
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiels–Alder reactions between cyclopentadiene analogs and benzoquinone in water and their application in the synthesis of polycyclic cage compounds†
Yijun Shi *ab,
Xuejing Liu*a,
Ying Hana,
Peng Yana,
Fusheng Biea and
Han Caoa
*ab,
Xuejing Liu*a,
Ying Hana,
Peng Yana,
Fusheng Biea and
Han Caoa
aEngineering and Technology Research Institute of Lunan Coal Chemical, Zaozhuang University, Beian Road, Zaozhuang 277160, Shandong Province, China. E-mail: 673230386@qq.com
bCollege of Chemistry, Chemical Engineering and Materials Science, Zaozhuang University, Beian Road, Zaozhuang 277160, Shandong Province, China
First published on 2nd January 2020
Abstract
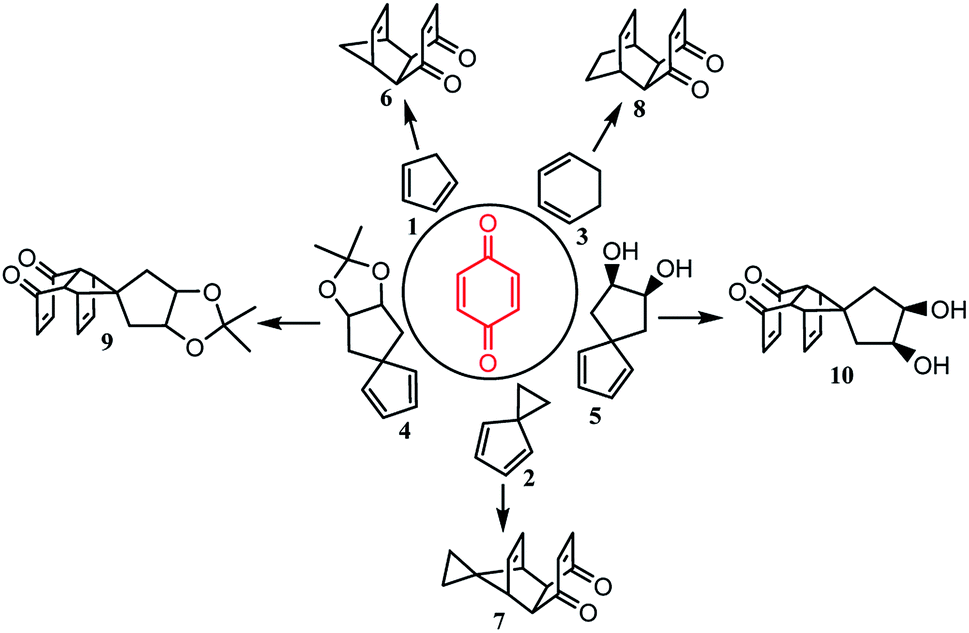
Diels–Alder reactions between cyclopentadiene analogs and p-benzoquinone were explored in water and yielded 83–97% product, higher than the results reported in water with a catalyst or cetrimonium bromide (CTAB) micelles. The novel adduct 10 was synthesized and further used to synthesize the bi-cage hydrocarbon 4,4′-spirobi[pentacyclo[5.4.0.02,6.03,10.05,9]undecane], which has a high density (1.2663 g cm−3) and a high volumetric heat of combustion (53.353 MJ L−1). Four novel bi-cage hydrocarbon compounds were synthesized in water using this method starting from 2,2′-bi(p-benzoquinone) and cyclopentadiene analogs.
Introduction
In recent years, water has been reported as a desirable solvent for many chemical reactions for reasons of cost, safety, and environmental concerns. Most notably, the Diels–Alder reaction has been found to be accelerated in dilute aqueous solution, and it is undoubtedly one of the most important reactions in the synthesis of polycyclic cage compounds.1–5 Polycyclic cage compounds have drawn more and more attention in the fields of medicinal chemistry and high energy density materials.6–56 The Diels–Alder reaction was developed by Diels and Alder in 1928 and the product of this reaction between cyclopentadiene and p-benzoquinone57 was further applied in the preparation of pentacyclo[5.4.0.02,6.03,10.05,9]undecane (PCUD) by Marchand53 (Scheme 1). | ||
| Scheme 1 Synthesis of PCUD. | ||
So far, several Diels–Alder reactions between cyclopentadiene analogs and p-benzoquinone have been carried out in water (Table 1) and the Diels–Alder adducts (Scheme 2) have been further applied in the synthesis of polycyclic cage compounds, but this required the addition of an organotungsten Lewis acid as a catalyst (entry 1)58 or cetrimonium bromide (CTAB) micelles (entry 2)59 and yielded lower than in organic solvents. Only the Diels–Alder reaction between 1,3-cyclohexadiene and p-benzoquinone was done in water without a catalyst or CTAB micelles, but the yield was 67%, which was lower than in toluene (entry 3).60 Apart from these, the other Diels–Alder reactions shown in Table 1 were done in organic solvents, and their yields were 76–97%.54,56,61–63
| Entry | Diene | Solvent | Conditions | Time/h | Yield/% |
|---|---|---|---|---|---|
| 1 | 1 | CH2Cl2 | 0–25 °C, organotungsten Lewis acid, rt | 2.25 | 94–97 |
| Water | 2 | 87 | |||
| 2 | 2 | Benzene | Reflux, CTAB micelles, rt | 15 | 82 |
| Water | 3 | 66 | |||
| 3 | 3 | Toluene | Reflux, rt | 24 | 76 |
| Water | 48 | 67 | |||
| 4 | 4 | MeOH | −10–25 °C | 5 | 76 |
 | ||
| Scheme 2 Diels–Alder reactions between cyclopentadiene analogs and p-benzoquinone. | ||
In this paper, Diels–Alder reactions between cyclopentadiene analogs and p-benzoquinone were further explored in water to improve the adduct yields. The new structure of adduct 10 is reported and used to synthesize the bi-cage hydrocarbon 4,4′-spirobi[pentacyclo[5.4.0.02,6.03,10.05,9]undecane], which has a high density (1.2663 g cm−3) and a high volumetric heat of combustion (53.353 MJ L−1).56 Furthermore, new polycyclic bi-cage scaffolds were synthesized in water using this method starting from 2,2′-bi(p-benzoquinone) and cyclopentadiene analogs (1 and 2).
