
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments on oximes to improve the blood brain barrier penetration for the treatment of organophosphorus poisoning: a review
Mohd Nor Faiz Norrrahim
a,
Mas Amira Idayu Abdul Razakb,
Noor Aisyah Ahmad Shahc,
Herdawati Kasimc,
Wan Yusmawati Wan Yusoffc,
Norhana Abdul Halimc,
Siti Aminah Mohd Norc,
Siti Hasnawati Jamalc,
Keat Khim Ongac,
Wan Md Zin Wan Yunus
d,
Victor Feizal Knight
 a and
Noor Azilah Mohd Kasim
*ac
a and
Noor Azilah Mohd Kasim
*ac
aResearch Centre for Chemical Defence, Universiti Pertahanan Nasional Malaysia, Kem Perdana Sungai Besi, 57000 Kuala Lumpur, Malaysia. E-mail: azilah@upnm.edu.my; Fax: +603-90514291; Tel: +6013-3906789
bDepartment of Defence Science, Faculty of Defence Science and Technology, Universiti Pertahanan Nasional Malaysia, Kem Perdana Sungai Besi, 57000 Kuala Lumpur, Malaysia
cDepartment of Chemistry and Biology, Centre for Defence Foundation Studies, Universiti Pertahanan Nasional Malaysia, Kem Perdana Sungai Besi, 57000 Kuala Lumpur, Malaysia
dResearch Centre for Tropicalisation, Universiti Pertahanan Nasional Malaysia, Kem Perdana Sungai Besi, 57000 Kuala Lumpur, Malaysia
First published on 27th January 2020
Abstract
Organophosphorus (OP) compounds are highly toxic synthetic compounds which have been used as pesticides and developed as warfare nerve agents. They represent a threat to both military and civilian populations. OP pesticides affect the nervous system and are thought to have caused at least 5 million deaths since their discovery in the 1930s. At present the treatment of OP nerve agent poisoning commonly involves the use of parenteral oximes. However, the blood brain barrier (BBB) remains a challenge in the delivery of oximes to the central nervous system (CNS). This is because almost all macromolecule drugs (including oximes) fail to pass through the BBB to reach the CNS structures. The presence of a permanent cationic charge in oximes has made these compounds inefficient in crossing the BBB. Thus, oximes are unable to reactivate acetylcholinesterase (AChE) in the CNS. Using current structural and mechanistic understanding of the BBB under both physiological and pathological conditions, it becomes possible to design delivery systems for oximes and other drugs that are able to cross the BBB effectively. This review summarises the recent strategies in the development of oximes which are capable of crossing the BBB to treat OP poisoning. Several new developments using oximes are reviewed along with their advantages and disadvantages. This review could be beneficial for future directions in the development of oxime and other drug delivery systems into the CNS.
 Mohd Nor Faiz Norrrahim | Mohd Nor Faiz Norrrahim has a PhD in Nanotechnology. He is working as a post-doctoral fellow at the Research Center for Chemical Defence, National Defence University of Malaysia. His research interests are nanotechnology, organic chemistry, materials science, biopolymers and biotechnology. |
 Wan Md Zin Wan Yunus | Wan Md Zin Wan Yunus is working as a Professor at the National Defence University of Malaysia. His research interests are materials, chemical synthesis, nanomaterials, polymers, advanced materials, and biocomposites. |
 Victor Feizal Knight | Victor Feizal Knight is working as a Professor at the National Defence University of Malaysia. He is also the director of the Research Center for Chemical Defence. His expertise is military medicine and health, and biomedical and chemical defence. |
 Noor Azilah Mohd Kasim | Noor Azilah Mohd Kasim is working as an Associate Professor at the Department of Defence Science, National Defence University of Malaysia. Her research interests are organic chemistry, chemistry, materials science and sensors development. |
Introduction
Organophosphorus (OP) compounds were first developed as insecticides in the 1930s before being later developed as a weapon of war. OP poisoning has been a frequent cause of admission of people to hospitals and Intensive Care Units (ICU) in developing countries.1 OP poisoning causes about 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 acute intoxications annually, 300000 of which lead to fatalities.2 OPs that were developed as chemical warfare nerve agents (CWAs) are all highly toxic and dangerous.3–5 OP nerve agents have been used and are most likely to be used in the future by terrorists and dictators around the world because of their relatively easy synthesis and availability of suitable delivery systems.6 Warfare and terrorist use of CWAs include the Aum Shinrikyo terrorist attack in the Tokyo subway in 1995, the 1980–1988 Iraq–Iran war where Iraq reportedly used nerve agents against Iranian troops and later on Kurd civilians and the murder of a family member of the North Korea Leader, Kim Jong Nam on February 2017 in Kuala Lumpur.7–9 Despite international efforts aimed at regulating and lessening the use of these environmentally toxic compounds, more than 100 different OP compounds are still being used intensively as pesticides, with only unreliable or sporadic monitoring of the environment and workers involved in their use.
