
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combustion of lean methane over Co3O4 catalysts prepared with different cobalt precursors†
Yifan Zhengab,
Yueqin Yuab,
Huan Zhoub,
Wanzhen Huangb and
Zhiying Pu *ab
*ab
aCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China
bResearch Center of Analysis and Measurement, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: zhiyingpu@zjut.edu.cn
First published on 28th January 2020
Abstract
To investigate the effect of catalyst precursors on physicochemical properties and activity of lean methane catalytic combustion, a series of Co3O4 catalysts were prepared via a precipitation method by using four different cobalt precursors: Co(C2H3O2)2, Co(NO3)2, CoCl2, and CoSO4. The catalysts were characterized by BET, XRD, SEM, Raman, XPS, XRF, O2-TPD and H2-TPR techniques. It was found that the different types of cobalt precursor had remarkable effects on the surface area, particle size, reducibility and catalytic performance. In contrast, the Co3O4-Ac catalyst showed a relatively small surface area, but its activity and stability were the highest. XPS, Raman, O2-TPD and H2-TPR results demonstrated that the superior catalytic performance of Co3O4-Ac was associated with its higher Co2+ concentration, more surface active oxygen species and better reducibility. In addition, the activity of the Co3O4-S catalyst reduced significantly due to the residual impurity SO42−, which could reduce the concentration of surface adsorbed active oxygen species and inhibit oxygen migration.
Introduction
Nowadays, there has been considerable attention focused on energy shortage and environmental protection issues. Recent decades have witnessed increasing requirements of natural gas for many fields owing to its abundant reserves.1–3 Nevertheless, the exhaust emission directly releases into the atmosphere because of the incomplete combustion of natural gas, which is a great threat to the environment. Methane, the main component of natural gas, is regarded as the second most damaging greenhouse gas, contributing approximately 20 times more to global warming than carbon dioxide.4,5 However, it is difficult for methane to be oxidized due to the strongest C–H bond among hydrocarbons.6 The main ways to perform complete oxidation of methane are flame combustion and catalytic combustion. But higher reaction temperature, emission of unburned hydrocarbons and more polluting environmental pollutants (NOx), as well as lower energy efficiency represent serious disadvantages of flame combustion.7 Catalytic combustion is considered to be one of the most efficient and promising technologies for the removal of methane due to its low operating temperature and high efficiency.8,9 Thus, it is crucial to develop a catalyst with outstanding catalytic ignition activity and high thermal stability for complete methane combustion.Supported noble metals catalysts, especially Pd and Pt, have been widely used in complete combustion of methane due to their superior performance.10–13 However, the inherent high cost and inferior anti-sintering of noble metals limit their extensive application in the commerce, motivating the investigation of alternative transition metal (Co, Mn, Fe, Cr, Cu, etc.) oxides catalysts.14–17 Among these candidate metal oxides, cobalt-based oxides are well known because of the structured and electronic properties of spinel type oxides with variable valence states (Co2+/Co3+) as well as the lower bonding energy of Co–O bonds, which exhibit high activity for catalytic combustion of lean methane.18 Therefore, the cobalt-based catalysts have extensive applications in sensors,19 lithium-ion batteries,20 magnetic materials,21 pigments22 and solar cells,23 which also show high activity for the oxidation of CH4, CO and VOCs due to their high redox ability and strong oxygen mobility.24,25
It is well known that the catalytic performance of catalysts is closely related to its structure and properties, which strongly depends on the catalyst synthesis method and the types of catalyst precursors.26,27 As reported by Li et al., SiO2-supported Co catalysts derived from Co(OH)2 and Co(NO3)2 exhibited higher reducibility and CO conversion and C5+ selectivity than those from CoCl2 and Co(C2H3O2)2.28 Jean-Sébastien Girardon and coworkers revealed that the catalyst derived from Co(NO3)2 showed higher reducibility than from Co(C2H3O2)2 and their catalytic activity in FT synthesis depended on the concentration of cobalt metal sites.29 However, only few pioneering studies have been reported to explore the influence of different cobalt precursors on the catalytic combustion of methane over Co3O4 catalysts.
In this work, a series of Co3O4 catalysts were synthesized by a precipitation method with different cobalt precursors. Multiple techniques including BET, XRD, SEM, Raman, XPS, XRF, O2-TPD and H2-TPR were applied to characterize the physical and chemical properties. The specific objectives were to evaluate the effect of cobalt precursors for methane combustion.
Experimental
Catalyst preparation
A series of Co3O4 catalysts were prepared by a precipitation method previous published.30,31 All chemicals were in analytical grade and used as received without purification. With the preparation of a Co3O4 sample by precipitating Co(C2H3O2)2 solution as an example here, Na2CO3 aqueous solution (1 mol L−1) was added drop by drop into Co(C2H3O2)2 solution (0.3 mol L−1), accompanied by continual magnetic stirring at 25 °C until the pH reached 9 since the Co3O4 catalyst prepared at pH = 9.0 showed the best catalytic activity according to our previous work.32 The precipitate was immediately vacuum-filtered and washed with deionized water. Afterwards, the precipitate was dried at 105 °C overnight. Finally, all the samples were calcined at 500 °C in static air for 4 h to get the final catalyst, which was named Co3O4-Ac. All the other Co3O4 samples were prepared basically as the same procedures by changing the following cobalt precursors: CoCl2, Co(NO3)2 and CoSO4. The achieved catalysts were denoted as Co3O4-Cl, Co3O4-N and Co3O4-S, respectively.Catalyst characterization
Nitrogen adsorption and desorption isotherms were measured on a micromeritics apparatus (ASAP 2020). All samples were outgassed under vacuum at 300 °C for 5 h prior to analysis. The surface areas of the samples were calculated using the Brunauer–Emmett–Teller (BET) method.Powder X-ray diffraction (XRD) patterns of the samples were recorded on PANalytical X'Pert PRO X-ray diffractometer using Cu Kα radiation (λ = 0.1541 nm) at 40 kV and 40 mA. The patterns were collected with the 2θ range from 10 to 80° at the step of 0.03°. The average crystallite sizes of the samples were calculated using the Scherrer equation based on the most intense hkl (3 1 1) diffraction peak of Co3O4.