Results and discussion
In order to improve the yields of Diels–Alder reactions between cyclopentadiene analogs and p-benzoquinone in water, the reaction between cyclopentadiene and p-benzoquinone at room temperature in water was chosen as the model reaction. We found that dissolving p-benzoquinone into cyclopentadiene was an instantly exothermic process, and the state of this reaction changed to solid from liquid along with a decrease in temperature.Based on this phenomenon and the solubility of p-benzoquinone in water, different methods of dropping were explored (Fig. 1). When water was added to p-benzoquinone (a), only a small amount of p-benzoquinone was dissolved in water (b). The system presented three phases because of the different densities after cyclopentadiene was added (c), and a large amount of p-benzoquinone was unreacted with cyclopentadiene after 24 h (d).
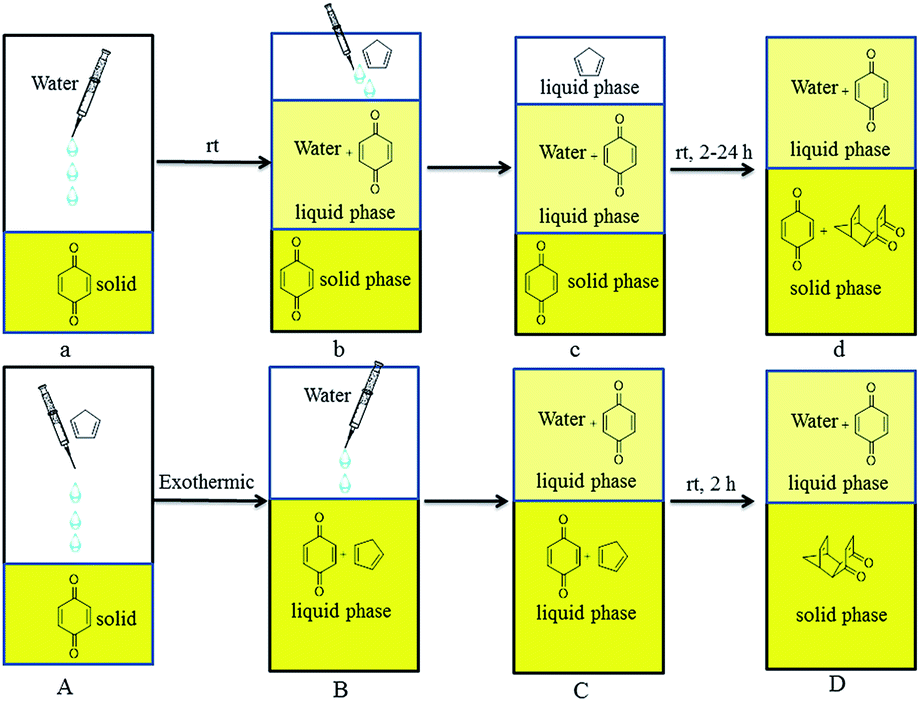 | ||
Fig. 1 Diels–Alder reactions between cyclopentadiene and p-benzoquinone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in water. :1) in water. | ||
When cyclopentadiene was added to p-benzoquinone (A), the state of this reaction changed to liquid from solid along with an increase in temperature. The system presented two liquid phases after water was added (C), and almost all of the product (6) had precipitated after 2 h. The product was simply obtained in 96% yield by filtration and recrystallization. Compared with the reaction in Table 1, this reaction was done in water without a catalyst and the yield was improved from 87% to 96%. Compared with the method of dropping in Fig. 1a and b, the method of dropping in Fig. 1A and B yielded more because it reacted more fully in two phases.
Based on the method shown in Fig. 1A–D, the results of Diels–Alder reactions between cyclopentadiene analogs and p-benzoquinone in water are shown in Table 2. The yields (90–97%, entries 1–4) were higher than those reported previously and the novel product 10 was synthesized in 83% yield. Products 6–9 have been reported to be important in the synthesis of polycyclic cage hydrocarbons, such as PCUD and bi-cage hydrocarbon 4,4′-spirobi[pentacyclo[5.4.0.02,6.03,10.05,9]undecane].
Further investigation was carried out to study the synthesis of a bi-cage hydrocarbon from adduct 10. Finally, the polycyclic cage hydrocarbon compound 12, which was important in the synthesis of 4,4′-spirobi[pentacyclo[5.4.0.02,6.03,10.05,9]undecane], was synthesized by intramolecular [2 + 2] photocyclization and Wolff–Kishner reduction from 10 (Scheme 3).
