
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe C–H activated controlled mono- and di-olefination of arenes in ionic liquids at room temperature†
Kaifeng Du b and
Tian Yao*a
b and
Tian Yao*a
aKey Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry and Sichuan Province, West China School of Pharmacy, Sichuan University, Chengdu, 610041, P. R. China. E-mail: yaotian90@scu.edu.cn
bDepartment of Pharmaceutical & Biological Engineering, School of Chemical Engineering, Sichuan University, Chengdu 610065, P. R. China
First published on 20th January 2020
Abstract
In this study, controlled mono and di-olefination of arenes was first realized at room temperature via the C–H bond activation in ionic liquids, probably due to the positive effects of ionic liquids. It is an energy-saving routes in industrial production without the need for heating equipment. Different catalysts were screened, and it was found that [Ru(p-cymene)Cl2]2 generated mono-olefinated products predominantly while [Cp*RhCl2]2 selectively gave di-olefinated products. These catalysts ([BMIM]NTf2 and [BMIM]PF6) as green and recyclable reaction media are highly efficient under mild conditions. This reaction process can avoid any volatile and environmentally toxic organic solvents, and is much safer without the need for pressure-tight equipment. A wide substrate scope with good yields and satisfactory selectivity was achieved. The reactions can be scaled up to gram-scale. Furthermore, an expensive rhodium/ruthenium catalytic system was recycled for at least 6 times with consistently high catalytic activity, which was economical and environmental friendly from an industrial point of view. According to the mechanistic study, the C–H bond cleavage was probably achieved via the concerted metalation–deprotonation. This technique can be applied in the synthesis of various valuable unsaturated aromatic compounds and shows a great potential for industrial production.
Introduction
Recently, transition metal-catalyzed C–H activation has emerged as one of the most atom-economical approach for the formation of C–C bonds and has made significant progress in the field of synthetic chemistry.1 It is straightforward, highly efficient and can avoid multi-step pre-activation with a rare production of unwanted by-products.2 The establishment of a wide range of new C–C, C–N, C–O, C–P bonds is assisted by suitable applications of Lewis-basic directing groups.3 Most of these transition metal catalysts, such as Pd, Ir, Ru and Rh, are expensive, earth-scarce, require harsh reaction conditions and environmentally toxic organic solvents. These drawbacks greatly limit their applications in the synthesis of functional organic compounds.4 Some researchers explored to find inexpensive earth-abundant transition metals to replace them as alternative catalysts. For example, Ackermann, Yoshikai and other groups discovered co-catalyzed C–H activation and applied in various chemical reactions, which were only catalyzed by Rh, Ru or Pd.5 Although this strategy reduces costs significantly, the catalytic conditions are tough in most cases, and toxic organic solvents can cause serious environmental pollution. Some other researchers have found some ways to achieve the recycling of these noble transition-metals without the loss in their content. Nano-functional materials or metal-immobilized macromolecules6 are most popular because they can be reused and recycled to lower the overall cost and reduce environmental contamination. However, unavoidable harsh catalytic conditions as well as tedious multiple steps for the synthesis of metal-loaded functional materials are still needed. Thus, it is highly desirable to develop new green catalytic systems with easy accessibility to solve these problems.Aromatic olefins are important chemical intermediates and widely applied in the synthesis of pharmaceutical intermediates, natural products, and functional materials.7 The olefination of unreactive aryl C–H bonds catalyzed by transition metals is among the most significant chemical transformations in organic syntheses.8 It has drawn considerable attention through these years for a single reaction step and a few side reactions compared with conventional methods.9 Most achievements in the olefination of arene via the C–H activation have been accomplished by using noble metal catalyst systems, such as ruthenium,10 rhodium,11 palladium,12 cobalt,5b and iridium,13 with high temperature and organic solvents. These catalysts generally show high reactivity and broad substrate scope. A systematic comparative study on the catalytic activities of different catalysts on the olefination of arenes is important and necessary.
Lately, the controllably selective catalysis of the mono- and di-olefination via C–H activation has been of great interest. For example, N. Umeda reported the selective mono and di-vinylation of 1-phenylpyrazoles by controlling the Cu(OAc)2 amount.14 Selective olefination was also controlled by changing the solvent, catalyst or substrate.15 However, all these expensive catalytic systems could be used only once and the reaction condition is harsh (Scheme 1a). Recently, our group realized the temperature-controlled mono- and di-olefination of arenes with good yield, excellent selectivity and satisfactory recyclability.16 However, the generation of di-olefinated products required a high temperature, which was energy-consuming and unsafe in an industrial production process. To the best of our knowledge, a protocol for the selective synthesis of mono- and di-olefinated products at room temperature with full control is yet to be developed.
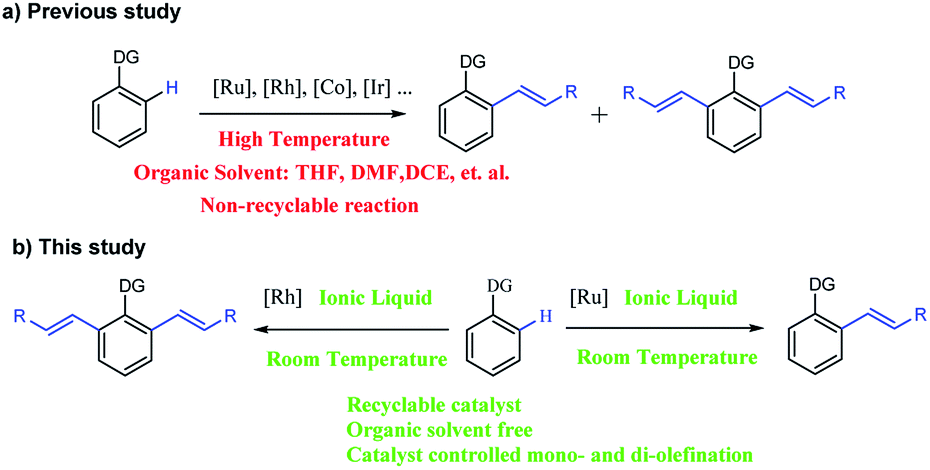 | ||
| Scheme 1 C–H activated olefination of arene. | ||
A solvent is generally used in large amounts in most reactions and plays a key role in an organic synthesis. Many volatile and environmentally toxic organic solvents are commonly applied in the C–H olefination. In recent years, many researchers have paid attention to the replacement of these harmful solvents with an eco-friendly medium to meet the requirement of green chemistry. Water and polyethylene glycol are extensively studied and successfully applied in some chemical transformations.17 However, their applications are significantly restricted by the low solubility of starting compounds and metal catalysts. Considering this, it is preferable to find an excellent medium to allow the C–H olefination to perform smoothly under mild conditions and reuse the metal catalyst. Ionic liquids (ILs), a class of non-molecular solvents at room temperature,18 which generally consist of an organic cation and a weak nucleophilic anion, have drawn widespread attention of researchers due to their superior properties such as negligible volatility, thermal and chemical stability, wide liquid range, non-flammability, and good solubility.19 They often replace hazardous organic solvents and serve as an environmental friendly, non-volatile and recyclable reaction medium. Furthermore, they are much safer in the high-temperature or high-pressure synthetic processes. Up to now, ILs have been successfully employed in the Suzuki reaction, cross-coupling reaction, Heck reaction, C–H activated olefination and so on.16,20 The employment of ILs in C–H activation is still rare and needs to be expanded.
