
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic study of N–H- and H–N-codoping of a TiO2 photocatalyst for efficient degradation of benzene under visible light†
Minghui Li ab,
Wulin Song*ab,
Lei Zenga,
Dawen Zenga,
Changsheng Xiea and
Qun Yangc
ab,
Wulin Song*ab,
Lei Zenga,
Dawen Zenga,
Changsheng Xiea and
Qun Yangc
aState Key Laboratory of Materials Processing and Die & Mould Technology, Huazhong University of Science and Technology, Wuhan, 430074, P. R. China. E-mail: wulins@126.com; Fax: +86 27 87792025; Tel: +86 27 87792025
bAnalytical and Testing Center, Huazhong University of Science and Technology, P. R. China
cWuhan Maritime Communication Research Institute, P. R. China
First published on 15th January 2020
Abstract
Non-metal codoping including nitrogen (N) and hydrogen (H) codoping has been emerging as an effective way to improve the performance of anatase TiO2 in solar cell, fuel conversion and pollutant degradation. However, the mechanism of the synergistic effect of N doping and H doping on TiO2 is still far from thorough. In this paper, N and H codoped TiO2 nanoparticles are obtained by N doping in ammonia and then H doping in hydrogen gas, which achieves substantially boosted efficiency and reaction rate in the photocatalytic degradation of benzene under visible light excitation. The superiority of the N–H–TiO2 photocatalyst was fully elaborated by comparing with H–N–TiO2, which was obtained by thermal treating in H2 and then NH3. The reaction rate of N–H–TiO2 in the photocatalytic degradation of benzene was nearly 2 times that of H–N–TiO2, ∼7 times higher than that of pristine TiO2. Furthermore, the cycling test revealed the high repeatability and stability of the N–H–TiO2 photocatalyst. The excellent performance N–H–TiO2 was attributed to an adequate concentration of NiHi defects occupying interstitial sites of the TiO2 structure and a disordered surface layer introduced by annealing in NH3 and H2 successively. The synergistic effect of N–H-codoping also increased the separation and migration of electron–hole pairs triggering a photoinduced redox reaction on the surface of TiO2.
1. Introduction
The breakthrough of water splitting on a TiO2 photoelectrode has triggered great enthusiasm to develop TiO2-based photocatalysts to wrestle with growing environment contamination and the energy crisis.1 TiO2 possesses many significant advantages in photocatalytic applications (such as, suitable band energy, non-toxicity, stable to corrosion, and low cost). Unfortunately, the TiO2 photocatalyst is extremely limited by its wide bandgap (anatase ∼3.2 eV, rutile ∼3.0 eV) as it can only absorb the UV fragment of sunlight and thus more than 95% of sunlight energy is not utilised.To overcome these obstacles, tremendous efforts have been devoted to developing visible light-responsive TiO2 by extrinsic element doping, intrinsic self-doping, semiconductor coupling, and noble metal modifications.2–5 Among these approaches, N doping and H doping have shown the great potential to improve the photocatalytic properties of TiO2 by enhancing visible light absorption. For example, Asahi et al. developed a visible-light responsive TiO2 photocatalyst by N doping,6 it ushered a new era of modulation of optical absorption and band structure of TiO2 by inter-bandgap element doping. This work revealed that the N doped at the substitutional sites of TiO2 could narrow the bandgap, which enhanced the visible light absorption of TiO2−xNx and thus improved the photocatalytic activity. Since then, N doping becomes one of the most desirable methods to achieve the better performance of TiO2 photocatalysts by forming the inter-bandgap states and enhancing the visible light absorption.7–10 It was reported that N doping facilitated the formation of oxygen vacancies in TiO2, which could introduce mid-gap states below conduction band.11 Previous researches also showed that the presence of N dopants benefited for the formation of oxygen vacancies, and meanwhile, the existence of oxygen vacancies might stabilize N impurities, which proved the correlation of N doping and defects formation.12,13 Besides N doping, H doping became more noteworthy as it was found the hydrogenated disorder-engineered TiO2 nanocrystals showed excellent optical absorption capability to visible and even infrared lights.14 The authors highlighted that the disordered surface layer was responsible for the enhanced light absorption and photocatalytic activity of TiO2. Since then, hydrogenation becomes a powerful approach to improve the performances of TiO2 nanomaterials.
However, N doping and H doping individually are not enough to obtain an efficient TiO2 photocatalyst. One issue is the localized states formed by N doping increase the recombination centers of photogenerated electrons and holes, and the other is that visible light activity after H doping is far from practical applications. Consequently, various investigations have been carried out to the development of visible light responsive TiO2 photocatalysts by N and H codoping, and then explain their advantages in enhancing light absorption, narrowing bandgap, improving the separation of electron–hole pairs, reducing the recombination centers and so on. For example, Hoang et al. suggested that the enhanced visible light activity of rutile (H, N)–TiO2 nanowire arrays was due to the interaction of substitutional N dopants and Ti3+.15 Meanwhile, Zhang et al. proposed that the photocatalytic activity of N–H-codoped TiO2 was improved by the synergistic effect of oxygen vacancies and N dopants.16 Moreover, Wang et al. suggested that hydrogenation could induced the defect energy states in TiO2 by forming Ti3+ and N dopants and combining with disordered structures, which contributed to the enhanced efficiency of hydrogenated N–F doped TiO2.17 Beyond doubt, previous investigations in N and H codoping TiO2 provide new insights into utilizing solar light and enlarging the scopes of applications. However, due to lack of comparison under a uniform standard, the mechanism of N and H codoping is still not really clarified. Moreover, though many efforts have been done to investigate the N and H codoping on photocatalytic applications, such as, water splitting and water purification, unsufficient attention has been paid to the applications of volatile organic compounds (VOCs).18 Benzene, especially, as a typical indoor contaminant and strong cancerogen, is a degradation-resistant VOC gas and may cause serious health concerns. Thus, more efficient photocatalyts to remove it to purify the environment is urgent to develop.19–21
Given these challenges, we developed a highly efficient N–H–TiO2 photocatalyst for benzene removal utilizing visible light. In this work, the synergetic effect of N and H codoping on TiO2 photocatalyst was deeply explored on the basis of the characterizations of structural and properties. The distinct performances of N–H–TiO2 and H–N–TiO2 in decomposing benzene under visible light were aroused and the mechanism was fully understood. The facile preparation and the inspiring performance of N–H–TiO2 photocatalyst in benzene degradation make us confident in its extensive applications.