Scanning electron microscopy (SEM) characterizations were performed on a Hitachi S-4700 instrument operated at 15 kV. The samples were covered with a thin layer (5 nm) of Pt by sputter coating before analysis.
Raman measurements were performed on a LABRAM-HR confocal laser Raman spectrometer using a 514 nm laser source, scanning from 100 to 800 nm. Before measurement, the powder samples were pressed to form disks with a homemade mold.
The content of impurity elements (S) in the investigated catalysts, in the form of pressed discs, was determined with the use of Wavelength-Dispersive XRF spectrometer (ThermoFisher Scientific, ADVANT'X 4200). The X-rays were generated with the Rh anode. For quantitative analysis, the calibration with a series of metallic standards and UniQuant software were used.
X-ray photoelectron spectroscopy (XPS) measurements were performed with a Kratos AXIS UItra DLD spectrometer using the Al X-ray source. The working voltage was 15 kV and the working current was 15 mA. The binding energies were calibrated using C 1s peak of contaminant carbon (284.8 eV) as standard. The spectra were performed with XPSPEAK software (ver. 4.1).
The O2 temperature programmed desorption (O2-TPD) was conducted on a chemisorption analyzer (AutoChem II 2920), 100 mg of sample was loaded in a quartz reactor, and cleaned in a He flow (30 mL min−1) at 200 °C for 30 min, followed by cooling down to 50 °C in the same flow. Afterward, a flow of 21% O2/N2 at a rate of 30 mL min−1 was passed through the sample for 1 h at 50 °C. Finally, the sample was heated to 420 °C at a rate of 10 °C min−1 for the desorption of the previous adsorbed oxygen in a flow of He (30 mL min−1).
The H2 temperature programmed reduction (H2-TPR) instrument equipped with a thermal conductivity detector (TCD) was used to investigate the reducibility of the samples. 30 mg of the samples was placed into a quartz tube, reduced in 5% H2–Ar gas mixture (40 mL min−1) raised to 700 °C with a heat rate of 10 °C min−1.
Catalytic performance evaluation
The activity test for methane combustion was conducted in a fixed-bed quartz tubular reactor (internal diameter = 6 mm) at atmospheric pressure, loaded with 200 mg of the catalyst sieved between 40 and 60 mesh. The temperature inside the reactor was measured by a multipoint K type thermocouple placed in the middle of the catalyst bed. The volume composition of the feed gas was 0.5% CH4, 8.0% O2 and 91.5% N2. The total flow rate of the feed gas is 30 mL min−1. The reactants and products were analysed on an Echrom A90 gas chromatograph equipped with hydrogen flame ionization detector (FID) and Ni catalyst convertor. The methane conversion was calculated from the inlet and outlet concentration of CH4. Arrhenius equation can be simplified since the composition of reactant gas remains essentially unchanged. Thus the activation energy (Ea, kJ mol−1) was evaluated according to the following equations:| r = N × X/W | (1) |
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) r = Ea/RT + C r = Ea/RT + C
| (2) |
Results and discussion
Structural and morphological properties
The BET surface areas of Co3O4 catalyst are listed in Table 1. The SBET of Co3O4-Ac, Co3O4-Cl, Co3O4-N and Co3O4-S are 15.4, 12.2, 25.2 and 25.6 m2 g−1, respectively. In contrast, the Co3O4-Ac and Co3O4-Cl exhibit relatively smaller surface areas than Co3O4-N and Co3O4-S. The results indicate that the cobalt precursors greatly affect the physical properties of these catalysts.| Catalysts | SBET (m2 g−1) | Crystallite size (nm) | I192/I684 |
|---|---|---|---|
| Co3O4-Ac | 15.4 | 46.8 | 0.18 |
| Co3O4-Cl | 12.2 | 57.0 | 0.16 |
| Co3O4-N | 25.2 | 39.7 | 0.16 |
| Co3O4-S | 25.6 | 35.9 | 0.17 |
XRD technique has been used to identify both the phase composition and the crystallite size of the Co3O4 samples prepared with different precursors, as displayed in Fig. 1. The peaks at 2θ = 19.0, 31.3, 36.8, 38.6, 44.8, 55.7, 59.4 and 65.2° are assigned to the (1 1 1), (2 2 0), (3 1 1), (2 2 2), (4 0 0), (4 2 2), (5 1 1) and (4 4 0) lattice planes of spinel Co3O4 oxide (ICDD: 01-078-1969).33 No any characteristic diffraction peaks for impurities are observed in the XRD patterns, illustrating the complete transition into Co3O4 or the impurities content is too low to be detected. The average crystallite size of the Co3O4 samples were estimated from XRD result using the Scherrer equation and listed in Table 1. The crystallite size of Co3O4-Ac and Co3O4-Cl catalysts are 46.8 and 57.0 nm, respectively, higher than that of the other two catalysts (39.7 and 35.9 nm), which is likely due to the precursors in the preparation process affecting the nucleation and growth rate of crystallite during precipitation. This is in good agreement with the BET results.
