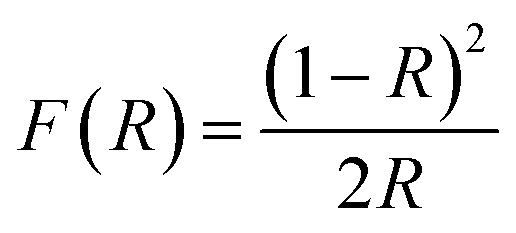DOI:
10.1039/C9RA09579A
(Paper)
RSC Adv., 2020,
10, 3796-3804
A reliable chemiresistive sensor of nickel-doped tin oxide (Ni-SnO2) for sensing carbon dioxide gas and humidity
Received
17th November 2019
, Accepted 26th December 2019
First published on 22nd January 2020
Abstract
Herein, we report the chemiresistive gas and humidity sensing properties of pristine and nickel-doped tin oxide (Ni-SnO2) gas sensors prepared by a microwave-assisted wet chemical method. The structural and optical properties are characterised using X-ray diffraction, scanning electron microscopy, scanning transmission electron microscopy, ultraviolet spectroscopy, Fourier transform infrared spectroscopy, and X-ray photoelectron spectroscopy. The structural elucidation and morphology analyses confirm a particle size of 32–46 nm, tetragonal rutile crystal structure and small cauliflower-type surface appearance. Nickel doping can tune the structure of NPs and morphology. The tested carbon dioxide gas and humidity sensing properties reveal a rapid sensing performance with high-to-moderate sensitivity. Also, the materials favour gas sensing because their sensitivity is enhanced with the increase in nickel concentration. The sensing results suggest that nickel is a vibrant metal additive to increase the gas sensitivity of the sensor. However, nickel doping decreases the electron density and increases the oxygen vacancies. Ultimately, the gas sensor produces highly rapid sensing with a response time of 4 s.
1. Introduction
In the field of agriculture and biotechnology, the detection and monitoring of humidity and carbon dioxide are very essential as they play a crucial role in the greenhouse effect.1,2 Also, they contribute to climate change. Generally, carbon dioxide mixes with the open atmosphere and is relevant to the indoor and outdoor air quality monitoring.3 Furthermore, the presence of carbon dioxide in the environment is steadily increasing as it is generated from motor vehicles, industries and other resources, which are logically responsible for increased global warming.4–6 The industrial revolution is one of the major factors responsible for increasing the carbon dioxide levels. Thus, the tracking and control of carbon dioxide is essentially important in food packaging and breath analysis technologies.7,8 On the other hand, humidity provides a critical response regarding human comfort and industry, which further influences human health, electrical circuits and manufacturing processes in industry and agriculture.9 Therefore, smart humidity sensors are needed to monitor the environment as well as control food preservation and packaging and in the processes of plantation and weather prediction.10 Several sensors are available in the market today. Among these, chemiresistive-type sensors have the ability to sense humidity and carbon dioxide gas at relatively low temperatures and under ambient operating conditions. In particular, nanomaterials demonstrate these chemiresistive features as they are semiconductors and have various band gap energies. They can be easily functionalized under a reduced operating temperature with the features of less power consumption and easy and safe operation. The distinct characteristic of nanomaterials is their high surface area/volume ratio, which eventually increases the gas sensitivity of the sensor.11
SnO2 being an n-type metal oxide is a chemiresistive sensor because of its large band gap (3.40 eV), d-10 electronic configuration, high reactivity against pollutant gases, moderate chemical stability, high reactive surface, and quantum confinement.12–14 It has been widely used in oxidation catalysts,15 gas sensors,16 conducting electrodes17 and opto-electronic devices.18 The electronic structure of tin oxide changes in accordance with doping, the influence of surfactants and thermal annealing processes.19 Also, the low electrical resistance of tin oxide favours better electrical conduction in the process of charge/electron/ion transportation.20 SnO2 has been considered as the most potent sensor material as far as sensitivity, selectivity and response are concerned.21,22 The doping of nickel according to its size, oxidation state and concentration in tin oxide alters the sensitivity and selectivity, and nickel ions can easily occupy the tin sites because of their close ionic radii: Ni2+ = 69 nm and Sn2+ = 71 nm. Furthermore, the doping level decreases the electron density and increases the oxygen vacancies, which elevates the sensitivity greatly.23,24 In the past, the specific area of SnO2 was found to increase upon the addition of additives such as Pt, Pd, Co, Fe and Ni. Of these additives, nickel has been widely employed to enhance the sensing performance of tin oxide sensors for various target gases.25
In this work, nickel-doped tin oxide (Ni-SnO2) nanoparticles were successfully synthesized using a microwave-assisted wet chemical method. The effects of Ni doping on the structural, morphological and sensing properties of the SnO2 nanoparticles with different concentrations were analysed and their sensor performance was studied and reported. The Ni-SnO2 sensor showed the highest sensitivity and response for CO2 gas, where the sensitivity increased with the concentration of Ni doping because the electron density decreased. On the other hand, the tested humidity performance also showed an increasing trend. From the results for the optimization of the sensor, the nickel-doped tin oxide sensor was found to be an appropriate material for carbon dioxide gas detection. The results for the gas sensor are compared to recently reported results, as shown in Table 1. The literature survey concludes that the present sensor demonstrates high sensitivity and rapid detection.
Table 1 Comparison of the Ni-SnO2 sensor with some recently reported CO2 sensors
| S. no. |
Materials |
Gas |
Gas concentration |
Operating temperature |
Response |
Sensitivity |
Ref. |
| 1 |
BaTiO3 |
CO2 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm 000 ppm |
550 °C |
— |
1.25 |
1 |
| 2 |
Ca-ZnO |
CO2 |
5000 ppm |
300 °C |
120 s |
10 |
6 |
| 3 |
CoAl2O4 |
CO2 |
100 ppm |
400 °C |
46 s |
0.43 |
62 |
| 4 |
SnO2 |
CO2 |
5000 ppm |
279 °C |
180 s |
48 |
63 |
| 5 |
La2O3 |
CO2 |
350 ppm |
321 °C |
105 s |
65 |
2 |
| 6 |
Pd-La2O3 |
CO2 |
500 ppm |
250 °C |
50 s |
28 |
64 |
| 7 |
Ni-SnO2 |
CO2 |
100 ppm |
275 °C |
04 s |
73.29 |
Present work |
2. Experimental method
Pristine SnO2 and Ni-SnO2 nanoparticle sensors were synthesized via a microwave-irradiation wet chemical method. Tin chloride (II, 99.995%) and nickel chloride (II, 99.995%) analytical grade salts were chosen for preparing the precursor solution in de-ionized water as a solvent. The as-obtained precursor solution was stirred for 2 h with each precursor solution of unique color. The nickel precursor solution (green) and the tin precursor solution (milky white) were mixed together and the solution revealed a slight change in the color, which was again stirred for 2 h. Once the solution reached pH 11, the addition of NaOH was stopped. The NaOH solvent acted as a precipitating agent, forming nanoparticles via nucleation. The solution was rigorously cleaned and used to extract the impurities and inorganic contaminants if there were any. The solution was rigorously dried in a microwave oven for 30 min. During microwave irradiation, a reaction took place, with the solution turning milky white and then, a black powder product was obtained. The dried powder was prepared via ball/physical milling for 2 h and air-annealed at 400 °C for 4 h.
2.1 Measurement details
The structure analysis of pristine SnO2 and Ni-SnO2 NPs was carried out using a PAN analytical X'Pert PRO powder X-ray diffractometer. The surface morphology was observed through scanning electron microscopy (Raith 150TWO Electron beam lithography system) and scanning transmission electron microscopy (FEI 200kV Titan Themis S-TEM). The Fourier transform infrared (FTIR) band transition was observed using an FT/IR-4600 model. For the optical properties, JASCO V-770 and JASCO FP8300 fluorescence spectrophotometers were utilized. The binding energies were obtained from the X-ray photoelectron spectroscopy (Kratos AXIS Nova) measurements. Humidity measurements were performed by placing the Ni-SnO2 sensor in a thermo-stabilized box consisting of a computerized humidity sprayer and a fan, allowing different levels of humidity. The gas influence measurements were performed through a controlled temperature box that allowed insertion of the test gases on demand.