2. Experimental
2.1 Chemicals
All chemicals applied in this experiment including tetra-n-butyl titanate (TTIP, 98%; Sinopharm Chemical Reagent Co., Ltd.), ethanol, ammonia gas (NH3, 99.9999%) and H2/Ar (5% H2/95% Ar) gas mixture were purchased and used without any further purification.2.2 Synthesis of pure TiO2
Typically, 2 mL TTIP was added dropwise to a beaker with 75 mL distilled water under vigorous magnetic stirring and then the final suspension solution was transferred into a 100 mL Teflon-lined autoclave to react at 160 °C for 12 h. Afterwards, the resulting product centrifuged and rinsed with absolute ethanol and distilled water for several times, and then dried in an oven at 80 °C overnight to acquire TiO2 nanoparticles.2.3 Preparation of N and H codoping TiO2 samples
The as-prepared pure TiO2 was placed in a quartz glass tube and followed by thermal treatment in a tube furnace. The tube was filled with the desired gas for 30 min to purge the residual air before thermal treatment. Two kinds of gases, hydrogen and ammonia were used in this experiment. The flow rate of ammonia and hydrogen was 200 mL min−1 controlled by rotormeter. After thermal treatment, the samples were naturally cooled to room temperature. All the samples were treated at 600 °C with a ramp rate 5 °C min−1. The details were as follows: firstly, the pure TiO2 was treated in H2/Ar in the former 2 h and sequentially switched to NH3 for the latter 2 h; the product was labeled as H–N–TiO2. In the same way, we exchanged the gas feed order of H2/Ar and NH3 in the process of thermal treatment and N–H–TiO2 was prepared. Secondly, to further investigate the importance of treating order and the effect of hydrogenation, N–H–TiO2-E was obtained by annealing in NH3 for 2 h and then held in H2 for 4 h. Similarly, H–N–TiO2-E was obtained by treating in H2 for 4 h and NH3 for 2 h.2.4 Characterizations
The structures and morphologies of the TiO2 before and after the thermal treatment were investigated by X-ray diffraction (XRD, X'Pert powder) with Cu radiation, high-resolution transmission electron microscopy (HRTEM, FEI TecnaiG2 F30). Raman spectroscopy (Raman, LabRAM HR800) was investigated from 100–1000 cm−1 by using a LabRAM HR800 spectrometer and operating at 532 nm. Fourier transform infrared (FT-IR, Bruker VERTEX 70) spectroscopy was investigated in the range of 400–4000 cm−1. Ultraviolet-visible diffuse reflectance spectra (UV-vis DRS, Shimadzu U-3010) was conducted using BaSO4 as reference. Photoluminescence (PL) emission spectra were acquired under an excitation at 325 nm by using an Edinburgh Instruments PLSP920 spectrometer. The X-ray photoemission spectroscopy (XPS, Shimadzu AXIS-ULTRA DLD-600W) studies were examined to obtain information on the chemical bonding of the samples. The binding energy of XPS spectrum was corrected by the C 1s level at 284.6 eV.2.5 Photocatalytic activity test
The performance evaluation of the samples in our experiment was conducted in photocatalysis testing system, which was comprised of a home–made reaction chamber with an injection hole to input benzene and a sample loading plate, a gas chromatograph (GC9560) with hydrogen and nitrogen gas supply systems. The light source was a 300W Xe-arc lamp (CEL-HXF300) with a UV cut-off filter (λ > 400 nm). The irradiance of the lamp in the visible region was 1200 mW cm−2. The test was carried out by monitoring the degradation of benzene and the yield of CO2 under visible light irradiation within 4 h to estimate the photocatalytic activity. Generally, 100 mg TiO2 catalyst was dispersed on the loading plate (50 × 50 mm2) and then injected benzene into the reaction chamber (2 L) when the GC attained the running state according to the operation directions. Finally, we turned on the light source and then examined the photocatalytic activity of TiO2 samples by monitoring the change of benzene concentration and the yield of CO2 every 15 min intervals in 4 hours. The degradation rate of benzene was calculated by C/C0, where C was the concentration of benzene in each time period, and C0 was the initial concentration of benzene after adsorption equilibrium but before irradiation. Meanwhile, the CO2 production was calculated by C–C0 (unit: ppm), where C was the concentration of CO2 in each time period, and C0 was the concentration of CO2 after adsorption equilibrium but before irradiation. A blank control test without photocatalyst was performed for reference.3. Results and discussion
3.1 Basic structural information
The influence of N doping and H doping on the structure of N–H–TiO2 and H–N–TiO2 were manifested by XRD and FTEM. High temperature annealing in NH3 and H2 improves the crystallinity of N–H–TiO2 and H–N–TiO2, as the XRD results shown in Fig. 1. It is obvious that all the TiO2 samples consist of anatase phase (JCPDS file no. 21-1272) and brookite phase (JCPDS file no. 03-0380). However, H–N–TiO2 obtained by H doping contains rutile phase (JCPDS file no. 21-1276). In addition, there is surface disordered layers outside the core of H2 treated TiO2 nanoparticles, as shown by the dotted line of FTEM images in Fig. 2. It confirms the special structure induced by hydrogenation effect. The increase in crystallinity and the unchanged phase structure of N–H–TiO2 indicate the distinct effect of N–H- and H–N-codoping on the phase transition of TiO2. XRD results confirm that N doping is beneficial to suppress the phase transformation from anatase to rutile, and the following H doping can induce a disordered surface layer.22 Instead, in H–N–TiO2, H doping first facilitates the phase transformation, and thus increases the numbers of crystal boundaries and bulk defects, which act as the recombination centers of photogenerated electron–hole pairs and thus deteriorate the photocatalytic performance of H–N–TiO2.23–25 Therefore, N doping followed by H doping is a favorable stratagem to reduce bulk defects and then improves the crystallinity and the stability of anatase TiO2, and it also infers that anatase TiO2 behaves better photocatalytic activity than rutile TiO2.26,27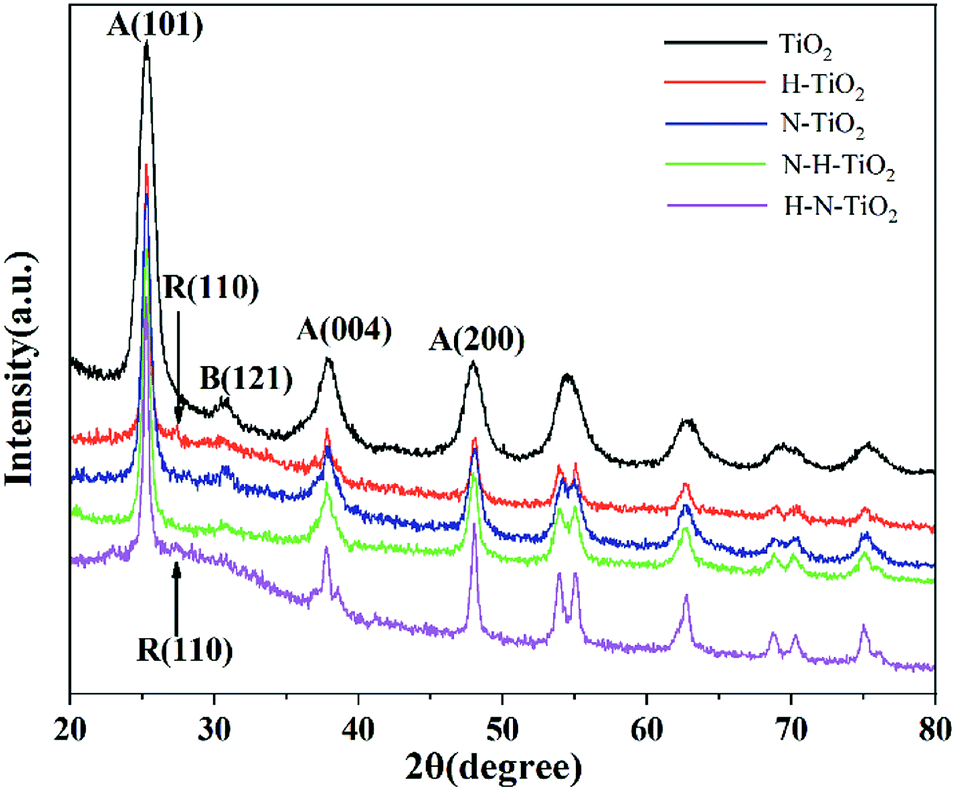 | ||
| Fig. 1 XRD patterns of TiO2 samples. | ||
 | ||
| Fig. 2 FTEM and HRTEM images of (a) TiO2, (b) H–TiO2, (c) N–TiO2, (d) N–H–TiO2, (e) H–N–TiO2, (f) N–H–TiO2-E. | ||
Additionally, the average particle sizes of TiO2, H–N–TiO2 and N–H–TiO2 are estimated to be 22.80, 44.21 and 37.70 nm respectively by the Scherrer formula, consistent with the FTEM images presented in Fig. 2. It is evident that the pristine TiO2 nanoparticles have the smallest size and the worst crystallinity. The average grain size and the crystallinity of H–N–TiO2 and N–H–TiO2 increase moderately, and the grain growth of N–H–TiO2 can be restrained by the ordering treatment in NH3 and then H2. Moreover, the H–N–TiO2 nanoparticles agglomerate severely which leads to its inferior performance. This may be ascribed to the thicker disordered surface layer and the worse bulk crystallinity of H–N–TiO2 than N–H–TiO2, as shown in Fig. 2d and e. According to the different microstructures of N–H–TiO2 and H–N–TiO2, it can be deduced that the formation process of disordered layer is different from each other. This special surface layer may be generated in the high temperature annealing process and finally formed in the cooling step. Thus, the different annealing orders in H2 result in the different bulk structures of N–H–TiO2 and H–N–TiO2. It could be because that H doping would introduce bulk defects, and after N doping, the formed disordered layer may restrain the migration of bulk defects and then lead to the inferior crystallinity of H–N–TiO2. However, N doping increases the crystallinity and decreases the bulk defects significantly, then after H doping, the H atoms may occupy the interstitial position (Hi) bonding with N atoms, which may maintain the advantages of both N doping and H doping.