000000 acute intoxications annually, 300000 of which lead to fatalities.2 OPs that were developed as chemical warfare nerve agents (CWAs) are all highly toxic and dangerous.3–5 OP nerve agents have been used and are most likely to be used in the future by terrorists and dictators around the world because of their relatively easy synthesis and availability of suitable delivery systems.6 Warfare and terrorist use of CWAs include the Aum Shinrikyo terrorist attack in the Tokyo subway in 1995, the 1980–1988 Iraq–Iran war where Iraq reportedly used nerve agents against Iranian troops and later on Kurd civilians and the murder of a family member of the North Korea Leader, Kim Jong Nam on February 2017 in Kuala Lumpur.7–9 Despite international efforts aimed at regulating and lessening the use of these environmentally toxic compounds, more than 100 different OP compounds are still being used intensively as pesticides, with only unreliable or sporadic monitoring of the environment and workers involved in their use.
OP poisonings involve the inhibition of enzyme acetylcholinesterase (AChE) that physiologically plays an important role in neurotransmission.10–12 This will result in the overstimulation of cholinergic receptors by acetylcholine (ACh) which will occur in the peripheral nervous system, central nervous system (CNS) and at neuromuscular junctions causing cholinergic crises, seizures, respiratory arrest, bronchorrhea and ultimately lead to fatal consequences.13,14 Current treatment of OP poisonings consists of the administration of a muscarinic receptor antagonist (e.g. atropine), which counteracts the excessive muscarinic stimulation, and also the administration of an AChE reactivator (pyridinium oximes), which reactivates the physiological function of AChE.3,15,16
Successful reactivation of AChE can terminate overstimulation of the muscarinic and nicotinic receptors by ACh and thus prevent further neurotoxicity. However, several reports on efficacy testing of experimental oximes claimed that the oximes have difficulty in reactivating the AChE located in the CNS.7,9 In addition to this potential lethality of this overstimulation, of great concern also is the fact that AChE inhibition by OPs in the CNS can lead to seizures occurring from an excess of ACh-mediated activation of the excitotoxic glutamatergic pathways which can prolong seizures and thus lead to brain damage.17,18 It has also been suggested that exposure to sub-lethal doses of OP can dramatically impact CNS mediated behavioural attributes such as decreased ability to perform tasks.12 All the currently available oxime reactivators are quaternary oximes. Quaternary pyridinium aldoximes have relatively contained permanent positive charges which provides little or no protection against the neurological effects of OP exposure in the CNS due to their poor blood brain barrier (BBB) penetration.19,20
The BBB is a highly selective permeable membrane system responsible for maintaining homeostasis by regulating the chemical environment, immune cell transport, and the entry of toxins into the CNS.21,22 The success rate of BBB penetration for drugs such as oximes is very low.23–25 There are several limitations imposed by the BBB which includes molecule size, hydrophilicity, polarity, substrate specificity, and active efflux mechanisms.26–29 These properties however, are lacking in most oxime structures. Therefore, the development of effective measures to counteract the problem of BBB penetration by oximes in order to treat OP poisoning remains a challenging issue. Investigations to develop oximes that can penetrate through the BBB has been reported as one of the current areas of research interest in the treatment of OP poisonings.30 To the best of our knowledge, there is lack of papers that discuss on the approaches formulated towards BBB penetration of oximes for OP poisoning treatment.