 | ||
| Fig. 1 XRD patterns of Co3O4 catalysts prepared with different cobalt precursors. | ||
The SEM images of the Co3O4 catalysts prepared with different cobalt precursors are presented in Fig. 2. Apparently, the catalyst particles have good dispersion and smooth surface with uniform spherical shape. There is almost no agglomeration occurred in Co3O4 catalysts and a large number of particles are generated in the size range 30 to 60 nm. It can be noticed that the Co3O4-Ac, Co3O4-Cl, Co3O4-N and Co3O4-S catalysts have the average size about 46, 53, 42 and 37 nm, respectively, measured and calculated by Nano Measurer software. That's to say, the Co3O4-Ac and Co3O4-Cl catalysts have relatively larger particle size than other samples. This is in accordance with the XRD and BET results, which further illustrates that the cobalt precursors highly influence the grain sizes and morphologies of Co3O4 catalysts.
 | ||
| Fig. 2 SEM images of Co3O4 catalysts prepared with different cobalt precursors. | ||
Surface analyses
Fig. 3 shows the Raman spectra of the Co3O4 catalysts. Five vibration modes at 192, 476, 516, 615 and 684 cm−1 are observed in the spectra at ambient conditions. The weak band located at 476 cm−1 can be assigned to the Eg symmetry, while the peaks at 516 and 615 cm−1 are assigned to the F2g symmetry. Furthermore, the band at 192 cm−1 is assigned to tetrahedral sites (CoO4), corresponding to F2g symmetry mode, whereas the band around 684 cm−1 is attributed to the characteristics of octahedral sites (CoO6) with A1g symmetry, respectively.34,35 The peak positions are in agreement with those reported for crystalline Co3O4,36 which further confirms the XRD results. According to the literature, Co3O4 is a transition metal oxide with a spinel structure in which the Co2+ ions occupy the tetrahedral sites while the Co3+ ions are situated at the octahedral sites due to the phonon symmetries of these Raman bands.37,38 Therefore, the intensity ratios of 192 cm−1 and 684 cm−1 (I192/I684) band, indicating the ratio of Co2+ and Co3+, were calculated and listed in Table 1. It can be clearly seen that I192/I684 of Co3O4-Ac catalyst is higher than the others. That is to say, Co3O4-Ac catalyst possesses the highest Co2+ concentration on the catalyst surface. In addition, it's worth noting that the Raman bands for the highly active Co3O4-Ac catalyst shift slightly to lower frequencies. It is reported that this phenomenon can be taken as a sensitive indication of a higher concentration of cobalt in low state.39 That is to say, Co3O4-Ac catalyst possesses the highest Co2+ concentration on the catalyst surface, which is also further confirmed by the XPS results described below. | ||
| Fig. 3 Raman spectra of the Co3O4 catalysts prepared with different cobalt precursors. | ||
The surface composition and the oxidation state of the prepared Co3O4 catalysts were investigated by X-ray photoelectron spectroscopy (XPS) analysis. Fig. 4(a) shows the full scan spectra of Co3O4 catalysts, in which the Co 2p, O 1s peaks can be observed. The corresponding high-resolution XPS spectra of Co 2p and O 1s spectra of Co3O4 catalysts prepared with different cobalt precursors are shown in Fig. 4(b) and (c). Generally, Co3O4 has two types of cobalt ions, containing Co3+ in octahedral coordination and Co2+ in tetrahedral coordination. As shown in Fig. 4(b), the Co 2p spectra have two major peaks centered at 780.0 (Co 2p3/2) and 794.9 eV (Co 2p1/2) with a spin–orbit splitting of ∼15.1 eV, indicating the existence of both Co(II) and Co(III).40 Satellite peak at about 786.0 eV is also a fingerprint for recognition of Co2+.41,42 The Co 2p3/2 and its satellites recorded from the catalysts were deconvoluted into five contributions. Detailed peak deconvolution and the peak assignment were listed in Table 2. The peaks at around 779.8, 779.1 and 782.2 eV can be ascribed to Co3+ in octahedral coordination, mixed Co(II,III), Co2+ in tetrahedral coordination, respectively.32,43,44 The area ratios of Co2+ and Co3+ are calculated and listed in Table 2. As presented in Table 2, the highest ratio of Co2+/Co3+ is 0.37 for Co3O4-Ac catalyst, implying that more Co2+ appeared on the catalyst surface, which is in accordance with the Raman results. According to the electroneutrality principle, an increase in the Co2+ concentration means a rise in the amount of oxygen vacancies,45 that is, surface oxygen vacancies are more easily formed on the surface of Co3O4-Ac nanoparticles. According to the literature, oxygen vacancies not only can activate adsorbed oxygen and provide the lattice sites of oxygen migration, resulting in the formation of highly active electrophilic oxygen species, but also can generate new defective states in the energy band gap of Co3O4, leading to enhancement of the electronic conductivity of Co3O4,46,47 which can be further confirmed by the high-resolution O 1s XPS spectra.