3.2 Raman spectroscopy
Fig. 3a shows the Raman spectra of TiO2 samples. Six typical Raman active modes of anatase TiO2 occurs at 144 cm−1 (Eg), 197 cm−1 (Eg), 397 cm−1 (B1g), 518 cm−1 (A1g + B1g), and 640 cm−1 (Eg).28 The enlarged Raman spectra in Fig. 3b shows the peak at 440 cm−1 in H–TiO2 and 443 cm−1 in H–N–TiO2 and N–H–TiO2, which can be ascribed to the Raman modes of rutile.29 More importantly, these peaks only appear in H doping samples. It proves the successful doping of H atoms after annealing in H2 atmosphere. In addition, the weak two-phonon scattering bands at about 324 cm−1 assigned to anatase Raman feature and the band near 248 cm−1 ascribed to A1g mode of rutile can be observed.30 Generally, the Raman band below 400 cm−1 is ascribed to the Ti–O–Ti bending type and those above belongs to the Ti–O stretching type vibrations.28 However, the new peak at 480 cm−1 has never been found in other researches. Because it has been confirmed that N doping play little influence on the Ti–O stretching by comparing the Raman spectra of H–TiO2 and H–N–TiO2, these emerging changes at 480 cm−1 in H–TiO2, N–H–TiO2 and H–N–TiO2 may be induced by H-doping effect. Thus, the sharp peak of N–H–TiO2 at 480 cm−1 provides an evidence for the better synergetic effect of N–H-codoping than H–N-codoping because its higher intensity than that of the N–H bond in N–H–TiO2 photocatalyst.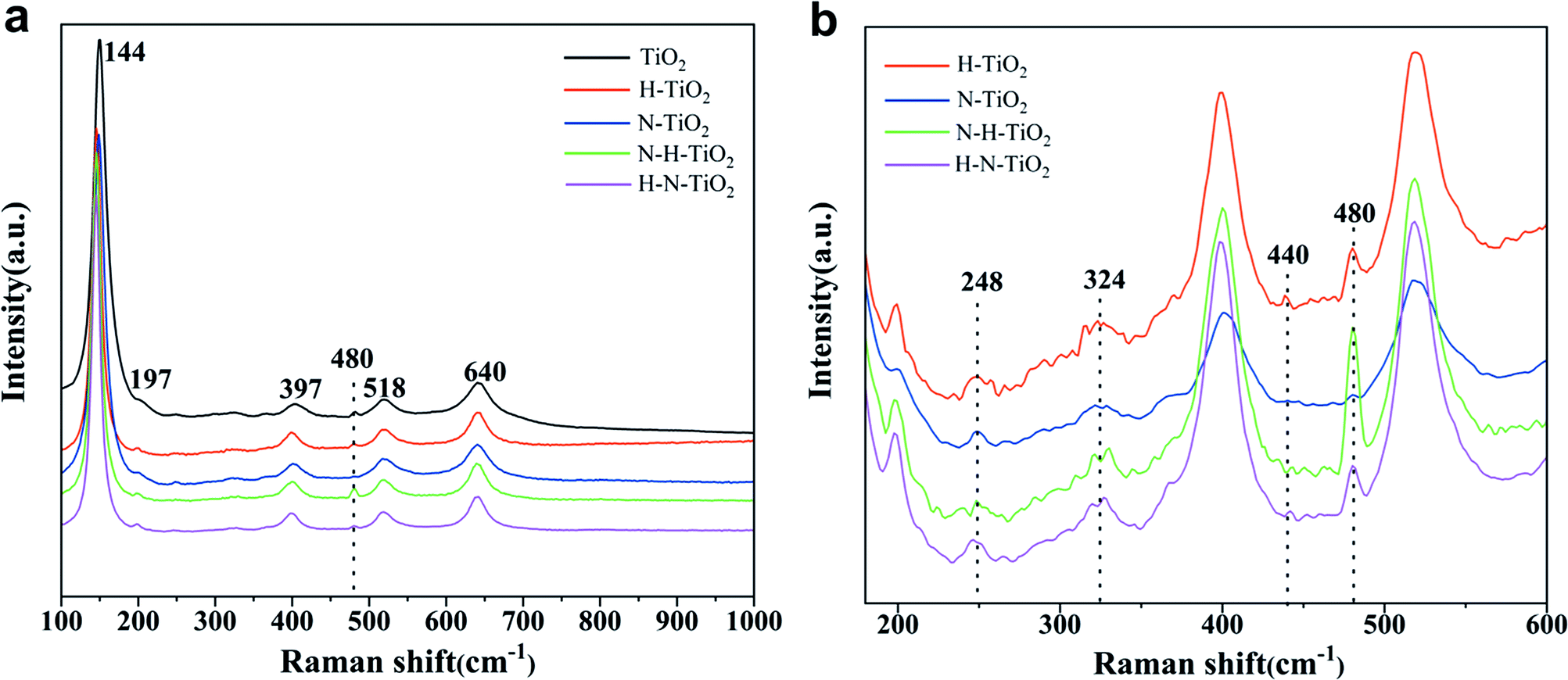 | ||
| Fig. 3 (a) Raman spectra of TiO2 samples; (b) enlarged Raman spectra from 180 to 600 cm−1. | ||
3.3 FTIR spectroscopy
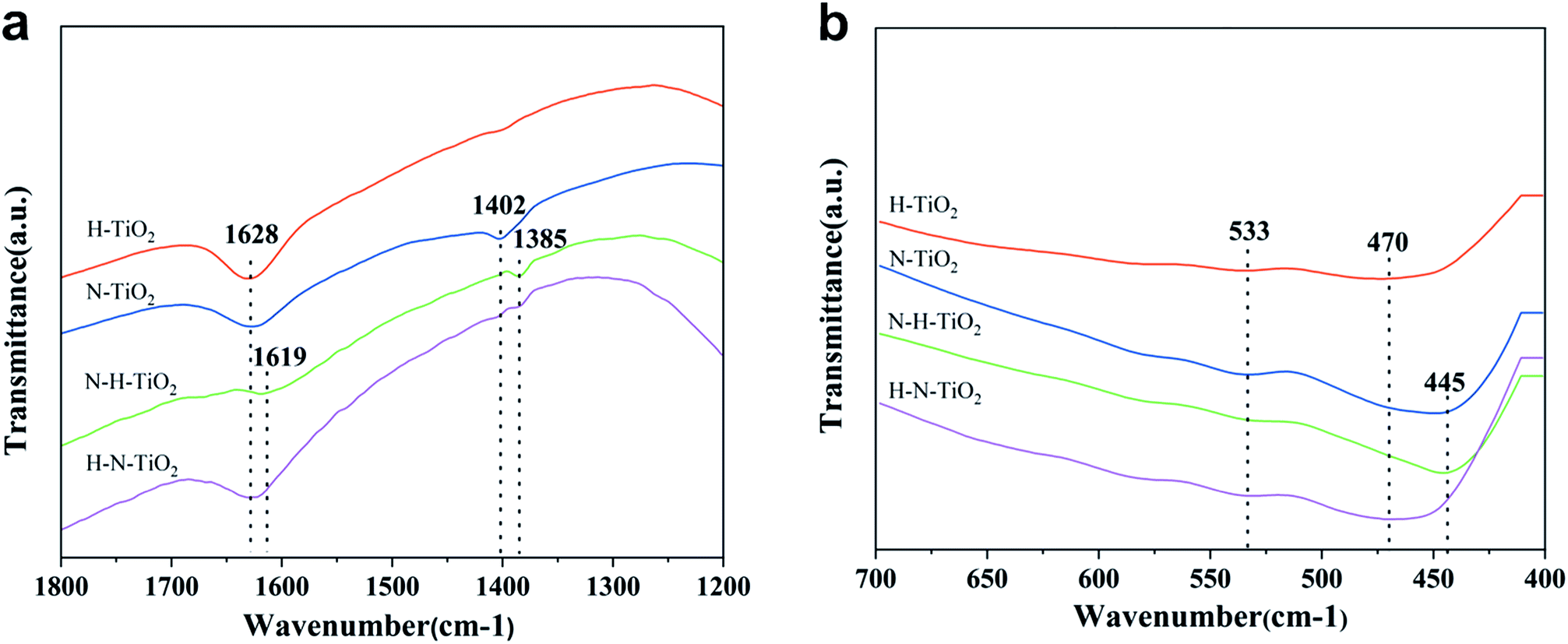
To investigate the chemical information on the surface functional groups of TiO2 samples, the FT-IR transmittance spectra were measured, as shown in Fig. 4. Besides, Fig. 5a and b are the enlarged spectra in the range of 1200–1800 cm−1 and 400–700 cm−1 respectively. Previous research has shown that the signals in the range of 400–1250 cm−1 are characteristic of O–Ti–O lattice and the peak near 1628 cm−1 is caused by Ti–O structure.29,31 In addition, the broad absorption presented at 2900–3600 cm−1 corresponds to O–H stretch.32 Generally, the typical infrared bands of NO2 are detected at 1618 cm−1.33 The NO2 band cannot be detected in N doping TiO2 but instead in N–H–TiO2. Furthermore, as shown in Fig. 5a, the absorption peak at 1402 cm−1 in N–TiO2 shifts to 1385 cm−1 in N and H codoping TiO2, which implies the effect of H doping. Considering the N doping effect, the peaks at about 1385 cm−1 in N–H–TiO2 and H–N–TiO2 infer the formation of impurity defects like N–H bond in the codoping TiO2 lattice. Moreover, Fig. 5b presents the consistent shift direction of N–TiO2 and N–H–TiO2 compared to H–TiO2 and H–N–TiO2. It indicates the distinct effect of N–H- and H–N-codoping on structure and defect states.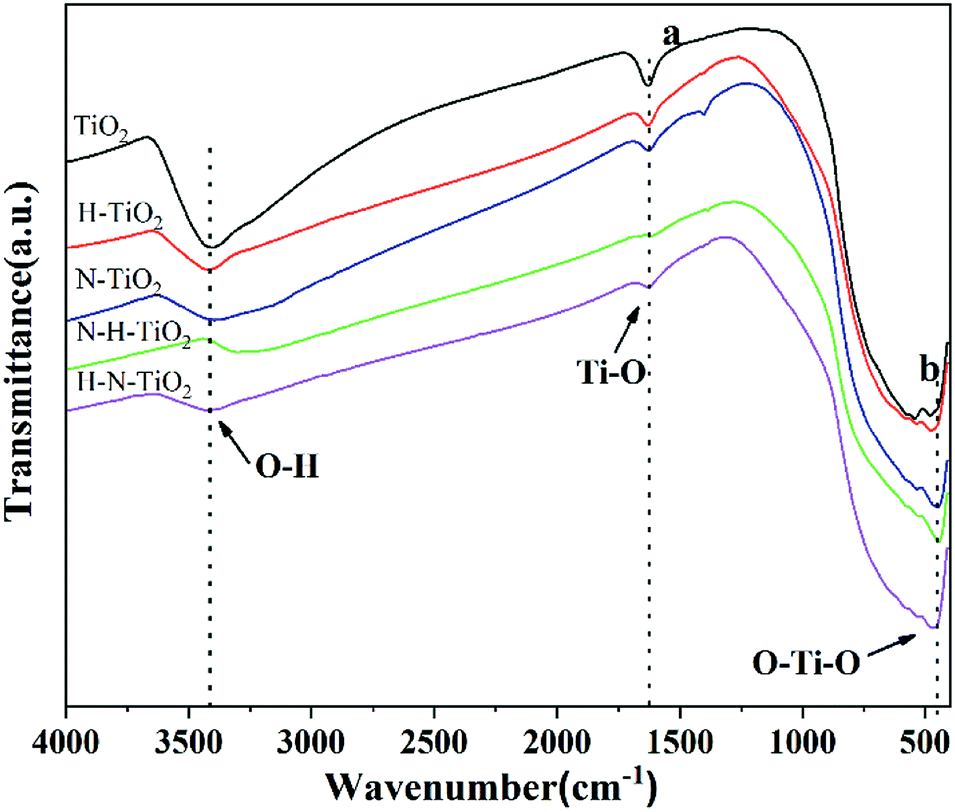 | ||
| Fig. 4 FT-IR patterns of TiO2 samples. | ||
 | ||
| Fig. 5 Enlarged FT-IR patterns of TiO2 samples at position (a) and (b) of Fig. 4. | ||
3.4 Chemical states
