
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtracellular electron transfer mediated by a cytocompatible redox polymer to study the crosstalk among the mammalian circadian clock, cellular metabolism, and cellular redox state†
Masahito Ishikawa *ad,
Kazuki Kawaia,
Masahiro Kanekob,
Kenya Tanakac,
Shuji Nakanishicd and
Katsutoshi Hori*a
*ad,
Kazuki Kawaia,
Masahiro Kanekob,
Kenya Tanakac,
Shuji Nakanishicd and
Katsutoshi Hori*a
aDepartment of Biomolecular Engineering, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: ishikawa.masahito@chembio.nagoya-u.ac.jp; khori@chembio.nagoya-u.ac.jp
bDepartment of Materials Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
cGraduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan
dResearch Center for Solar Energy Chemistry, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan
First published on 9th January 2020
Abstract
The circadian clock is an endogenous biological timekeeping system that controls various physiological and cellular processes with a 24 h rhythm. The crosstalk among the circadian clock, cellular metabolism, and cellular redox state has attracted much attention. To elucidate this crosstalk, chemical compounds have been used to perturb cellular metabolism and the redox state. However, an electron mediator that facilitates extracellular electron transfer (EET) has not been used to study the mammalian circadian clock due to potential cytotoxic effects of the mediator. Here, we report evidence that a cytocompatible redox polymer pMFc (2-methacryloyloxyethyl phosphorylcholine-co-vinyl ferrocene) can be used as the mediator to study the mammalian circadian clock. EET mediated by oxidized pMFc (ox-pMFc) extracted intracellular electrons from human U2OS cells, resulting in a longer circadian period. Analyses of the metabolome and intracellular redox species imply that ox-pMFc receives an electron from glutathione, thereby inducing pentose phosphate pathway activation. These results suggest novel crosstalk among the circadian clock, metabolism, and redox state. We anticipate that EET mediated by a redox cytocompatible polymer will provide new insights into the mammalian circadian clock system, which may lead to the development of new treatments for circadian clock disorders.
Introduction
The circadian clock is a biological system that generates an approximately 24 hour cell-autonomous rhythm for the purpose of anticipating periodic changes in the environment and enabling organisms to adapt to such predictable changes.1,2 The mammalian circadian clock is driven by transcriptional–translational feedback loops composed of clock genes. In the core feedback loop, the transcription factors BMAL1 and CLOCK, or the closely related homolog NPAS2, activate expression of the genes Period (Per1 and Per2) and Cryptochrome (Cry1 and Cry2). After translation and nuclear localization, PER and CRY proteins inhibit the function of either the BMAL1/CLOCK or BMAL1/NPAS2 heterodimer, closing the negative feedback loop. Because BMAL1/CLOCK and BMAL1/NPAS2 also regulate the transcription of other genes, various physiological and cellular processes, including metabolism, exhibit circadian rhythms. Studies have implied that metabolic rhythm is not only a simple output of circadian regulation, but also provides important input to the circadian clock, which is essential for maintaining the robustness of the circadian clock.2,3 Because metabolism is a network of biochemical reactions accompanied by electron transfers and the intracellular redox state is closely related to cellular metabolism, the intracellular level and balance of redox species also show circadian rhythm. Studies have implied that cellular redox cycles are integrated between circadian clock and metabolism.4–7 Therefore, in order to achieve a comprehensive understanding of the circadian clock system, it is very important to elucidate the crosstalk among the circadian clock, cellular metabolism, and cellular redox state.Redox/metabolic perturbations that modulate the circadian clock are useful for a good understanding of the crosstalk among the circadian clock, metabolism, and redox state. Redox-active molecules or inhibitors against metabolic enzymes have been employed for redox/metabolic perturbations; such perturbations have provided new insights into how the redox state and metabolism are related to the circadian clock system.5,8–11 Therefore, the use of chemical compounds capable of inducing redox state and metabolic alternations has been identified as a promising approach for studying the circadian clock system. The discovery and development of such chemical compound will contribute to a comprehensive understanding of the circadian clock system, and may lead to new treatments for diseases caused by circadian clock disorders.
The construction of an extracellular electron transfer (EET) pathway, in which the intracellular electrons are exchanged with an extracellular electron donor/acceptor across the cell membrane, is an effective approach for altering intracellular redox balance and cellular metabolism.12–14 The EET pathway can be constructed using a membrane-permeable redox-active compound, namely, an electron mediator.15,16 Modifications of intracellular redox balance via EET have been demonstrated in many microbial species.17–19 Nevertheless, the use of EET as a redox/metabolic perturbation has been limited in studies on the mammalian circadian clock possibly due to the cytotoxic effects of electron mediators. To date, we have demonstrated that 2-methacryloyloxyethyl phosphorylcholine (MPC)-based redox polymers are cytocompatible electron mediators that can alter the intracellular redox states and metabolism of several microbial species.20–23 For example, the metabolism of Saccharomyces cerevisiae was altered by extracting intracellular electrons with the oxidized form of pMFc (ox-pMFc, where pMFc is poly MPC-co-vinyl ferrocene, EM = +0.5 V vs. SHE).22 Hence, we assumed that EET mediated by ox-pMFc can also alter the intracellular redox state and metabolism of mammalian cells, thereby altering the mammalian circadian clock (Fig. 1).
 | ||
| Fig. 1 Schematic of the concept in this study. The oxidized form of pMFc (ox-pMFc) crosses the plasma membrane of a living U2OS cell by a simple diffusion process and accepts an electron from intracellular redox species, resulting in the alternation of the cellular redox state. The redox potential of pMFc is higher than those of various intracellular redox species such as NAD(P)H, the reduced form of glutathione (GSH), and quinone derivatives. Thus, the electron transfer from these species to ox-pMFc is thermodynamically allowed. This redox perturbation affects the circadian clock either directly, indirectly, or both ways through the metabolic alternation induced by the redox state alternation. | ||
Here, we report for the first time, evidence that EET, facilitated by a cytocompatible electron mediator based on MPC, can be used to study the mammalian circadian clock.