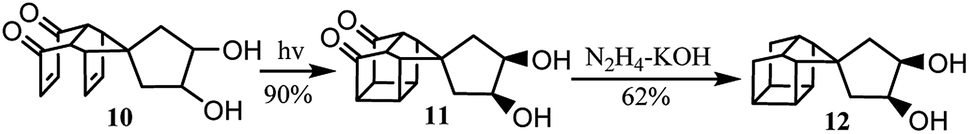 | ||
| Scheme 3 Synthesis of 12 from 10. | ||
To expand the scope of this method in water, we found that the structure of 2,2′-bi(p-benzoquinone) was similar to that of p-benzoquinone and can be synthesized by oxidation of 2,2′-biphenol.64 According to the structure of 2,2′-bi(p-benzoquinone) and the high reactivity of p-benzoquinone with cyclopentadiene, inspiration was drawn and a convenient synthetic strategy to construct two cages simultaneously in the synthesis of bi-cage hydrocarbon compounds was designed (Scheme 4). In this scheme, the Diels–Alder reaction in water was applied and bi-cage hydrocarbon compounds HV-1 and HV-2 were synthesized.
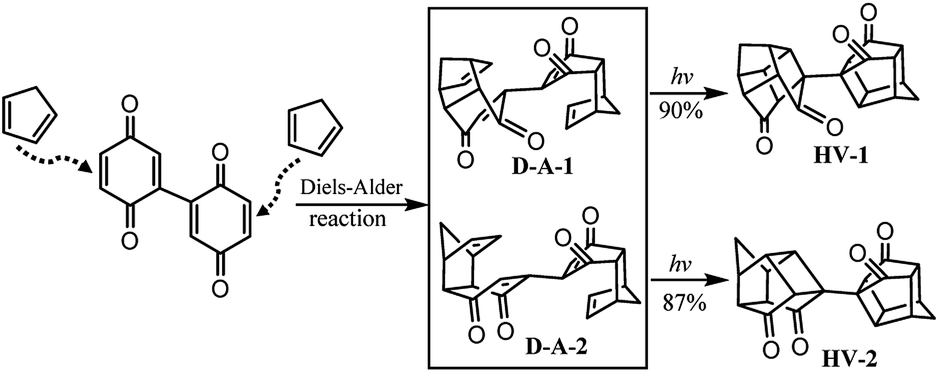 | ||
| Scheme 4 Synthesis of bi-cage hydrocarbon compounds HV-1 and HV-2. | ||
Following Scheme 4, two isomers (D-A-1:D-A-2 = 32:28) were obtained by the Diels–Alder reaction of 2,2′-bi(p-benzoquinone) with cyclopentadiene with an overall yield of 60% and separated. The intramolecular [2 + 2] photocyclization of D-A-1 at room temperature with a medium-pressure mercury immersion lamp formed HV-1 in 90% yield and HV-2 was obtained in 87% yield by the intramolecular [2 + 2] photocyclization of D-A-2.
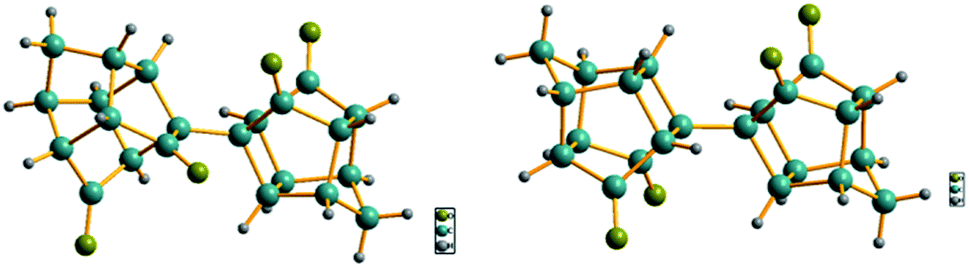
To confirm the structures of the bi-cage hydrocarbon compounds HV-1 and HV-2, crystals of compounds HV-1 and HV-2 suitable for X-ray diffraction were obtained by recrystallization. In the crystal structures of compounds HV-1 and HV-2 (Fig. 2), two polycyclic cages were clearly connected with a single C–C bond, which directly confirmed the bi-cage structures.
Based on Scheme 4, bi-cage hydrocarbon compounds HV-3 and HV-4 were synthesized as shown in Scheme 5. Two isomers (D-A-3:D-A-4 = 38:34) were obtained by the Diels–Alder reaction of 2,2′-bi(p-benzoquinone) with 2 with an overall yield of 72% and separated. The intramolecular [2 + 2] photocyclization of D-A-3 at room temperature with a medium-pressure mercury immersion lamp formed HV-3 in 88% yield and HV-4 was obtained in 80% yield by the intramolecular [2 + 2] photocyclization of D-A-4.
 | ||
| Scheme 5 Synthesis of bi-cage hydrocarbon compounds HV-3 and HV-4. | ||
To confirm the structures of the bi-cage hydrocarbon compounds HV-3 and HV-4, only a crystal of compound HV-4 suitable for X-ray diffraction was obtained by recrystallization. The crystal structure of compound HV-4 (Fig. 3) is very similar to the structure of compound HV-2 and the two cyclopropyl structures share two spiro-carbons with the bi-cage structures.