3. Results and discussion
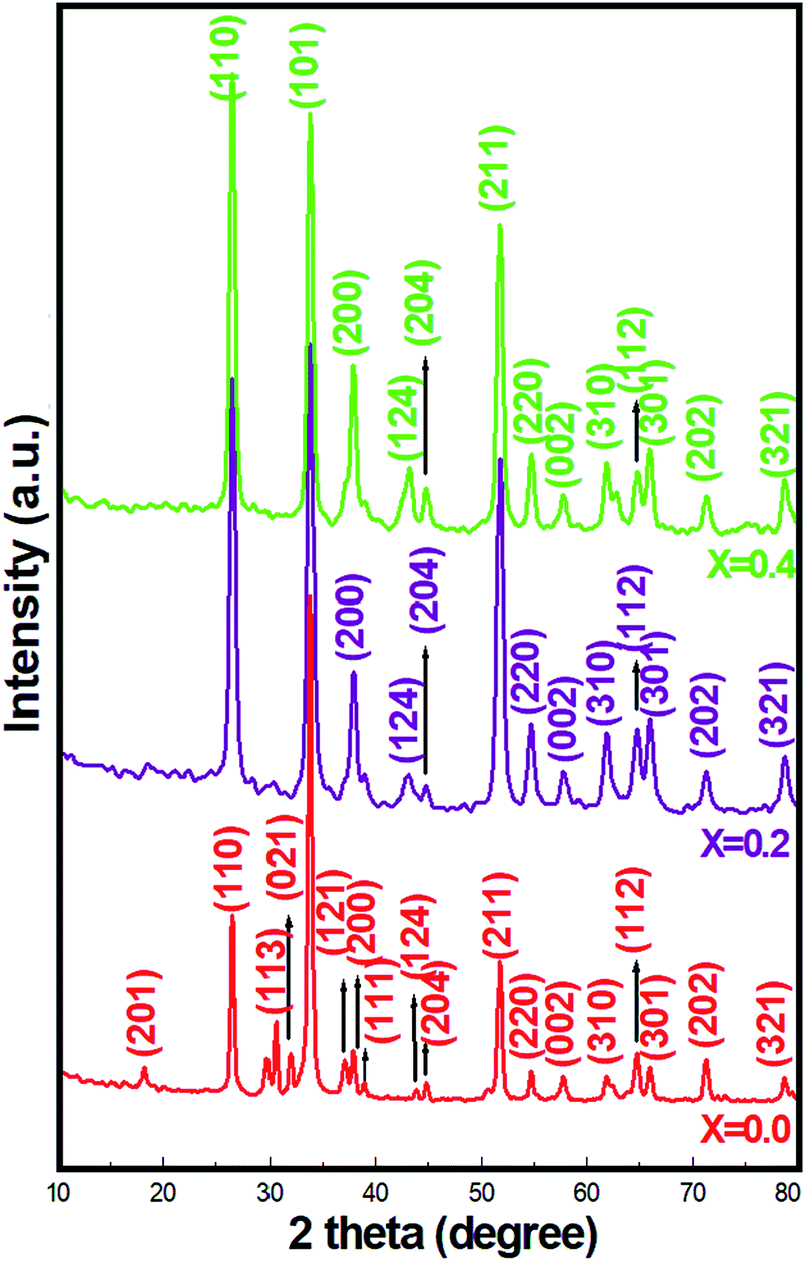
Fig. 1 presents the XRD patterns of pristine SnO2 and Ni-SnO2 NPs obtained at different concentrations of Ni. The emissions of all the reflection planes were attributed to the tetragonal rutile structure. The diffraction angles showed small deviation only after Ni doping, suggesting that the tetragonal rutile structure of SnO2 could not be rebuilt after Ni doping. NPs presented the (110), (101), (200), (211), (220), (310) and (301) prime reflection peaks, which were free from impurity peaks.21 The average ∼32 (±5) nm particle size, calculated using the Scherrer formula, did not substantially change as nickel doping intercepted the particle growth and caused lattice distortion, which increased the oxygen vacancies.26 Also, the full width at half maximum values of SnO2 increased with the Ni-doping level as there was a charge imbalance between tin and nickel ions, which occurred via electron holes during the air-annealing process, and this, in general, causes oxygen vacancies in materials.20,27 Thus, the particle size decreased. Ni doping caused a minor lattice distortion with the introduction of microstrain as by virtue of their similar ionic radii, the Ni (125 pm) ions can easily occupy the Sn (118 pm) sites.23 Overall, Ni-SnO2 NPs remained as a tetragonal rutile phase structure. In addition to this, the decreased d-spacing after Ni doping was due to the slight difference in the ionic radii, suggesting the deviation of the diffraction angles at the relatively lower side and the incorporation of Ni in the SnO2 lattice. The particle sizes are listed in Table 2.
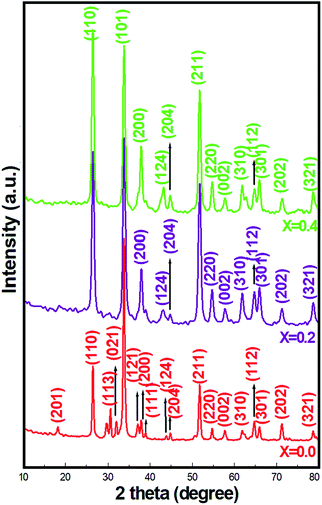 |
| | Fig. 1 XRD patterns of pristine SnO2 and Ni-SnO2 NPs. | |
Table 2 XRD and sensing parameters of pristine and nickel-doped tin oxide nanoparticles
| Material Ni-SnO2 |
CO2 gas sensor |
Humidity sensor |
| Concentration |
Particle size (nm) |
Res. time (s) |
Rec. time (s) |
Sensitivity (Rg/Ra) |
Res. time (s) |
Rec. time (s) |
Sensitivity |
| SnO2 (x = 0.0) |
46.70 |
29 |
40 |
62.31 |
41 |
68 |
81.98 |
| Ni-SnO2 (x = 0.2) |
32.17 |
21 |
24 |
71.53 |
49 |
71 |
78.11 |
| Ni-SnO2 (x = 0.4) |
42.32 |
04 |
13 |
73.29 |
54 |
84 |
72.03 |
Fig. 2a–c highlight the SEM images of the pristine SnO2 and Ni-SnO2 surfaces at different Ni-doping levels. The surface of pristine SnO2 is like brick stone; after Ni doping, it changes to a cauliflower type. This indicated that the morphology of the Ni-doped SnO2 NPs significantly changed. Pristine nanoparticles had no interesting morphology, but the defined shape of a cauliflower structure with a fine gap was observed due to the addition of nickel, which enabled the target gas to simply penetrate through the fine gap, yielding a high resistance change in sensor analysis. Fig. 3a–f show the STEM images of pristine SnO2 and Ni-SnO2 NPs, where the brick stone-like surface of high-surface-energy pristine sample is observed. The surface energy was reduced due to Ni doping and the particles showed a few needles in addition to chain formation. However, nickel acted as an energy barrier and this energy barrier reduced the surface energy, leading to good crystallization. The selected area electron diffraction patterns of the Ni-SnO2 nanoparticles afford a continuous ring pattern without additional diffraction spots, suggesting the nanocrystalline feature of Ni-SnO2.