Results and discussion
EET via pMFc in U2OS cells
The all experiments in this study were conducted in the soluble concentration range of pMFc polymers. To verify the feasibility of our approach shown in Fig. 1, we began by determining if pMFc can cross the plasma membrane of human osteosarcoma U2OS cells, which is a well-characterized cell line for circadian clock research. Since the MPC-based amphiphilic phospholipid polymers have been reported to shuttle the interior and the exterior of the cells by a simple diffusion process,24,25 pMFc was also expected to cross the plasma membrane of U2OS cells. The membrane permeability of pMFc was evaluated using rhodamine-tagged pMFc (rho-pMFc).25 After staining with MitoTracker Green FM, U2OS cells were incubated in the presence of rho-pMFc and then observed with a confocal laser scanning microscopy (CLSM). The fluorescence from the rhodamine unit of rho-pMFc was detected in U2OS cells, indicating that pMFc crossed the plasma membrane and was internalized in U2OS cells (Fig. 2). Colocalization of rho-pMFc and MitoTracker Green FM were also observed in the merged image, suggesting the interaction of pMFc with mitochondria. | ||
| Fig. 2 Confocal images of U2OS cells treated with rhodamine-tagged pMFc and MitoTracker Green FM. DIC, a differential-interference-contrast image; Rho-pMFc, the fluorescence of rhodamine-tagged pMFc (rho-pMFc) excited at 559 nm; MitoTracker, the fluorescence of MitoTracker Green FM excited at 473 nm; Merge, a merged image of DIC, rho-pMFc, and MitoTracker. Scale bars indicate 20 μm. | ||
Next, we determined if ox-pMFc can accept an electron from U2OS cells. The UV/vis absorption spectrum of ox-pMFc in the medium displays a characteristic peak at 620 nm, in contrast to the reduced form of pMFc (red-pMFc) in the medium (Fig. 3a). Hence, the abundance of ox-pMFc in the medium can be determined by the absorbance of the medium at 620 nm (A620). We exchanged the cultivation medium of confluent U2OS cells with the medium containing 1 mM ox-pMFc, incubated at 37 °C supplemented with CO2, and measured the A620 of the medium at the indicated time points (Fig. 3b). The abundance of ox-pMFc gradually decreased, indicating the reduction of ox-pMFc by accepting an electron from reductive species. Although the reduction of ox-pMFc was also observed in the absence of U2OS cells, the complete reduction of ox-pMFc was faster in the presence of U2OS cells than in their absence; complete reduction required 24 h in the presence of U2OS cells and 72 h in their absence. This result means that ox-pMFc molecules accept electrons not only from cells but also from redox species in the medium. Commercial media generally contain numerous reductive compounds as antioxidants, such as vitamins (e.g. choline chloride, D-calcium pantothenate, folic acids, niacinamide, pyridoxine hydrochloride, riboflavin, thiamine hydrochloride, and i-inositol). Since the redox potential of pMFc is higher than those of such vitamins, we deduced that these compounds donate electrons to ox-pMFc molecules. The abundance of the ox-pMFc was 84.5% in the presence compared with 89.2% the absence of U2OS cells after the first hour of incubation. Therefore, 4.7% ox-pMFc (47.7 μM) accepted electrons from the U2OS cells during the first 1 h.
 | ||
| Fig. 3 Reactivity of ox-pMFc. (a) UV-vis spectra of 1 mM ox-pMFc and red-pMFc in DMEM for recording bioluminescence rhythms. Baseline indicates the UV-vis spectrum of the DMEM containing no pMFc. (b) Change in ox-pMFc abundance with time. One millimolar ox-pMFc in the DMEM was incubated with or without U2OS cells at 37 °C supplemented with 5% CO2. The abundance of pMFc was calculated from the absorbance at 620 nm (A620) of the medium. Data are shown as means ± SD (n = 3). (c) Reactivity of ox-pMFc to intracellular redox compounds. One millimolar ox-pMFc was mixed with either the reduced form of glutathione (GSH), the oxidized form of glutathione (GSSG), NADH, NAD+, NADP+, or NADPH. The A620 of the mixtures was measured at 1 s intervals. The concentration of pMFc corresponds to that of the vinyl ferrocene unit. | ||
Next, we tested if the ox-pMFc can accept an electron from representative intracellular redox species in vitro, namely the reduced form of glutathione (GSH), NADH, and NADPH (Fig. 3c). As controls, we used the oxidized form of glutathione (GSSG), NAD+, and NADP+. Immediately after 1 mM of each compound was mixed with a 1 mM ox-pMFc solution, A620 of the mixture was measured with a spectrophotometer over time. We observed a decrease in A620 when ox-pMFc was mixed with the reduced forms of these redox species, whereas A620 did not change when ox-pMFc was mixed with its oxidized forms, indicating that the ox-pMFc can accept electrons from the reduced form of these compounds in living cells.