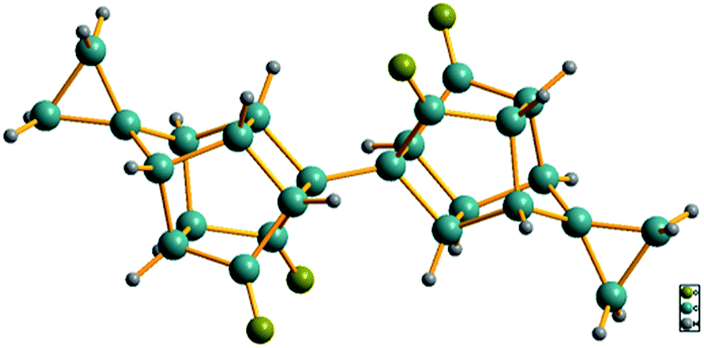 | ||
| Fig. 3 X-ray crystal structure of HV-4 (CCDC 1936543). | ||
Conclusions
In summary, the Diels–Alder reactions between cyclopentadiene analogs and p-benzoquinone were done in water used the method of dropping shown in Fig. 1A and B and yielded more than previously reported in water with a catalyst or CTAB micelles. The novel adduct 10 was synthesized and further used to synthesize the bi-cage hydrocarbon 4,4′- spirobi[pentacyclo[5.4.0.02,6.03,10.05,9]undecane]. Furthermore, four bi-cage hydrocarbon compounds were synthesized in water using this method starting from 2,2′-bi(p-benzoquinone) and cyclopentadiene analogs (1 and 2). Additionally, further work on the synthesis of more polycyclic cage (one cage or two cages) compounds is underway in our laboratory.Experimental
General procedures
All chemicals were purchased from commercial sources and used without further purification. Melting points were measured using an INESA (SGWX-4B) melting point detector and are uncorrected. NMR spectra were recorded with Bruker Avance 400M or Bruker Avance 500M spectrometers. MS data were recorded with a Micromass-GC/TOF mass spectrometer GCT (UK) and a Thermo-LTQ ORBITRAP XL. IR data were recorded with a NICOLET 6700 FT-IR. The X-ray single-crystal diffraction analysis was performed on an Agilent Supernova CCD diffractometer instrument.Synthesis and analytical data for 2
2 was prepared by the procedure described by Marchand.54 1H NMR (400 MHz, CDCl3) δ 6.59–6.49 (m, 2H), 6.19–6.10 (m, 2H), 1.66 (s, 4H).Synthesis and analytical data for 4
4 was prepared by the procedure described by Semmelhack.65 1H NMR (500 MHz, CDCl3) δ 6.86–6.78 (m, 1H), 6.30–6.27 (m, 1H), 6.21–6.14 (m, 1H), 6.08–5.99 (m, 1H), 4.88–4.81 (m, 2H), 2.12–2.03 (m, 2H), 1.77–1.72 (m, 2H), 1.60 (s, 3H), 1.34 (s, 3H).Synthesis and analytical data for 5
5 was prepared by the procedure described by Semmelhack.65 1H NMR (400 MHz, CDCl3) δ 6.39–6.35 (m, 1H), 6.25–6.20 (m, 1H), 6.20–6.13 (m, 2H), 4.29 (t, J = 4.4 Hz, 2H), 3.67 (s, 1H), 3.60 (s, 1H), 1.99–1.94 (m, 4H).Synthesis and analytical data for 2,2′-bi(p-benzoquinone)
2,2′-bi(p-benzoquinone) was prepared by the procedure described by Bouaziz.64 1H NMR (400 MHz, CDCl3) δ 6.85 (d, J = 15.8 Hz, 6H).Synthesis and analytical data for 6
0.50 g (4.63 mmol) of p-benzoquinone and 0.31 g (4.70 mmol) of cyclopentadiene were added sequentially to a 25 mL flask equipped with a magnetic stirrer bar. Water (5 mL) was added and the mixture was stirred at room temperature for 2 h. The precipitate was filtered and recrystallized from n-hexane to yield 6 (0.77 g, 4.42 mmol, 96%) as yellow needles. Mp: 76–77 °C. IR (film): 2986, 2950, 2926, 1659, 1604, 1298, 1280, 1141, 1066, 915, 872, 852, 725, 709 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.55 (s, 2H), 6.05 (d, J = 1.3 Hz, 2H), 3.53 (d, J = 1.1 Hz, 2H), 3.20 (d, J = 1.2 Hz, 2H), 1.52 (dd, J = 8.7, 1.4 Hz, 1H), 1.41 (d, J = 8.7 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 199.46 (C), 142.06 (CH), 135.30 (CH), 48.77 (CH), 48.71 (CH), 48.34 (CH2).Synthesis and analytical data for 7
7 was prepared with the same procedure as 6 starting with p-benzoquinone (0.50 g, 4.63 mmol), 2 (0.44 g, 4.78 mmol) and water (5 mL). The mixture was stirred at room temperature for 4 h. The precipitate was filtered and recrystallized from n-hexane to yield 7 (0.83 g, 4.15 mmol, 90%) as yellow needles. Mp: 100–102 °C. IR (film): 2980, 1662, 1606, 1301, 1282, 1131, 1028, 956, 929, 897, 874, 848, 734 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.58 (s, 2H), 6.14 (d, J = 1.6 Hz, 2H), 3.38 (d, J = 1.4 Hz, 2H), 2.89 (s, 2H), 0.60 (dd, J = 9.3, 5.7 Hz, 2H), 0.49 (dd, J = 9.5, 5.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 199.31 (C), 142.32 (CH), 135.34 (CH), 53.66 (C), 49.21 (CH), 44.56 (CH), 7.96 (CH2), 6.94 (CH2).Synthesis and analytical data for 8