 |
| | Fig. 2 (a) SEM images of pristine SnO2 and (b and c) Ni-SnO2 NPs. | |
 |
| | Fig. 3 (a and b) STEM images of pristine SnO2 and (c–f) Ni-SnO2 NPs. | |
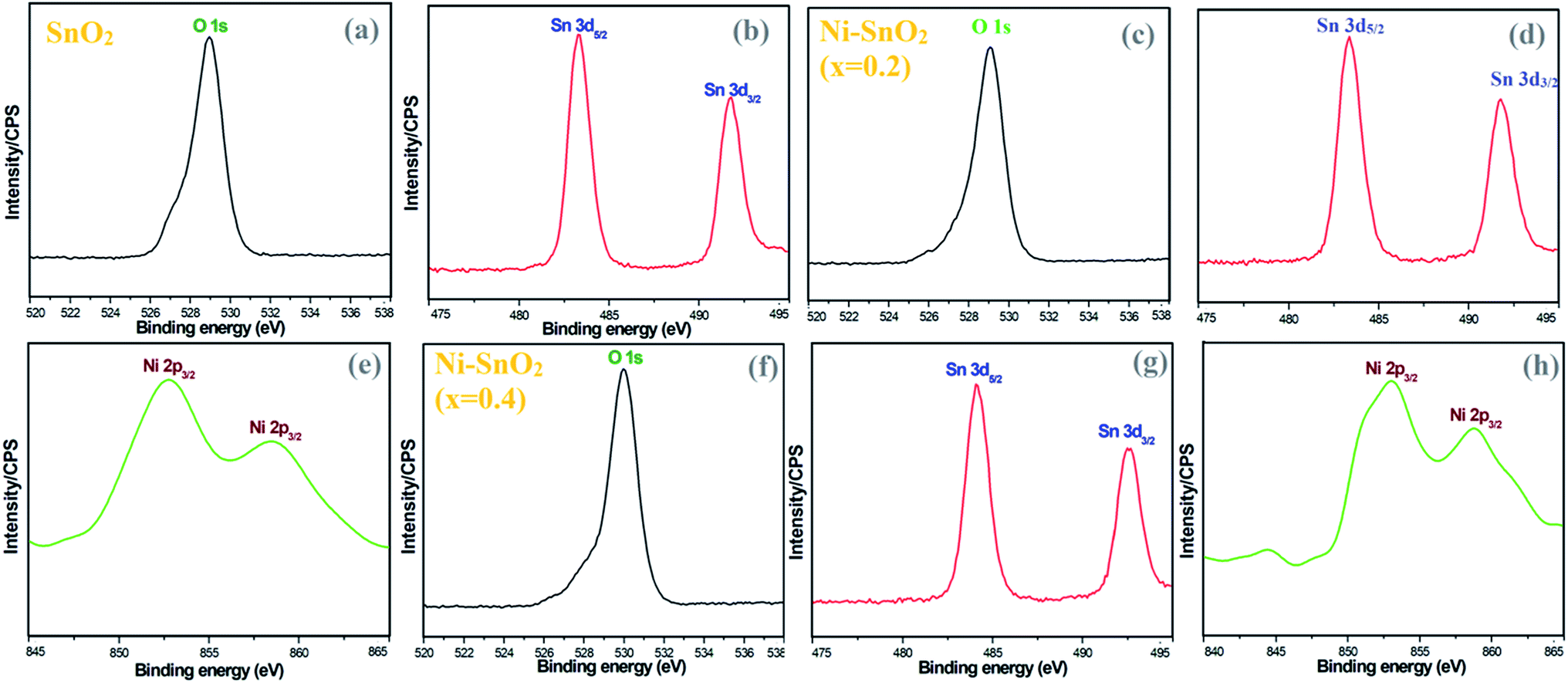
To verify the presence of chemical elements, XPS analysis was carried out for pristine SnO2 and Ni-SnO2 NPs. The binding energies of the O 1s peak of oxygen are centred at 528.94, 529.07 and 529.96 eV (Fig. 4). From the binding energies of O 1s, a shift in the position of a small peak due to the doping of nickel ions was confirmed.28,29 The increase in binding energy is associated with OH in the oxygen-deficient regions.30 Also, it causes O2− adsorption on the surface of the sensor and increases oxygen adsorption as a result of nickel doping, which would eventually enhance the gas-sensing performance.30,31 In Fig. 4, the XPS spectrum of tin shows similar peaks. The binding energies of Sn in pristine SnO2 NPs were compared with the binding energies of Ni-SnO2 NPs. From this comparison, the obtained binding energies of Ni-SnO2 showed an extremely small shift due to oxygen deficiencies caused by nickel doping.32,33 The binding energies of Ni 2p3/2 were 852.76 and 858.58 eV for a concentration of x = 0.2 and 853.03 and 858.81 eV for a concentration of x = 0.4 with a small peak shift. These results suggest that nickel ions can be doped into the tin lattice without structural deviation.34,35
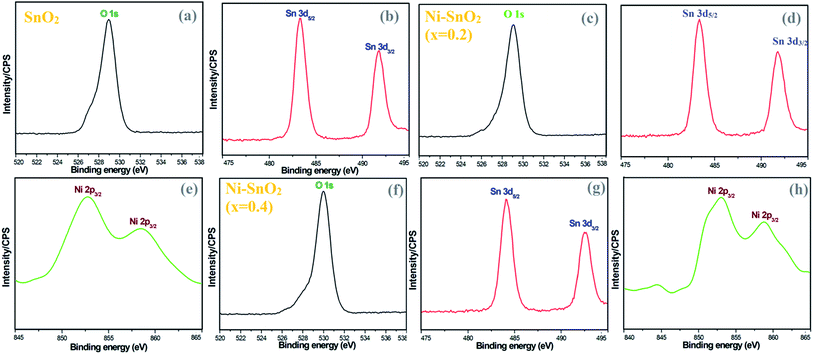 |
| | Fig. 4 (a and b) XPS spectra of pristine SnO2 and (c–h) Ni-SnO2 NPs. | |
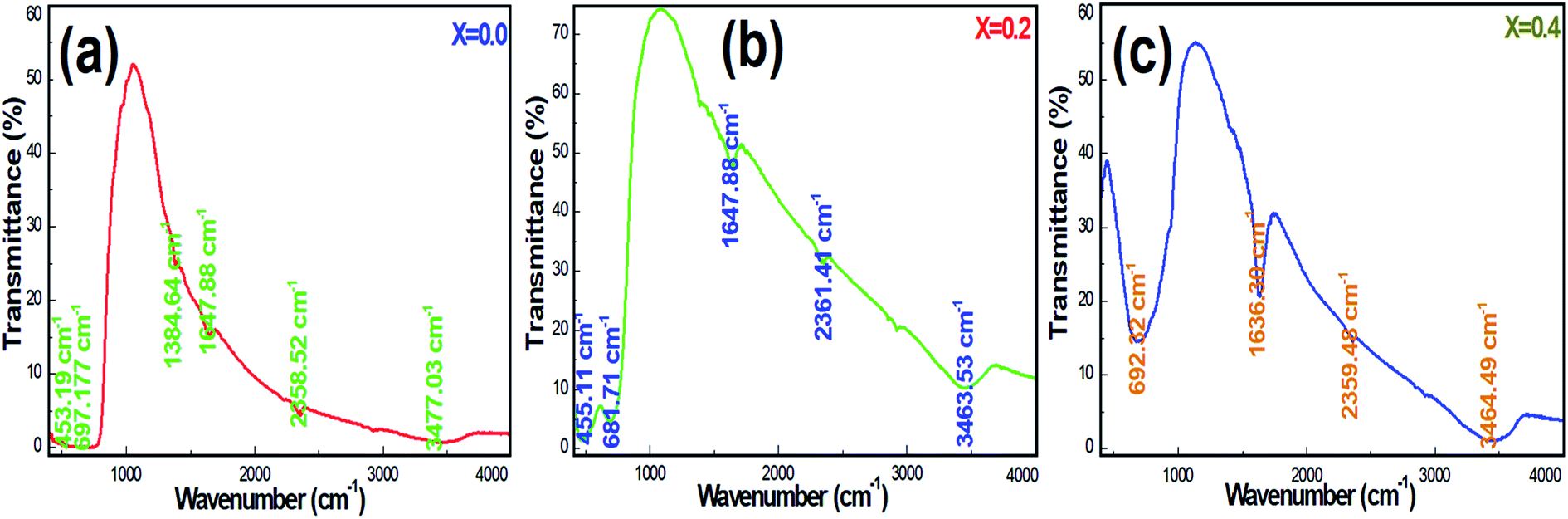
Fig. 5a–c display the FTIR transmittance spectra of Ni-SnO2 NPs containing different concentrations of nickel. The transmittance spectra of NPs present changes in the position and shape of the IR bands with respect to the concentration, suggesting the incorporation of nickel in the SnO2 lattice. The IR band at 450 cm−1 can be assigned to the O–Sn–O antisymmetric stretching vibrations and that at 693 cm−1 can be assigned to the symmetric stretching vibrations.36 The IR band at around 1380–1647 cm−1 is due to the C–O bonds.37 The O–H stretching of water molecules contributed to bands at 2558–3465 cm−1.38 Pristine SnO2 NPs contributed to IR bands and these were compared to that of Ni-SnO2, where a shift in the IR band as a result of the particle size and shape change was evidenced by nickel substitution. Due to the variation in bond length on nickel doping, the band shift extended the position of the IR band towards a higher wavenumber. The intensities of the IR bands increased due to the increase in the nickel doping level, which decreased the particle size due to the high surface area and the continuous adsorption of water molecules from the atmosphere.39 In all the samples, the IR bands related to the O–H bonds suggested the presence of active sites, fast adsorption and surface modification.