Herein, we report the first catalyst-controlled selective mono and di-olefination of arenes via C–H activation at room temperature. The notable features of our methodology include: (a) the selective mono- and di-olefination of arenes was realized at room temperature for the first time, which was energy-saving in industrial production without the need for heating equipment. (b) The catalysts play a key role in controlling the mono- and di-olefination, where [Ru(p-cymene)Cl2]2 gave mono-olefination products predominantly, while [Cp*RhCl2]2 afforded the diolefination products with high selectivity. (c) Instead of an organic solvent, [BMIM]NTf2 and [BMIM]PF6 served as excellent solvents to allow the recycling of the noble metal catalytic system for at least 6 times, which was economical and environmental friendly from an industrial point of view, and the process was much safer without the need of pressure-tight equipment. (d) The reaction tolerated a broad substrate with satisfactory yields and excellent selectivity even in gram-scale (Scheme 1b).
Results and discussion
Our initial experiment was performed with 2-phenylpyridine (1a) and styrene (2a) in the ionic liquid-[BMIM]NTf2 at room temperature. Different types of catalysts were investigated (entry 1–9) at the beginning. A catalyst with a pentamethylcyclo-pentadienyl (Cp*) ligand tended to have catalytic activity. Cp*Co(CO)I2, [Cp*CoCl2]2, and [Cp*Co(MeCN)3][SbF6]2 were effective under these conditions, but only less than 30% conversion was achieved. The above successes encouraged us to explore more transition-metal complexes. To our delight, [Cp*Rh(MeCN)3][SbF6]2 and Cp*Rh(OAc)2 demonstrated nearly 50% conversion of the starting compound. Surprisingly, [Cp*RhCl2]2 could achieve 97% conversion of 1a and di-olefinated (3a′) product was obtained with an isolated yield of 91%. When [Ru(p-cymene)Cl2]2 was employed as the catalyst, 87% isolated yield of the mono-olefinated product was obtained. RhCl3, [Cp*IrCl2]2, Pd(OAc)2, RuCl3 and Ru3(CO)12 could not afford any products. As a result, selective mono- and di-olefination was achieved at room temperature controlled by the catalyst. Inspired by the preliminary success, we screened several other reaction parameters, and the results are listed in Table 1. In the [Ru(p-cymene)Cl2]2 catalyzed reaction systems, different amounts of 2a were then investigated (entry 10, 13 and 14). 1 equimolar of 2a reduced the conversion of 1a to 66%, while 2 equiv. of 2a could increase the conversion of 1a up to 89% and afforded satisfactory yield of mono-olefinated product 3a (85%) with very few di-olefinated products (3a′). Cu(OAc)2 served as an oxidant in this catalytic system and 2 equiv. of Cu(OAc)2 or more could afford a good yield of the mono-olefinated product (entry 10 and 13). To save the starting compound, 2a and Cu(OAc)2 were both determined as 2 equiv. It demonstrated that selectivity was not controlled by the amount of the starting compound. Further, the effects of different additives were explored (entry 15–18). Various species of Ag salts were screened and all of them had satisfactory performances except that no reaction occurred when an Ag salt was not added. It revealed that the Ag salt as an additive was essential in the C–H activated olefination. The anion of the additive is not important and has little impact on the reaction efficiency. Thus, Ag+ might be crucial in the catalytic process. Furthermore, reaction performances were evaluated with different ILs, organic solvents and water (entry 19–27). Traditional organic solvents such as toluene, CH2Cl2, t-AmOH, HFIP and DMF could not make the reaction happen. When reaction was performed in water, no transformation occurred either. Four different ILs were investigated, and all of them were effective. [BMIM]BF4 and [BMIM]OTf had a low conversion of 1a with poor selectivity, while [BMIM]NTf2 and [BMIM]PF6 had more than 80% conversion of 1a with satisfactory selectivity. This phenomenon is probably because [BMIM]NTf2 and [BMIM]PF6 were much more hydrophobic than the other ILs. They could better dissolve the transition-metal complex and the hydrophobic starting compounds. Thus, sufficient interactions of starting compounds with catalysts could make the reaction happen more easily. 0.1 equiv. amount of the catalyst could not improve the yield obviously (entry 28), so 0.05 equiv. amount of the catalyst was enough. Moreover, increasing the reaction temperature to 80 °C had no positive effect on the conversion of 1a, and the yield of 3a investigated (entry 29 and 30). Subsequently, the experiment was conducted under air (entry 31). Only 35% yield of mono-olefinated product was afforded and most of the starting compound was converted to impurity. So, the argon atmosphere was essential. In the [Cp*RhCl2]2 catalyzed reaction systems, when 2a or Cu(OAc)2 was less than 4 equiv. in amount, the conversion of 1a decreased sharply and selectivities were poor with both mono-(3a) and di-olefinated (3a′) products generated (entry 32 and 33). 4 equiv. of 2a and Cu(OAc)2 could afford excellent yield of di-olefinated product (91%). Increasing the catalyst amount or the reaction temperature had no positive effect (entry 34–37). Employing [BMIM]BF4 or [BMIM]OTf as the reaction solvent could make the conversion of 1a higher than 40% but with poor selectivities. [BMIM]NTf2 and [BMIM]PF6 also demonstrated excellent performances with the conversion of 1a higher 90% (entry 38–40). Similar to the [Ru(p-cymene)Cl2]2 catalytic system, argon atmosphere was necessary for the reaction. Accordingly, the reaction conditions of selective mono-olefination was optimized as follows: 1a (0.2 mmol), 2a (0.4 mmol), [Ru(p-cymene)Cl2]2 (0.01 mmol), Cu(OAc)2 (0.4 mmol) and AgNTf2 (0.02 mmol) in [BMIM]NTf2 or [BMIM]PF6 (0.6 mL) under argon at room temperature for 24 h in a sealed tube. The reaction conditions of the selective di-olefination was optimized as follows: 1a (0.2 mmol), 2a (0.8 mmol), [Cp*RhCl2]2 (0.01 mmol), Cu(OAc)2 (0.8 mmol) and AgNTf2 (0.02 mmol) in [BMIM]NTf2 or [BMIM]PF6 (0.6 mL) under argon at room temperature for 24 h in a sealed tube.