8 was prepared with the same procedure as 6 starting with p-benzoquinone (0.50 g, 4.63 mmol), 3 (0.38 g, 4.75 mmol) and water (5 mL). The mixture was stirred at room temperature for 4 h. The precipitate was filtered and recrystallized from n-hexane to yield 8 (0.84 g, 4.47 mmol, 97%) as yellow needles. Mp: 88–90 °C. IR (film): 2947, 2866, 1662, 1611, 1289, 1277, 1257, 1176, 1163, 1107, 1044, 1018, 877, 798, 722, 710 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.63 (s, 2H), 6.29–6.11 (m, 2H), 3.20 (d, J = 1.1 Hz, 2H), 2.98 (s, 2H), 1.80–1.60 (m, 2H), 1.47–1.17 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 198.36 (C), 140.98 (CH), 132.45 (CH), 48.32 (CH), 34.32 (CH), 23.72 (CH2).Synthesis and analytical data for 9
9 was prepared with the same procedure as 6 starting with p-benzoquinone (0.50 g, 4.63 mmol), 4 (0.89 g, 4.63 mmol) and water (5 mL). The mixture was stirred at room temperature for 4 h. The precipitate was filtered and recrystallized from n-hexane to yield 9 (1.26 g, 4.20 mmol, 91%) as yellow needles. Mp: 158–160 °C. IR (film): 2974, 2943, 2908, 1666, 1604, 1384, 1372, 1283, 1208, 1119, 1052, 1022, 988, 877, 843, 728 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.57 (s, 2H), 6.04 (ddd, J = 30.2, 5.7, 2.8 Hz, 2H), 4.57 (dt, J = 32.1, 6.0 Hz, 2H), 3.67 (s, 1H), 3.43 (dd, J = 8.4, 4.2 Hz, 1H), 3.17 (dd, J = 8.4, 4.0 Hz, 1H), 2.96 (s, 1H), 2.03 (dd, J = 14.9, 1.9 Hz, 1H), 1.75 (dd, J = 14.6, 1.6 Hz, 1H), 1.62–1.47 (m, 5H), 1.26 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 199.43 (C), 199.23 (C), 142.58 (CH), 141.97 (CH), 136.89 (CH), 134.37 (CH), 109.33 (C), 80.54 (CH), 79.65 (CH), 68.18 (C), 57.42 (CH2), 53.66 (CH2), 47.99 (CH), 47.69 (CH), 37.77 (CH), 36.68 (CH), 26.36 (CH3), 23.52 (CH3).Synthesis and analytical data for 10
10 was prepared with the same procedure as 6 starting with p-benzoquinone (0.50 g, 4.63 mmol), 5 (0.71 g, 4.67 mmol) and water (5 mL). The mixture was stirred at room temperature for 4 h. The precipitate was filtered and subjected to chromatography on silica gel to give 10 (1.00 g, 3.84 mmol, 83%) as yellow needles. Mp: 160–162 °C. IR (film): 3348, 2968, 1712, 1659, 1325, 1124, 1075, 956 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.58 (s, 2H), 6.04 (dd, J = 9.3, 2.0 Hz, 2H), 4.04 (d, J = 35.0 Hz, 2H), 3.36 (s, 1H), 3.27 (ddd, J = 18.1, 8.3, 3.9 Hz, 2H), 3.11 (s, 1H), 2.40 (s, 1H), 2.27 (s, 1H), 1.85–1.60 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 199.40 (C), 199.26 (C), 142.45 (CH), 142.24 (CH), 136.18 (CH), 135.59 (CH), 73.62 (CH), 72.86 (CH), 64.22 (C), 57.75 (CH2), 56.80 (CH2), 48.18 (CH), 47.99 (CH), 37.08 (CH), 36.76 (CH). HRMS (EI): C15H16O4 [M]+ calcd, 260.1049; found, 260.1054.Synthesis and analytical data for 11
A solution of 10 (1.00 g, 3.84 mmol) in EtOAc (200 mL) was irradiated with a 400 W medium-pressure mercury immersion lamp for 3 h. The reaction mixture was concentrated in vacuo, and the residue was subjected to chromatography on silica gel to give 11 (0.90 g, 3.46 mmol, 90%) as a white solid. Mp: 200–202 °C. IR (film): 3343, 2952, 2925, 1732, 1194, 1076, 1049 cm−1. 1H NMR (400 MHz, DMSO) δ 4.40 (s, 2H), 3.85 (s, 2H), 3.17 (s, 2H), 2.74 (s, 4H), 2.53 (d, J = 38.4 Hz, 2H), 1.78–1.43 (m, 4H). 13C NMR (101 MHz, DMSO) δ 213.19 (C), 73.12 (CH), 73.02 (CH), 59.80 (C), 54.50 (CH2), 54.46 (CH2), 54.43 (CH), 53.33 (CH), 43.90 (CH), 43.87 (CH), 38.39 (CH), 38.34 (CH), 37.41 (CH), 33.64 (CH). HRMS (EI): C15H16O4 [M]+ calcd, 260.1049; found, 260.1057.Synthesis and analytical data for 12
A solution of 11 (1.00 g, 3.84 mmol) and hydrazine monohydrate (3.3 mL, 80%, 54.31 mmol) in diethylene glycol (3 mL) was heated at 135 °C for 20 h and the solution was distilled until the temperature of the distillate reached 190 °C. Solid potassium hydroxide (1.51 g, 26.96 mmol) was added portionwise and the solution was heated at 200 °C for 20 h, allowed to cool, and diluted with water (50 mL). The precipitate was filtered and recrystallized from EtOAc to yield 12 (0.55 g, 2.37 mmol, 62%) as a white solid. Mp: 156–158 °C. IR (film): 3266, 2933, 2857, 1088, 1031 cm−1. 1H NMR (400 MHz, DMSO) δ 4.18 (dd, J = 11.7, 4.2 Hz, 2H), 3.85–3.68 (m, 2H), 2.50 (d, J = 30.5 Hz, 4H), 2.28 (s, 2H), 1.88–1.60 (m, 4H), 1.47–1.26 (m, 4H), 0.86 (d, J = 11.9 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 73.34 (CH), 73.26 (CH), 57.17 (CH2), 56.11 (CH2), 51.80 (C), 43.12 (CH), 42.97 (CH), 41.33 (CH), 41.16 (CH), 38.58 (CH), 37.48 (CH), 35.39 (CH), 35.35 (CH), 27.14 (CH2), 27.06 (CH2).Synthesis and analytical data for D-A-1 and D-A-2