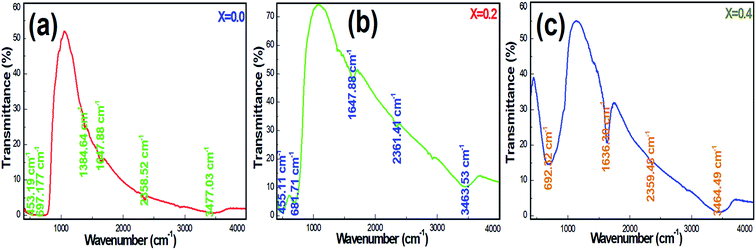 |
| | Fig. 5 (a–c) FT-IR spectra of pristine and Ni-SnO2 NPs. | |
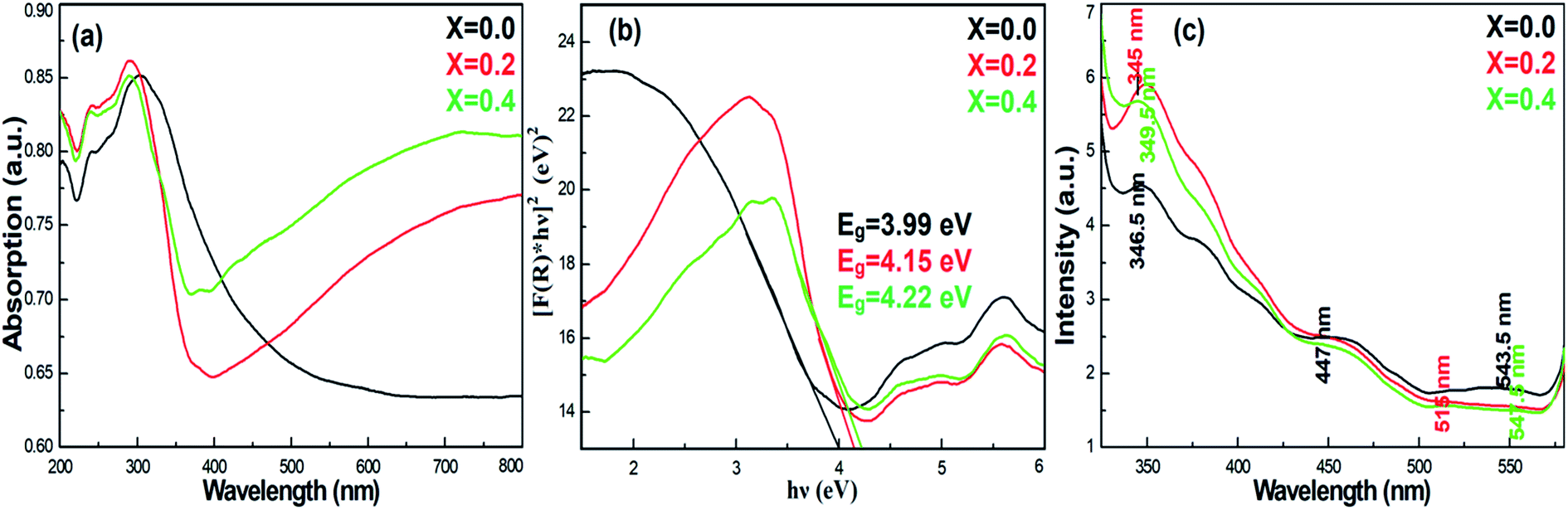
To understand the impact of nickel doping on the optical properties of SnO2 at various concentrations, the UV-Vis absorption and photoluminescence spectra were recorded (Fig. 6a). Upon nickel doping, the absorption band of SnO2 was deduced. In particular, due to photo-excitation, the absorption band at 300 nm shifted to 291 nm for Ni-SnO2.40 Also, the absorption spectra showed an ultra-violet cut-off around 275–325 nm. With photo-excitation, the energy of the atoms was lifted to a certain level by the absorption of photons of a particular wavelength. In this case, the energy of the atoms decreased due to Ni doping as the excited electrons attained transition from the valence band to the conduction band. Usually, the absorption spectrum of wide-band materials depends on oxygen deficiencies, surface roughness and impurities. In addition, the absorption decreased with the increase in the nickel concentration. The optical energy band gap obtained from the diffuse reflectance spectra using the Kubelka–Munk function F(R) is as follows:41
| |
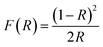 | (1) |
Here,
R describes the percentage of reflectance. The optical energy band gaps of the Ni-SnO
2 sensors were determined by [
F(
R) ×
hν]
2 versus the incident photon energy and the extrapolated linear part of [
F(
R) ×
hν]
2 to 0. The band gap energies of all the Ni-SnO
2 sensors are shown in
Fig. 6b. The band gap energy of SnO
2 increased on Ni doping from 3.99 to 4.22 eV, which could be due to the particle size change.
20 The charge transition between nickel and tin and the modification of the electronic structure could lead to an increase in the band gap energy of SnO
2.
30,42 Photoluminescence is a powerful tool to explore the optical properties of NPs.
Fig. 6c presents the photoluminescence spectra of pristine SnO
2 and Ni-SnO
2 NPs. The excitation wavelength of each spectrum was 300 nm. From
Fig. 6c, a strong emission at 345 nm and weak emissions at 447, 515 and 547 nm are noted. The strong emission at ∼345 nm was due to the recombination of the band-to-acceptor and donor–acceptor pairs and a direct recombination of the conduction electrons in the 4p band and holes in the oxygen 2p valence band.
43 The emission at 477 nm is weak due to the electron energy transition from tin vacancies to interstitial sites, the so-called surface defects. The weak emissions at 515 and 547 nm belonged to green emissions originating from the oxygen vacancies. Also, the emissions of all NPs were in the visible region. The intensity of the photoluminescence spectra increased due to the increase in the nickel concentration and it was directly related to the oxygen vacancies.
30 Furthermore, the oxygen vacancies increased due to the increase in the intensity of the spectra. In particular, the intensity of the photoluminescence spectra was high for nickel at a concentration of
x = 0.2, which was due to the charge transition between the tin and nickel atoms.