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (0.05 equiv.) | 2a (equiv.) | Oxidant (equiv.) | Additive (0.1 equiv.) | Solvent | Temp. (°C) | Yieldb (%) 3a/3a′ | Conversion of 1ac (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (certain equivalent), catalyst (0.01 mmol), Cu(OAc)2 (oxidant) and additive (0.02 mmol) in a solvent (0.6 mL) were stirred under argon at a certain temperature for 24 h in a sealed tube.b Yield of the product isolated after the preparative thin layer chromatography.c Conversion based on the yield of the recovered 1a.d [Ru(p-cymene)Cl2]2 (0.10 equiv.).e Reaction performed under air for 24 h.f [Cp*RhCl2]2 (0.10 equiv.).g [Cp*RhCl2]2 (0.15 equiv.).h Cp* = pentamethylcyclopentadienyl; HFIP = hexafluoroisopropanol; t-AmOH = tertiary amyl alcohol; r.t. = room temperature. | ||||||||
| 1 | Cp*Co(CO)I2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 8/4 | 15 |
| 2 | [Cp*CoCl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 14/13 | 30 |
| 3 | [Cp*Co(MeCN)3][SbF6]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 10/11 | 23 |
| 4 | [Cp*Rh(MeCN)3][SbF6]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 18/29 | 49 |
| 5 | RhCl3 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | —/— | — |
| 6 | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 5/91 | 97 |
| 7 | Cp*Rh(OAc)2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 8/37 | 47 |
| 8 | [Cp*IrCl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | —/— | — |
| 9 | Pd(OAc)2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | —/— | — |
| 10 | [Ru(p-cymene)Cl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 87/3 | 92 |
| 11 | RuCl3 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | —/— | — |
| 12 | Ru3(CO)12 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | —/— | — |
| 13 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | [BMIM]NTf2 | r.t. | 85/3 | 89 |
| 14 | [Ru(p-cymene)Cl2]2 | 1 | 2 | AgNTf2 | [BMIM]NTf2 | r.t. | 61/3 | 66 |
| 15 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgSbF6 | [BMIM]NTf2 | r.t. | 83/3 | 87 |
| 16 | [Ru(p-cymene)Cl2]2 | 2 | 2 | Ag2SO4 | [BMIM]NTf2 | r.t. | 82/5 | 88 |
| 17 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgOAc | [BMIM]NTf2 | r.t. | 80/3 | 85 |
| 18 | [Ru(p-cymene)Cl2]2 | 2 | 2 | — | [BMIM]NTf2 | r.t. | —/— | — |
| 19 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | [BMIM]BF4 | r.t. | 15/7 | 24 |
| 20 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | [BMIM]PF6 | r.t. | 76/4 | 82 |
| 21 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | [BMIM]OTf | r.t. | 19/10 | 32 |
| 22 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | H2O | r.t. | —/— | — |
| 23 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | Toluene | r.t. | —/— | — |
| 24 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | t-AmOH | r.t. | —/— | — |
| 25 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | CH2Cl2 | r.t. | —/— | — |
| 26 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | DMF | r.t. | —/— | — |
| 27 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | HFIP | r.t. | —/— | — |
| 28d | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | [BMIM]NTf2 | r.t. | 84/3 | 89 |
| 29 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | [BMIM]NTf2 | 60 | 87/3 | 91 |
| 30 | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | [BMIM]NTf2 | 80 | 86/3 | 90 |
| 31e | [Ru(p-cymene)Cl2]2 | 2 | 2 | AgNTf2 | [BMIM]NTf2 | r.t. | 35/7 | 87 |
| 32 | [Cp*RhCl2]2 | 2 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 16/55 | 72 |
| 33 | [Cp*RhCl2]2 | 2 | 2 | AgNTf2 | [BMIM]NTf2 | r.t. | 27/31 | 61 |
| 34f | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 5/90 | 97 |
| 35g | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 5/92 | 98 |
| 36 | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | 60 | 4/92 | 97 |
| 37 | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | 80 | 5/92 | 98 |
| 38 | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]PF6 | r.t. | 7/81 | 90 |
| 39 | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]BF4 | r.t. | 11/27 | 41 |
| 40 | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]OTf | r.t. | 13/31 | 46 |
| 41e | [Cp*RhCl2]2 | 4 | 4 | AgNTf2 | [BMIM]NTf2 | r.t. | 7/29 | 92 |
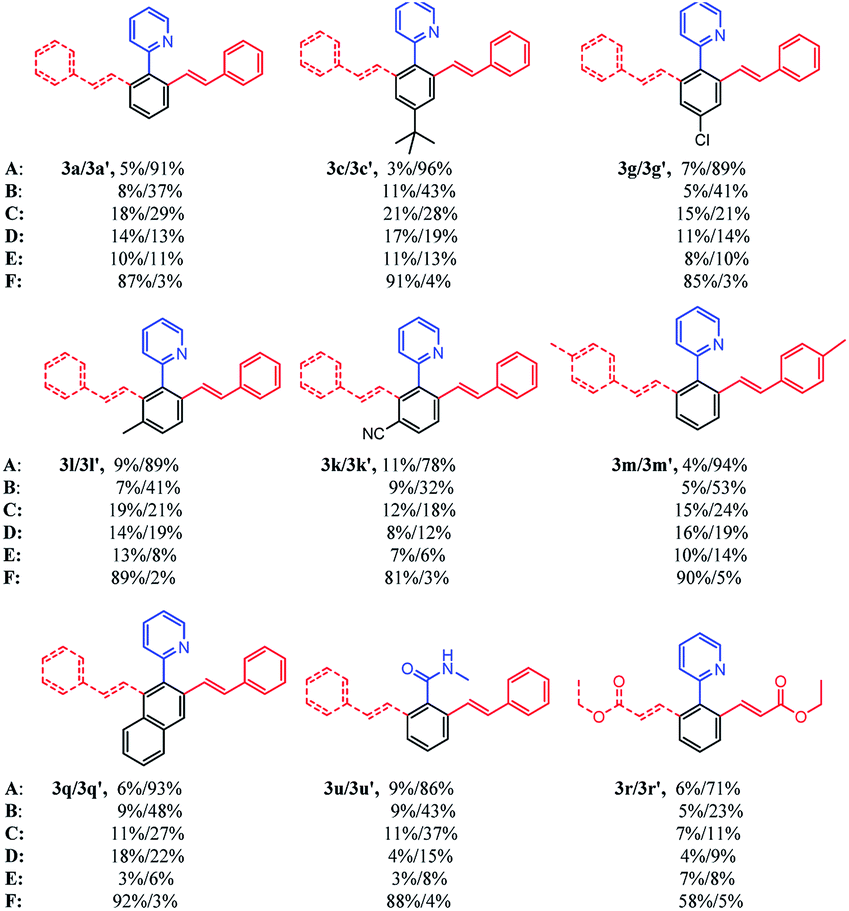
Based on the above results, we then compared the reaction scope of different transition-metal catalysts under the optimal conditions. As shown in Table 2, different substituted substrates at different positions of the arenes and substrates with different directing groups were studied in detail. Both electron-donating and electron-withdrawing groups were selected as the substituent. In addition, 2-(naphthalen-2-yl) pyridine as well as ethyl acrylate were also explored. It is obvious that all 6 catalysts were effective for this kind of reaction. Cp*Rh(OAc)2, [Cp*Rh(MeCN)3][SbF6]2, [Cp*CoCl2]2 and [Cp*Co(MeCN)3][SbF6]2 were able to make reactions happen, but the catalytic activities were not satisfactory and the selectivity of the products were poor. On the contrary, [Cp*RhCl2]2 and [Ru(p-cymene)Cl2]2 revealed excellent catalytic activities and wonderful selectivity for all kinds of substrates. In particular, the yield of 3r or 3r′ was relatively lower than that of other products, so ethyl acrylate had lower reactivity than styrene. Consequently, [Cp*RhCl2]2 and [Ru(p-cymene)Cl2]2 were chosen to further explore the substrate scope of selective mono- and di-olefination.