0.50 g (2.34 mmol) 2,2′-bi(p-benzoquinone) and 0.31 g (4.70 mmol) cyclopentadiene were added one after another to a 25 mL flask equipped with a magnetic stirrer bar. Water (5 mL) was added and the mixture was stirred at room temperature for 4 h. The precipitate was filtered and subjected to chromatography on silica gel to give D-A-1 (0.26 g, 0.75 mmol, 32%) as a yellow microcrystalline solid and D-A-2 (0.23 g, 0.66 mmol, 28%) as a yellow microcrystalline solid.Synthesis and analytical data for HV-1
A solution of D-A-1 (0.51 g, 1.47 mmol) in EtOAc (30 mL) was irradiated with a 400 W medium-pressure mercury immersion lamp for 2 h. The reaction mixture was concentrated in vacuum, and the residue was subjected to chromatography on silica gel to give HV-1 (0.46 g, 1.33 mmol, 90%) as a white solid. Mp: 310–313 °C. IR (film): 2962, 2944, 1742, 1731, 1079, 1042 cm−1. 1H NMR (400 MHz, CDCl3) δ 3.15 (d, J = 6.2 Hz, 2H), 2.99 (d, J = 28.5 Hz, 6H), 2.71 (d, J = 11.6 Hz, 6H), 2.03 (d, J = 11.3 Hz, 2H), 1.89 (d, J = 11.3 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 211.85 (C), 210.54 (C), 55.17 (C), 55.08 (CH), 52.36 (CH), 46.26 (CH), 44.72 (CH), 44.14 (CH), 42.50 (CH), 40.88 (CH), 37.47 (CH2). HRMS (EI): C22H18O4 [M]+ calcd, 346.1205; found, 346.1212.Synthesis and analytical data for HV-2
A solution of D-A-2 (0.46 g, 1.33 mmol) in EtOAc (30 mL) was irradiated with a 400 W medium-pressure mercury immersion lamp for 2 h. The reaction mixture was concentrated in vacuum, and the residue was subjected to chromatography on silica gel to give HV-2 (0.40 g, 1.16 mmol, 87%) as white solid. Mp: 306–309 °C. IR (film): 2991, 2935 1742, 1718, 1223, 1083 cm−1. 1H NMR (400 MHz, CD2Cl2) δ 3.22–3.04 (m, 2H), 3.03–2.73 (m, 6H), 2.62 (s, 4H), 2.40 (s, 2H), 1.96 (d, J = 11.2 Hz, 2H), 1.82 (d, J = 11.2 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 210.66 (C), 209.57 (C), 54.00 (C), 53.73 (CH), 50.81 (CH), 45.69 (CH), 43.23 (CH), 42.85 (CH), 40.56 (CH), 39.85 (CH), 36.54 (CH2). HRMS (EI): C22H18O4 [M]+ calcd, 346.1205; found, 346.1206.Synthesis and analytical data for D-A-3 and D-A-4
D-A-3 and D-A-4 were prepared with the same procedure as D-A-1 and D-A-2 starting with 2,2′-bi(p-benzoquinone) (0.50 g, 2.34 mmol), 2 (0.44 g, 4.78 mmol) and water (5 mL). The mixture was stirred at room temperature for 4 h. The precipitate was filtered and subjected to chromatography on silica gel to give D-A-3 (0.35 g, 0.88 mmol, 38%) as a yellow microcrystalline solid and D-A-4 (0.32 g, 0.80 mmol, 34%) as a yellow microcrystalline solid.Synthesis and analytical data for HV-3
HV-3 was prepared with same procedure as HV-1 and yielded in 88% as white solid. Mp: 260–262 °C. IR (film): 2985, 1734, 1182, 1080, 1048, 956, 942, 922, 876 cm−1. 1H NMR (400 MHz, CDCl3) δ 3.31 (dd, J = 13.9, 6.2 Hz, 2H), 3.19–3.05 (m, 2H), 2.93 (s, 4H), 2.79 (d, J = 6.2 Hz, 2H), 2.38 (s, 2H), 2.32–2.23 (m, 2H), 0.77–0.56 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 211.87 (C), 210.52 (C), 55.63 (CH), 55.56 (CH), 53.11 (CH), 50.34 (CH), 49.68 (CH), 46.95 (C), 42.72 (C), 37.70 (CH), 37.50 (CH), 5.50 (CH2), 4.05 (CH2). HRMS (EI): C26H22O4 [M]+ calcd, 398.1518; found, 398.1527.Synthesis and analytical data for HV-4
HV-4 was prepared with the same procedure as HV-1 and yielded 80% as a white solid. Mp: 318–320 °C. IR (film): 3003, 2985, 2962, 1727, 1221, 1181, 1083, 974, 955, 942, 761 cm−1. 1H NMR (400 MHz, CDCl3) δ 3.48–3.34 (m, 2H), 3.24 (s, 2H), 3.00–2.85 (m, 4H), 2.43 (s, 2H), 2.29–2.15 (m, 4H), 0.77–0.56 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 210.65 (C), 209.48 (C), 54.48 (CH), 54.18 (CH), 51.50 (CH), 48.86 (CH), 48.45 (CH), 46.36 (C), 40.77 (C), 36.77 (CH), 36.52 (CH), 4.43 (CH2), 3.06 (CH2). HRMS (EI): C26H22O4 [M]+ calcd, 398.1518; found, 398.1522.Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Windmon and V. Dragojlovic, Green Chem. Lett. Rev., 2008, 1, 155–163 CrossRef CAS.