Effect of ox-pMFc-mediated EET on the circadian clock in U2OS cells
To investigate the effect of EET mediated by ox-pMFc on the mammalian circadian clock, we performed a cell-based luminescence assay using U2OS cell lines harboring a mBmal1–dLuc reporter.26,27 The bioluminescence was monitored with 0.25–1.5 mM ox-pMFc (Fig. S1†). Although ox-pMFc caused period lengthening in a dose-dependent manner, it became difficult to extract peaks or troughs from the bioluminescence rhythm at high concentrations (1.25–1.50 mM). Because the reporter activity of mBmal1–dLuc is dependent on cell viability, a high concentration of ox-pMFc may be cytotoxic towards U2OS cells. Thus, we determined the effect of less than 1 mM ox-pMFc on the circadian clock of U2OS cells. The detrended bioluminescence rhythm clearly indicated that the ox-pMFc lengthened the period of the circadian clock in U2OS cells (Fig. 4a). Since the abundance of ox-pMFc in the medium decreased gradually as shown in Fig. 3b, the effect of ox-pMFc-mediated EET should change with duration. The period changes relative to the control were analyzed as a period of each day, namely, a period between one trough and the next trough, or one peak and the next peak (Fig. 4b). For the first two days after ox-pMFc treatment, the periods lengthened in a dose-dependent manner. Thereafter, the periods of cells treated with lower concentrations of ox-pMFc (0.25 and 0.5 mM) were restored to their original length, whereas those at higher concentrations (0.75 mM and 1 mM) maintained an approximately 1 h extension. When U2OS cells were treated with red-pMFc, which cannot accept electrons from cells, the circadian period was hardly altered (Fig. 4c and d). These remarkably different results obtained with different redox forms of pMFc suggest that EET induced the observed period lengthening. | ||
| Fig. 4 Effect of pMFc-mediated EET on the circadian clock in U2OS cells. Representative curves of the detrended bioluminescence rhythms of U2OS:mBaml1–dLuc cells in the presence of (a) ox-pMFc or (c) red-pMFc, or (e) in the medium oxidized by ox-pMFc. (b, d and f) Changes in the circadian period length relative to 10 mM HEPES buffer control. The period length was calculated as the time between either one trough and the next, or between one peak and the next. Each concentration of pMFc corresponds to that of the vinyl ferrocene unit. Data are expressed as means ± SD (n = 3). | ||
As shown in Fig. 3b, ox-pMFc accepts an electron not only from intracellular redox species in U2OS cells but also from redox-active components in the medium. We assumed that oxidation of the medium by ox-pMFc affects the circadian clock of U2OS cells. To confirm this, we determined if the medium oxidized by ox-pMFc lengthens the circadian period. The medium for measuring bioluminescence rhythm was mixed with each concentration of ox-pMFc in the absence of U2OS cells, and incubated at 37 °C supplemented with 5% CO2 for 3 d. The complete reduction of ox-pMFc was verified by measuring the A620 of the medium. The bioluminescence rhythm of U2OS cells was monitored after exchanging the cultivation medium with these oxidized media. The graph in Fig. 4e seems to show little difference between the control and samples treated with the oxidized media. When the change in period relative to the control was measured, we observed an approximately 1 h longer period lengthening only in U2OS cells treated with medium oxidized by 1 mM ox-pMFc (Fig. 4f). Other oxidized media did not alter the U2OS circadian period, indicating that the oxidation of media components affects the circadian period of U2OS to a lesser extent than EET via ox-pMFc.
Metabolic and redox alternations induced by ox-pMFc-mediated EET
To investigate which metabolic alternations were induced by the ox-pMFc-mediated EET, metabolites were extracted from U2OS cells after 24 h treatment with either 1 mM ox- or red-pMFcs and were analyzed comprehensively. We assumed that the metabolic profiles after the 24 h treatment should be altered because the abundance of ox-pMFc remains at about 12% (Fig. 3b) and the period was lengthened by approximately 4 h (Fig. 4b). Following our metabolomic analysis, 116 peaks (52 cations and 64 anions) were detected by the anion and cation modes of CE-TOFMS and CE-QqQMS. The results of principal component analysis (PCA) and hierarchical cluster analysis (HCA) are shown in Fig. S2.† The PCA revealed that PC1 (horizontal axis) distinctly separated the samples treated with 1 mM ox-pMFc from the samples treated with 1 mM red-pMFc and untreated samples (control). PC2 (vertical axis) clarified the differences between the same samples, suggesting that there is an outlier among the red-pMFc-treated samples. HCA revealed that the metabolic patterns of 1 mM red-pMFc-treated cells and untreated cells were similar, whereas those of 1 mM ox-pMFc-treated cells was distinct, implying that the metabolic alternation was induced by ox-pMFc-mediated EET. The metabolic pathways and metabolites associated with glycolysis/gluconeogenesis, the pentose phosphate pathway (PPP), and the tricarboxylic acid (TCA) cycle are illustrated in Fig. 5. The concentrations of some metabolites associated with PPP increased significantly in cells treated with 1 mM ox-pMFc, whereas those with glycolysis and TCA cycle decreased significantly. The results of metabolomic analysis indicate the activation of PPP by EET via ox-pMFc. | ||
| Fig. 5 Metabolic alternation of U2OS cells induced by ox-pMFc-mediated EET for 24 h. The metabolic pathways and metabolites associated with glycolysis/gluconeogenesis, the pentose phosphate pathway, and the tricarboxylic acid (TCA) cycle are shown. The vertical axis in the graphs indicates the absolute concentration of metabolites (mol per 106 cells). Significant differences were examined by Welch's t-test with Bonferroni correction. Different letters (A and B) with one or two asterisks represent approached significance (p < 0.10) or significant difference (p < 0.05), respectively. For the same letter, the difference is not statistically significant. The concentration of pMFc corresponds to that of vinyl ferrocene unit. Data are shown as means ± SD (n = 3). | ||
PPP is a pathway to provide NADPH, which serves as reducing power for regenerating GSH from GSSG. We assumed that the ratios of NADPH:NADP+ and GSH:GSSG should be altered by EET via the ox-pMFc. Fig. 6 shows time courses of the GSH:GSSG and NADPH:NADP+ ratios in U2OS cells treated with either 1 mM ox- or red-pMFc and in untreated cells (control). For the first 3 h, the GSH:GSSG ratio in ox-pMFc-treated cells was significantly lower than those in red-pMFc-treated cells and control cells. The GSH:GSSG ratio in ox-pMFc-treated cells subsequently became the same level as that in the other cells at 6 h before finally becoming more reductive than that in the other cells (Fig. 6). On the other hand, the NADPH:NADP+ ratio in ox-pMFc-treated cells was always higher than that of the other samples.