XPS analysis is carried out to determine the chemical states of TiO2 samples. N 1s peaks of N or (N, H) doping TiO2 are presented in Fig. 6. The concentrations of N dopants calculated by N 1s peak area are listed in Table 1. It is generally believed that the doped N species results in the narrowed bandgap and the enhanced photocatalytic activity of TiO2, in which the N 1s peak at a binding energy around 396 eV is the characteristic of Ns occupying substitutional sites in the form of TiN but the peak at a binding energies above 399 eV suggests the Ni dominating interstitial position attributed to NO or NO2.10,34,35 Obviously, only two peaks located at 399.7 eV and 402.5 eV in N 1s XPS spectral region in N–H–TiO2, indicating the interstitial nitrogen dopants. More importantly, the content of Ni in N–H–TiO2 is higher than N–TiO2 and H–N–TiO2. It is highly likely that the interaction between N and O is strong for the interstitial N doping in a type of NOx species.36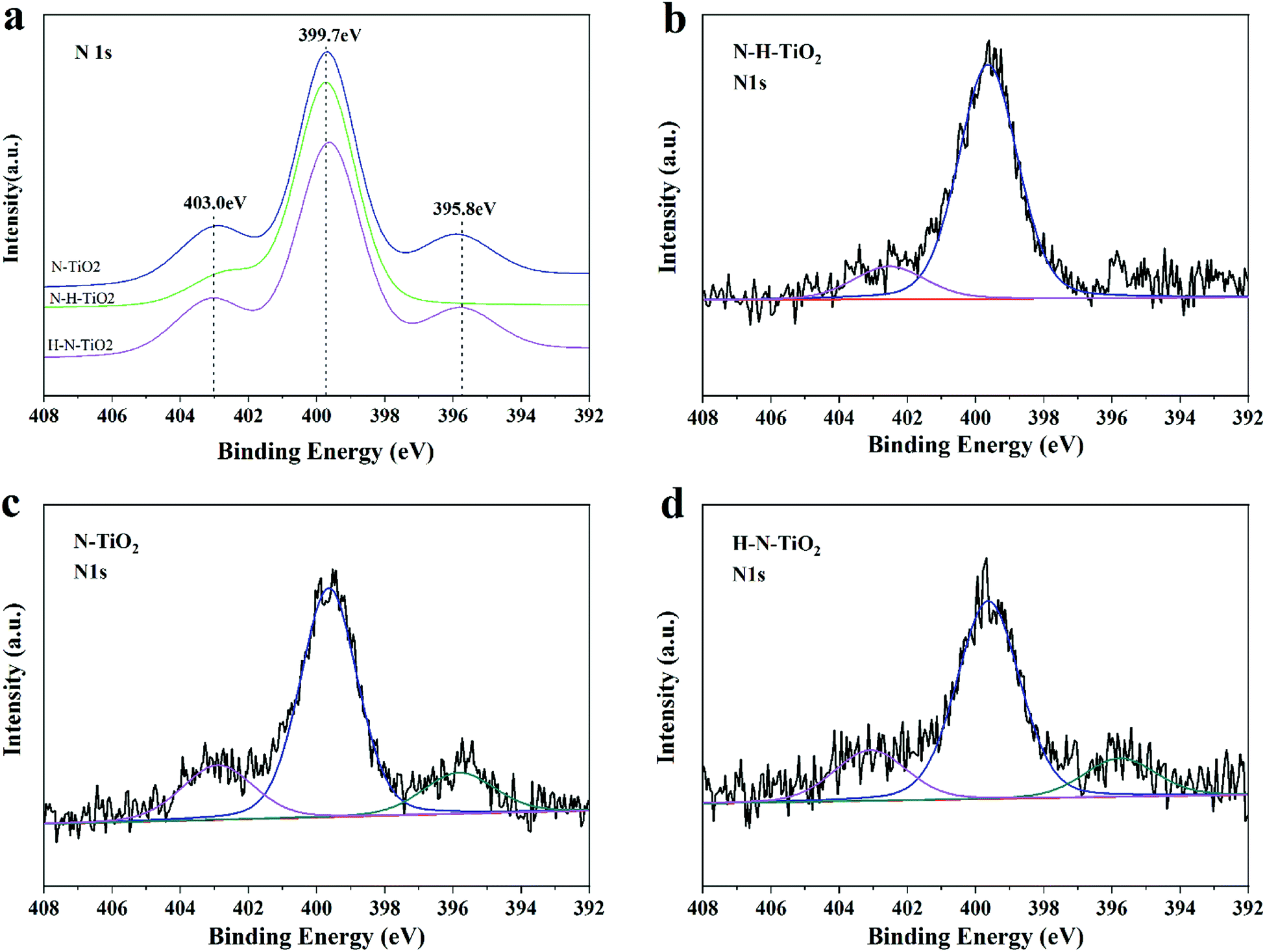 | ||
| Fig. 6 (a–d) N 1s XPS spectra of N–TiO2, N–H–TiO2 and H–N–TiO2. | ||
| Sample | Ns@396 eV | Ni@400 eV | Ni@403 eV | Total |
|---|---|---|---|---|
| N–TiO2 | 0.08 | 0.37 | 0.11 | 0.56 |
| N–H–TiO2 | — | 0.61 | 0.11 | 0.72 |
| H–N–TiO2 | 0.09 | 0.40 | 0.11 | 0.6 |
However, one more peak located at 396 eV appeared in H–N–TiO2 and N–TiO2, which is usually attributed to Ns species.6,37 Previous theoretical calculations showed that the different treatment conditions could tend to result in the different doping positions of N atoms in TiO2, such as excess oxygen and nitrogen, and the doped N would contribute to the formation of Ni, and otherwise the strong reduction atmosphere would benefit the introduction of Ns and oxygen vacancies.38 The theoretical inference is well consist with our experimental results. In our work, the Ni doping is definitely preferred after the annealing treatment in ammonia, while the Ns species coupled with oxygen vacancies is favoured under the highly reducing conditions such as the annealing treatment in H2. That's why more interstitial nitrogen species in N–H–TiO2 when the N doping is firstly applied, while H–N–TiO2 exists more substitutional nitrogen species when the H doping is firstly applied. Besides, the lower formation energy of Ni than Ns implies that Ni is more favorable in N–TiO2; but when phase transformation from anatase to rutile, it becomes more and more difficult to form Ni while the Ns doping becomes easier.39 Thus, there are more Ns atoms in H–N–TiO2 than N–H–TiO2 because of phase transformation, which is consistent with the results of XRD and Raman spectroscopy. In other words, the H doping benefits the formation of Ns. Although Ns dopants can lead to narrowed bandgap, the resultant localized states may decrease the mobility of charge carriers and then lead to more recombination centres. From the perspective of energy, the N species in TiO2 occupy the interstitial sites after the N doping.34 However, when interstitial position reaches saturation, the excess N atoms may enter into substitutional sites to replace O atoms. Thus, N–TiO2 contains both Ni and Ns dopants. After continuous hydrogenation, the surface of the N–H–TiO2 nanoparticles becomes amorphous and the O atoms escape from the original lattice to form the surface oxygen vacancies, which makes the Ns atoms escape from the primary position and then enter the interstitial position of TiO2 lattice again.