- P. A. Grieco, K. Yoshida and P. Garner, J. Org. Chem., 1983, 48, 3137–3139 CrossRef CAS.
- M. C. Pirrung and K. D. Sarma, Tetrahedron, 2005, 61, 11456–11472 CrossRef CAS.
- Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492–5502 CrossRef CAS PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- J. Wlochal, R. D. M. Davies and J. Burton, Org. Lett., 2014, 16, 4094 CrossRef CAS PubMed.
- W. J. Geldenhuys, S. F. Malan, T. Murugesan, C. J. Van der Schyf and J. R. Bloomquist, Bioorg. Med. Chem., 2004, 12, 1799 CrossRef CAS PubMed.
- W. J. Geldenhuys, S. F. Malan, J. R. Bloomquist, A. P. Marchand and C. Van der Schyf, Med. Res. Rev., 2005, 25, 21 CrossRef CAS PubMed.
- A. Mdzinarishvili, W. J. Geldenhuys, T. J. Abbruscato, U. Bickel, J. Klein and C. Van der Schyf, Neurosci. Lett., 2005, 383, 49 CrossRef CAS PubMed.
- D. W. Oliver and S. F. Malan, Med. Chem. Res., 2008, 17, 137 CrossRef CAS.
- H. Gunosewoyo, J. L. Guo, M. R. Bennett, M. J. Coster and M. Kassiou, Bioorg. Med. Chem. Lett., 2008, 18, 3720 CrossRef CAS PubMed.
- J. Hao, A. Mdzinarishvili, T. J. Abbruscato, J. Klein, W. J. Geldenhuys, C. J. Van der Schyf and U. Bickel, Brain Res., 2008, 1196, 113 CrossRef CAS PubMed.
- W. J. Geldenhuys, L. M. Bezuidenhout and D. E. Dluzen, Eur. J. Pharmacol., 2009, 619, 38 CrossRef CAS PubMed.
- C. J. Van der Schyf and W. J. Geldenhuys, Neurotherapeutics, 2009, 6, 175 CrossRef CAS PubMed.
- J. Joubert, S. van Dyk, I. R. Green and S. F. Malan, Eur. J. Med. Chem., 2011, 46, 5010 CrossRef CAS PubMed.
- J. Wang, C. Ma, V. Balannik, L. H. Pinto, R. A. Lamb and W. F. DeGrado, ACS Med. Chem. Lett., 2011, 2, 307 CrossRef CAS PubMed.
- H. J. R. Lemmer, J. Joubert, S. van Dyk, F. H. van der Westhuizen and S. F. Malan, Med. Chem., 2012, 8, 361 CrossRef CAS.
- J. Joubert, W. J. Geldenhuys, C. J. Van der Schyf, D. W. Oliver, H. G. Kruger, T. Govender and S. F. Malan, ChemMedChem, 2012, 7, 375 CrossRef CAS PubMed.
- J. A. Lockman, W. J. Geldenhuys, M. R. Jones-Higgins, J. D. Patrick, D. D. Allen and C. Van der Schyf, Brain Res., 2012, 1489, 133 CrossRef CAS PubMed.
- A. S. Sklyarova, V. N. Rodionov, C. G. Parsons, G. Quack, P. R. Schreiner and A. A. Fokin, Med. Chem. Res., 2013, 22, 360 CrossRef CAS.
- J. Joubert, R. Sharma, M. Onani and S. F. Malan, Tetrahedron Lett., 2013, 54, 6923 CrossRef CAS.
- C. Beinat, S. D. Banister, J. Hoban, J. Tsanaktsidis, A. Metaxas, A. D. Windhorst and M. Kassiou, Bioorg. Med. Chem. Lett., 2014, 24, 828 CrossRef CAS PubMed.
- J. Joubert, H. Samsodien, Q. R. Baber, D. L. Cruickshank, M. R. Caira and S. F. Malan, J. Chem. Crystallogr., 2014, 44, 194 CrossRef CAS.
- S. M. Wilkinson, H. Gunosewoyo, M. L. Barron, A. Boucher, M. McDonnell, P. Turner, D. E. Morrison, M. R. Bennett, I. S. McGregor, L. M. Rendina and M. Kassiou, ACS Chem. Neurosci., 2014, 5, 335 CrossRef CAS PubMed.
- F. T. Zindo, Q. R. Barber, J. Joubert, J. J. Bergh, J. P. Petzer and S. F. Malan, Eur. J. Med. Chem., 2014, 80, 122 CrossRef CAS PubMed.
- A. O. Egunlusi, S. F. Malan and J. Joubert, ChemMedChem, 2015, 10, 1259 CrossRef CAS PubMed.
- F. T. Zindo, J. Joubert and S. F. Malan, Future Med. Chem., 2015, 7, 609 CrossRef CAS PubMed.
- J. Joubert, E. Kapp, D. Taylor, P. J. Smith and S. F. Malan, Bioorg. Med. Chem. Lett., 2016, 26, 1151 CrossRef CAS PubMed.
- A. d. Meijere, S. Redlich, D. Frank, J. Magull, A. Hofmeister, H. Menzel, B. Konig and J. Svoboda, Angew. Chem., Int. Ed., 2007, 46, 4574 CrossRef PubMed.