 | ||
| Fig. 4 XPS spectra of Co3O4 catalysts prepared with different cobalt precursors: full scan spectra (a), Co 2p (b), O 1s (c) and S 2p (d). | ||
| Catalysts | Oads/OLatt | Co2+/Co3+ | Binding energy of Co 2p2/3 (eV) | S content (wt%) | ||
|---|---|---|---|---|---|---|
| Co3O4-Ac | 0.52 | 0.37 | 779.8 | 781.0 | 782.2 | — |
| Co3O4-Cl | 0.47 | 0.33 | 779.7 | 781.0 | 782.3 | — |
| Co3O4-N | 0.45 | 0.31 | 779.6 | 780.9 | 782.3 | — |
| Co3O4-S | 0.23 | 0.34 | 779.9 | 781.2 | 782.6 | 0.324 |
| Attribution | — | — | Co3+ | Co3+; Co2+ | Co2+ | — |
The O 1s spectra are frequently used to identify the types of oxygen species over the surface of catalysts. The chemical environment of oxygen in metal oxide catalysts plays a critical role in their catalytic properties. The XPS spectra of O 1s over the surface of Co3O4 catalysts prepared with different cobalt precursors are shown in Fig. 4(c). The asymmetrical O 1s XPS profiles of the catalysts can be deconvoluted into four components. The peaks at 529.9, 530.9, 532.0 and 533.2 eV can be ascribed to the lattice oxygen, surface adsorbed oxygen (O22−, O2−, O−), hydroxyl (OH−) and/or carbonate species (CO32−), as well as adsorbed water, respectively.48–50 In general, the electrophilic oxygen species (O22−, O2−, O−) are vital for the oxidation of methane.51 Therefore, the ratio of the surface adsorbed oxygen and the lattice oxygen (Oads/OLatt) was calculated and summarized in Table 2. According to early studies, the higher Oads/OLatt ratio indicates the richer active oxygen species on the catalysts surface. In addition, it is well known that the adsorbed oxygen species concentration is associated with the oxygen vacancies density. For an oxygen-deficient material, more oxygen vacancies give a higher oxygen species concentration.52 As shown in Table 2, the Co3O4-Ac catalyst has higher Oads/OLatt ratio of 0.52 than others, indicating the Co3O4-Ac catalyst possesses higher adsorbed oxygen species concentration. Therefore, it is reasonable to propose that CH4 molecule might have more sufficient contact with the surface active sites of this Co3O4-Ac catalyst in comparison with other catalysts, thus it could show better methane combustion activity.
For Co3O4-S catalyst, although the surface Co2+ concentration is higher than that of Co3O4-Cl and Co3O4-N, the concentration of oxygen species adsorbed on the surface of Co3O4-S catalysts is the lowest. Considering the deposition of impurity sulfate may cause the catalyst poisoning due to unclean washing,53 thus the content of S was measured by XRF (Table 2). It is found the content of S in Co3O4-S is 0.324 wt%, and no impurity residual is detected in other samples. To further explore the state of S, the S 2p XPS spectrum of the Co3O4-S sample was performed and presented in Fig. 4(d). It is worth noting the binding energy of S 2p (168.7 eV) is assigned to S(VI) oxidation state,54 which demonstrates that there are still non-negligible sulfates on the catalyst surface. These residual impurities SO42− may cover the active sites on the surface of the catalyst, reduce concentration of surface adsorbed active oxygen species, weaken the oxygen mobility, and thus reducing the catalytic performance. It is further confirmed by the catalytic activity of methane combustion.
Fig. 5 shows the O2 temperature programmed desorption (O2-TPD) profiles. It is reported that the peak below 300 °C is ascribed to the desorption of adsorbed oxygen species such as O2−, O− in Co3O4 while the desorption peak of lattice oxygen generally appears above 350 °C.55 Additionally, the desorption temperature and the peak intensity are related to both the properties and the amount of surface adsorbed oxygen, which might improve the catalytic activity.56,57 Notably, the intensity of desorption peak decreases and the peak gradually shifts to high temperature range follow the sequence of Co3O4-Ac > Co3O4-Cl > Co3O4-N > Co3O4-S, indicating that the most active Co3O4-Ac catalyst with lager peak area and lower peak temperature is more preferable for O2 dissociative adsorption and easier to produce active oxygen species on the surface.58 Therefore, it can be concluded that the amount and the mobility of adsorbed oxygen might be enhanced by changing the cobalt precursors, which is also consistent with the XPS result.
 | ||
| Fig. 5 O2-TPD patterns of Co3O4 catalysts prepared with different cobalt precursors. | ||
Reduction properties
The H2 temperature-programmed reduction measurements (H2-TPR) were carried out over the Co3O4 catalysts prepared with different cobalt precursors to investigate the reduction behavior. Fig. 6 shows the H2-TPR profiles of the catalysts. Distinguished from the reduction peak shapes, it is obvious that the change of cobalt precursors has influence on the redox property of the prepared Co3O4 catalysts. All the Co3O4 samples display two main reduction peaks at 250–550 °C. The low temperature peak (α) and the high temperature peak (β1 and β2) are assigned to stepwise the reduction of Co3O4 to CoO and subsequently CoO to Co,59,60 respectively. The α peaks of Co3O4-Ac, Co3O4-N and Co3O4-Cl catalysts are very similar with the lowest reduction temperatures. In contrast, the reduction temperature of α peak for Co3O4-S catalyst shifts obviously to higher temperature region, implying the Co3O4-S catalyst is hard to be reduced. It was reported that the hydrogen consumption peak intensity of the poisoning catalyst would be obviously weakened, and the reduction peaks would shift toward the high temperature.53 The XPS results mentioned above also show that SO42− exists on the surface of Co3O4-S catalyst. Therefore, it can be deduced that the reducibility of the Co3O4-S catalyst is significantly weakened due to the residue SO42− impurity. The TPR profiles were integrated and calibrated with an appropriate hydrogen consumption to quantitatively measure the total hydrogen consumption and the results are shown in Table 3. The theoretical H2 consumption is 16.6 mmol g−1 when Co2+ and Co3+ are entirely reduced to metallic Co.61 By comparing the four catalysts, it can be found that their actual hydrogen consumption is close to the theoretical one, indicating that Co3O4 catalysts have been almost completely reduced to Co, except for Co3O4-S catalyst. Taking into account the factors mentioned above, we can deduce that the less hydrogen consumption of Co3O4-S is due to the presence of impurities, which is hard to be reduced. This result is in accordance with the composition of the catalysts. | ||