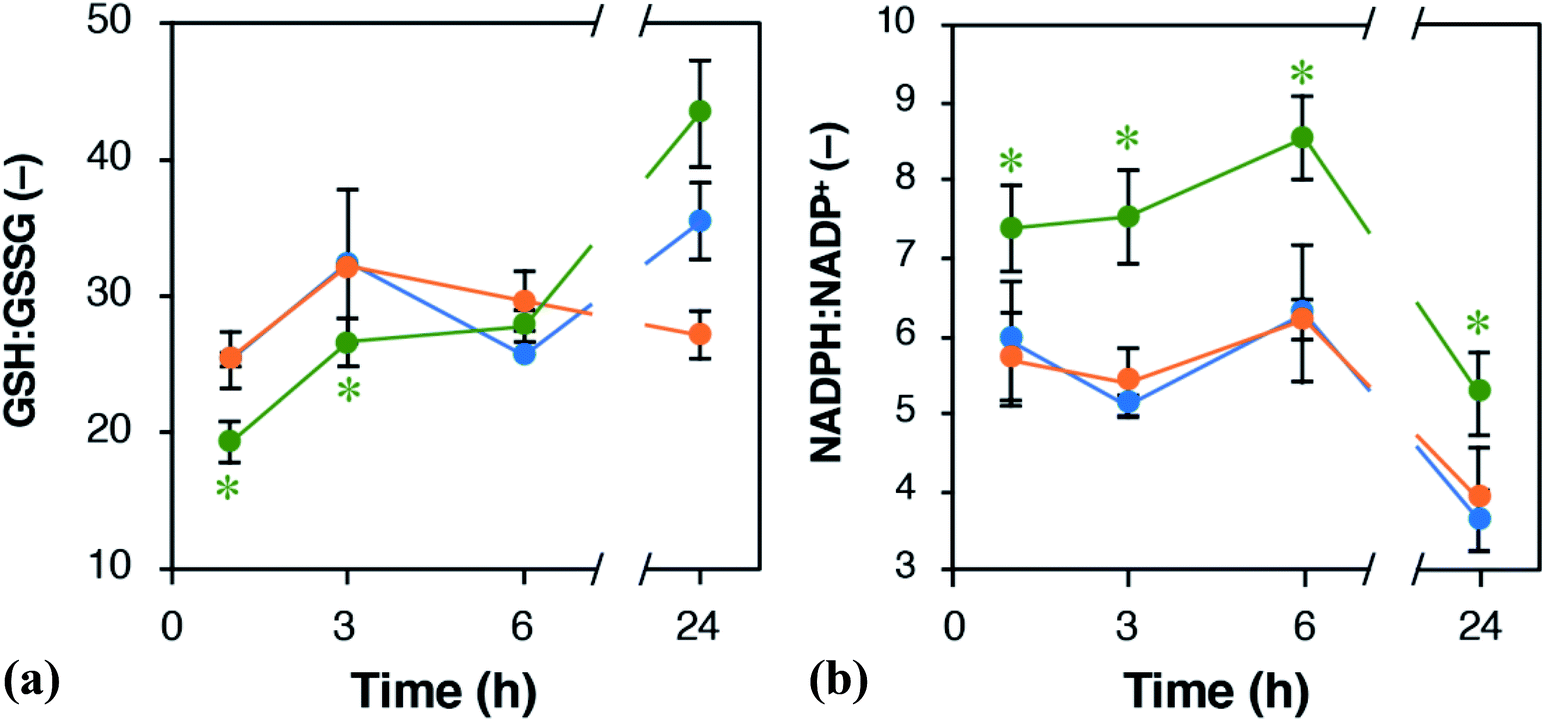 | ||
| Fig. 6 Time course alternations of the ratios of (a) GSH:GSSG and (b) NADPH:NADP+ in U2OS cells. Green, orange, and blue symbols represent U2OS cells treated with either 1 mM ox- or red-pMFc and untreated cells (control), respectively. Data are expressed as mean ± SD (n = 3). Asterisks indicate the significant difference (Welch's t-test, control verses the pMFc-treated cells, p < 0.05). | ||
The alternations of GSH:GSSG and NADPH:NADP+ ratios suggested that ox-pMFc accepted an electron mainly from intracellular GSH. As GSH is more abundant in the cytosol than other redox species such as NADH and NADPH,28 it is the most likely redox molecule that pMFc molecules passing through the membrane will encounter first. U2OS cells whose GSH:GSSG ratio became oxidative are thought to activate their PPP in order to maintain the redox balance, resulting in a higher ratio of NADPH:NADP+. As the abundance of ox-pMFc gradually decreased and stayed low for 24 h (Fig. 3b), little reduction in GSH should have occurred at 24 h. According to the result of metabolomic analysis (Fig. 5), PPP remained activated in ox-pMFc-treated cells after 24 h. Hence, the GSH:GSSG ratio in ox-pMFc-treated U2OS cells were thought to recover to the same level as the other samples at 6 h and become more reductive than other samples at 24 h.