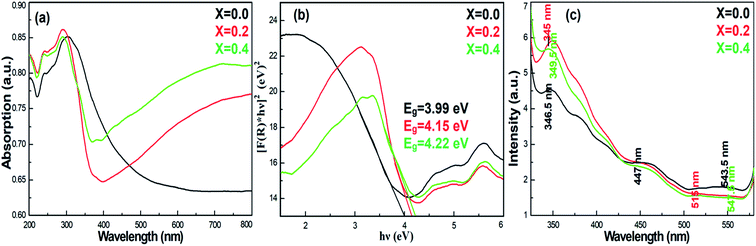 |
| | Fig. 6 (a) Absorption, (b) band gap energy and (c) photoluminescence spectra of pristine SnO2 and Ni-SnO2 NPs. | |
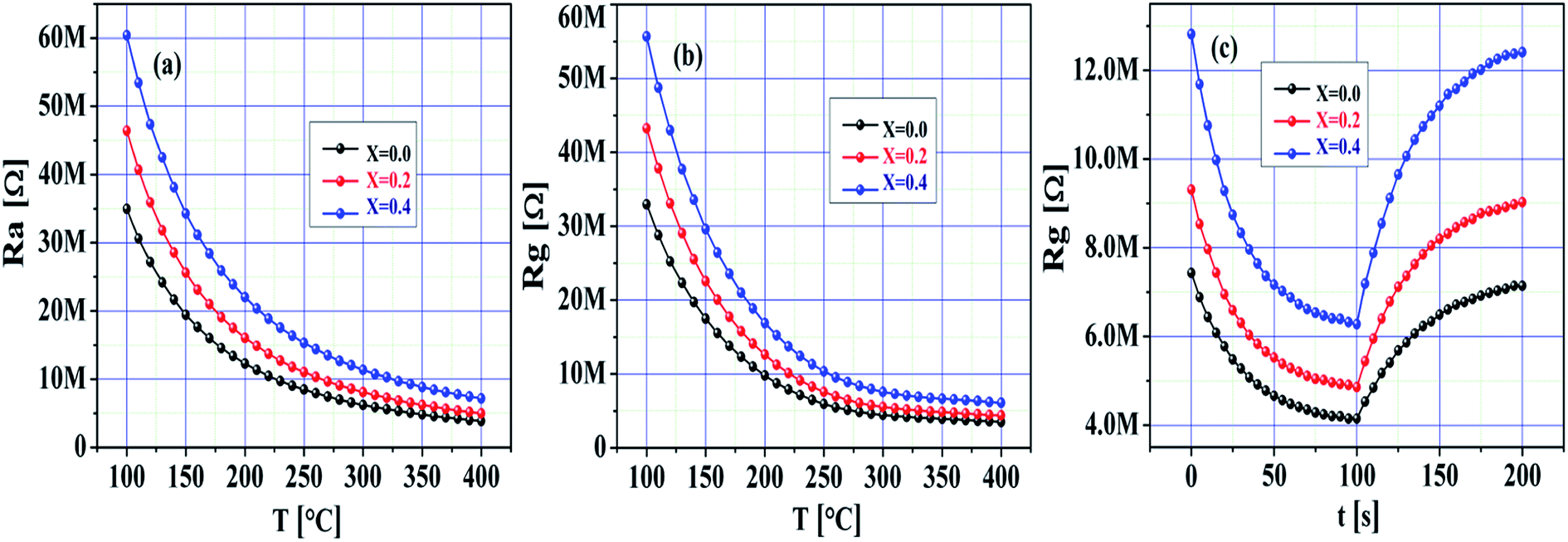
Fig. 7a–c demonstrate the quick response of the SnO2 and Ni-SnO2 sensors for CO2 gas from 0 to 200 ppm. To gain a first insight into the electrical behaviour of the pristine SnO2 and Ni-SnO2 sensors, the resistance change of the sensor with respect to various operating temperatures, i.e., 100–400 °C was measured in dry air. An increase in resistance with respect to the operating temperature was observed in all sensors, which was adopted for stabilizing the resistance (Ra).1,44 The decrease in the sensor resistance (Rg) of the Ni-SnO2 sensor on exposure to CO2 was evidenced. Both the resistance values, i.e., Ra and Rg increased with the Ni-doping level. However, the sensitivity was very resistive due to the decrease in conductivity. Also, the resistance values of the sensor in air and gas decreased with the increase in the operating temperature as the band gap of the sensor increased with the increase in nickel concentration; this suggested that electrons get sufficient thermal energy to excite from the valence band to the conduction band, thus continuously decreasing the resistance.45 The sensor confirmed rapid detection of the gas with an ultra-fast response time (Fig. 7c). The response times of the pristine SnO2 and Ni-SnO2 sensors were 29, 21 and 04 s, whereas the recovery time values were 40, 24 and 13 s. Both the response time and recovery time decreased with the increase in the Ni-doping level, which could be the result of a faster reaction between CO2 and the chemisorbed oxygen molecules.46
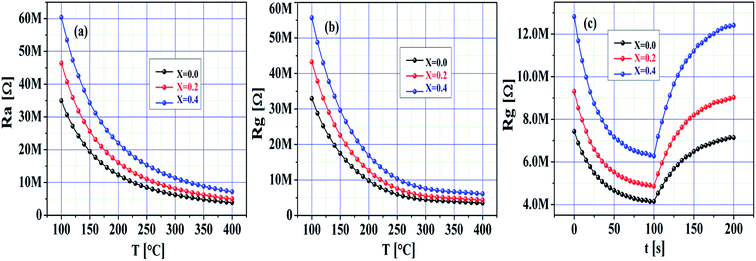 |
| | Fig. 7 (a and b) Resistance of pristine SnO2 and Ni-SnO2 sensors in air and gas versus temperatures. (c) Dynamic response of pristine SnO2 and Ni-SnO2 sensors. | |
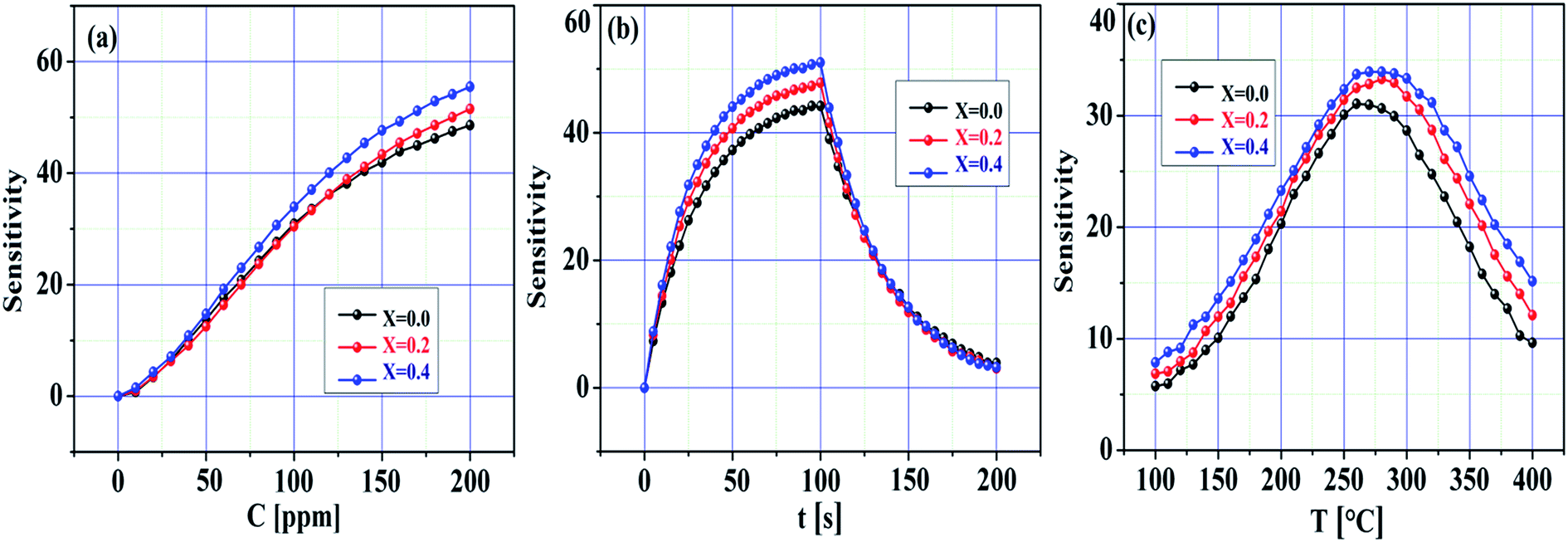
Fig. 8a–c demonstrate the sensitivity of the pristine SnO2 and Ni-SnO2 sensors obtained at various Ni-doping levels versus the concentration of the CO2 gas, time and operating temperature. The sensitivity of the pristine SnO2 and Ni-SnO2 sensors increased with the increase in the CO2 gas concentration. Here, oxygen vacancies play a leading role in sensitivity estimation as they increase with the increase in gas concentration, which increases the sensitivity.47,48 Fig. 8b demonstrates the sensitivity curves with respect to time. From this figure, it can be seen that the sensitivity of the Ni-SnO2 sensors increases with the nickel-doping level, suggesting a moderate repeatability signature.13,49 The sensitivity of the pristine SnO2 and Ni-SnO2 sensors with the operating temperature increased and then decreased (Fig. 8c). Initially, the sensitivity increased up to 275 °C and then decreased, which depended on the adsorption and desorption of gas molecules on the sensor surface. In this case, the sensitivity might be controlled through adsorption from 100 to 275 °C, whereas desorption started from 275–400 °C. At a particular temperature, adsorption and desorption were equal and the equilibrium position was reached.2,50 In the present case, the optimum sensing temperature of the pristine SnO2 and Ni-SnO2 sensors for CO2 gas was 275 °C. The sensitivity varied from 62 to 73.29 and the maximum sensitivity with a 4 s response time was obtained when x = 0.4 for 100 ppm CO2 gas, which was comparable with other values reported recently (Table 1).