- K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis and R. Priefer, Chem. Rev., 2015, 115, 6719 CrossRef CAS PubMed.
- H. N. Lim and G. Dong, Org. Lett., 2016, 18, 1104 CrossRef CAS PubMed.
- A. P. Marchand, M. N. Deshpande and G. M. Reddy, Trans. ASME: J. Energy Resour. Technol., 1989, 34, 946 CAS.
- C. Segal and W. Shyy, Trans. ASME, 1996, 118, 180 CrossRef.
- C. Segal, S. Pethe and K. R. Williams, Combust. Sci. Technol., 2001, 163, 229 CrossRef CAS.
- L. Qiu, D. Ye, W. Wei, K. Chen, J. Hou, J. Zheng, X. Gong and M. Xiao, J. Mol. Struct.: THEOCHEM, 2008, 866, 63 CrossRef CAS.
- S. Lal, S. Rajkumar, A. Tare, S. Reshmi, A. Chowdhury and I. N. N. Namboothiri, Chem.–Asian J., 2014, 9, 3533 CrossRef CAS PubMed.
- L. Pan, R. Feng, H. Peng, X. E, J. Zhou, L. Wang and X. Zhang, RSC Adv., 2014, 4, 50998 RSC.
- C. Segal, M. J. Friedauer, H. S. Udaykumar, W. Shyy and A. P. Marchand, J. Propul. Power, 1997, 13, 246 CrossRef CAS.
- R. J. Stedman, L. S. Miller, L. D. Davis and J. R. E. Hoover, J. Org. Chem., 1970, 35, 4169 CrossRef.
- A. P. Marchand and R. W. Allen, J. Org. Chem., 1974, 39, 1596 CrossRef CAS.
- P. G. Gassman and R. Yamaguchi, J. Org. Chem., 1978, 43, 4654 CrossRef CAS.
- A. P. Marchand, Chem. Rev., 1989, 89, 1011 CrossRef CAS.
- G. Mehta, S. Padma and S. R. Karra, J. Org. Chem., 1989, 54, 1342 CrossRef CAS.
- G. W. Griffin, Chem. Rev., 1989, 89, 997 CrossRef CAS.
- M. A. Forman and W. P. Dailey, J. Org. Chem., 1993, 58, 1501 CrossRef CAS.
- H. Takeshita, H. Kawakami, Y. Ikeda and A. Mori, J. Org. Chem., 1994, 59, 6490 CrossRef CAS.
- A. d. Meijere, C. Lee, B. Bengtson, E. Pohl, S. I. Kozhushkov, P. R. Schreiner, R. Boese and T. Haumann, Chem.–Eur. J., 2003, 9, 5481 CrossRef PubMed.
- A. M. Kenwright and J. D. Sellars, Magn. Reson. Chem., 2012, 50, 803 CrossRef CAS PubMed.
- A. P. Marchand, P. Jin and M. N. Deshpande, Acta Crystallogr., 1988, 44, 1617 Search PubMed.
- A. P. Marchand, G. M. Reddy, M. N. Deshpande, W. H. Watson, A. Nagl, O. S. Lee and E. Osawa, J. Am. Chem. Soc., 1990, 112, 3521 CrossRef CAS.
- A. P. Marchand, P. Jin and K. Siam, J. Mol. Struct.: THEOCHEM, 1990, 204, 209 CrossRef.
- A. P. Marchand and V. Vidyasagar, J. Org. Chem., 1991, 56, 282 CrossRef CAS.
- A. P. Marchand, A. Zope, F. Zaragoza and G. Bott, Tetrahedron, 1994, 50, 1687 CrossRef CAS.
- A. P. Marchand, V. D. Sorokin, W. H. Watson, T. F. Carlson and M. Krawiec, Struct. Chem., 1994, 5, 367 CrossRef CAS.
- Y. Shi, J. Jiang, L. Ma, J. Wang and W. Li, Tetrahedron Lett., 2017, 58, 1376 CrossRef CAS.
- Y. Shi, J. Jiang, J. Wang and Y. Wang, Tetrahedron Lett., 2015, 56, 6704 CrossRef CAS.
- C. C. Nawrat and C. J. Moody, Angew. Chem., Int. Ed., 2014, 53, 2056 CrossRef CAS PubMed.
- I. Chen, J. Young and S. Yu, Tetrahedron, 2004, 60, 11903 CrossRef CAS.
- V. K. Singh, B. N. S. Raju and P. T. Deota, Synth. Commun., 1986, 16, 1731 CrossRef CAS.
- T. Sunakawa and C. Kuroda, Molecules, 2005, 10, 244 CrossRef CAS PubMed.
- O. Masaji, K. Takeshi, O. Tomoaki and E. Tetsuya, Org. Synth., 1996, 73, 253 CrossRef.
- D. Mal and S. Ray, Eur. J. Org. Chem., 2008, 3014 CrossRef CAS.
- Y. Ishii, S. Ito, Y. Saito, D. Uno and T. Oba, Tetrahedron, 2015, 71, 8892 CrossRef CAS.
- Z. Bouaziz, A. Gherardi, F. Regnier, M. Sarciron, X. Bertheau, B. Fenet, N. Walchshofer and H. Fillion, Eur. J. Org. Chem., 2002, 1834 CrossRef CAS.
- M. F. Semmelhack, J. S. Foos and S. Katz, J. Am. Chem. Soc., 1973, 95, 7325 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1936540, 1936541 and 1936543. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09745g |
| This journal is © The Royal Society of Chemistry 2020 |