
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA self-healing supramolecular hydrogel with temperature-responsive fluorescence based on an AIE luminogen†
Botian Li *,
Yichi Zhang,
Bo Yan,
Da Xiao,
Xue Zhou,
Junwei Dong and
Qiong Zhou*
*,
Yichi Zhang,
Bo Yan,
Da Xiao,
Xue Zhou,
Junwei Dong and
Qiong Zhou*
Department of Materials Science and Engineering, China University of Petroleum-Beijing, Beijing, 102249, People's Republic of China. E-mail: botian.li@cup.edu.cn; zhouqiong_cn@163.com
First published on 17th February 2020
Abstract
In this work, an AIE luminogen-based hydrogel with temperature-responsive fluorescence was designed and synthesized. The polymeric hydrogel consisted of a supramolecular network through coordination and ionic interactions. When the temperature was decreased, due to the motion restriction of the polyacrylic acid macromolecular segments and the enhancement in ionic interaction, the hydrogel exhibited a blue-shift in the fluorescence emission peak and increase in the fluorescence intensity, resulting in the visualization of fluorescence changes. The hydrogel network benefitted from non-covalent crosslinking and thus possessed self-healing properties at room temperature with good toughness and resiliency. Therefore, this fluorescent supramolecular hydrogel might be used as a temperature-responsive material.
Introduction
In the last decade, supramolecular hydrogels have attracted significant interest due to their unique physical properties and wide applications in many areas, including sensors,1,2 drug delivery,3,4 and biomaterials.5–7 Generally, these kinds of hydrogels are composed of a supramolecular network formed by low-molecular-weight gelators (LMWGs) or physically crosslinked polymers; thus, they can be categorized as the so-called supramolecular hydrogels based on LMWGs8–10 and supramolecular polymeric hydrogels.11,12 The supramolecular interactions are dynamic and reversible, imparting the hydrogels with responsive properties to external stimuli, such as light, temperature, redox, pH, and mechanical force.13,14 Also, the dynamic linkages in the network endow the hydrogels with prominent self-healing characteristics, which allow the gels to self-mend the damage intrinsically and automatically.15–17Aggregation-induced emission (AIE) has been considered as a great discovery worldwide.18–21 According to the mechanism of the restriction of intramolecular motion (RIM) proposed by Tang and coworkers,22 the AIE luminogens with a “propeller-shape” molecular structure exhibit a restricted intramolecular motion in the aggregated state, causing enhancement in the fluorescence emission by the reduction of the nonradiative transition. Recent research has shown that many AIE phenomena rely on surface adsorption, self-assembly, crystallization, polymerization, etc.23–28 Besides, some groups reported the monitoring of the transition temperature by introducing AIE units into polymer chains, which illustrated the potential application of the AIE units as fluorescent probes. For example, Bao et al. developed a sensitive and reliable approach for the detection of the glass transition temperature (Tg) of polymers using tetraphenylethene derivatives as fluorescent sensors;29 Yang and coworkers proposed an efficient method to measure the topology freezing transition temperature (Tv) of a vitrimer by the fluorescence change in the AIE molecules;30 Wang et al. reported enhanced fluorescence emission around the lower critical solution temperature (LCST) by importing the AIE property into the thermo-responsive polymer.31 These studies employed the characteristics of AIE for the indication of the polymer phase transition; however, there are few examples where AIE luminogens are utilized as fluorescent indicators to characterize supramolecular interactions and macromolecular motions without aggregation.
In order to develop the fluorescence indication of AIE in macromolecular dissolved systems, we designed a supramolecular polymeric hydrogel based on ionic interaction and coordination by using tetra(4-(pyridin-4-yl)phenyl) ethylene (TPPE) as the AIE luminogen. The hydrogel not only featured self-healing performance, but also exhibited temperature-responsive fluorescence with thermochromism. This was different from the traditional AIE behavior as the fluorescence response was not induced in the aggregation state of the AIE molecules but in the transparent gel and solution. Therefore, this study might help us to gain a deeper understanding of the details of the polymer chain motion and supramolecular interaction through the fluorescence indication of the AIE luminogen; therefore, the hydrogel might have potential applications as a smart fluorescent material.
Experimental
Materials
Tetra(4-(pyridin-4-yl)phenyl) ethylene (TPPE) was purchased from Yukang Biotechnology (Shanghai). Acrylic acid (AA), ammonium persulfate (APS), poly(acrylic) acid (PAA), and 2-acrylamido-2-methylpropane sulfonic acid (AMPS) were purchased from Aladdin Reagent (Shanghai). Anhydrous methanol and zinc oxide were supplied from Beijing Chemical. All chemicals were used directly without further purification.Gel preparation
Zinc oxide (0.3 g, 3.7 mmol) and AA (1 g, 13.9 mmol) were dissolved in deionized water (4 g); TPPE (1 mg, 1.56 μmol) and AA (1 g, 13.9 mmol) were dissolved in deionized water (4 g). The solutions were mixed in a vial and stirred to produce a light-yellow solution. Then, 40 mg of APS was added, and the vial was placed in a 60 °C oven for 8 hours. After the reaction was completed, the vial was taken out, and the formation of the TPPE–PAA hydrogel was confirmed by the inversion test.Zinc oxide (0.3 g, 3.7 mmol), AMPS (2 g, 9.7 mmol), TPPE (1 mg, 1.56 μmol) were dissolved in deionized water (8 g); the solutions were mixed in a vial and stirred to produce a light-yellow solution. Then, 40 mg of APS was added, and the vial was placed in a 60 °C oven for 8 hours. After the reaction was completed, the vial was taken out, and the formation of the TPPE–PAMPS hydrogel was confirmed by the inversion test.