|
|---|
| a The reaction conditions: 1x (0.2 mmol), 2x (0.8 mmol), catalyst (0.01 mmol), Cu(OAc)2 (0.8 mmol) and AgNTf2 (0.02 mmol) in [BMIM]NTf2 (0.8 mL) were stirred under argon at r.t. for 24 h in a sealed tube.b Isolated yield.c The catalysts are: A ([Cp*RhCl2]2); B (Cp*Rh(OAc)2); C ([Cp*Rh(MeCN)3][SbF6]2); D ([Cp*CoCl2]2); E ([Cp*Co(MeCN)3][SbF6]2); F ([Ru(p-cymene)Cl2]2). |
 |
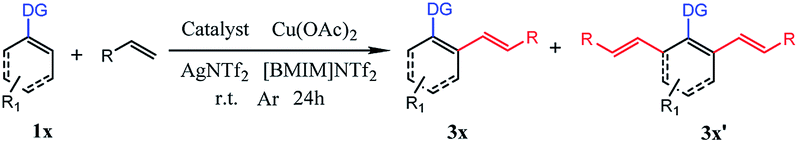
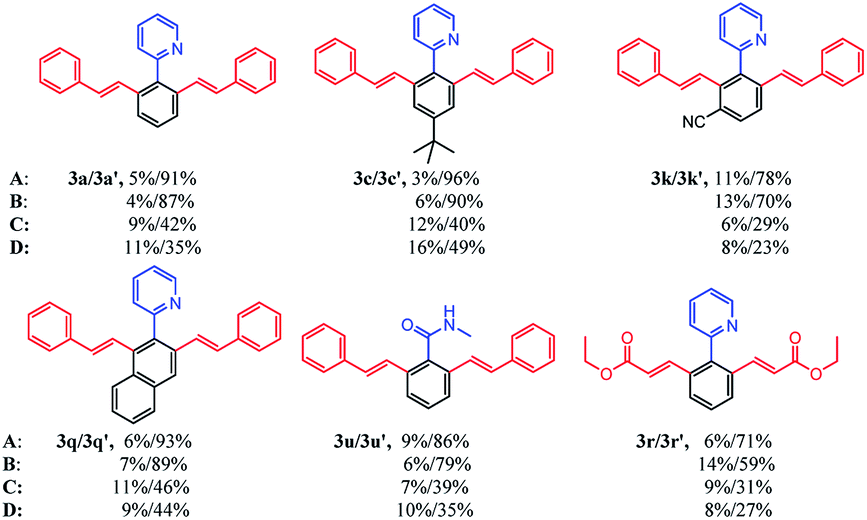
To examine the effects of ILs on the reaction activity, we compared the reaction scope of different ILs in the [Cp*RhCl2]2 catalytic system. As listed in Table 3, six different kinds of substrates were investigated. To our delight, applying these four ILs as reaction media could generally give the corresponding products in moderate to good yields for all substrates. Surprisingly, for all substrates, [BMIM]NTf2 and [BMIM]PF6 could afford excellent yield of the product with satisfactory selectivity compared with [BMIM]BF4 and [BMIM]OTf. Theoretically, [BMIM]NTf2 and [BMIM]PF6 were much more hydrophobic than another two ILs. They could better dissolve the transition-metal complex and the hydrophobic starting compounds. As a result, the interactions of starting compounds with catalysts were more sufficient, and the conversion of starting compounds was higher. Then, [BMIM]NTf2 and [BMIM]PF6 were applied to further explore the substrate scope of selective mono- and di-olefination.
|
|---|
| a The reaction conditions: 1x (0.2 mmol), 2x (0.8 mmol), [Cp*RhCl2]2 (0.01 mmol), Cu(OAc)2 (0.8 mmol) and AgNTf2 (0.02 mmol) in a certain ionic liquid (0.8 mL) were stirred under argon at r.t. for 24 h in a sealed tube.b Isolated yield.c The ionic liquids are: A ([BMIM]NTf2); B ([BMIM]PF6); C ([BMIM]BF4); D ([BMIM]OTf). |
 |
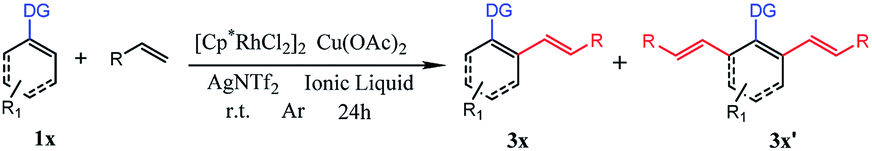
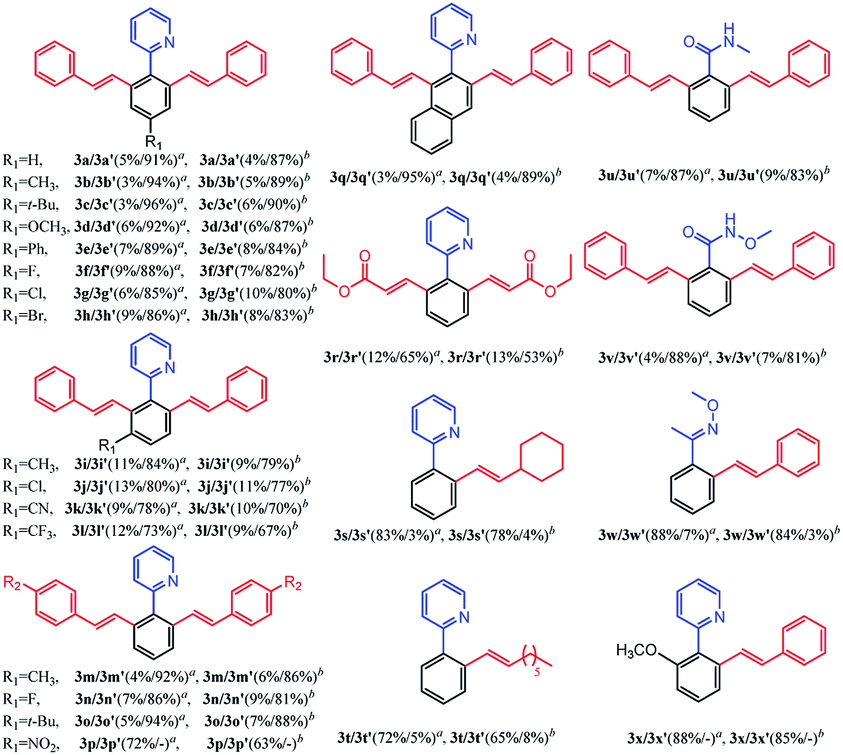
With the optimized reaction conditions in hand, we investigated the substrate scope for the selective mono-olefination of different arenes via C–H activation catalyzed by [Ru(p-cymene)Cl2]2. Both [BMIM]NTf2 and [BMIM]PF6 were employed for the substrate scope expansion. As shown in Table 4, various substituted 2-aryl-pyridine could afford mono-olefinated products in high yields with ideal selectivity at room temperature. Unsubstituted 2-phenylpyridine 1a gave an 85% isolated yield of 3a in [BMIM]NTf2 and a 79% yield of 3a in [BMIM]PF6, respectively. Electron donating group substituted 2-aryl-pyridine, such as 4′-Me, 4′-t-Bu, 4′-OCH3, 4′-Ph and 5′-Me derivatives, had slightly increased yields of 3b, 3c, 3d, 3e and 3i for both ILs reaction systems, respectively. Although electron-withdrawing group substituted 2-aryl-pyridine (CF3 or CN) have a relatively lower conversion of the substrate and lower yield of products, these groups were able to exhibit fine performances. As for halogens (F, Cl, and Br), they were satisfactorily tolerated irrespective of their position (para- or meta-), and obtained 82% yield of 3f, 84% yield of 3g, 86% yield of 3h, 83% yield of 3j in [BMIM]NTf2, and could achieve 81% yield of 3f, 78% yield of 3g, 80% yield of 3h, 79% yield of 3j in [BMIM]PF6. Interestingly, 3q had an excellent yield probably due to the increased conjugation of the naphthalene ring. Different types of olefins were also reacted with 1a. Similarly, Me, t-Bu and halogen-substituted styrene had excellent performances, and NO2 substituted styrene only had about 60% yield of the mono-olefinated product in both ILs. Surprisingly, ethyl acrylate and unreactive 1-octene even vinylcyclohexane could react with 1a with ideal selectivity. Moreover, altering the directing group to N-methylamide, N-methoxylamide and oxime ether could also selectively afford the mono-olefinated products in fine yields.
|
|---|
| a The reaction conditions: 1a (0.25 mmol), 2a (0.50 mmol), [Ru(p-cymene)Cl2]2 (0.0125 mmol), Cu(OAc)2 (0.50 mmol) and AgNTf2 (0.025 mmol) in [BMIM]NTf2 (0.8 mL) were stirred under argon at r.t. for 24 h in a sealed tube.b The reaction conditions: 1a (0.25 mmol), 2a (0.50 mmol), [Ru(p-cymene)Cl2]2 (0.0125 mmol), Cu(OAc)2 (0.50 mmol) and AgNTf2 (0.025 mmol) in [BMIM]PF6 (0.8 mL) were stirred under argon at r.t. for 24 h in a sealed tube.c Isolated yield. |
 |
Finally, ortho-substituted 2-phenylpyridine could satisfactorily give the mono-olefinated product. This mono-olefinated strategy demonstrated wide substrate adaptability, good reactivity and high selectivity. Both [BMIM]NTf2 and [BMIM]PF6 could serve as an excellent reaction medium, and [BMIM]NTf2 had slightly better performances than [BMIM]PF6 for all substrates.