| Fig. 6 H2-TPR patterns of Co3O4 catalysts prepared with different cobalt precursors. | ||
| Catalysts | Total H2 consumption (mmol g−1) | T50 (°C) | T90 (°C) | Ea (kJ mol−1) |
|---|---|---|---|---|
| Co3O4-Ac | 16.6 | 318 | 385 | 24.94 |
| Co3O4-Cl | 16.2 | 332 | 400 | 27.21 |
| Co3O4-N | 17.0 | 344 | 416 | 37.14 |
| Co3O4-S | 13.8 | 367 | 429 | 38.75 |
In addition, the high temperature peak β do not take the shape of a single peak, but instead is divided into two different contributions, which confirms the existence of Co2+ species with different reducibility. The peak area of β1 for the samples follows the sequence of Co3O4-Ac > Co3O4-Cl > Co3O4-N > Co3O4-S, while the sequence of β2 is opposite. The order of β1 peaks in Co3O4-Ac, Co3O4-Cl and Co3O4-N catalysts is consistent with that of surface Co2+/Co3+ in XPS results. In other words, the higher the ratio of Co2+/Co3+, the stronger the reduction peak of β1. Co3O4-S, as a special case, displays not only the highest reduction temperatures of α and β peak, but also the lowest peak area of β1, although its Co2+/Co3+ concentration is high. It is likely due to the presence of residue SO42− impurity on the surface of the catalyst, which covers the active sites on the surface of the catalyst, resulting in the reduction of reaction activity. The lowest surface adsorbed oxygen species concentration in XPS results also confirms this conclusion. According to the literature, the reduction temperature of surface Co2+ is lower than that of bulk Co2+, that is to say, surface Co2+ is easier to be reduced.62 Combined with our previous XPS results, it can be inferred that β1 is assigned to the reduction of surface Co2+ and β2 is assigned to the reduction of bulk Co2+. The higher the concentration of Co2+/Co3+ and adsorbed oxygen species on the catalysts surface, the easier it is to be reduced.
Catalytic activity
Fig. 7 represents the activity for methane combustion over Co3O4 catalysts prepared with different cobalt precursors. The catalytic activity of Co3O4 catalysts decreases in the order of Co3O4-Ac > Co3O4-Cl > Co3O4-N > Co3O4-S. As shown in Table 3, the characteristic temperatures T50 and T90 correspond to the initiation of the oxidation, 50% conversion and 90% conversion of CH4. It is clear that Co3O4-Ac catalyst presents the best activity. In general, catalysts with smaller crystallite size and higher specific surface area expose more active sites and facilitate the catalytic oxidation process. However, no obvious relationship between catalytic activity and surface area was found. The most important thing for the catalyst to enhance the catalytic performance is to decrease the activation energy (Ea) of the reaction, making the catalyst easier to be initiated.63 Arrhenius plots for Co3O4 catalysts are shown in Fig. S1 (ESI†). The Ea can be obtained from the slope of the linear plot of lnr versus 1/T according to eqn (2) and the data are listed in Table 3.64 It is obvious that the sequence of the Ea is Co3O4-Ac < Co3O4-Cl < Co3O4-N < Co3O4-S, which is in well agreement with the activity. The Co3O4-Ac catalyst possesses the lower activation energy (Ea is 24.94 kJ mol−1) and a better performance with a lower reaction temperature at 318 °C for the 50% methane conversion, while the conversion is only 16% for the Co3O4-S sample at this temperature, as comparison. The results imply that some other factors play a more decisive role than surface areas to improve catalytic activity.
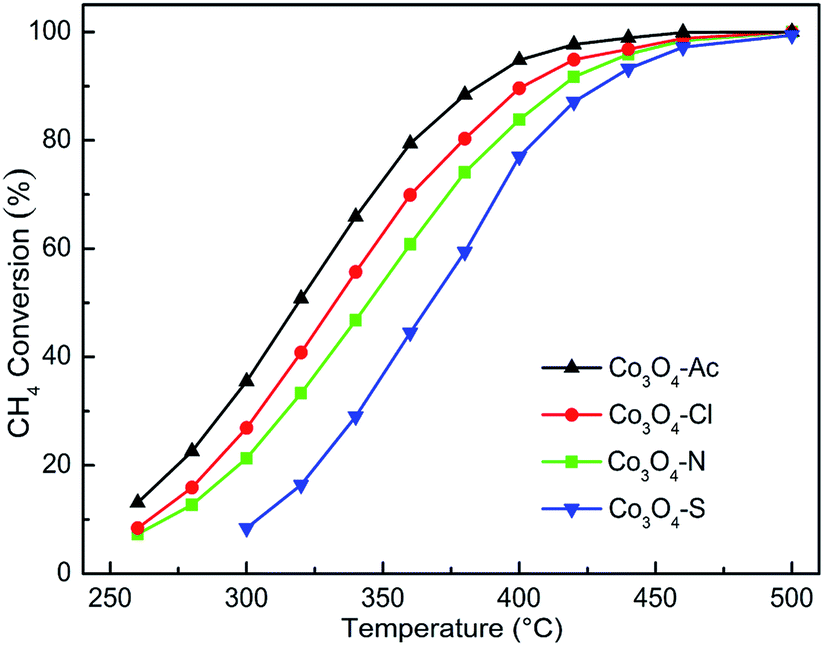 | ||
| Fig. 7 Catalytic activity for methane combustion over Co3O4 catalysts prepared with different cobalt precursors. | ||
From the XPS and several other characterization results mentioned above, it can be found the ratio of Co2+/Co3+ maybe strictly correlated to the performance. Moreover, the higher the concentration of Co2+, the higher the concentration of oxygen species adsorbed on the surface and thus improving the catalytic activity. The Co3O4-Ac catalyst possesses the highest concentration of Co2+/Co3+ and surface absorbed oxygen which enable the enhancement of its catalytic activity. As a special case, although the surface Co2+ concentration of the worst active Co3O4-S catalyst is higher than that of Co3O4-Cl and Co3O4-N catalysts, the concentration of oxygen species adsorbed on the surface of Co3O4-S catalyst is the lowest due to the influence of residual impurity SO42−, which can cause catalyst poisoning and inhibit oxygen migration, thus reducing its methane catalytic performance. Therefore, it can be concluded that the performance of catalytic combustion for lean methane is closely related to the Co2+ concentration and surface adsorbed oxygen species, which is consistent with our previous research. The result has proved strongly that by tuning the cobalt precursors, Co3O4 with optimal redox property and the highest methane combustion activity can be obtained. It indicates that cobalt precursor play a vital role in controlling structural properties of the catalysts, resulting in a significant difference in physicochemical properties and catalytic performance.