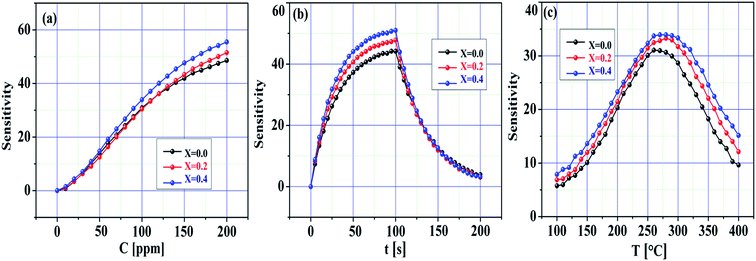 |
| | Fig. 8 Sensitivity change of pristine SnO2 and Ni-SnO2 sensors with (a) gas concentration, (b) time and (c) temperature. | |
The fabricated pristine SnO2 and Ni-SnO2 sensors were basically chemiresistive, where the sensor resistance was controlled by the CO2 gas molecules and the chemisorbed oxygen molecules. The free electrons from the sensor surface were arrested using the adsorbed oxygen molecules and then converted to oxygen anions, such as O2 or O−, as follows:51
| | |
O2(ads) + e− ↔ O2(ads)−
| (3) |
| | |
O2(ads)− + e− ↔ 2O(ads)−
| (4) |
The adsorption of oxygen molecules was influenced by the operating temperature. Also, the oxygen vacancies played a leading role at the chemisorption sites under the influence of oxygen. The concern oxygen molecules were adsorbed on the sensor surface by a chemisorption process. During adsorption, bonding occurred between the chemisorbed oxygen and the surface of the sensor. Also, weak bonding was observed at a low operating temperature with a low response. However, a strong bond produced at higher operating temperatures allows a higher response under the influence of a gas.52 Carbonate ions were formed during the exposure of the CO2 gas to the SnO2 and Ni-SnO2 sensors, and the adsorbed gas molecules could act as acceptors as per the following reaction:53
| | |
2CO2(gas) + 2O2(ads)− → 2CO3(gas)− + O2−
| (5) |
The resistance returned to its original value due to the removal of carbon dioxide gas, whereas the oxygen molecules (air) were adsorbed on the surface of the sensor again.54
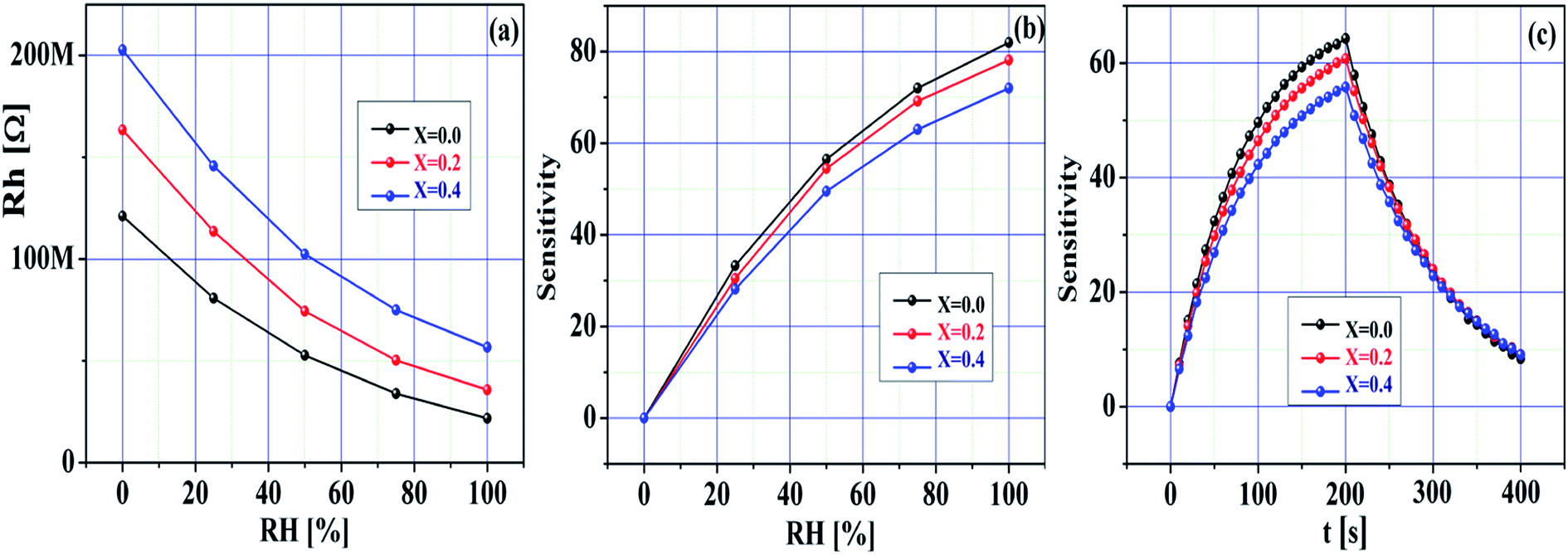
Fig. 9a–c demonstrate the humidity sensing response of the pristine SnO2 and Ni-SnO2 sensors measured at room temperature (27 °C). The humidity measurement was implemented in the range of 0–100% RH. The resistance of the sensor changed in accordance with the humidity (Fig. 7a). Here, the resistance to humidity increased with the Ni-doping level, suggesting that the sensor response occurred in a good manner with respect to humidity. Fig. 9b demonstrates the sensitivity of the sensor in accordance with the humidity. From this figure, the sensitivity of each sensor can be seen to have improved with the humidity. Here, the sensitivity of the pristine SnO2 sensor was high compared to that of the Ni-SnO2 sensor. Notably, the sensitivity of the sensor to humidity decreased after Ni doping with the increase in the elemental concentration (Ni); this can be used to support the use of the material as a gas sensor (CO2) because the sensitivity to gas increases with the increase in the elemental concentration (Ni). Also, their sensitivity followed a descending order with Ni doping. Usually, humidity changes the sensitivity of metal-oxide-based sensors. In this case, the carbon dioxide gas molecules adsorbed on the surface of the sensor previously, thus limiting the availability of the active sites for the adsorption of water molecules. As a consequence of the limited active sites, the sensitivity was reduced since the active sites are used for gas sensing.6,55 Although, the pristine SnO2 sensor surface could hold water vapour for a long duration during the exposure to humidity, which resulted in high sensitivity, the Ni-SnO2 sensor failed to hold water vapour for a long duration under humidity, thus reducing the sensitivity.56 On further increasing Ni doping, the surface of the sensor failed to hold water vapour again, thus decreasing the overall sensitivity.56 Fig. 9c demonstrates the response time and recovery time performance of the pristine SnO2 and Ni-SnO2 sensors, where a fast response and quite long recovery to humidity are noted. The response time and recovery time of the pristine and Ni-SnO2 sensors were 41, 49, 54 and 68, 71, 84 s, respectively, due to Ni doping. The response time for adsorption and the recovery time for desorption were relatively longer.57 Moreover, in each case, the response time was lower than the recovery time because of the fact that the adsorption could be faster than the desorption of adsorbed water molecules.58,59 The figure presents the repeatability curves for all the sensors, which signify their good sensing ability for humidity. The humidity sensing mechanism was based on a Grotthuss chain reaction. Humidity sensing was characterized by the change in resistance in accordance with the humidity. Initially, a few water vapour molecules could chemically adsorb on the surface of the sensor, yielding hydroxyl groups on the sensor surface.60 The surface hydroxyl groups underwent hydrogen bonding with the physisorption of water molecules; the primary adsorbed layer was then localized using hydrogen bonds to the hydroxyl group and hydronium ions were produced due to the proton transfer from the hydroxyl groups to the water molecules. More water molecules formed a liquid network similar to a chain-type network.61 Also, the hydrogen ions increased with the increase in the moisture content. Thus, the hydrogen ions easily moved to the liquid network, which produced a change in resistance in accordance with the variation in humidity.