The metabolic profile of the 1 mM ox-pMFc-treated U2OS cells reflected the homeostasis for an intracellular redox balance. Homeostatic changes to metabolism were thought to induce the period-lengthening of circadian rhythm in U2OS cells. Based on in vitro experiments using the purified transcriptional factors, Rutter et al. reported that NADPH enhanced DNA binding of CLOCK:BMAL1 and NPAS2:BMAL1 heterodimers.29 Hence, the increase in NADPH:NADP+ ratio by PPP activation might enhance the DNA-binding activity of CLOCK:BMAL1 and NPAS2:BMAL1 heterodimers in U2OS cells. Rey et al. reported that PPP inhibition induces an oxidative NADPH:NADP+ ratio and lengthens the circadian period in U2OS cells. They thereby proposed that PPP is an important regulator of the circadian rhythm.8 Conversely, our results show that the circadian period was lengthened in U2OS cells whose NADPH:NADP+ ratio was increased by PPP activation. Although our results also indicate that PPP is important for regulating the circadian clock, the intracellular redox state and metabolism of U2OS cells that showed period-lengthening differ from those reported by Rey et al. Therefore, our study suggests the existence of an undefined mechanism underlying the regulation of the circadian clock by the cellular redox state and metabolism. Elucidation of this mechanism will require further study.
Conclusion
We have demonstrated that ox-pMFc-mediated EET induced redox and metabolic alternations, resulting in lengthening of the circadian period in U2OS cells. Our results suggest that MPC-based redox polymers such as pMFc are applicable to studies of the mammalian circadian clock. Because MPC-based polymers are flexible in their design, electron mediators with either different redox potentials or reactivities can be synthesized.22,23,30 We anticipate that the use of various MPC-based redox polymers will provide new insights to improve understanding of the crosstalk among the circadian clock, cellular metabolism, and cellular redox state, which may subsequently impact the development of new treatments for diseases caused by circadian clock disorders.Experimental section
Chemicals
NAD+, NADH, NADP+, and NADPH were purchased from Oriental Yeast Co., Ltd. (Tokyo, Japan). GSH and GSSG were purchased from Nacalai Tesque (Kyoto, Japan). MPC was purchased from NOF Co., Ltd., (Tokyo, Japan). VFc and α,α′-azobisisobutyronitrile (AIBN) were purchased from Wako Pure Chemicals Co., Ltd. (Osaka, Japan) and Kanto Chemical Co., Inc. (Tokyo, Japan), respectively. AIBN was recrystallized in methanol before use. pMFc was synthesized using 3.54 g MPC and 1.70 g VFc by conventional free radical polymerization with 164 mg AIBN as an initiator in 20 mL ethanol. Polymerization was conducted in a test tube at 65 °C for 48 h under an argon gas atmosphere. After the polymerization reaction, the polymer solution was precipitated with a mixed solvent composed of diethyl ether/chloroform (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v). The polymer precipitate was filtered and dried in a vacuum overnight. The polymer was subsequently dissolved in distilled water and dialysis was performed for 4 d. The polymers were freeze-dried, and the resulting yellow powder was obtained as reduced pMFc polymer. Molecular weight was measured by gel permeation chromatography with poly(ethylene glycol) as a standard. The composition was determined by UV/vis spectroscopy. To prepare ox-pMFc, red-pMFc was dissolved in 20 mM HEPES buffer at final concentration of 4 mM (corresponds to the concentration of the VFc unit in pMFc) and electrochemically oxidized at +0.6 V for 20 h, with stirring. Oxidization of red-pMFc was confirmed by measuring A620. As shown in Fig. S3,† 4 mM ox-pMFc was diluted to different concentrations (0.5–3.0 mM) with 20 mM HEPES buffer (pH 7.0) to prepare DMEM for recoding bioluminescence rhythms, which is composed of DMEM (D2902, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS), 3.5 g L−1 D-glucose, 0.35 g mL−1 sodium hydrogen carbonate, 100 μg mL−1 penicillin, 100 μg mL−1 streptomycin, 0.1 mM luciferin, 100 nM dexamethasone, 10 mM HEPES (pH 7.0), and ox-pMFc at the different concentrations (0.25–1.5 mM). Dexamethasone was contained for the synchronization of U2OS cells.27