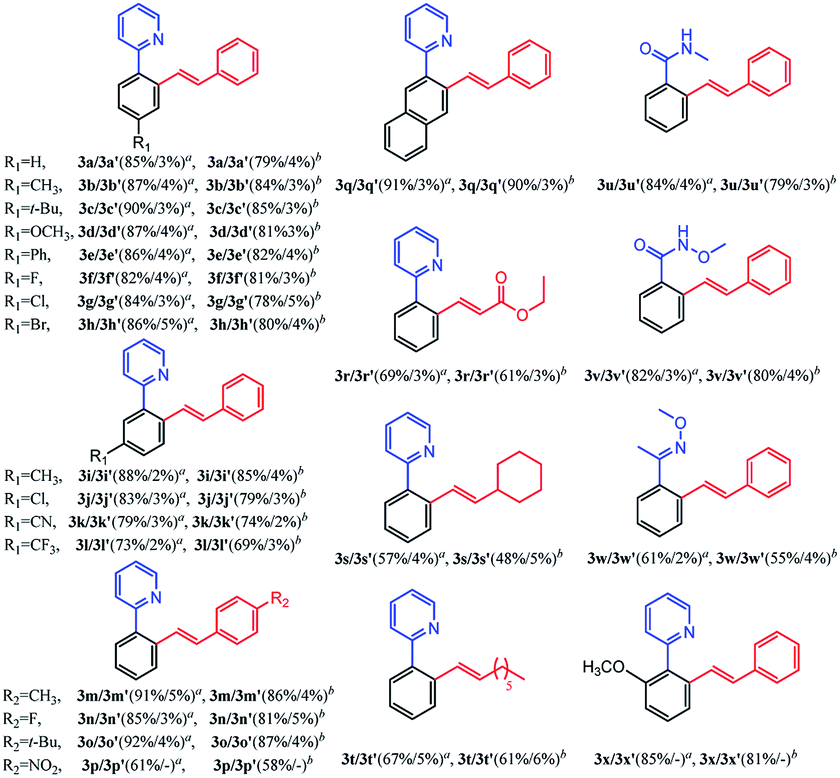
Subsequently, we further explored the substrate scope of the selective di-olefination of different arenes via C–H activation catalyzed by [Cp*RhCl2]2. Both [BMIM]NTf2 and [BMIM]PF6 were employed for the substrate scope expansion. As demonstrated in Table 5, most of the substituted 2-aryl-pyridine could selectively give di-olefinated products in high yields at room temperature and had similar electronic effects as above. Exceptionally, the NO2-substituted substrate only gave mono-olefinated product at this condition, probably because the extremely strong electron-withdrawing effects deactivate the aromatic ring towards the second olefination. Notably, meta-substituted 2-aryl-pyridine had a relatively lower yield of di-olefinated products compared to para-substituted substrates. It was probably because the steric hindrance of the meta-substituted group interferes with the formation of the di-olefinated product. Ethyl acrylate performed well to selectively give a di-olefinated product with good yield. Then, 2-phenylpyridine was subjected to react with unreactive 1-octene and vinylcyclohexane, and mono-olefinated products were predominantly afforded. It was possibly due to the low reactivity of the unconjugated olefins. When applying the naphthalene ring to increase conjugation, 3q exhibited an ideal yield of di-olefinated products. Moreover, changing the directing group to N-methylamide and N-methoxylamide could also selectively afford the di-olefinated products in satisfactory yields. However, less reactive aryl oxime ether only gave mono-olefinated products at this condition. It can be concluded that aryl pyridine and aryl amides have a much higher reactivity than aryl oxime ether towards the second olefination. The ortho-substituted 2-phenylpyridine had higher yields of mono-olefinated products compared with the Ru catalytic system. This di-olefination strategy also showed wide substrate adaptability, good reactivity and high selectivity. As a result, the selective mono- and di-olefination of arenes was achieved by controlling the catalyst at room temperature. [BMIM]NTf2 and [BMIM]PF6 both served as ideal reaction solvents, and [BMIM]NTf2 performed slightly better than [BMIM]PF6 for all substrates.
|
|---|
| a The reaction conditions: 1a (0.25 mmol), 2a (1.00 mmol), [Cp*RhCl2]2 (0.0125 mmol), Cu(OAc)2 (1.00 mmol) and AgNTf2 (0.025 mmol) in [BMIM]NTf2 (0.8 mL) were stirred under argon at r.t. for 24 h in a sealed tube.b The reaction conditions: 1a (0.25 mmol), 2a (1.00 mmol), [Cp*RhCl2]2 (0.0125 mmol), Cu(OAc)2 (1.00 mmol) and AgNTf2 (0.025 mmol) in [BMIM]PF6 (0.8 mL) were stirred under argon at r.t. for 24 h in a sealed tube.c Isolated yield. |
 |
Selective olefination was also realized in organic solvents with high yield of mono- and di-olefinated products. However, high reaction temperatures and environmentally toxic organic solvents were required.15a,b Zhang15c realized high yield of selective olefination in water, but a high temperature of more than 378 K was required for the reaction. Compared with the previous literatures, this study reported comparably high yield of mono- and di-olefinated products with ionic liquids as a green solvent at room temperature.
Recyclability could reduce the cost of noble metal and avoid environmental contamination. Therefore, it is very important to investigate the recyclability of these catalytic systems. The reactions of 2-phenylpyridine with styrene were selected as a model for the reusability evaluation. The recyclability performances are shown in Fig. 1. After the completion of the reaction, the mixture was extracted with an equivoluminal amount of diethyl ether 4 times. The upper diethyl ether layers, containing the product were combined and evaporated, and then directly used for purification simply through a preparative thin layer chromatography to get the pure product. The lower layer (reaction system) was further treated by vacuum evaporation to completely remove the miscible diethyl ether. Then, this catalytic system was added with new starting materials and used for the next cycle. It was found that the desired products were obtained in high yields with only a slight drop after 6 cycles for all four different catalytic systems. These results strongly indicate that [BMIM]NTf2 and [BMIM]PF6 were indeed excellent recyclable reaction media for the selective mono- and di-olefination.
 | ||
Fig. 1 Recycling performances of the reaction of 2-phenylpyridine with styrene at room temperature:  isolated yield of di-olefinated product in the [Cp*RhCl2]2/[BMIM]NTf2 catalytic system; isolated yield of di-olefinated product in the [Cp*RhCl2]2/[BMIM]NTf2 catalytic system;  isolated yield of di-olefinated product in the [Cp*RhCl2]2/[BMIM]PF6 catalytic system; isolated yield of di-olefinated product in the [Cp*RhCl2]2/[BMIM]PF6 catalytic system;  isolated yield of mono-olefinated product in the [Ru(p-cymene)Cl2]2/[BMIM]NTf2 catalytic system; isolated yield of mono-olefinated product in the [Ru(p-cymene)Cl2]2/[BMIM]NTf2 catalytic system;  isolated yield of mono-olefinated product in the [Ru(p-cymene)Cl2]2/[BMIM]PF6 catalytic system. isolated yield of mono-olefinated product in the [Ru(p-cymene)Cl2]2/[BMIM]PF6 catalytic system. | ||
Next, we investigated the selective mono- and di-olefination in gram-scale with 2-phenylpyridine and styrene as an example. Scheme 2 demonstrates that satisfactory yields were obtained at both circumstances. Therefore, this method is economical, green and shows a great potential in industrial production.