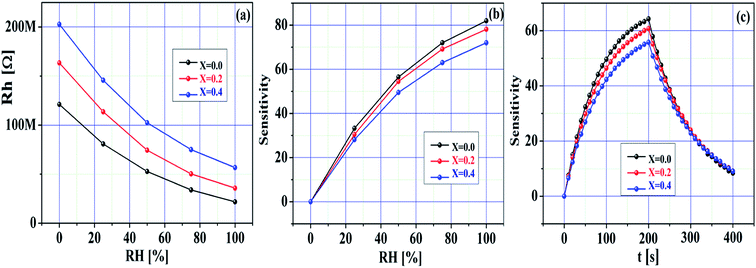 |
| | Fig. 9 (a) Resistance towards humidity, (b) sensitivity towards RH (%), (c) sensitivity towards time of pristine SnO2 and Ni-SnO2 NPs. | |
In summary, the fabricated sensor played a dual role in gas and humidity sensing. In accordance with sensitivity, the sensor was favourable for gas sensing as the sensitivity increased with the increase in the Ni-doping level, whereas it decreased under humidity tests.
4. Conclusion
Pristine SnO2 and Ni-SnO2 sensors were successfully prepared using a simple microwave-assisted wet chemical method. This method resulted in a tiny cauliflower-type surface of the sensor. XRD analysis confirmed a particle size of 32–42 nm and chain of particles, as seen in the STEM measurement. XPS analysis confirmed the purity of samples and oxygen deficiencies. In the sensing studies, the sensitivity was enhanced with respect to the concentration and the optimum sensing temperature was 275 °C. The pristine SnO2 sensor showed a high response for humidity, which decreased with the increase in the Ni-doping level. The fabricated sensor worked for CO2 gas as well as humidity. From the sensing results, the Ni-SnO2 sensors are promising materials for effective CO2 sensing applications as the Ni-SnO2 sensors exhibit high sensitivity with rapid detection.
Conflicts of interest
The authors declare there are no conflicts of interest.
Acknowledgements
The authors thank De Ming Zhu for his assistance in the XPS analysis.
References
- P. Keller, Sens. Actuators, B, 1999, 39–46 CrossRef CAS.
- A. A. Yadav, A. C. Lokhande, J. H. Kim and C. D. Lokhande, J. Ind. Eng. Chem., 2017, 49, 76–81 CrossRef CAS.
- N. B. Tanvir, O. Yurchenko and G. Urban, Procedia Eng., 2015, 120, 667–670 CrossRef CAS.
- C. Willa, A. Schmid, D. Briand, J. Yuan and D. Koziej, ACS Appl. Mater. Interfaces, 2017, 9, 25553–25558 CrossRef CAS PubMed.
- J. D. Shakun, P. U. Clark, F. He, S. A. Marcott, A. C. Mix, Z. Liu, B. Otto-Bliesner, A. Schmittner and E. Bard, Nature, 2012, 484, 49–54 CrossRef CAS.
- R. Dhahri, S. G. Leonardi, M. Hjiri, L. El Mir, A. Bonavita, N. Donato, D. Iannazzo and G. Neri, Sens. Actuators, B, 2017, 239, 36–44 CrossRef CAS.
- P. Puligundla, J. Jung and S. Ko, Food Control, 2012, 25, 328–333 CrossRef CAS.
- S. Wiegärtner, G. Hagen, J. Kita, R. Moos, M. Seufert, E. Glaser, K. Grimmel, A. Bolz, C. Schmaus and A. Kießig, IEEE Sensors Proceedings, 2011, 1014–1016 Search PubMed.
- F. Tudorache, I. Petrila, S. Condurache-Bota, C. Constantinescu and M. Praisler, Superlattices Microstruct., 2015, 77, 276–285 CrossRef CAS.
- J. Shah, R. K. Kotnala, B. Singh and H. Kishan, Sens. Actuators, B, 2007, 128, 306–311 CrossRef CAS.
- F. J. Arregui, Sensors Based Nanostructured Mater, 2009, pp. 1–317 Search PubMed.
- F. H. Aragón, J. A. H. Coaquira, P. Hidalgo, S. W. Da Silva, S. L. M. Brito, D. Gouvêa and P. C. Morais, J. Raman Spectrosc., 2011, 42, 1081–1086 CrossRef.
- V. Manikandan, X. Li, R. S. Mane and J. Chandrasekaran, J. Electron. Mater., 2018, 47, 3403–3408 CrossRef CAS.
- M. M. Rahman, A. Jamal, S. B. Khan and M. Faisal, Biosens. Bioelectron., 2011, 28, 127–134 CrossRef CAS.
- K. Sekizawa, H. Widjaja, S. Maeda, Y. Ozawa and K. Eguchi, Appl. Catal., A, 2000, 200, 211–217 CrossRef CAS.
- N. Sergent, P. Gélin, L. Périer-Camby, H. Praliaud and G. Thomas, Sens. Actuators, B, 2002, 84, 176–188 CrossRef CAS.
- T. T. Emons, J. Li and L. F. Nazar, J. Am. Chem. Soc., 2002, 124, 8516–8517 CrossRef CAS PubMed.
- F. Gu, S. F. Wang, M. K. Lü, Y. X. Qi, G. J. Zhou, D. Xu and D. R. Yuan, Opt. Mater., 2004, 25, 59–64 CrossRef CAS.
- A. S. Ahmed, M. Shafeeq M., M. L. Singla, S. Tabassum, A. H. Naqvi and A. Azam, J. Lumin., 2011, 131, 1–6 CrossRef CAS.