:10, v/v). The polymer precipitate was filtered and dried in a vacuum overnight. The polymer was subsequently dissolved in distilled water and dialysis was performed for 4 d. The polymers were freeze-dried, and the resulting yellow powder was obtained as reduced pMFc polymer. Molecular weight was measured by gel permeation chromatography with poly(ethylene glycol) as a standard. The composition was determined by UV/vis spectroscopy. To prepare ox-pMFc, red-pMFc was dissolved in 20 mM HEPES buffer at final concentration of 4 mM (corresponds to the concentration of the VFc unit in pMFc) and electrochemically oxidized at +0.6 V for 20 h, with stirring. Oxidization of red-pMFc was confirmed by measuring A620. As shown in Fig. S3,† 4 mM ox-pMFc was diluted to different concentrations (0.5–3.0 mM) with 20 mM HEPES buffer (pH 7.0) to prepare DMEM for recoding bioluminescence rhythms, which is composed of DMEM (D2902, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS), 3.5 g L−1 D-glucose, 0.35 g mL−1 sodium hydrogen carbonate, 100 μg mL−1 penicillin, 100 μg mL−1 streptomycin, 0.1 mM luciferin, 100 nM dexamethasone, 10 mM HEPES (pH 7.0), and ox-pMFc at the different concentrations (0.25–1.5 mM). Dexamethasone was contained for the synchronization of U2OS cells.27
Mammalian cell culture
Human osteosarcoma U2OS cell line harboring a mBmal1–dLuc reporter26 was kindly provided by T. Nishiwaki-Ohkawa and T. Yoshimura from Nagoya University. This was obtained by the transfection with the pGL4.11 plasmid harboring the promoter region of mBmal1.31 Cells were maintained at 37 °C under 5% CO2 and 95% air in DMEM for cultivation, which is composed of DMEM (1199560, Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with 100 μg mL−1 penicillin, 100 μg mL−1 streptomycin, and 10% FBS. Some experiments in this study were conducted after replacing the DMEM for cultivation with the DMEM for recoding bioluminescence rhythms as shown in Fig. S4.†To determine ox-pMFc reactivity towards cells, U2OS reporter cells were cultivated to confluence on a 35 mm dish with a 9 cm2 surface area (MS-11350, Sumitomo Bakelite Co., Ltd., Tokyo, Japan) in the DMEM for cultivation. The medium was replaced with 2.6 mL of the DMEM for recoding bioluminescence rhythms. The abundance of ox-pMFc was estimated by measuring the A620 of the medium.
For real-time monitoring of the cellular bioluminescence rhythms, U2OS reporter cells were cultivated to confluence on a 60 mm dish with 21 cm2 surface area (CELLSTAR, Greiner Bio-One International GmbH, Kremsmünster, Germany). The medium was replaced with 6 mL of the DMEM for recoding the bioluminescence rhythms. When required, the medium contained either ox- or red-pMFc at the different concentrations (0.25–1.5 mM). Bioluminescence signals of the cultured cells were recorded at intervals of 1 h at 37 °C in air with a Gene Light 55 GL-100A luminometer (Microtec Co., Ltd., Chiba, Japan).
Membrane permeability of rhodamine-tagged pMFc in U2OS cells
The membrane permeability of pMFc was evaluated as described previously.24,25 Briefly, U2OS cells were seeded in a glass-bottomed dish (3911-035, Asahi Glass Co., Ltd., Shizuoka, Japan) at 1.5 × 105 cells per dish in 2 mL of the DMEM for cultivation and incubated for 24 h at 37 °C in 5% CO2. Then, the cells were incubated with 100 nM MitoTracker Green FM (Thermo Fisher Scientific Inc.) for 1 h at 37 °C in 5% CO2. After rinsing with the DMEM for cultivation three times, the cells were placed in 2 mL of fresh DMEM for cultivation containing 1 mg mL−1 rho-pMFc. After 1 h-incubation at 37 °C in 5% CO2, the cells were observed with a CLSM (FV-1000, OLYMPUS, Tokyo, Japan).Calculation of circadian period
Raw data obtained by measuring bioluminescence rhythms were detrended by subtracting their 24 h moving averages. To calculate the period length for each day, the detrended 24 h data was fitted to a cosine curve with the following equationwhere m is the gradient of the baseline, c is the y offset, k describes the damping rate, α is the amplitude, r is the phase, and p is the period. Curve-fitting was performed by the least squares method using the Solver function of Microsoft Excel.