Characterization
Fluorescence spectra and fluorescence lifetime were measured on a Spectrofluorometer FS5 UV fluorescence spectrophotometer (Edinburgh Instruments); the self-healing of the hydrogel was observed through a DM 2500P spectroscopic microscope. The rheological experiments were conducted on a parallel plate rheometer (Anton Paar, Physica MCR-300). The tensile properties were tested using the AGS-X tensile tester (Shimadzu) at a 100 mm min−1 velocity at room temperature. 1H NMR spectra were obtained on a Japan JEOL JNM-ECA400 spectrometer (400 MHz) using CD3OD and D2O as a mixed solvent.Results and discussion
Design and synthesis of hydrogel
As shown in Scheme 1, after the polymerization initiated by ammonium persulfate (APS), non-covalent crosslinking was formed between the macromolecular chains through the coordination of Zn2+ and COO− ions. Also, the protonated TPPE cations associated with COO− in polyacrylic acid (PAA) chains by ionic interactions in the aqueous solution were the ion crosslinking points with fluorescence. The supramolecular crosslinking network provided effective energy dissipation capability for hydrogels. In the tensile test, the polymer hydrogel with 20% PAA exhibited excellent stretchability with the fracture strain at 5600%, which was much higher than that for the hydrogel containing 30% PAA (Fig. S1†). Therefore, in the following discussion, the hydrogels with 20% PAA were employed.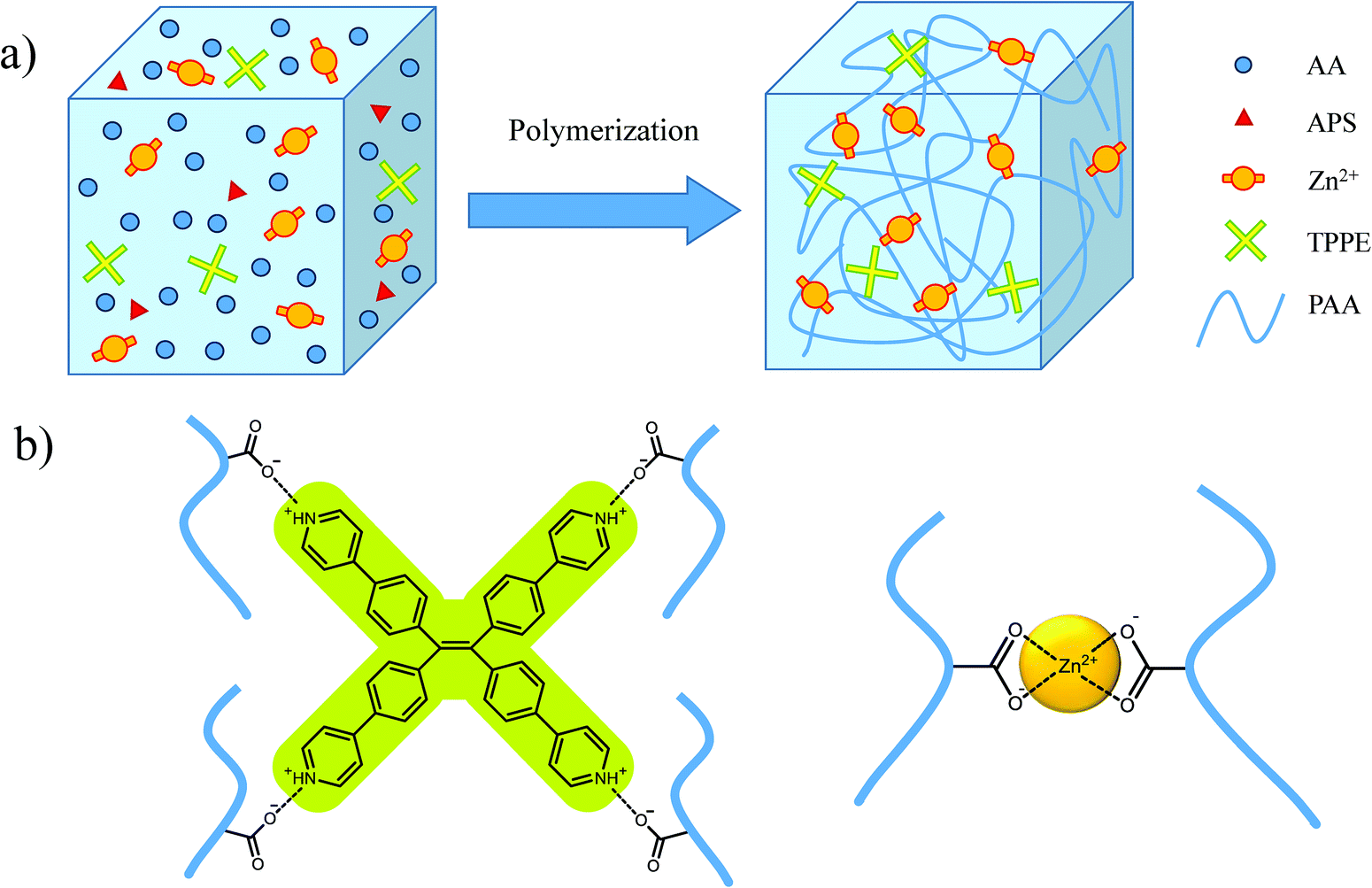 | ||
| Scheme 1 (a) Three-dimensional network structure of hydrogel; (b) non-covalent crosslinker points in hydrogel. | ||
The TPPE solid showed bright blue fluorescence and had typical AIE characteristics; nevertheless, the absolutely transparent TPPE–PAA hydrogel emitted tender yellow fluorescence under 365 nm UV light (Fig. 1), and the fully dissolved TPPE–PAA solution had exactly the same fluorescence. The yellow fluorescence of the TPPE–PAA hydrogel should be attributed to the characteristic emission spectrum of the TPPE cations due to its larger Stokes shift caused by protonation.32 Notably, herein, the AIE luminogen was emissive in the solution state. Several contrast experiments were designed to explore this anomalous AIE phenomenon. First, by comparing the TPPE acetic acid solution with the TPPE–PAA gel (Fig. S2†), it was found that the TPPE acetic acid solution was hardly fluorescent; only when combined to the macromolecular chain, TPPE could emit strong fluorescence. According to the RIM mechanism, we assumed that the ionic interaction between the TPPE cation and PAA chains (Scheme 1b) constrained the intramolecular motion of TPPE as the adjacent macromolecular anions were less motional.33,34 Second, when a small amount of HCl was added to the TPPE–PAA solution, one could immediately observe that the fluorescence significantly decreased (Fig. S3†). It was reasoned that the increase in the H+ concentration suppressed the ionization of COOH, thus reducing the macromolecular anions; therefore, the protonated TPPE cations could not bond with the polymer chain and consequently, the fluorescence of TPPE diminished with the free intramolecular motion.