- A. Azam, A. S. Ahmed, M. S. Ansari, M. Shafeeq M and A. H. Naqvi, J. Alloys Compd., 2010, 506, 237–242 CrossRef CAS.
- K. P. Gattu, K. Ghule, A. A. Kashale, V. B. Patil, D. M. Phase, R. S. Mane, S. H. Han, R. Sharma and A. V. Ghule, RSC Adv., 2015, 5, 72849–72856 RSC.
- Y. Chen, L. Yu, D. Feng, M. Zhuo, M. Zhang, E. Zhang, Z. Xu, Q. Li and T. Wang, Sens. Actuators, B, 2012, 61–67 CrossRef.
- Q. Zhou, W. Chen, L. Xu, R. Kumar, Y. Gui, Z. Zhao, C. Tang and S. Zhu, Ceram. Int., 2018, 44, 4392–4399 CrossRef CAS.
- W. T. Li, X. D. Zhang and X. Guo, Sens. Actuators, B, 2017, 244, 509–521 CrossRef CAS.
- S. Singkammo, A. Wisitsoraat, C. Sriprachuabwong, A. Tuantranont, S. Phanichphant and C. Liewhiran, ACS Appl. Mater. Interfaces, 2015, 7, 3077–3092 CrossRef CAS PubMed.
- Z. Lin, N. Li, Z. Chen and P. Fu, Sens. Actuators, B, 2017, 239, 501–510 CrossRef CAS.
- N. Lavanya, S. Radhakrishnan and C. Sekar, Biosens. Bioelectron., 2012, 36, 41–47 CrossRef CAS PubMed.
- W. Xia, H. Wang, X. Zeng, J. Han, J. Zhu, M. Zhou and S. Wu, CrystEngComm, 2014, 16, 6841–6847 RSC.
- H. Chen, L. Ding, W. Sun, Q. Jiang, J. Hu and J. Li, RSC Adv., 2015, 5, 56401–56409 RSC.
- K. Srinivas, S. Manjunath and P. V. Reddy, Structural, Nanaoscale, 2011, 3, 642–653 RSC.
- Y. Zheng, J. Wang and P. Yao, Sens. Actuators, B, 2011, 156, 723–730 CrossRef CAS.
- T. Jia, W. Wang, F. Long, Z. Fu, H. Wang and Q. Zhang, J. Phys. Chem. C, 2009, 21, 9071–9077 CrossRef.
- F. Zhang, Y. Lian, M. Gu, J. Yu and T. B. Tang, J. Phys. Chem. C, 2017, 29, 16006–16011 CrossRef.
- V. Inderan, M. M. Arafat, S. Kumar, A. S. M. A. Haseeb, Z. Jiang, M. Altarawneh and H. L. Lee, Nanotechnology, 2017, 28, 265702 CrossRef.
- V. Manikandan, I. Petrila, S. Vigneselvan, R. Dharmavarapu, S. Juodkazis, S. Kavita and J. Chandrasekaran, J. Mater. Sci.: Mater. Electron., 2018, 29, 18660–18667 CrossRef CAS.
- J. Kaur, J. Shah, R. K. Kotnala and K. Chand, Ceram. Int., 2012, 38, 5563–5570 CrossRef CAS.
- T. Ahmad, S. Khatoon and K. Coolahan, J. Am. Ceram. Soc., 2016, 4, 1207–1211 CrossRef.
- Y. Xin and G. Jun, Inorg. Chem. Commun., 2003, 6, 882–885 CrossRef.
- D. Singh, V. Singh and A. S. Maan, J. Mol. Struct., 2015, 1100, 562–569 CrossRef CAS.
- P. P. Dorneanu, A. Airinei, M. Grigoras, N. Fifere, L. Sacarescu, N. Lupu and L. Stoleriu, J. Alloys Compd., 2016, 668, 65–72 CrossRef.
- S. Bhuvana, H. B. Ramalingam, G. Thilakavathi and K. Vadivel, Mater. Technol., 2016, 7857 Search PubMed.
- B. Nandan, B. Venugopal, S. Amirthapandian, B. K. Panigrahi and P. Thangadurai, J. Nanopart. Res., 2013, 15, 1999 CrossRef.
- K. Sakthiraj and K. Balachandrakumar, J. Magn. Magn. Mater., 2015, 395, 205–212 CrossRef CAS.
- A. Prim, E. Pellicer, E. Rossinyol, F. Peiró, A. Cornet and J. R. Morante, Adv. Funct. Mater., 2007, 17, 2957–2963 CrossRef CAS.
- Z. Li, D. Ding, Q. Liu and C. Ning, Sensors, 2013, 13, 8393–8402 CrossRef CAS PubMed.
- S. Basu, Y. H. Wang, C. Ghanshyam and P. Kapur, Bull. Mater. Sci., 2013, 36, 521–533 CrossRef CAS.
- J. H. Lee, Sens. Actuators, B, 2009, 140, 319–336 CrossRef CAS.
- G. Korotcenkov, V. Brinzari and B. K. Cho, Microchim. Acta, 2016, 183, 1033–1054 CrossRef CAS.
- V. Manikandan, M. Singh, B. C. Yadav and J. C. Denardin, Journal of Science: Advanced Materials and Devices, 2018, 3, 145–150 Search PubMed.
- V. Manikandan, J.-H. Kim, A. Mirzaei, S. S. Kim, S. Vigneselvan, M. Singh and J. Chandrasekaran, J. Mol. Struct., 2019, 1177, 485–490 CrossRef CAS.
- A. A. Yadav, V. C. Lokhande, R. N. Bulakhe and C. D. Lokhande, Microchim. Acta, 2017, 184, 3713–3720 CrossRef CAS.
- S. Choi, G. Ankonina, D. Youn, S. Oh and J. Hong, ACS Nano, 2009, 9, 2623–2631 CrossRef.
- A. Marsal, G. Dezanneau, A. Cornet and J. R. Morante, Sens. Actuators, B, 2003, 95, 266–270 CrossRef CAS.
- A. Marsal, A. Cornet and J. R. Morante, Sens. Actuators, B, 2003, 94, 324–329 CrossRef CAS.
- C. Wang, L. Yin, L. Zhang, D. Xiang and R. Gao, Sensors, 2010, 10, 2088–2106 CrossRef CAS.
- S. C. Nagaraju, A. S. Roy, J. B. P. Kumar, K. R. Anilkumar and G. Ramagopal, J. Eng., 2014, 925020 Search PubMed.
- C. L. Cao, C. G. Hu, L. Fang, S. X. Wang, Y. S. Tian and C. Y. Pan, J. Nanomater., 2011, 707303 Search PubMed.
- T. Şaşmaz Kuru and E. Şentürk, Sens. Actuators, A, 2016, 249, 62–67 CrossRef.
- P. Li, X. Zheng, Y. Zhang, M. Yuan, B. Jiang and S. Deng, Ceram. Int., 2015, 41, 14251–14257 CrossRef CAS.
- Z. Jing-Li, L. Yue-Dong, W. Guo-Biao and L. Biao-Rong, Sens. Actuators, A, 1991, 29, 43–47 CrossRef.
- A. Cavalieri, T. Caronna, I. Natali Sora and J. M. Tulliani, Ceram. Int., 2012, 38, 2865–2872 CrossRef CAS.
- C. R. Michel, Sens. Actuators, B, 2010, 147, 635–641 CrossRef CAS.
- U. Hoefer, G. Kühner, W. Schweizer, G. Sulz and K. Steiner, Sens. Actuators, B, 1994, 22, 115–119 CrossRef CAS.
- A. A. Yadav, A. C. Lokhande, J. H. Kim and C. D. Lokhande, RSC Adv., 2016, 6, 106074–106080 RSC.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Iulian Petrilab,
S. Vigneselvanc,
R. S. Mane
*a,
Iulian Petrilab,
S. Vigneselvanc,
R. S. Mane