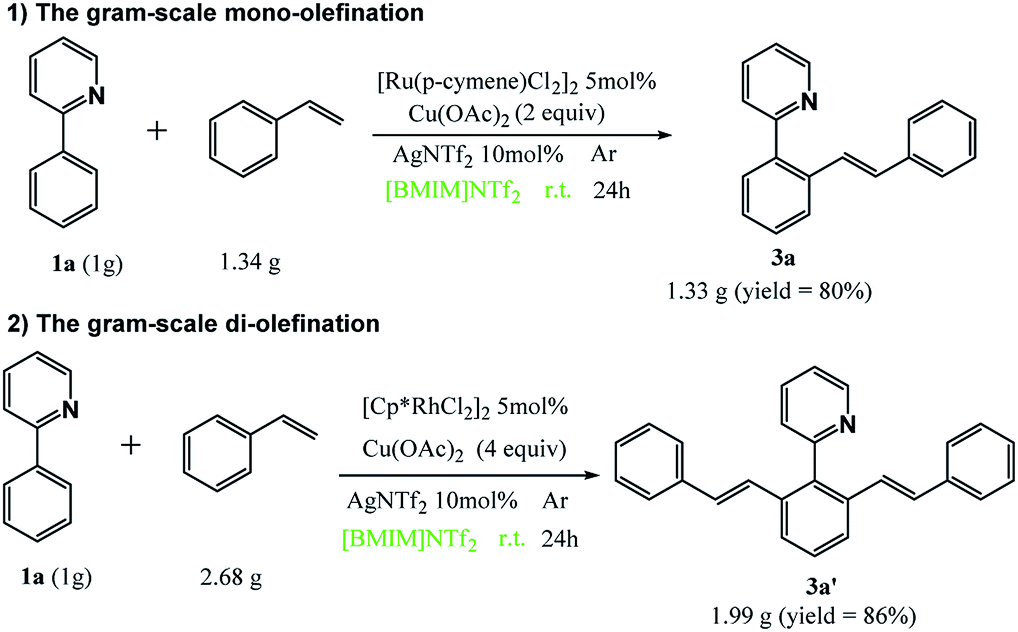 | ||
| Scheme 2 Gram-scale mono- and di-olefination of 1a. | ||
To further gain a mechanistic understanding of the reaction, a series of experiments were performed, and the results are demonstrated in Scheme 3. First, the H/D exchange experiments were performed by treating 1a with CD3OD in [BMIM]NTf2. The 1H NMR spectra revealed that 43% ortho C–H for the [Ru(p-cymene)Cl2]2 system and 37% ortho C–H for the [Cp*RhCl2]2 system were deuterated after a 4 h reaction, and it suggested that the C–H bond activation was reversible. Furthermore, the intermolecular competition experiments were conducted between 1b and 1k for both catalytic systems. The products were separately obtained with isolated yields of 3b/3k = 63%/37% for the [Ru(p-cymene)Cl2]2 system and 3b′/3k′ = 81%/19% for the [Cp*RhCl2]2 system. The results reveal that the electron-rich substrate had much higher reactivity over the electron-deficient substrate and is in accordance with previous literatures.
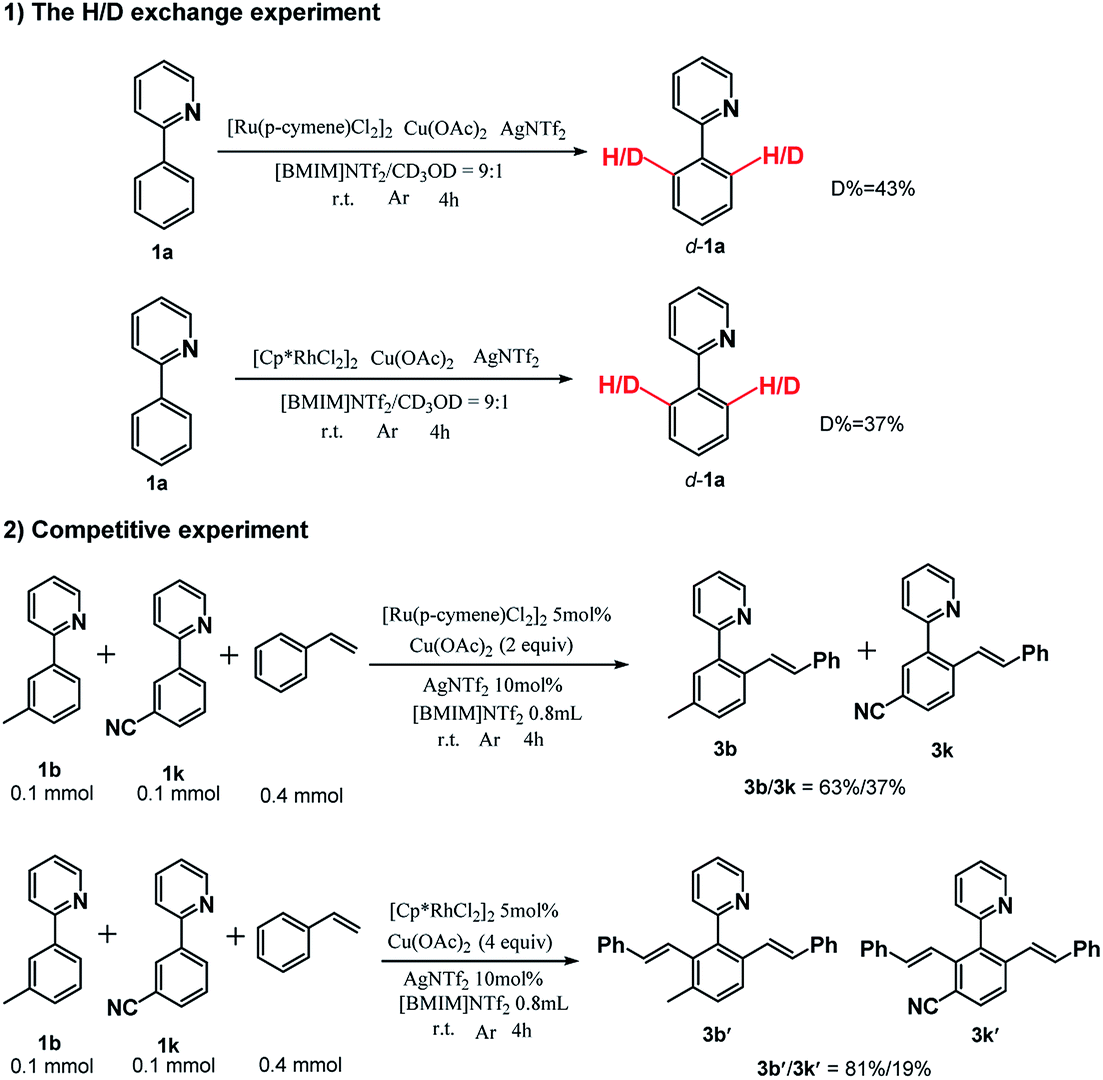 | ||
| Scheme 3 Mechanistic study. | ||
Based on the above results and some previous related reports,13,21 a possible mechanism has been proposed and demonstrated in Fig. 2. First, chloride ions in the catalyst combined with silver ion formed a precipitate. The Rh or Ru catalyst dimer splits into monomers. The active catalytic species I for the Rh system (or I′ for Ru system) were formed by exchanging the ligand with Cu(OAc)2. Then, this active species combined with 2-aryl-pyridine was used to activate the C–H bond through concerted metalation deprotonation to afford the intermediate II or II′. Further, a molecule of acetic acid was eliminated to produce coordinate intermediate III or III′, followed by further coordination with styrene to form IV or IV′. After the β-H elimination, mono-olefinated product was produced.