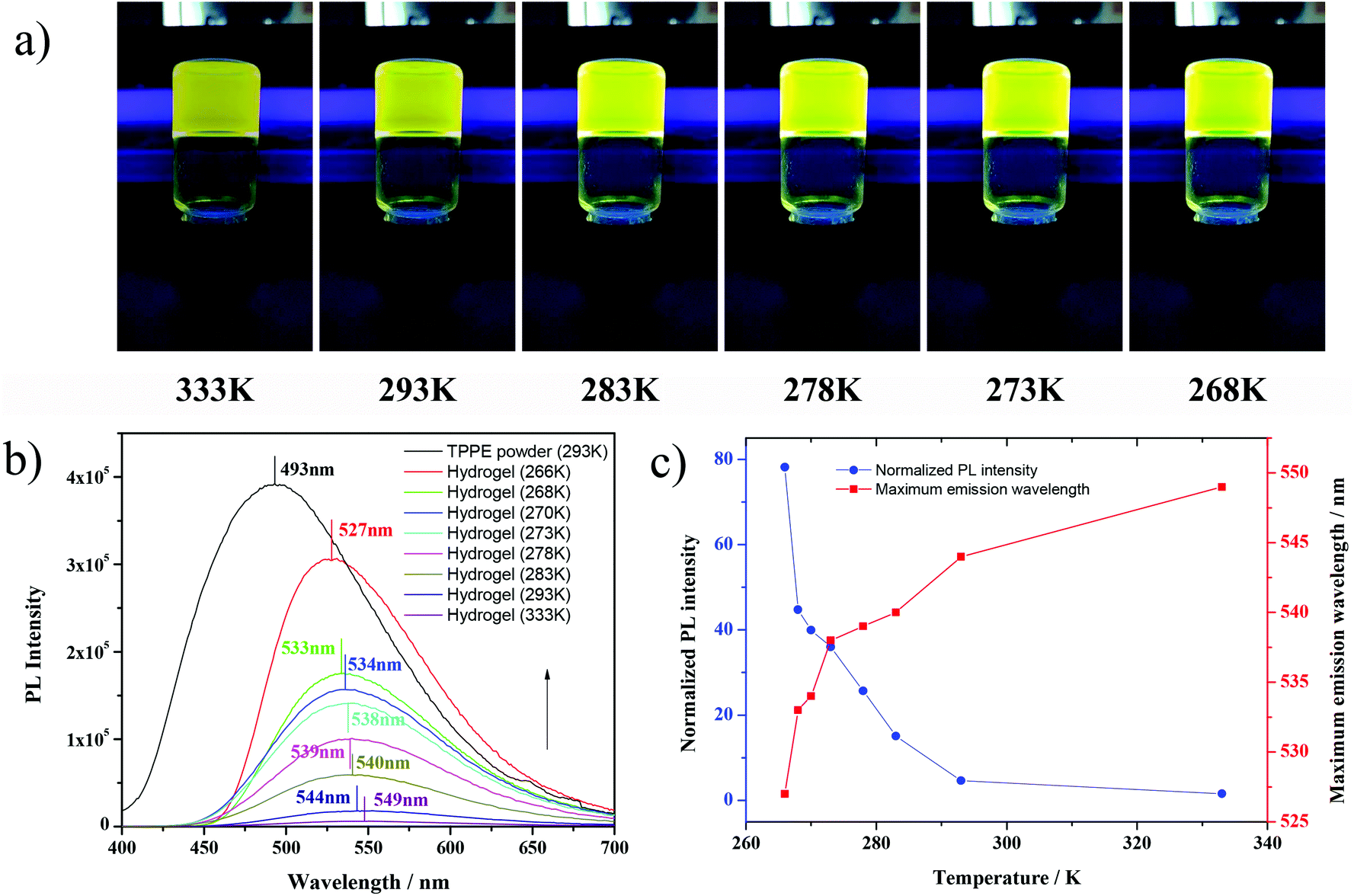 | ||
| Fig. 1 (a) Fluorescence images of TPPE–PAA hydrogel at different temperatures under 365 nm UV light; (b) fluorescence emission spectra of TPPE–PAA hydrogel under 365 nm UV light; (c) temperature dependency of fluorescence of TPPE–PAA hydrogel. | ||
Temperature-responsive fluorescence
More interestingly, it was found that under the cooling process, the TPPE–PAA hydrogel obviously changed its color from tender yellow to yellow green (Fig. 1a), and the fluorescence was intensified by dozens of times. The fluorescence emission spectra of the TPPE powder and TPPE–PAA hydrogel at different temperatures are shown in Fig. 1b. As the temperature decreased from 333 K to 266 K, the maximum emission wavelength of the hydrogel shifted from 549 nm to 527 nm; nevertheless, it was still higher than the maximum emission wavelength (493 nm) of the TPPE original state. The temperature-responsive fluorescence with thermochromism was visually obvious, especially when the temperature was lower than 278 K. As displayed in Fig. 1c, the fluorescence of the hydrogel shows a non-linear correlation with the temperature. Similarly, under 448 nm UV excitation (the maximum excitation wavelength of the hydrogel), in the cooling procedure, the fluorescence emission of the TPPE–PAA hydrogel blue-shifted and enhanced (Fig. 2).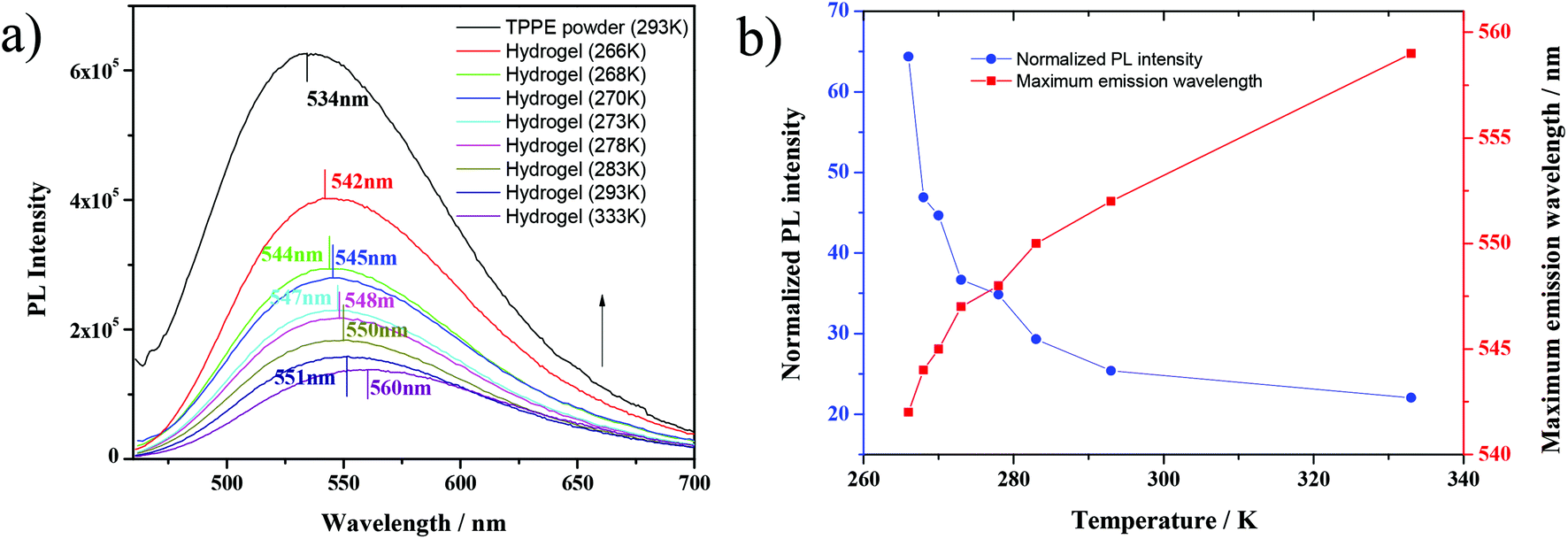 | ||
| Fig. 2 (a) Fluorescence emission spectra of TPPE–PAA hydrogel and (b) temperature dependency of fluorescence of TPPE–PAA hydrogel under 448 nm UV light. | ||
To study the fluorescence shift for the TPPE–PAA hydrogel responding to temperature, a contrast test was performed, where 2-acrylamido-2-methylpropane sulfonic acid (AMPS) was used to replace the AA monomer, and the TPPE–PAMPS hydrogel was prepared. In Fig. S4,† under 365 nm UV excitation, the TPPE–PAMPS hydrogel displays similar tender yellow fluorescence, but its color remains unchanged during cooling, implying that the dissociation of the protonated TPPE cations and macromolecular anions plays an important role in the change in fluorescence. Since AMPS is a strong electrolyte, the ion pair of TPPE–PAMPS can largely dissociate in the aqueous solution. However, TPPE–PAA should be assumed as the macromolecular salt of a weak acid and weak base, and the dissociation of this salt in solution is greatly affected by the temperature.35 Being an endothermic process, the dissociation of TPPE–PAA was restrained under cooling, and the ionic interactions between the TPPE cations and PAA anions became stronger, causing a blue-shift in fluorescence.