Stability and cyclicity
The stability and cyclicity of catalysts play important indicators in the application of catalysts. Thus, the stability, moisture resistance and recycle performance were investigated. The long term stability and moisture were examined at 360 °C and shown in Fig. 8. For each stability test, the methane combustion was carried out using a dry feed for the first 24 h. Afterwards, 5.0% water vapor was added into the reaction feed. After stabilized for 30 min, the first injection started and continued for 6 h. Then the water vapor was taken off until reacting for 36 h. As shown in this figure, methane conversion for Co3O4-Ac, Co3O4-Cl and Co3O4-N catalysts remained, no remarkable loss of catalytic activity is observed in the first 24 h, except for Co3O4-S catalyst, which shows the worst catalytic performance, decreased from 43% to 37% in the initial period. After 24 h tests, the CH4 conversion of Co3O4-Ac catalyst tends to decline from ∼78% to ∼74% with the moisture (5.0 vol%) adding into the reaction feed for 6 h. After interruption of moisture, the CH4 conversion has a recovery in 1 h, stabilized at ∼76%, slightly lower than the initial conversion, which implying that the presence of moisture slightly inhibits the catalytic activity. The moisture resistance (at 360 °C) of all the catalysts decrease as follows: Co3O4-Ac ≈ Co3O4-Cl ≈ Co3O4-N > Co3O4-S. By comparison, the activity of Co3O4-S catalyst has rebounded to some extent, but it can be obviously found a pronounced downward within 6 h after interruption of moisture, which may be due to the presence of impurity sulfate. In summary, Co3O4 catalysts exhibit good long-term stability and moisture resistance in the complete combustion of lean methane if impurities can be effectively avoided. | ||
| Fig. 8 On-stream methane combustion and moisture resistance over Co3O4 catalysts at 360 °C. | ||
Fig. 9 shows four consecutive runs curves of methane combustion over Co3O4 catalysts prepared with different cobalt precursors. It can be seen from the figure that the conversion of the second run curve decreases slightly compared with the fresh sample (first run), but the last three runs curves possess well overlapping, indicating they are effective and stable catalysts for long-term operations, except for Co3O4-S catalyst with relatively poor recycle performance. Moreover, the used catalysts (after four consecutive runs) were characterized by XRD and Raman techniques, the results are shown in Fig. S2 and S3.† No any other characteristic signals are observed except for the characteristic diffraction peaks and vibration modes of Co3O4, indicating that the physical–chemical properties of the Co3O4 catalysts have not changed after the reaction.
 | ||
| Fig. 9 CH4 conversion during four consecutive runs for Co3O4 catalysts prepared with different cobalt precursors. | ||
Conclusions
A series of Co3O4 catalysts have been prepared with a simple precipitation method by changing the cobalt precursors and tested for lean methane combustion. It demonstrated that different cobalt precursors had great influence on the microstructure, surface properties, reducibility, catalytic activity and stability of Co3O4 catalysts. Co3O4-Ac catalyst showed the highest performance for lean methane combustion, although it exhibited relatively small BET surface area. The results of Raman and XPS showed that the extraordinary performance was closely related to the concentration of Co2+ and surface active oxygen species. As a special case, although the surface Co2+ concentration of the worst active Co3O4-S catalyst was high, the concentration of oxygen species adsorbed on the surface of Co3O4-S catalyst was the lowest due to the residual impurity SO42−, which could cause catalyst poisoning and inhibit oxygen migration. Therefore, the negative effects of residual impurity on catalytic performance should be considered and avoided. Indeed, the information achieved in this study might provide some novel insight for people to design cheaper and more applicable methane combustion catalysts without noble metals for real emission control applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- C. Ding, W. Liu, J. Wang, P. Liu, K. Zhang, X. Gao, G. Ding, S. Liu, Y. Han and X. Ma, Fuel, 2015, 162, 148–154 CrossRef CAS.
- F. Wang, L. Zhang, L. Xu, Z. Deng and W. Shi, Fuel, 2017, 203, 419–429 CrossRef CAS.
- H. Zhang, P. Li, N. Hui, J. Liang, Y. Ding and T. Liu, J. Alloys Compd., 2017, 712, 567–572 CrossRef CAS.
- M. Jeong, N. Nunotani, N. Moriyama and N. Imanaka, Catal. Sci. Technol., 2017, 7, 1986–1990 RSC.
- Y. Wang, H. Arandiyan, J. Scott, M. Akia, H. Dai, J. Deng, K.-F. Aguey-Zinsou and R. Amal, ACS Catal., 2016, 6, 6935–6947 CrossRef CAS.