3.5 PL emission
The room temperature PL spectra of TiO2 samples are showed in Fig. 7. The PL spectra have been widely used to investigate the dynamic of electron–hole pairs in semiconductors because the PL emission is caused by the recombination of free carriers.40 The results clearly indicate that a substantial increase of PL emission occurs in H–TiO2, which may result from its tremendous recombination centers including bulk oxygen vacancies and thicker disordered layer after the H2 annealing. However, as N–TiO2 has a good crystallinity after N doping, its bulk defect density should be dramatically reduced compared with the pristine TiO2. In addition, higher PL emission intensity of H–N–TiO2 suggests it owns more recombination centres than N–H–TiO2 because the recombination of charge carriers usually occurs at the grain boundary of two phases or in the bulk of nanomaterials.41 Specially, H–N–TiO2 exhibits a higher emission than N–TiO2, and N–H–TiO2 exhibits a lower emission than H–TiO2. It further demonstrates that the H doping may lead to create more recombination centres due to the formation of bulk defects. Besides, it is generally accepted that N doping facilitates the formation of oxygen vacancies in TiO2,11 and N doping first may reduce the formation energy of oxygen vacancies and in turn the formed O vacancies can stabilize the N impurities.38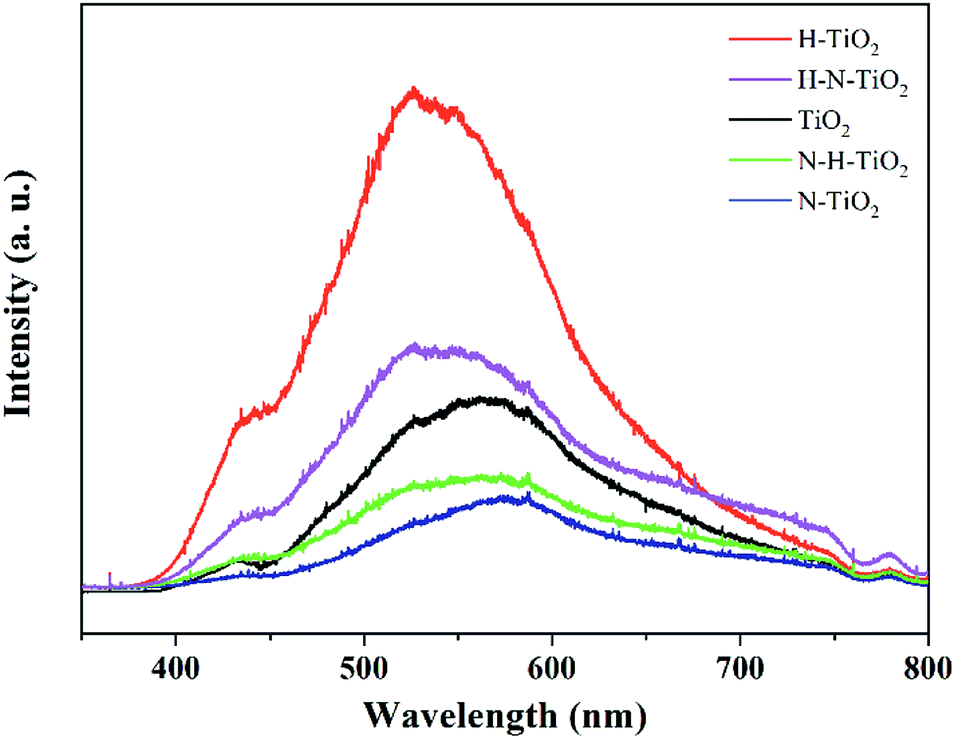 | ||
| Fig. 7 PL of TiO2 samples. | ||
Thus, as to N–H–TiO2, the N doping could improve the stability of the H doping-introduced oxygen vacancies, and then facilitate the separation and the transfer of photogenerated charge carriers to participate in the surface photocatalytic reactions. However, when it comes to H–N–TiO2, much more defects are imported by the first H doping, but these defects cannot exist stably and then become recombination centers. Therefore, the higher PL intensity of H–N–TiO2 can be assigned to its inferior bulk crystallinity and the phase transition from anatase to rutile. Moreover, it does not necessarily that the more defects, the better photocatalytic activity because some surface defects can serve as charge carrier traps and absorption sites to prevent electron–hole pairs from recombination while some bulk defects tend to act as recombination sites to annihilate photogenerated electron–hole pairs.42 It has been calculated that H doping may replace oxygen to form Ho dopants.43 Though subsequent N doping can narrow the bandgap and then increase the visible light absorption, the deep energy states formed by NiHo in H–N–TiO2 may promote the recombination of electron–hole pairs. On the contrary, more NiHi defects in N–H–TiO2 would generate the shallow states above the top of valence band, which is beneficial to the separation of charge carriers.