To evidence the above-mentioned assumption, we introduced methanol as a weak ionizing solvent to regulate the polarity of the solvent and control the dissociation of TPPE–PAA. Intriguingly, a green fluorescent solution was obtained after dissolving dry TPPE–PAA in anhydrous methanol (Fig. 3a). With the addition of H2O, the fluorescent color of the solution changed from green to yellow, and the maximum emission wavelength red-shifted from 532 nm to 543 nm with the fluorescent intensity reduced by half (Fig. 3b and c). The increase in the water content in the mixed solvent could also enhance the solvation of the TPPE–PAA ion pair, promoting the dissociation of the TPPE cations; accordingly, its freer intramolecular motion lowered the fluorescence intensity and increased the maximum emission wavelength.36
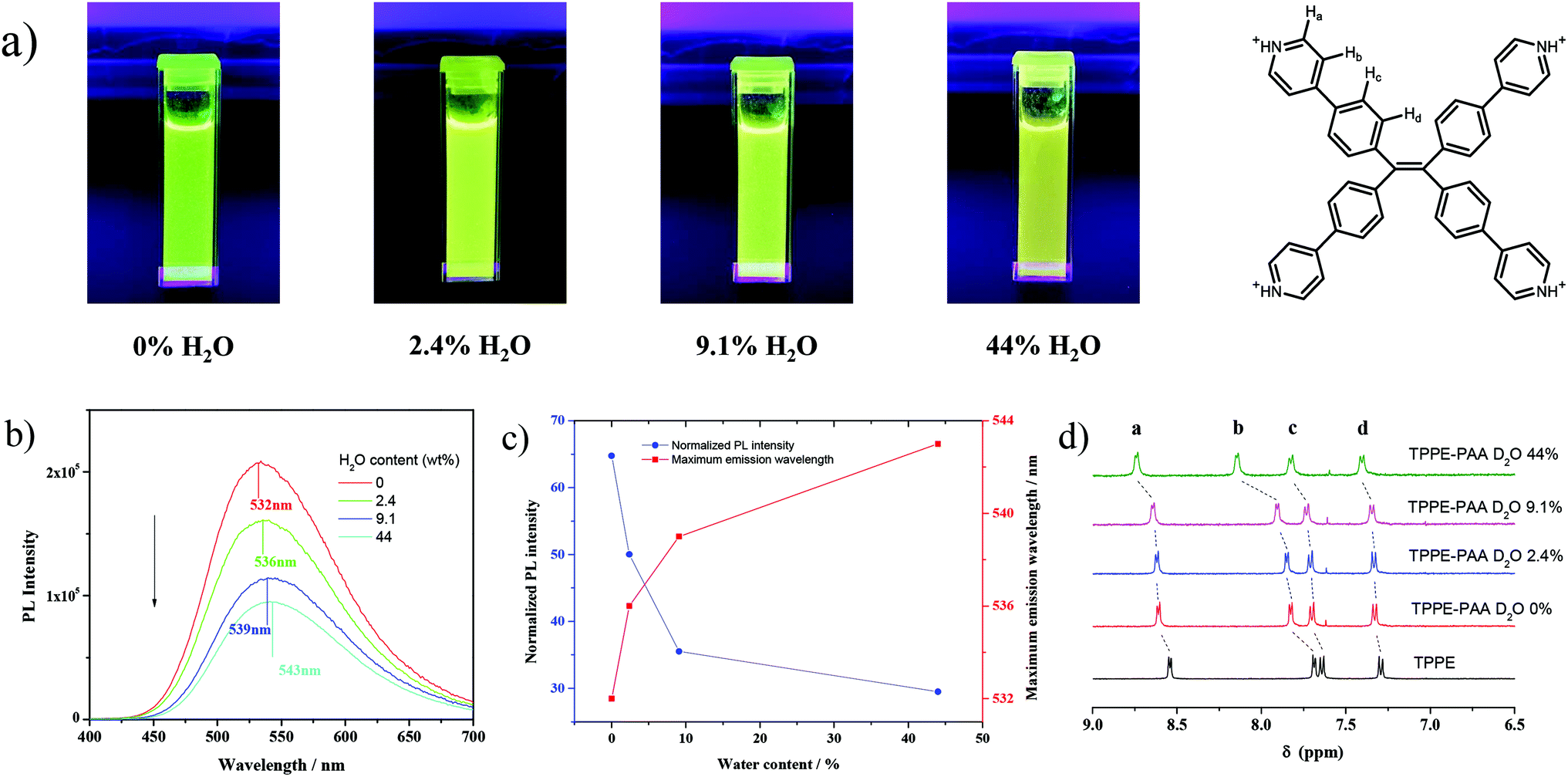 | ||
| Fig. 3 (a) Fluorescence of TPPE–PAA solid and its MeOH–H2O solutions under 365 nm UV light; (b) fluorescence emission spectra of TPPE–PAA solid and its MeOH–H2O solutions under 365 nm UV light; (c) solvent dependency of fluorescence of TPPE–PAA solution; (d) 1H NMR spectra of TPPE and TPPE–PAA. | ||
Furthermore, 1H NMR analysis was implemented to characterize the electrolytic dissociation at the molecular level. From the clear and strong signal in the 1H NMR spectrum, it is confirmed that TPPE is in the cationic form without aggregation. As shown in Fig. 3d, in anhydrous CD3OD, the protons on the pyridine and benzene rings of TPPE display four signals. After the addition of PAA, the pyridine groups on TPPE were all protonated along with positive charge; due to this, the peaks of aryl H moved to the lower field, wherein the signal of the pyridine meta hydrogen remarkably shifted from 7.69 to 7.83 ppm. After the addition of D2O, the four peaks gradually shifted towards the lower field, and this indicated the increase in the positive charge on protonated TPPE, especially on the pyridine ring. Thus, this evidenced that the COO− anions were dissociated from the TPPE cations when the solvent polarity was increased. Similarly, with the increase in the temperature, the positive charge on the protonated TPPE was enhanced due to the dissociation of COO− according to the result of 1H NMR (Fig. S4†). Meanwhile, TPPE–PAA dissolved in the mixed solvent of MeOH and H2O exhibited the same fluorescence change as the TPPE–PAA hydrogel as heating triggered a red-shift in fluorescence emission and reduction in fluorescence intensity (Fig. S5†).