- Z. Chen, S. Wang, W. Liu, X. Gao, D. Gao, M. Wang and S. Wang, Appl. Catal., A, 2016, 525, 94–102 CrossRef CAS.
- J. Li, R. Hu, J. Zhang, W. Meng, Y. Du, Y. Si and Z. Zhang, Fuel, 2016, 178, 148–154 CrossRef CAS.
- J. Ma, Y. Lou, Y. Cai, Z. Zhao, L. Wang, W. Zhan, Y. Guo and Y. Guo, Catal. Sci. Technol., 2018, 8, 2567–2577 RSC.
- J. Jin, C. Li, C.-W. Tsang, B. Xu and C. Liang, RSC Adv., 2015, 5, 102147–102156 RSC.
- H. Geng, L. Zhang, Z. Yang, Y. Yan and J. Ran, Int. J. Hydrogen Energy, 2018, 43, 11069–11078 CrossRef CAS.
- W. Qi, J. Ran, Z. Zhang, J. Niu, P. Zhang, L. Fu, B. Hu and Q. Li, Appl. Surf. Sci., 2018, 435, 776–785 CrossRef CAS.
- Z. Zhao, B. Wang, J. Ma, W. Zhan, L. Wang, Y. Guo, Y. Guo and G. Lu, Chin. J. Catal., 2017, 38, 1322–1329 CrossRef CAS.
- Y. Liu, S. Wang, D. Gao, T. Sun, C. Zhang and S. Wang, Fuel Process. Technol., 2013, 111, 55–61 CrossRef CAS.
- I. Popescu, N. Tanchoux, D. Tichit and I.-C. Marcu, Appl. Catal., A, 2017, 538, 81–90 CrossRef CAS.
- X. Yuan, S. Chen, H. Chen and Y. Zhang, Catal. Commun., 2013, 35, 36–39 CrossRef CAS.
- K. Zhang, X. Peng, Y. Cao, H. Yang, X. Wang, Y. Zhang, Y. Zheng, Y. Xiao and L. Jiang, Mater. Res. Bull., 2019, 111, 338–341 CrossRef CAS.
- J. Chen, X. Zhang, H. Arandiyan, Y. Peng, H. Chang and J. Li, Catal. Today, 2013, 201, 12–18 CrossRef CAS.
- S. Zhang, J. J. Shan, Y. Zhu, A. I. Frenkel, A. Patlolla, W. Huang, S. J. Yoon, L. Wang, H. Yoshida, S. Takeda and F. F. Tao, J. Am. Chem. Soc., 2013, 135, 8283–8293 CrossRef CAS PubMed.
- Z. Zhang, Z. Wen, Z. Ye and L. Zhu, RSC Adv., 2015, 5, 59976–59982 RSC.
- H. Wang, Y. Song, Y. Li, M. Wang, Q. Ma, W. Yu, D. Li, X. Dong, J. Wang and G. Liu, RSC Adv., 2018, 8, 30794–30801 RSC.
- A. Diallo, T. B. Doyle, B. M. Mothudi, E. Manikandan, V. Rajendran and M. Maaza, J. Magn. Magn. Mater., 2017, 424, 251–255 CrossRef CAS.
- Y. Liu, J. Xie, M. Luo, S. Jian, B. Peng and L. Deng, Infrared Phys. Technol., 2017, 83, 88–93 CrossRef CAS.
- M. W. Khan, J. Yao, K. Zhang, X. Zuo, Q. Yang, H. Tang, K. M. U. Rehman, H. Zhang, G. Li, S. Jin and M. Wu, J. Electroanal. Chem., 2019, 844, 142–154 CrossRef CAS.
- J. González-Prior, R. López-Fonseca, J. I. Gutiérrez-Ortiz and B. de Rivas, Appl. Catal., B, 2016, 199, 384–393 CrossRef.
- L. Bai, F. Wyrwalski, M. Safariamin, R. Bleta, J.-F. Lamonier, C. Przybylski, E. Monflier and A. Ponchel, J. Catal., 2016, 341, 191–204 CrossRef CAS.
- S. Dey, G. C. Dhal, R. Prasad and D. Mohan, Resour.-Effic. Technol., 2017, 3, 293–302 Search PubMed.
- G. Ercolino, A. Grodzka, G. Grzybek, P. Stelmachowski, S. Specchia and A. Kotarba, Top. Catal., 2016, 60, 333–341 CrossRef.
- X. Li, H. Almkhelfe, N. M. Bedford, T. C. Back, K. L. Hohn and P. B. Amama, Catal. Today, 2019, 338, 40–51 CrossRef CAS.
- J. Girardon, A. Lermontov, L. Gengembre, P. Chernavskii, A. Gribovalconstant and A. Khodakov, J. Catal., 2005, 230, 339–352 CrossRef CAS.
- H. Wu, G. Pantaleo, G. Di Carlo, S. Guo, G. Marcì, P. Concepción, A. M. Venezia and L. F. Liotta, Catal. Sci. Technol., 2015, 5, 1888–1901 RSC.
- X. Xu, H. Han, J. Liu, W. Liu, W. Li and X. Wang, J. Rare Earths, 2014, 32, 159–169 CrossRef CAS.
- Y. Zheng, Y. Liu, H. Zhou, W. Huang and Z. Pu, J. Alloys Compd., 2018, 734, 112–120 CrossRef CAS.
- X. Du, C. Huang and X. Zhang, J. Alloys Compd., 2019, 809, 151821 CrossRef CAS.