3.6 Optical property
Fig. 8 displays the light absorption ability and the estimated band gaps of TiO2 samples. Compared to the pure TiO2, N–TiO2 exhibits typical characteristic of N doping with the absorption from 400 nm to 500 nm and even extending to 550 nm.13 In addition, the absorption edge of H–TiO2 extending to infrared wavelength confirms the hydrogenation effect on the enhanced light absorption.44 After the N and H codoping, the samples behave the visible light absorption and the light harvesting of N–H–TiO2 in UV region is better than H–N–TiO2, while their abilities of light harvesting are opposite in visible light region. The reason is perhaps due to that the H doping promotes the interstitial Hi bonding with the interstitial Ni and then form the NiHi defects in N–H–TiO2,39,43 and thus, the visible light absorption is partially eliminated compared with the sample doped only with N species.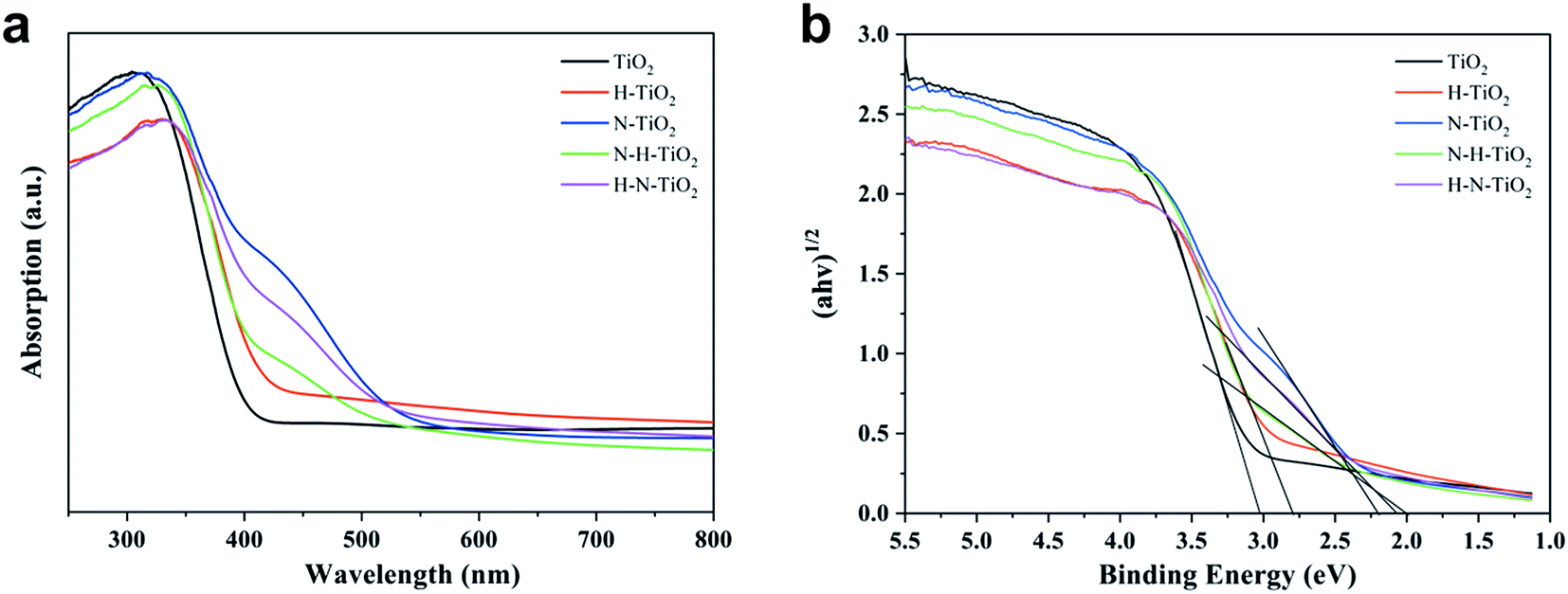 | ||
| Fig. 8 (a) UV-vis DRS and (b) estimated band gap of TiO2. | ||
However, compared with N–H–TiO2, the concentration of N atoms in H–N–TiO2 tends to become lower due to the surface disordered structure. Therefore, there are fewer NiHi bonds are formed in H–N–TiO2, however, compared with and finally it leads to the stronger visible light harvesting than N–H–TiO2. The results are also confirmed by XPS and PL. The bandgaps of N–H–TiO2 and H–N–TiO2 are estimated by Kubelka–Munk equation according to the UV-vis DRS spectrum. As shown in Fig. 8b, both their bandgaps are narrowed. It is because the new states formed by an interaction of defect states introduced by N doping and H doping that changes the band structure of N–H–TiO2, which not only ensures the visible light absorption but also increases the photogenerated electron–hole separation and facilitates the electron migration to surface.43 However, although with the relative higher light absorption, the deep energy level and the thicker surface disordered layer caused by H–N-codoping may result in more recombination of photoformed charge carriers in H–N–TiO2 and lead to poorer activity than N–H–TiO2.
3.7 Evaluation of photocatalytic activity
Fig. 9 shows the photocatalytic activity of TiO2 samples when degrading benzene within 4 hours under visible light. In detail, Fig. 9a and b showed the degradation of benzene and the production of CO2. It reveals that N–H–TiO2 exhibit the best photocatalytic activity. The excellent reaction rate constant and the stability of N–H–TiO2 are presented in Fig. 9c and d, respectively. Generally, the photocatalytic degradation of pollution obeys the first-order kinetics, and the reaction rate constant could be calculated by the following equation: −ln(C/C0) = kappt, where C/C0 is the normalized benzene concentration, t is the reaction time, and k is the reaction rate constant (10−3 min−1).45 As shown in Fig. 9c, the rate constant of N–H–TiO2 towards benzene is calculated to be 0.0193 min−1, which is more than twice the rate constant of H–N–TiO2, and especially, about 7 times higher than that of pristine TiO2. For the practical application of photocatalysts, the stability of photocatalyst is one of the key issues, such as the decrease of active sites on the surface of photocatalyst or the unsufficient photocatalytic activity after long time storage.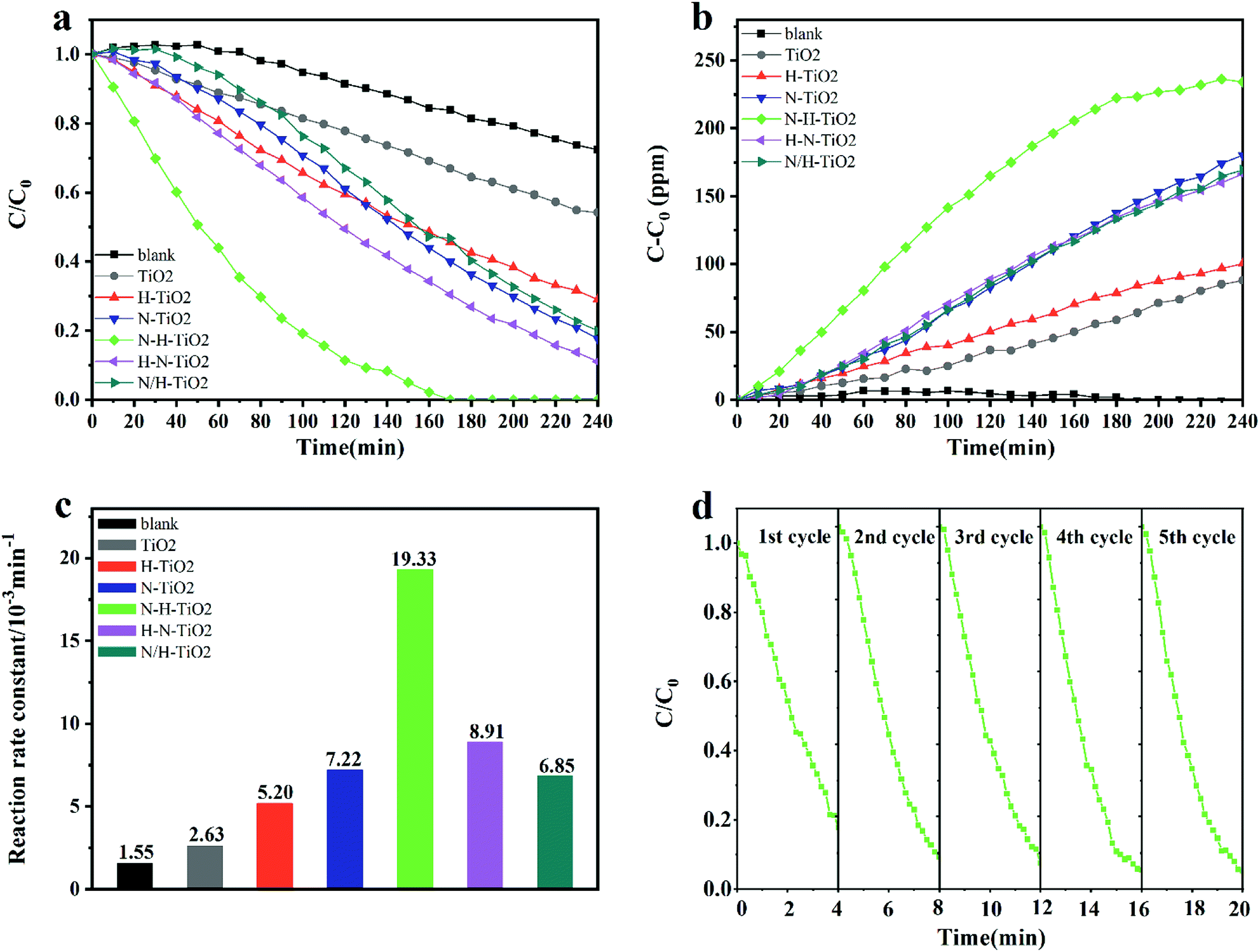 | ||
| Fig. 9 (a) Photocatlytic degradation of benzene (C/C0) by TiO2 samples under visible light; (b) CO2 production (C–C0) corresponding to Fig. 1a; (c) the reaction rate constants of all samples; (d) the cycling test of N–H–TiO2 under visible light. | ||
To evaluate the stability of N–H–TiO2, recycle experiments under same conditions were conducted. According results shown in Fig. 9d, N–H–TiO2 photocatalyst exhibits excellent repeatability and stability. The slightly fluctuated data may be on account of the variation of the initial concentration of benzene. The different activity of N–H–TiO2 and H–N–TiO2 indicates that TiO2 annealing in different atmospheres plays obviously different roles in the final products. The characterizations of microstructures and chemical states demonstrate that phase transformation, surface disordered structure, and concentration of N species have influence on the performance of N–H–TiO2 and H–N–TiO2, especially defect states introduced by N and H codoping. Moreover, stronger visible light absorption of N doping than N–H- or H–N-codoping reveals that property of N and H codoping is not the simple superposition of N doping and H doping. The excellent performance N–H–TiO2 was attributed to adequate concentration of NiHi defects occupying interstitial sites of TiO2 structure and disordered surface layer introduced by annealing in NH3 and H2 successively. The synergistic effect of N–H-codoping also increased the separation and migration of electron–hole pairs to trigger photoinduced redox reaction on surface of TiO2, confirmed by the PL results.