Moreover, the fluorescence lifetime was determined from the fluorescence decay profile to further investigate the supramolecular interaction on fluorophores. Generally, the lifetime is influenced by the environment of the AIE luminogen; a prolonged fluorescence lifetime reflects a more immobilized state and stronger interactions surrounding the AIE luminogen.37–39 Fig. 4a displays the decay profiles of TPPE–PAA in MeOH and H2O; the average lifetime value (τ) in MeOH (4.31 ns) was longer than that in H2O (2.95 ns). Meanwhile, the decrease in the temperature from 298 K to 278 K resulted in a longer lifetime (3.34 ns) in H2O (Fig. 4b). These findings evidenced that the ionic interaction between TPPE and PAA was enhanced by introducing a low-polarity solvent (MeOH) and cooling.
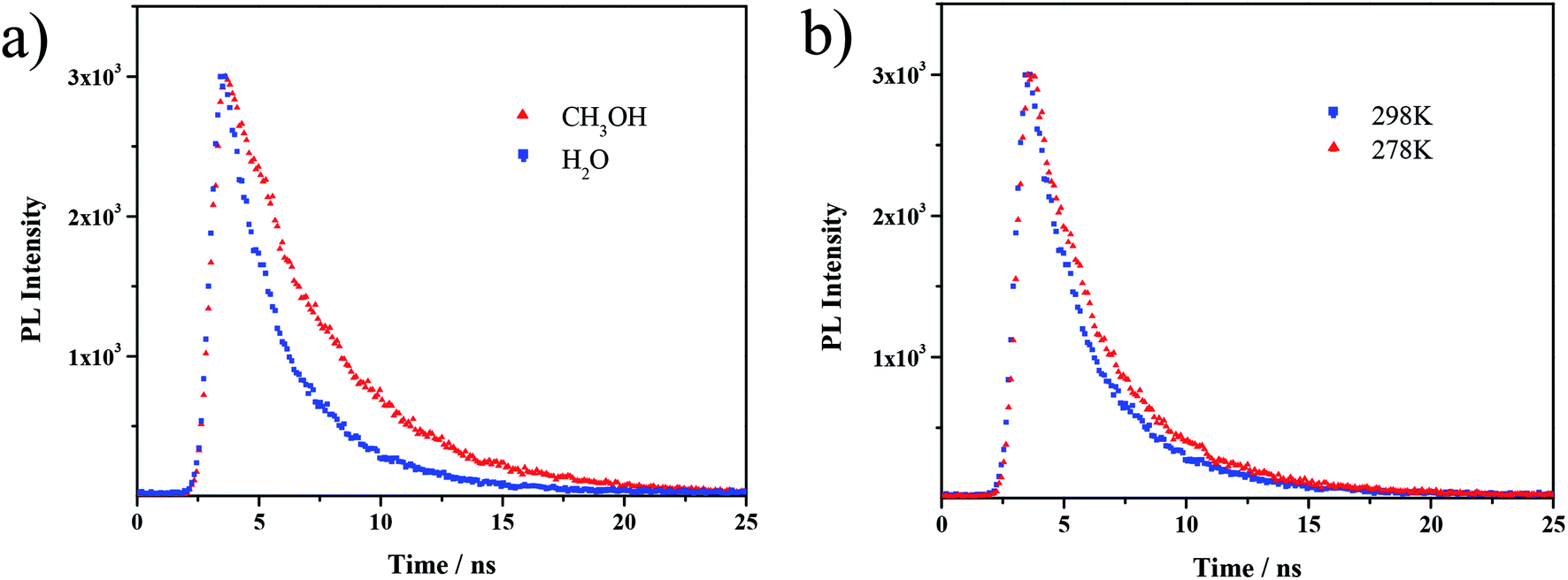 | ||
| Fig. 4 Fluorescence decay curves of (a) TPPE–PAA in MeOH and H2O (298 K) and (b) TPPE–PAA in H2O (298 K and 278 K). | ||
Based on these results, it was concluded that the blueshift in the fluorescence of the TPPE–PAA hydrogel under cooling was ascribed to the weakening of the dissociation of the macromolecule salts and the strengthening of ionic interactions. The fluorescent color was related to the dissociation of the TPPE–PAA salt, which could be regulated by solvent polarity and temperature.
The deduction presented above could also explain the results that the fluorescence intensity of TPPE–PAA increased as the temperature was decreased (Fig. 1 and 2). On the one hand, the motion of the PAA chain segment was confined under a low temperature;40 thus, the intramolecular motion of TPPE was restricted by the adjacent PAA chain, resulting in fluorescence enhancement. On the other hand, the ionic interactions between PAA and TPPE became stronger under cooling, and this simultaneously restrained the intramolecular motion of TPPE, leading to the increase in fluorescence. Comparatively, the fluorescence intensity of the TPPE–PAMPS hydrogel was lower than that of the TPPE–PAA hydrogel at 293 K (Fig. S4†) because the ionic interactions between TPPE and the PAMPS chain were relatively weak as they were largely dissociated in the solution, resulting in the increase in the motional freedom of TPPE than that in the TPPE–PAA hydrogel. In accordance with some recent studies,27–30 this work showed that the fluorescence enhancement of the AIE luminogen had a close relationship with the macromolecule motion ability and the supramolecular interaction; thus, AIE luminogens might be used as the indicator of the thermo-motion of polymer chains in the solution state.