In vitro reaction of ox-pMFc with NADH, NADPH, and GSH
One hundred milliliters of 1 mM ox-pMFc (corresponds to the concentration of the VFc unit in pMFc) in 10 mM HEPES buffer was placed into a microcuvette and then set on a spectrometer (UV1850, Shimadzu Corporation, Kyoto, Japan) equipped with a microcuvette holder (Shimadzu). Immediately after mixing with 1 mM NADH, NAD+, NADPH, NADP, GSH, or GSSG, the change in A620 in the ox-pMFc solution with time was measured at 1 s intervals.Measurement of intracellular NADPH:NADP+ and GSH:GSSG ratios
Intracellular NADPH:NADP+ and GSH:GSSG ratios were measured using an NADP+/NADPH-Glo Assay kit (Promega) and a GSH/GSSG-Glo Assay kit (Promega), respectively. U2OS cells were prepared and treated with either red- or ox-pMFc using the same procedure for monitoring cellular bioluminescence rhythm. Luminescence was measured with an ARVO X3 (PerkinElmer).Metabolite extraction
U2OS cells were prepared and treated with either ox- or red-pMFc using the same procedure for monitoring the bioluminescence rhythm. As a control, 10 mM HEPES was added to the medium instead of pMFc. After 24 h incubation at 37 °C under 5% CO2, the culture medium was aspirated from a dish. Cells were washed twice with 5% mannitol and treated with 400 μL methanol. The cell extract was treated with 275 μL pure water containing internal standards (H3304-1002, Human Metabolome Technologies (HMT), Tsuruoka, Yamagata, Japan) and left to rest for another 30 s. Cell debris was removed by centrifugation at 200g at 4 °C for 5 min and 350 μL of the supernatant was filtered by centrifugation through a Millipore 5 kDa cutoff filter (Ultrafree MC-PLHCC, HMT). Filtrate was concentrated by centrifugation and re-suspended in 50 μL deionized water for metabolomic analysis at HMT.Metabolome analysis
Metabolomic analysis was performed using the C-SCOPE package of HMT. Capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS) was used for cation analysis and CE-tandem mass spectrometry (CE-MS/MS) for anion analysis as described previously.32,33 Briefly, CE-TOFMS and CE-MS/MS analysis were carried out using an Agilent CE capillary electrophoresis system equipped with an Agilent 6210 time-of-flight mass spectrometer (Agilent Technologies, Waldbronn, Germany) and Agilent 6460 Triple Quadrupole LC/MS, respectively. The systems were controlled by Agilent G2201AA ChemStation software version B.03.01 for CE (Agilent Technologies) and connected by a fused silica capillary (50/μm i.d. × 80 cm total length) with a commercial electrophoresis buffer (H3301-1001 and I3302-1023 for cation and anion analyses, respectively, HMT) as the electrolyte. The time-of-flight mass spectrometer was scanned from m/z 50 to 1000 (ref. 33) and the triple quadrupole mass spectrometer was used to detect compounds in dynamic MRM mode. Peaks were extracted using MasterHands, automatic integration software (Keio University, Tsuruoka, Yamagata, Japan)34 and MassHunter Quantitative Analysis B.04.00 (Agilent Technologies) in order to obtain peak information, including m/z, peak area, and migration time (MT). Signal peaks were annotated according to the HMT metabolite database based on their m/z values with the MTs. The peak area of each metabolite was normalized with respect to the area of the internal standard and metabolite concentration was determined by standard curves with three-point calibrations using each standard compound. Hierarchical cluster analysis (HCA) and principal component analysis (PCA) were performed by HMT's proprietary software, PeakStat and SampleStat, respectively. Any metabolites detected were plotted on metabolic pathway maps using VANTED software.35Author contribution
M. I. and K. H. designed the research. K. K. mainly conducted the experiments. M. K., K. T., and S. N. synthesized pMFc. All authors analyzed and discussed the data. M. I. wrote the paper. All authors reviewed the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Taeko Nishiwaki-Ohkawa and Takashi Yoshimura (Nagoya University) for the mBmal1–dLuc reporter U2OS cell line and their helpful discussion. We also thank Kazuhiko Yagita (Kyoto Prefectural University of Medicine) for useful discussion. We are deeply grateful to Kazuhito Hashimoto (National Institute of Materials Science) for giving us the opportunity to start this study and warm encouragement. This work was supported by Research Foundation for the Electrotechnology of Chubu (M. I.) and the HMT Research Grant for Young Leaders in Metabolomics 2016 (M. K.) from Human Metabolome Technologies Inc.References
- J. C. Dunlap, Cell, 1999, 96, 271–290 CrossRef CAS PubMed.
- J. Bass and J. S. Takahashi, Science, 2010, 330, 1349–1354 CrossRef CAS PubMed.
- H. Reinke and G. Asher, Nat. Rev. Mol. Cell Biol., 2019, 20, 227–241 CrossRef CAS PubMed.
- K. Imamura, H. Yoshitane, K. Hattori, M. Yamaguchi, K. Yoshida, T. Okubo, I. Naguro, H. Ichijo and Y. Fukada, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3646–3651 CrossRef CAS PubMed.
- Y. Nakahata, S. Sahar, G. Astarita, M. Kaluzova and P. Sassone-Corsi, Science, 2009, 324, 654–657 CrossRef CAS PubMed.
- R. S. Edgar, E. W. Green, Y. Zhao, G. van Ooijen, M. Olmedo, X. Qin, Y. Xu, M. Pan, U. K. Valekunja, K. A. Feeney, E. S. Maywood, M. H. Hastings, N. S. Baliga, M. Merrow, A. J. Millar, C. H. Johnson, C. P. Kyriacou, J. S. O'Neill and A. B. Reddy, Nature, 2012, 485, 459–464 CrossRef CAS PubMed.
- C. B. Peek, A. H. Affinati, K. M. Ramsey, H. Y. Kuo, W. Yu, L. A. Sena, O. Ilkayeva, B. Marcheva, Y. Kobayashi, C. Omura, D. C. Levine, D. J. Bacsik, D. Gius, C. B. Newgard, E. Goetzman, N. S. Chandel, J. M. Denu, M. Mrksich and J. Bass, Science, 2013, 342, 1243417 CrossRef PubMed.
- G. Rey, U. K. Valekunja, K. A. Feeney, L. Wulund, N. B. Milev, A. Stangherlin, L. Ansel-Bollepalli, V. Velagapudi, J. S. O'Neill and A. B. Reddy, Cell Metab., 2016, 24, 462–473 CrossRef CAS PubMed.