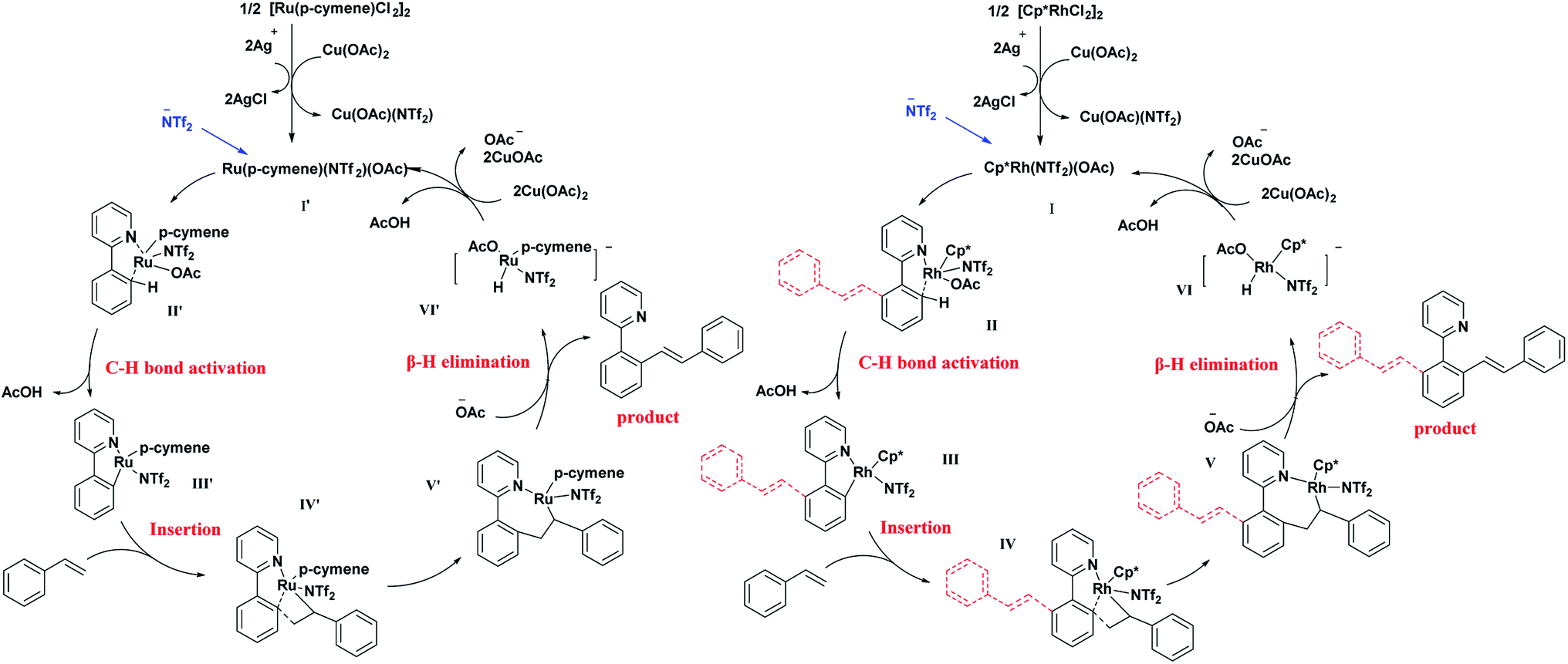 | ||
| Fig. 2 Possible mechanism. | ||
Acetate ion was trapped to form the catalyst intermediate VI or VI′. It was then oxidised by Cu(OAc)2 and eliminated a molecule of AcOH to afford the original active catalytic species. In the Rh system, the di-olefinated product was generated by another catalytic cycle.
Conclusions
In summary, we reported the transition-metal controlled mono- and di-olefination of arenes via the C–H activation in [BMIM]NTf2 and [BMIM]PF6 at room temperature. This is the first time to realize selective mono- and di-olefination at room temperature, probably due to the positive effects of ionic liquids. It is an energy-saving route in industrial production without the need of heating equipment. After the detailed screening of catalysts, it was found that the mono- and di-olefinated products can be selectively afforded by ruthenium and rhodium catalytic systems, respectively. The reported reactions demonstrated a broad substrate scope and gave the desired products in excellent yields with satisfactory selectivity even in gram-scale. In particular, the employment of non-volatile and chemically stable ionic liquid was environmental friendly without the use of organic solvents and made the catalytic process much safer without the need of pressure-tight equipment. Furthermore, the recyclability study revealed the consistent high activity of the catalytic systems even after recycling for 6 time. This advantage saves costs significantly and reduces environmental contamination caused by transition metals from an industrial point of view. The mechanistic study revealed that the C–H bond cleavage was probably achieved via the concerted metalation–deprotonation. This technique shows a great potential in the industrial production of various valuable unsaturated aromatic compounds.Experimental
General procedure for the synthesis of mono-olefinated product (taking 3a as an example)
To a 15 mL sealed tube, 2-phenylpyridine 1a (38.75 mg, 0.25 mmol), styrene 2a (52 mg, 0.50 mmol), [Ru(p-cymene)Cl2]2 (7.655 mg, 0.0125 mmol), AgNTf2 (9.7 mg, 0.025 mmol), Cu(OAc)2 (91 mg, 0.50 mmol), and [BMIM]NTf2 (0.8 mL) were added in sequence. The mixture was stirred at room temperature for 24 h and monitored by TLC. Afterwards, the mixture was extracted with diethyl ether and then the upper phase (diethyl ether) was evaporated in vacuo. The residue was further purified via preparative thin layer chromatography by silica gel (developing solvent, acetone/petroleum ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 with several drops of triethylamine), affording the product 3a.
:10 with several drops of triethylamine), affording the product 3a.
General procedure for the synthesis of di-olefinated product (taking 3a′ as an example)
To a 15 mL sealed tube, 2-phenylpyridine 1a (38.75 mg, 0.25 mmol), styrene 2a (104 mg, 1.00 mmol), [Cp*RhCl2]2 (7.725 mg, 0.0125 mmol), AgNTf2 (9.7 mg, 0.025 mmol), Cu(OAc)2 (182 mg, 1.00 mmol), and [BMIM]NTf2 (0.8 mL) were added in sequence. The mixture was stirred at room temperature for 24 h and monitored by TLC. Afterwards, the mixture was extracted with diethyl ether and then the upper phase (diethyl ether) was evaporated in vacuo. The residue was further purified via preparative thin layer chromatography by silica gel (developing solvent, acetone/petroleum ether = 1:10 with several drops of triethylamine), affording the product 3a′.
Abbreviations
| [BMIM]NTf2 | 3-Butyl-1-methyl-1H-imidazolium bis((trifluoromethyl)sulfonyl) amide |
| [BMIM]PF6 | 1-Butyl-3-methylimidazolium hexafluorophosphate |
| [BMIM]BF4 | 3-Butyl-1-methyl-1H-imidazolium tetrafluoroborate |
| [BMIM]OTf | 3-Butyl-1-methyl-1H-imidazolium trifluoromethanesulfonate |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81803486, 21676170), China Postdoctoral Science Foundation (Grant No. 2018M633385) and Sichuan Science and Technology Program (Grant No. 2019YJ0030).Notes and references
- (a) S. Zhang, F. Zhang and Y. Tu, Chem. Soc. Rev., 2011, 40, 1937 RSC; (b) J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003 RSC; (c) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- (a) B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2013, 42, 5744 RSC; (b) J. Wencel-Delord, T. Droege, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (d) B. Zhao, Z. Shi and Y. Yuan, Chem. Rec., 2016, 16, 886 CrossRef CAS PubMed.
