUS DSX1000, (C and D) visualizations of the depth and width of the gap, photos from OLYMPUS DSX1000. | |
Conclusions
In the present paper, novel naphthalene-based compounds are proposed as photosensitizers in photopolymerization processes, in conjunction with iodonium salt, under low power ecological UV-A and visible LEDs as sources of light. The synthesis and properties of naphthalene derivatives were described. The investigated compounds are versatile sensitizers under UV and visible LEDs irradiation. Versatility in this approach is related to the usefulness of the proposed bimolecular photoinitiating systems for various types of photopolymerization processes, including cationic photopolymerization of epoxy and vinyl monomers, free radical photopolymerization of acrylates and thiol–ene polymerization. The developed photoinitiating systems were also adapted for the synthesis of interpenetrating polymer networks, based on epoxy/acrylate blends. Additionally, an investigated bimolecular photoinitiating system based on naphthalene skeleton compounds can be applied in 3D printing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the National Science Centre – Project SONATA [Grant No. UMO-2012/07/D/ST5/02300, 2012]. The authors are also grateful to the Foundation for Polish Science (Warsaw, Poland) TEAM TECH project Grant No. TEAM TECH/2016-2/15 (POIR.04.04.00-00-204B/16-00) for financing the purchase of equipment for 3D printing applications. The computational procedures were done by using open infrastructure resources PLGrid Infrastructure. The authors are also grateful to the Olympus Company and Mr Mateusz Pernak for presentation the optical microscope DSX1000 and possibility made an observation of patterns written during the photopolymerization of composition under air using new bimolecular photoinitating systems by this type of microscope.
Notes and references
- W. Schnabel, Polymers and Light, Fundamentals and Technical Applications, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007 Search PubMed.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- J. del Barrio and C. Sánchez-Somolinos, Adv. Opt. Mater., 2019, 7, 1900598 CrossRef.
- Z. Czech, A. Kowalczyk, J. Ortyl and J. Swiderska, Pol. J. Chem. Technol., 2013, 15(1), 12–14 CAS.
- Z. Czech, A. Kowlaski and J. Ortyl, Coating, 2011, 44(1), 22–25 CAS.
- B. Khurana, P. Gierlich, A. Meindl, L. C. Gomes-da-Silva and M. O. Senge, Photochem. Photobiol. Sci., 2019, 18, 2613–2656 RSC.
- C. Mendes-Felipe, J. Oliveira, I. Etxebarria, J. Luis Vilas-Vilela and S. Lanceros-Mendez, Adv. Mater. Technol., 2019, 4, 1800618 CrossRef.
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Adv. Mater., 2019, 1903850 CrossRef.
- A. Bagheri and J. Jin, ACS Appl. Polym. Mater., 2019, 1(4), 593–611 CrossRef CAS.
- J. Zhang and P. Xiao, Polym. Chem., 2018, 9, 1530–1540 RSC.
- W. Tomal, M. Pilch, A. Chachaj-Brekiesz and J. Ortyl, Catalysts, 2019, 9, 10 CrossRef.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- A. Javadi, H. Shokouhi Mehr, M. Sobani and M. D. Soucek, Prog. Org. Coat., 2016, 100, 2–31 CrossRef CAS.
- J. Ortyl and R. Popielarz, Polymers, 2012, 57(7–8), 510–517 CAS.
- H. Mokbel, J. Toufaily, T. Hamieh, F. Dumur, D. Campolo, D. Gigmes, J. P. Fouassier, J. Ortyl and J. Lalevée, J. Appl. Polym. Sci., 2015, 132, 46 CrossRef.
- J. Kabatc, J. Ortyl and K. Kostrzewska, RSC Adv., 2017, 7(66), 41619–41629 RSC.
- B. Husár, S. Clark Ligon, H. Wutzel, H. Hoffmann and R. Liska, Prog. Org. Coat., 2014, 77, 1789–1798 CrossRef.
- C. Belon, X. Allonas, C. Croutxé-Barghorn and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2462–2469 CrossRef CAS.
- S. Clark Ligon, B. Husár, H. Wutze, R. Holman and R. Liska, Chem. Rev., 2014, 114(1), 557–589 CrossRef PubMed.
- J. Ortyl, in Photopolymerisation Initiating Systems, ed. J. LalCvee and J. P. Fouassier, The Royal Society of Chemistry, 2018, ch. 3, pp. 74–130 Search PubMed.
- S. Shia, C. Croutxé-Barghorn and X. Allonas, Prog. Polym. Sci., 2017, 65, 1–41 CrossRef.
- M. Sangermano, Pure Appl. Chem., 2012, 84(10), 2089–2101 CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- X. Ge, Q. Ye, L. Song, A. Misra and P. Spencer, Macromol. Chem. Phys., 2015, 216, 856–872 CrossRef CAS PubMed.
- E. Hola, M. Pilch, M. Galek and J. Ortyl, Polym. Chem., 2020, 11, 480–495 RSC.
- M. Topa, F. Petko, M. Galek, K. Machowski, M. Pilch, P. Szymaszek and J. Ortyl, Polymers, 2019, 11(11), 1756 CrossRef CAS PubMed.
- D. Nowak, J. Ortyl, I. Kamińska-Borek, K. Kukuła, M. Topa and R. Popielarz, Polym. Test., 2018, 67, 144–150 CrossRef CAS.
- D. Nowak, J. Ortyl, I. Kamińska-Borek, K. Kukuła, M. Topa and R. Popielarz, Polym. Test., 2017, 64, 313–320 CrossRef CAS.
- J. Ortyl, P. Fiedor, A. Chachaj-Brekiesz, M. Pilch, E. Hola and M. Galek, Sensors, 2019, 19(7), 1668 CrossRef CAS.
- J. P. Fouassier and J. Lalevée, Polymers, 2014, 6(10), 12588–12610 CrossRef.
- K. Taki and K. Sawa, J. Photopolym. Sci. Technol., 2018, 31(6), 753–757 CrossRef CAS.
- C. Dietlin, S. Schweizer, P. Xiao, J. Zhang, F. Morlet-Savary, B. Graff, J. P. Fouassier and J. Lalevée, Polym. Chem., 2015, 6, 3895–3912 RSC.
- K. D. Jandt and R. W. Mills, Dent. Mater., 2013, 29(6), 605–617 CrossRef CAS PubMed.
- A. Al Mousawi, C. Dietlin, B. Graff, F. Morlet-Savary, J. Toufaily, T. Hamieh, J. P. Fouassier, A. Chachaj-Brekiesz, J. Ortyl and J. Lalevée, Macromol. Chem. Phys., 2016, 217(17), 1955–1965 CrossRef.
- E. Hola, J. Ortyl, M. Jankowska, M. Pilch, M. Galek, F. Morlet-Savary, B. Graff, C. Dietlin and J. Lalevée, Polym. Chem., 2020, 11, 922–935 RSC.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- P. Romańczyk and S. Kurek, Electrochim. Acta, 2017, 255, 482–485 CrossRef.
- B. Strehmel, S. Ernst, K. Reiner, D. Keil, H. Lindauer and H. Baumann, Z. Phys. Chem., 2014, 228(2–3), 129–153 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of sensitizers, cyclic voltammograms, singlet state energy determination, HOMO and LUMO orbitals, fluorescence quenching results, steady state photolysis with and without iodonium salt, patterns obtained after 3D printing experiment. See DOI: 10.1039/c9ra10212d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Maciej Pilcha,
Paweł Fiedora,
Mariusz Galekc and
Joanna Ortyl
b,
Maciej Pilcha,
Paweł Fiedora,
Mariusz Galekc and
Joanna Ortyl