- T. Tamaru, M. Hattori, Y. Ninomiya, G. Kawamura, G. Vares, K. Honda, D. P. Mishra, B. Wang, I. Benjamin, P. Sassone-Corsi, T. Ozawa and K. Takamatsu, PLoS One, 2013, 8, e82006 CrossRef PubMed.
- K. A. Feeney, L. L. Hansen, M. Putker, C. Olivares-Yanez, J. Day, L. J. Eades, L. F. Larrondo, N. P. Hoyle, J. S. O'Neill and G. van Ooijen, Nature, 2016, 532, 375–379 CrossRef CAS PubMed.
- M. Putker, P. Crosby, K. A. Feeney, N. P. Hoyle, A. S. H. Costa, E. Gaude, C. Frezza and J. S. O'Neill, Antioxid. Redox Signaling, 2018, 28, 507–520 CrossRef CAS PubMed.
- C. I. Torres, A. K. Marcus, H. S. Lee, P. Parameswaran, R. Krajmalnik-Brown and B. E. Rittmann, FEMS Microbiol. Rev., 2010, 34, 3–17 CrossRef CAS PubMed.
- M. E. Hernandez and D. K. Newman, Cell. Mol. Life Sci., 2001, 58, 1562–1571 CrossRef CAS PubMed.
- F. Kracke, B. Lai, S. Yu and J. O. Kromer, Metab. Eng., 2018, 45, 109–120 CrossRef CAS PubMed.
- K. Watanabe, M. Manefield, M. Lee and A. Kouzuma, Curr. Opin. Biotechnol., 2009, 20, 633–641 CrossRef CAS PubMed.
- B. Huang, S. Gao, Z. Xu, H. He and X. Pan, Curr. Microbiol., 2018, 75, 99–106 CrossRef CAS PubMed.
- K. Sasaki, D. Sasaki, K. Kamiya, S. Nakanishi, A. Kondo and S. Kato, Curr. Opin. Biotechnol., 2018, 50, 182–188 CrossRef CAS PubMed.
- S. Kato, Microbes Environ., 2015, 30, 133–139 CrossRef PubMed.
- B. E. Logan and K. Rabaey, Science, 2012, 337, 686–690 CrossRef CAS PubMed.
- K. Nishio, Y. Kimoto, J. Song, T. Konno, K. Ishihara, S. Kato, K. Hashimoto and S. Nakanishi, Environ. Sci. Technol. Lett., 2013, 1, 40–43 CrossRef.
- Y. Lu, K. Nishio, S. Matsuda, Y. Toshima, H. Ito, T. Konno, K. Ishihara, S. Kato, K. Hashimoto and S. Nakanishi, Angew. Chem., Int. Ed., 2014, 53, 2208–2211 CrossRef CAS PubMed.
- M. Kaneko, M. Ishikawa, K. Hashimoto and S. Nakanishi, Bioelectrochemistry, 2017, 114, 8–12 CrossRef CAS PubMed.
- M. Kaneko, M. Ishikawa, J. Song, S. Kato, K. Hashimoto and S. Nakanishi, Electrochem. Commun., 2017, 75, 17–20 CrossRef CAS.
- T. Goda, Y. Goto and K. Ishihara, Biomaterials, 2010, 31, 2380–2387 CrossRef CAS PubMed.
- M. Kaneko, M. Ishikawa, K. Ishihara and S. Nakanishi, Biomacromolecules, 2019, 20, 4447–4456 CrossRef CAS PubMed.
- T. Oshima, I. Yamanaka, A. Kumar, J. Yamaguchi, T. Nishiwaki-Ohkawa, K. Muto, R. Kawamura, T. Hirota, K. Yagita, S. Irle, S. A. Kay, T. Yoshimura and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 7193–7197 CrossRef CAS PubMed.
- M. Izumo, T. R. Sato, M. Straume and C. H. Johnson, PLoS Comput. Biol., 2006, 2, e136 CrossRef PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS PubMed.
- J. Rutter, M. Reick, L. C. Wu and S. L. McKnight, Science, 2001, 293, 510–514 CrossRef CAS PubMed.
- K. Nishio, T. Pornpitra, S. Izawa, T. Nishiwaki-Ohkawa, S. Kato, K. Hashimoto and S. Nakanishi, Plant Cell Physiol., 2015, 56, 1053–1058 CrossRef CAS PubMed.
- Y. B. Kiyohara, S. Tagao, F. Tamanini, A. Morita, Y. Sugisawa, M. Yasuda, I. Yamanaka, H. R. Ueda, G. T. van der Horst, T. Kondo and K. Yagita, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10074–10079 CrossRef CAS PubMed.
- T. Ooga, H. Sato, A. Nagashima, K. Sasaki, M. Tomita, T. Soga and Y. Ohashi, Mol. BioSyst., 2011, 7, 1217–1223 RSC.
- Y. Ohashi, A. Hirayama, T. Ishikawa, S. Nakamura, K. Shimizu, Y. Ueno, M. Tomita and T. Soga, Mol. BioSyst., 2008, 4, 135–147 RSC.
- M. Sugimoto, D. T. Wong, A. Hirayama, T. Soga and M. Tomita, Metabolomics, 2010, 6, 78–95 CrossRef CAS PubMed.
- B. H. Junker, C. Klukas and F. Schreiber, BMC Bioinf., 2006, 7, 109 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10023g |
| This journal is © The Royal Society of Chemistry 2020 |