- A. Choya, B. de Rivas, J. R. González-Velasco, J. I. Gutiérrez-Ortiz and R. López-Fonseca, Appl. Catal., B, 2018, 237, 844–854 CrossRef CAS.
- X. Wang, Y. Liu, T. Zhang, Y. Luo, Z. Lan, K. Zhang, J. Zuo, L. Jiang and R. Wang, ACS Catal., 2017, 7, 1626–1636 CrossRef CAS.
- Q. Jiao, M. Fu, C. You, Y. Zhao and H. Li, Inorg. Chem., 2012, 51, 11513–11520 CrossRef CAS.
- S. Mo, S. Li, H. Xiao, H. He, Y. Xue, M. Zhang, Q. Ren, B. Chen, Y. Chen and D. Ye, Catal. Sci. Technol., 2018, 8, 1663–1676 RSC.
- S. K. Tripathy, M. Christy, N.-H. Park, E.-K. Suh, S. Anand and Y.-T. Yu, Mater. Lett., 2008, 62, 1006–1009 CrossRef CAS.
- B. de Rivas, R. López-Fonseca, C. Jiménez-González and J. I. Gutiérrez-Ortiz, J. Catal., 2011, 281, 88–97 CrossRef CAS.
- Z. Zhu, G. Lu, Z. Zhang, Y. Guo, Y. Guo and Y. Wang, ACS Catal., 2013, 3, 1154–1164 CrossRef CAS.
- G. Li, L. Li, J. Shi, Y. Yuan, Y. Li, W. Zhao and J. Shi, J. Mol. Catal. A: Chem., 2014, 390, 97–104 CrossRef CAS.
- M. I. Shilina, T. N. Rostovshchikova, S. A. Nikolaev and O. V. Udalova, Mater. Chem. Phys., 2019, 223, 287–298 CrossRef CAS.
- A. Amri, X. Duan, C.-Y. Yin, Z.-T. Jiang, M. M. Rahman and T. Pryor, Appl. Surf. Sci., 2013, 275, 127–135 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- S. Xie, H. Dai, J. Deng, Y. Liu, H. Yang, Y. Jiang, W. Tan, A. Ao and G. Guo, Nanoscale, 2013, 5, 11207–11219 RSC.
- Q. Wang, J. Liu, Y. Li, Z. Zhao, W. Song and Y. Wei, RSC Adv., 2017, 7, 18592–18600 RSC.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- N. A. Merino, B. P. Barbero, P. Eloy and L. E. Cadús, Appl. Surf. Sci., 2006, 253, 1489–1493 CrossRef CAS.
- K. Sutthiumporn and S. Kawi, Int. J. Hydrogen Energy, 2011, 36, 14435–14446 CrossRef CAS.
- Y. Liu, H. Dai, J. Deng, S. Xie, H. Yang, W. Tan, W. Han, Y. Jiang and G. Guo, J. Catal., 2014, 309, 408–418 CrossRef CAS.
- N. Yang, S. Ni, Y. Sun and Y. Zhu, Mol. Catal., 2018, 452, 28–35 CrossRef CAS.
- Z. Pu, Y. Liu, H. Zhou, W. Huang, Y. Zheng and X. Li, Appl. Surf. Sci., 2017, 422, 85–93 CrossRef CAS.
- K. Zhuang, Y. Zhang, T. Huang, B. LU and K. Shen, J. Fuel Chem. Technol., 2017, 45, 1356–1364 CrossRef CAS.
- A. Škvarlová, M. Kaňuchová, Ľ. Kozáková, E. Valušová, M. Holub and J. Škvarla, Microporous Mesoporous Mater., 2019, 284, 283–288 CrossRef.
- C. Ma, Z. Mu, J. Li, Y. Jin, J. Cheng, G. Lu, Z. Hao and S. Qiao, J. Am. Chem. Soc., 2010, 132, 2608–2613 CrossRef CAS PubMed.
- X. Li, X. Li, X. Zeng and T. Zhu, Appl. Catal., A, 2019, 572, 61–70 CrossRef CAS.
- R. Huang, K. Kim, H. J. Kim, M. G. Jang and J. W. Han, ACS Appl. Nano Mater., 2019 DOI:10.1021/acsanm.9b02056.
- S. Lu, F. Wang, C. Chen, F. Huang and K. Li, J. Rare Earths, 2017, 35, 867–874 CrossRef CAS.
- F. Teng, M. Chen, G. Li, Y. Teng, T. Xu, Y. Hang, W. Yao, S. Santhanagopalan, D. D. Meng and Y. Zhu, Appl. Catal., B, 2011, 110, 133–140 CrossRef CAS.
- T. Baidya, T. Murayama, S. Nellaiappan, N. K. Katiyar, P. Bera, O. Safonova, M. Lin, K. R. Priolkar, S. Kundu, B. Srinivasa Rao, P. Steiger, S. Sharma, K. Biswas, S. K. Pradhan, N. Lingaiah, K. D. Malviya and M. Haruta, J. Phys. Chem. C, 2019, 123, 19557–19571 CrossRef CAS.
- W. Zhang, F. Wu, J. Li and Z. You, Appl. Surf. Sci., 2017, 411, 136–143 CrossRef CAS.
- X. Fan, L. Li, X. Yang, Z. Guo, F. Jing and W. Chu, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.09.007.
- X. Li, F. Dong, L. Zhang, Q. Xu, X. Zhu, S. Liang, L. Hu and H. Xie, Chem. Eng. J., 2019, 372, 516–525 CrossRef CAS.
- Y. Shen, J. Yu, X. Xiao, X. Guo, D. Mao, H. Huang and G. Lu, J. Catal., 2017, 352, 466–479 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09544f |
| This journal is © The Royal Society of Chemistry 2020 |