Self-healing property
The TPPE–PAA hydrogel possessed a supramolecular crosslinking network, which endowed it with excellent self-healing performance at room temperature. As shown in Fig. 5a, the hydrogel can “self-repair” the scratching damage by regenerating a new network through the reversible coordination of zinc ions, and the self-healing process is completed within 12 hours. The self-healed hydrogel showed prominent mechanical strength in the stretching test (Fig. 5b); after the removal of the external force, it could recover to its original size, which demonstrated that the hydrogel based on supramolecular crosslinking had great toughness and perfect resiliency. | ||
| Fig. 5 (a) Self-healing of TPPE–PAA hydrogel at room temperature under microscopy; (b) self-healing of spliced hydrogel and stretching-recovery test. | ||
As shown in Fig. 6a, the recovery test of the gel has been conducted by rheological measurements upon applying alternating strains. When a high strain was applied (500%), the gel was disrupted, as indicated by the lower value of the storage modulus (G′) than that of the loss modulus (G′′). After applying a small strain (0.1%), the recovery of elasticity (G′ > G′′) was observed. This recovery behavior could be repeated in several cycles, demonstrating that the network of the gel was self-healing. Also, the tensile curves of the hydrogel (Fig. 6b) prove the completion of self-healing after 12 h as both the fracture strain and fracture stress of the self-healed hydrogel achieve the original value.
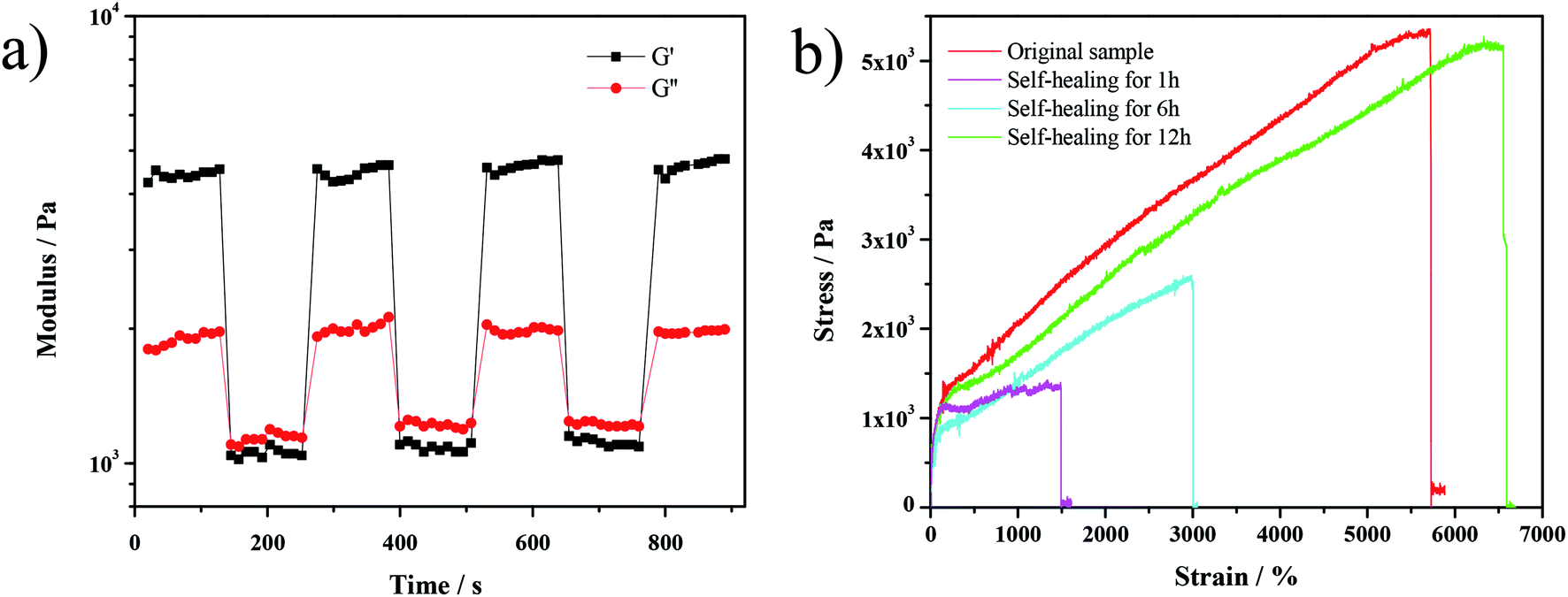 | ||
| Fig. 6 (a) Step strain measurements of hydrogel, alternate step strain switched from a small strain (0.1%) to a large strain (500%) at a fixed frequency (1 Hz); (b) tensile curves of hydrogels with different self-healing times. | ||
Conclusions
In summary, the TPPE–PAA hydrogel constituting a supramolecular network with Zn2+ and an AIE luminogen as the crosslinking point was prepared, and its temperature-responsive fluorescence was characterized. Due to the motion restriction of the polymer segments and the strengthening of ionic interactions, the hydrogel underwent a blue-shift in fluorescence emission and enhancement in fluorescence intensity as the temperature decreased. Also, the hydrogel featured self-healing properties at room temperature. This hydrogel might be considered as a potential self-healing smart material with thermo-sensitive fluorescence.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51903250) and the Science Foundation of China University of Petroleum-Beijing (No. 2462017YJRC008).Notes and references
- C. Y. Shao, M. Wang, L. Meng, H. L. Chang, B. Wang, F. Xu, J. Yang and P. B. Wan, Chem. Mater., 2018, 30, 3110–3121 CrossRef CAS.
- X. J. Wang, C. W. Wei, J. H. Su, B. He, G. B. Wen, Y. W. Lin and Y. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3504–3508 CrossRef CAS PubMed.
- W. Zheng, G. Yang, N. N. Shao, L. J. Chen, B. Ou, S. T. Jiang, G. S. Chen and H. B. Yang, J. Am. Chem. Soc., 2017, 139, 13811–13820 CrossRef CAS PubMed.
- C. B. Rodell, N. N. Dusaj, C. B. Highley and J. A. Burdick, Adv. Mater., 2016, 28, 8419–8424 CrossRef CAS PubMed.