Furthermore, the contrast tests of N–H–TiO2-E and H–N–TiO2-E in Fig. S1† confirm the special influence of annealing atmosphere orders on the photocatalytic activity of TiO2. N–H–TiO2-E shows a better performance compared with H–N–TiO2-E in accordance with the results of N–H–TiO2 and H–N–TiO2. The positive effect of H doping is confirmed by the contrast of H–N–TiO2 and H–N–TiO2-E, as well as the difference of N–H–TiO2 and N–H–TiO2-E, as shown in Fig. S2.† These results demonstrate that the visible light activity of TiO2 photocatalyst and the corresponding mineralization rate of benzene increase with the extension of annealing time in hydrogen gas. It proves the positive effect of H doping on the photocatalytic properties of TiO2 nanoparticles.
3.8 Mechanism discussion
A schematic diagram has been established to explain the photocatalytic reaction paths of the N–H–TiO2 nanoparticles, as presented in Fig. 10. It clearly shows the related processes that finally decide the reduction and oxidation reactions on N–H–TiO2 photocatalyst under the visible light excitation. The crystallinity of N–H–TiO2 nanoparticles is greatly enhanced and the internal defects are dramatically reduced after N doping, meanwhile the surface oxygen vacancies are introduced and stably exist after the H doping.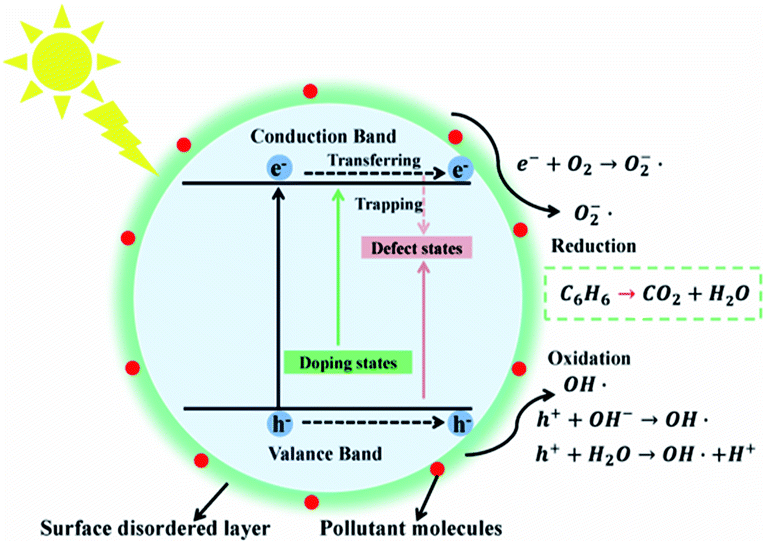 | ||
| Fig. 10 Schematic diagram of energy states and photocatalytic reaction paths on N–H–TiO2 nanoparticles. | ||
More importantly, the surface defective structure introduced by the H doping (defect states) stabilizes the N-doped structure and leads to the increase in the trapping sites and the surface reactive sites, which is beneficial to the photocatalytic reactions by reducing the recombination of charge carriers and increasing the absorption of small molecules like H2O and O2, so as to produce hydroxyl radicals (OH˙) or active oxygen (O2˙−) to degrade organic pollutants like benzene, as shown in Fig. 10.
The bandgap of N–H–TiO2 is mainly engineered by the new emerged NiHi states (doping states) above the valence band edge according to the N–H-codoping. The existence of Ni is beneficial to the incorporation of Hi and will facilitate the formation of NiHi defects in N–H–TiO2, which have been confirmed by Raman and FT-IR spectra. The NiHi defects in N–H–TiO2 give rise to the shallow states above VBM.40 However, as to H–N–TiO2, the structure of disordered layer on the surface is formed by the H doping, which is similar to a fence on the surface and suppresses the subsequent Ni doping. Consequently, the concentration of interstitial Ni in H–N–TiO2 is less than that in N–H–TiO2, which is confirmed by XPS results. In addition, the surface amorphous layer has a localized effect on the internal defects of nanoparticles, which blocks the charge carriers to transfer to the surface to participate in the following reactions. Therefore, the performance of H–N–TiO2 is not fully improved after the H–N-codoping. In fact, the pristine TiO2 has many defects because of its poor crystallinity; H may replace the lattice oxygen to form the Ho defects and then leave the bulk oxygen vacancies after the H doping. The N doping after H doping may cause the reduction of bulk defects due to the increased crystallinity, while the newly generated NiHo defects may result in the formation of deep states above valence band maximum (VBM), which not only increase the recombination centers of photogenerated charge carriers but also eliminate the positive effect on the H doping. Thus, the good cyrstallinity, higher concentration of NiHi dopants, lower recombination of photogenerated charge carriers, and the beneficial surface disordered layer synergistically contribute to the better photocatalytic activity of N–H–TiO2 in degrading benzene under visible light.