- (a) G. Chen, W. Gong and Z. Zhuang, Science, 2016, 353, 1023 CrossRef CAS PubMed; (b) R. Sang, Y. Zheng and H. Zhang, et al., Org. Chem. Front., 2018, 5, 648 RSC; (c) C. Zhang, Y. Pu and Z. Yu, et al., Org. Chem. Front., 2018, 5, 1288 RSC; (d) H. Zhang, L. Jing and Y. Zheng, et al., Eur. J. Org. Chem., 2018, 2018, 723 CrossRef CAS.
- L. Ilies, Q. Chen, X. Zeng and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 5221 CrossRef CAS PubMed.
- (a) L. Ackermann, J. Org. Chem., 2014, 79, 8948 CrossRef CAS PubMed; (b) P. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 17283 CrossRef CAS PubMed; (c) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed.
- (a) F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522 CrossRef CAS PubMed; (b) A. Del Zotto and D. Zuccaccia, Catal. Sci. Technol., 2017, 7, 3934 RSC; (c) R. Ye, A. V. Zhukhovitskiy, C. V. Deraedt, F. D. Toste and G. A. Somorjai, Acc. Chem. Res., 2017, 50, 1894 CrossRef CAS PubMed.
- (a) A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109(3), 897 CrossRef CAS PubMed; (b) A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef; (c) S. R. Marder, B. Kippelen, A. K. Jen and N. Peyghambarian, Nature, 1997, 388, 845 CrossRef CAS.
- (a) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407 CrossRef CAS PubMed; (b) K. S. Kanyiva, N. Kashihara and Y. Nakao, et al., Dalton Trans., 2010, 39, 10483 RSC; (c) X. G. Li, K. Liu, G. Zou and P. N. Liu, Eur. J. Org. Chem., 2014, 2014, 7878 CrossRef CAS.
- (a) M. Kim, S. Sharma, N. K. Mishra, S. Han, J. Park, M. Kim, Y. Shin, J. H. Kwak, S. H. Han and I. S. Kim, Chem. Commun., 2014, 50, 11303 RSC; (b) R. Manikandan and M. Jeganmohan, Org. Biomol. Chem., 2016, 14, 7691 RSC; (c) L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728 CrossRef CAS PubMed.
- Y. Hashimoto, K. Hirano, T. Satoh, F. Kakiuchi and M. Miura, Org. Lett., 2012, 14, 2058 CrossRef CAS PubMed.
- (a) T. Katagiri, T. Mukai, T. Satoh, K. Hirano and M. Miura, Chem. Lett., 2008, 38, 118 CrossRef; (b) Y. Shibata, Y. Otake, M. Hirano and K. Tanaka, Org. Lett., 2009, 11, 689 CrossRef CAS PubMed.
- (a) M. Ahlquist, G. Fabrizi, S. Cacchi and P. Norrby, J. Am. Chem. Soc., 2006, 128, 12785 CrossRef CAS PubMed; (b) N. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 5636 CrossRef CAS PubMed.
- J. Kim, S. Park, M. Baik and S. Chang, J. Am. Chem. Soc., 2015, 137, 13448 CrossRef CAS PubMed.
- N. Umeda, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7094 CrossRef CAS PubMed.
- (a) T. Patra, R. Watile, S. Agasti, T. Naveen and D. Maiti, Chem. Commun., 2016, 52, 2027 RSC; (b) S. Mochida, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 3024 CrossRef CAS PubMed; (c) H. Zhang, Z. Yang, Q. Ma, J. Liu, Y. Zheng, M. Guan and Y. Wu, Green Chem., 2018, 20, 3140 RSC.
- T. Yao and K. Du, ACS Sustain. Chem. Eng., 2019, 7, 6068 CrossRef CAS.
- (a) C. Kuai, L. Wang, B. Li, Z. Yang and X. Cui, Org. Lett., 2017, 19, 2102 CrossRef CAS PubMed; (b) P. Nareddy, F. Jordan and M. Szostak, Org. Lett., 2017, 20, 341 CrossRef PubMed; (c) C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383 CrossRef CAS PubMed.
- (a) T. Yao and S. Yao, J. Chromatogr. A, 2017, 1481, 12 CrossRef CAS PubMed; (b) T. Yao, S. Yao, C. Pan, X. Dai and H. Song, Energy Fuels, 2016, 30, 4740 CrossRef CAS.
- (a) T. Yao, S. Yao, D. Tang, L. Jing, D. Wang and H. Song, RSC Adv., 2016, 6, 52898 RSC; (b) L. Xie, S. Peng, J. Tan, R. Sun, X. Yu, N. Dai, Z. Tang, X. Xu and W. He, ACS Sustain. Chem. Eng., 2018, 6, 16976 CrossRef CAS; (c) C. Wu, L. Lu, A. Peng, G. Jia, C. Peng, Z. Cao, Z. Tang, W. He and X. Xu, Green Chem., 2018, 20, 3683 RSC; (d) L. Lu, Z. Wang, W. Xia, P. Cheng, B. Zhang, Z. Cao and W. He, Chin. Chem. Lett., 2019, 30, 1237 CrossRef CAS; (e) T. Yao, H. Yao, S. Yao, X. Dai and H. Song, J. Mol. Liq., 2018, 263, 72 CrossRef CAS; (f) T. Yao, X. Huang, H. Zang, H. Song and S. Yao, J. Mol. Liq., 2017, 231, 411 CrossRef CAS.
- (a) H. M. Savanur, R. G. Kalkhambkar and K. K. Laali, Appl. Catal., A, 2017, 543, 150 CrossRef CAS; (b) R. G. Soengas, V. L. Silva, J. Pinto, H. Rodríguez Solla and A. M. Silva, Eur. J. Org. Chem., 2016, 2016, 99 CrossRef CAS; (c) F. Wang, S. Tang, H. Ma, L. Wang, X. Li and B. Yin, Chin. J. Chem., 2014, 32, 1225 CrossRef CAS; (d) S. Lv, Y. Li, T. Yao, X. Yu, C. Zhang, L. Hai and Y. Wu, Org. Lett., 2018, 20, 4994 CrossRef CAS PubMed; (e) W. Zielinski, R. Kukawka, H. Maciejewski and M. Smiglak, Molecules, 2016, 21, 1115 CrossRef PubMed; (f) M. Khanmirzaei, S. Ramesh and K. Ramesh, J. Nanosci. Nanotechnol., 2020, 20, 2423 CrossRef CAS PubMed; (g) Q. Wang, T. Zhang and S. Zhang, Sep. Purif. Technol., 2020, 231, 115923 CrossRef CAS; (h) Y. Fu, H. Yin and A. Wang, Ind. Eng. Chem. Res., 2015, 54, 6619 CrossRef CAS; (i) A. Wang, Y. Jiang and W. Chen, J. Ind. Eng. Chem., 2012, 18, 237 CrossRef CAS; (j) L. shen, X. Zhou and H. Yin, et al., Braz. J. Chem. Eng., 2018, 35, 659 CrossRef CAS.
- (a) M. Brasse, J. Cámpora, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2013, 135, 6427 CrossRef CAS PubMed; (b) B. Liu, Y. Fan, Y. Gao, C. Sun, C. Xu and J. Zhu, J. Am. Chem. Soc., 2012, 135, 468 CrossRef PubMed; (c) X. Qi, Y. Li, R. Bai and Y. Lan, Acc. Chem. Res., 2017, 50, 2799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09736h |
| This journal is © The Royal Society of Chemistry 2020 |