- Y. C. Choi, J. S. Choi, Y. J. Jung and Y. W. Cho, J. Mater. Chem. B, 2014, 2, 201–209 RSC.
- L. Y. Shi, F. L. Wang, W. Zhu, Z. P. Xu, S. Fuchs, J. Hilborn, L. J. Zhu, Q. Ma, Y. J. Wang, X. S. Weng and D. A. Ossipov, Adv. Funct. Mater., 2017, 27, 1700591 CrossRef.
- L. Y. Shi, H. Carstensen, K. Holzl, M. Lunzer, H. Li, J. Hilborn, A. Ovsianikov and D. A. Ossipov, Chem. Mater., 2017, 29, 5816–5823 CrossRef CAS.
- W. Xiong, H. T. Zhou, C. Zhang and H. Lu, Chin. Chem. Lett., 2017, 28, 2125–2128 CrossRef CAS.
- F. Wang and C. L. Feng, Angew. Chem., Int. Ed., 2018, 57, 5655–5659 CrossRef CAS PubMed.
- P. F. Duan, H. Cao, L. Zhang and M. H. Liu, Soft Matter, 2014, 10, 5428–5448 RSC.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- L. Y. Shi, P. H. Ding, Y. Z. Wang, Y. Zhang, D. Ossipov and J. Hilborn, Macromol. Rapid Commun., 2019, 40, 1800837 CrossRef PubMed.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- S. Bera, A. Chakraborty, S. Karak, A. Halder, S. Chatterjee, S. Saha and R. Banerjee, Chem. Mater., 2018, 30, 4755–4761 CrossRef CAS.
- J. H. Jin, L. L. Cai, Y. G. Jia, S. Liu, Y. H. Chen and L. Ren, J. Mater. Chem. B, 2019, 7, 1637–1651 RSC.
- M. Häring and D. D. Díaz, Chem. Commun., 2016, 52, 13068–13081 RSC.
- C. H. Li and J. L. Zuo, Adv. Mater., 2019, e1903762, DOI:10.1002/adma.201903762.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- S. Y. Zhou, H. B. Wan, F. Zhou, P. Y. Gu, Q. F. Xu and J. M. Lu, Chin. J. Polym. Sci., 2019, 37, 302–326 CrossRef CAS.
- T. Y. Wu, J. B. Huang and Y. Yan, Chem.–Asian J., 2019, 14, 730–750 CrossRef CAS PubMed.
- Y. C. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- D. D. Li and J. H. Yu, Small, 2016, 12, 6478–6494 CrossRef CAS PubMed.
- C. W. Zhang, B. Ou, S. T. Jiang, G. Q. Yin, L. J. Chen, L. Xu, X. P. Li and H. B. Yang, Polym. Chem., 2018, 9, 2021–2030 RSC.
- Q. Lin, B. Sun, Q. P. Yang, Y. P. Fu, X. Zhu, T. B. Wei and Y. M. Zhang, Chem.–Eur. J., 2014, 20, 11457–11462 CrossRef CAS PubMed.
- G. X. Huang, Y. Q. Jiang, S. F. Yang, B. S. Li and B. Z. Tang, Adv. Funct. Mater., 2019, 29, 1900516 CrossRef.
- Y. Y. Bao, E. Guegain, V. Nicolas and J. Nicolas, Chem. Commun., 2017, 53, 4489–4492 RSC.
- S. J. Liu, Y. H. Cheng, H. K. Zhang, Z. J. Qiu, R. T. K. Kwok, J. W. Y. Lam and B. Tang, Angew. Chem., Int. Ed., 2018, 57, 6274–6278 CrossRef CAS PubMed.
- S. P. Bao, Q. H. Wu, W. Qin, Q. L. Yu, J. Wang, G. D. Liang and B. Z. Tang, Polym. Chem., 2015, 6, 3537–3542 RSC.
- Y. Yang, S. Zhang, X. Q. Zhang, L. C. Gao, Y. Wei and Y. Ji, Nat. Commun., 2019, 10, 3165 CrossRef PubMed.
- X. M. Wang, K. Y. Xu, H. C. Yao, L. M. Chang, Y. Wang, W. J. Li, Y. L. Zhao and J. L. Qin, Polym. Chem., 2018, 9, 5002–5013 RSC.
- X. Y. Yao, X. Ma and H. Tian, J. Mater. Chem. C, 2014, 2, 5155–5160 RSC.
- X. G. Gu, G. X. Zhang and D. Q. Zhang, Analyst, 2012, 137, 365–369 RSC.
- C. H. Li, T. Wu, C. Y. Hong, G. Q. Zhang and S. Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 455–459 CrossRef CAS PubMed.
- Y. J. Xu, J. Chem. Thermodyn., 2013, 64, 126–133 CrossRef CAS.
- Y. L. Su, H. S. Ren and X. Y. Li, Chem. Phys. Lett., 2019, 732, 136640 CrossRef CAS.
- S. J. Chen, Y. N. Hong, Y. Zeng, Q. Q. Sun, Y. Liu, E. G. Zhao, G. X. Bai, J. N. Qu, J. H. Hao and B. Z. Tang, Chem.–Eur. J., 2015, 21, 4315–4320 CrossRef CAS PubMed.
- M. X. Gao, Y. N. Hong, B. Chen, Y. N. Wang, W. J. Zhou, W. W. H. Wong, J. Zhou, T. A. Smith and Z. J. Zhao, Polym. Chem., 2017, 8, 3862–3866 RSC.
- L. Espinar-Barranco, M. Meazza, A. Linares-Perez, R. Rios, J. M. Paredes and L. Crovetto, Sensors, 2019, 19, 4932 CrossRef PubMed.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, Oxford, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10092j |
| This journal is © The Royal Society of Chemistry 2020 |