4. Conclusion
In summary, a highly efficient photocatalyst N–H–TiO2 for benzene degradation driven by visible light has been successfully synthesized by annealing in NH3 and H2 atmospheres. We have illustrated that the reverse annealing orders of NH3 and H2 induce the different structures of N–H–TiO2 and H–N–TiO2, and thus results in their different photocatalytic performances. The excellent photocatalytic performance of N–H–TiO2 is a final embodiment of the synergistic effect of good crystallinity, adequate concentration of NiHi defects and disordered surface layer, which lead to the lowered recombination of photogenerated charge carriers, increased amount of surface active sites and more surface absorption sites in N–H–TiO2. Through the investigation on the characteristics of N–H- and H–N-codoping, we provide a fundamentally new perspective to elucidate the role of codoping effect on TiO2 photocatalyst. The as-prepared N–H–TiO2 photocatalyst by a facile method with low energy consumption is meaningful for expanding the application prospect in environmental improvements, especially in the elimination of volatile organic compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Basic Research Program of China (Grant No. 2009CB939705). The authors are also grateful to Analytical and Testing Center of Huazhong University of Science and Technology.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- Y. Wu, J. Zhang, L. Xiao and F. Chen, Appl. Catal., B, 2009, 88, 525–532 CrossRef CAS.
- J. Pan, Z. Dong, B. Wang, Z. Jiang, C. Zhao, J. Wang, C. Song, Y. Zheng and C. Li, Appl. Catal., B, 2019, 242, 92–99 CrossRef CAS.
- Y. Cao, Z. Xing, Y. Shen, Z. Li, X. Wu, X. Yan, J. Zou, S. Yang and W. Zhou, Chem. Eng. J., 2017, 325, 199–207 CrossRef CAS.
- R. Asahi, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- G. Barolo, S. Livraghi, M. Chiesa, M. C. Paganini and E. Giamello, J. Phys. Chem. C, 2012, 116, 20887–20894 CrossRef CAS.
- Y. Chen, X. Cao, B. Gao and B. Lin, Mater. Lett., 2013, 94, 154–157 CrossRef CAS.
- M. Khan and W. Cao, J. Mol. Catal. A: Chem., 2013, 376, 71–77 CrossRef CAS.
- T. Boningari, S. N. R. Inturi, M. Suidan and P. G. Smirniotis, Chem. Eng. J., 2018, 350, 324–334 CrossRef CAS.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- S. Livraghi, M. C. Paganini, E. Giamello, A. Selloni, C. Di Valentin and G. Pacchioni, J. Am. Chem. Soc., 2006, 128, 15666–15671 CrossRef CAS PubMed.
- Y. Chen, X. Cao, B. Lin and B. Gao, Appl. Surf. Sci., 2013, 264, 845–852 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134, 3659–3662 CrossRef CAS PubMed.
- Z. Zhang, J. Long, X. Xie, H. Zhuang, Y. Zhou, H. Lin, R. Yuan, W. Dai, Z. Ding, X. Wang and X. Fu, Appl. Catal., A, 2012, 425–426, 117–124 CrossRef CAS.
- W. Wang, C. Lu, Y. Ni, M. Su and Z. Xu, Appl. Catal., B, 2012, 127, 28–35 CrossRef CAS.
- W. Lin, X. Xie, X. Wang, Y. Wang, D. Segets and J. Sun, Chem. Eng. J., 2018, 349, 708–718 CrossRef CAS.
- J. Wang, H. Ruan, W. Li, D. Li, Y. Hu, J. Chen, Y. Shao and Y. Zheng, J. Phys. Chem. C, 2012, 116, 13935–13943 CrossRef CAS.
- H. Huang, D. Li, Q. Lin, W. Zhang, Y. Shao, Y. Chen, M. Sun and X. Fu, Environ. Sci. Technol., 2009, 43, 4164–4168 CrossRef CAS PubMed.
- P. Devaraji, N. K. Sathu and C. S. Gopinath, ACS Catal., 2014, 4, 2844–2853 CrossRef CAS.
- M. Li, W. Song, L. Zeng, D. Zeng and C. Xie, Mater. Lett., 2014, 136, 258–261 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- S.-D. Mo and W. Y. Ching, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13023–13032 CrossRef CAS PubMed.
- T. Luttrell, S. Halpegamage, J. Tao, A. Kramer, E. Sutter and M. Batzill, Sci. Rep., 2014, 4, 4043 CrossRef PubMed.
- K. Nagaveni, M. S. Hegde, N. Ravishankar, G. N. Subbanna and G. Madras, Langmuir, 2004, 20, 2900–2907 CrossRef CAS PubMed.
- B. Sun, P. G. Smirniotis and P. Boolchand, Langmuir, 2005, 21, 11397–11403 CrossRef CAS PubMed.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321–324 CrossRef.
- X. Chen, Y.-B. Lou, A. C. S. Samia, C. Burda and J. L. Gole, Adv. Funct. Mater., 2005, 15, 41–49 CrossRef CAS.
- G. A. Tompsett, G. A. Bowmaker, R. P. Cooney, J. B. Metson, K. A. Rodgers and J. M. Seakins, J. Raman Spectrosc., 1995, 26, 57–62 CrossRef CAS.
- H. Li, J. Li and Y. Huo, J. Phys. Chem. B, 2006, 110, 1559–1565 CrossRef CAS PubMed.
- M. Zheng, M. Gu, Y. Jin and G. Jin, Mater. Sci. Eng., B, 2000, 77, 55–59 CrossRef.
- T. Shimanouchi, J. Phys. Chem. Ref. Data, 1977, 6, 993–1102 CrossRef CAS.
- C. Di Valentin, E. Finazzi, G. Pacchioni, A. Selloni, S. Livraghi, M. C. Paganini and E. Giamello, Chem. Phys., 2007, 339, 44–56 CrossRef CAS.
- J. Ananpattarachai, P. Kajitvichyanukul and S. Seraphin, J. Hazard. Mater., 2009, 168, 253–261 CrossRef CAS PubMed.
- J. A. Rodriguez, T. Jirsak, J. Dvorak, S. Sambasivan and D. Fischer, J. Phys. Chem. B, 2000, 104, 319–328 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef CAS.
- C. Di Valentin, G. Pacchioni, A. Selloni, S. Livraghi and E. Giamello, J. Phys. Chem. B, 2005, 109, 11414–11419 CrossRef CAS PubMed.
- H. Pan, Y.-W. Zhang, V. B. Shenoy and H. Gao, J. Phys. Chem. C, 2011, 115, 12224–12231 CrossRef CAS.
- H. Tang, K. Prasad, R. Sanjinès, P. E. Schmid and F. Lévy, J. Appl. Phys., 1994, 75, 2042–2047 CrossRef CAS.
- L. Zeng, Z. Lu, J. Yang, M. Li, W. Song and C. Xie, Appl. Catal., B, 2015, 166–167, 1–8 CrossRef CAS.
- M. Kong, Y. Li, X. Chen, T. Tian, P. Fang, F. Zheng and X. Zhao, J. Am. Chem. Soc., 2011, 133, 16414–16417 CrossRef CAS PubMed.
- M. Vasilopoulou, N. Kelaidis, E. Polydorou, A. Soultati, D. Davazoglou, P. Argitis, G. Papadimitropoulos, D. Tsikritzis, S. Kennou, F. Auras, D. G. Georgiadou, S.-R. G. Christopoulos and A. Chroneos, Sci. Rep., 2017, 7, 17839 CrossRef PubMed.
- O. Elbanna, M. Fujitsuka, S. Kim and T. Majima, J. Phys. Chem. C, 2018, 122, 15163–15170 CrossRef CAS.
- Z. Lu, L. Zeng, W. Song, Z. Qin, D. Zeng and C. Xie, Appl. Catal., B, 2017, 202, 489–499 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09661b |
| This journal is © The Royal Society of Chemistry 2020 |