Organophosphorus nerve agents
The G-series and V-series are the two major classes of OP nerve agents.31 Tabun was the first of the G-series OP nerve agents developed in 1936 by a German scientist named Gerhard Schroeder.32 This compound was developed as a pesticide for the chemical manufacturer IG Farbenindustrie. Its lethal properties lead to its use as a weapon against humans as well as for the development of other weaponised OPs. These were the G-series nerve agents and the letter ‘G’ referred to agents developed by the Germans. Later on, by the end of World War II, German scientists had developed several other OP nerve agents such as tabun (GA), soman (GD), sarin (GB) and cyclosarin (GF) which were synthesised in certain quantities that could be used as weapons of war. After World War II, the V-series of OP nerve agents, with the initial agent being known as VX was discovered by Dr Ranajit Ghosh in 1949, a chemist working in England. He was working on insecticide synthesis and discovered an extremely potent nerve agent. A similar V-series agent was produced by Russian military, namely Russian VX. VX and Russian VX nerve agents have been reported as the most abundant and most toxic chemical warfare agents when compared to the G-series nerve agents.33 Fig. 1 shows the chemical structure for all the G- and V-series OP nerve agents.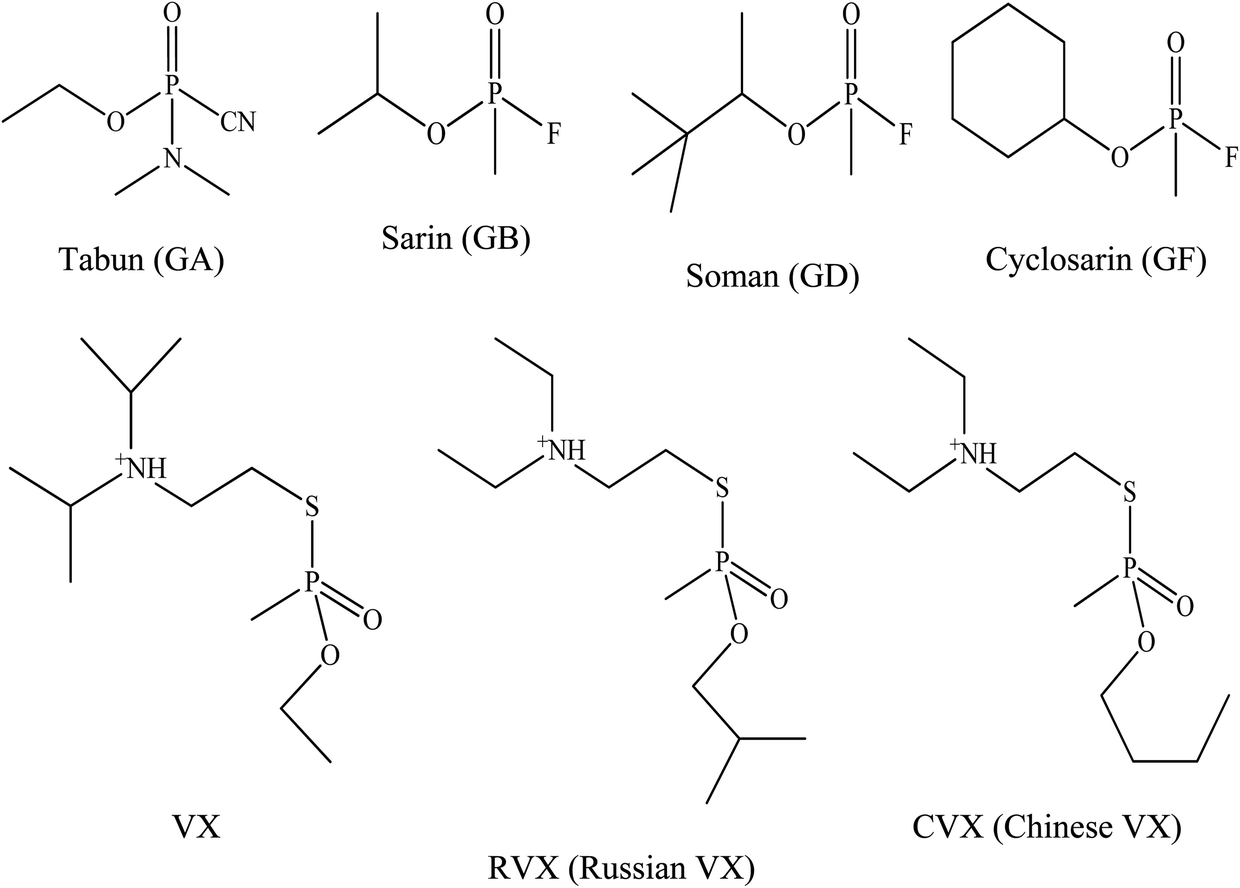 | ||
| Fig. 1 Chemical structures of organophosphorus nerve agents. | ||
OP nerve agents need to be recognised promptly. OP nerve agents are usually described as clear, colourless, tasteless, and odourless liquids and gases.34 They have demonstrated their ability to penetrate normal clothing and the skin. Furthermore, these agents are highly toxic when compared to biological agents with a far more rapid onset of signs and symptoms. Most G-series nerve agents are highly volatile and can be dispersed as aerosols that are inhaled by victims which in turn, absorbed into the body through the pulmonary blood circulation. Nerve agents, especially the V-series may also be disseminated in liquid form. Thus, the treatment for dermal exposure from the liquid dissemination begins with rapid topical decontamination.
The overall treatment approach to nerve agent exposure focuses on airway and ventilatory support, the aggressive use of antidotes, prompt control of seizures, and decontamination which are deemed necessary.
The normal mechanism of AChE is shown in Fig. 2(A). During its normal function, the active site of AChE catalyses the hydrolysis of ACh into choline and acetic acid. However, inhibition of AChE function leads to excessive amount of ACh, accumulating at the neuron synapse. This increased amount of ACh causes several signs and symptoms of poisoning as described in the introduction section. The mechanism of toxicity is identical to the one observed in OP exposure.35
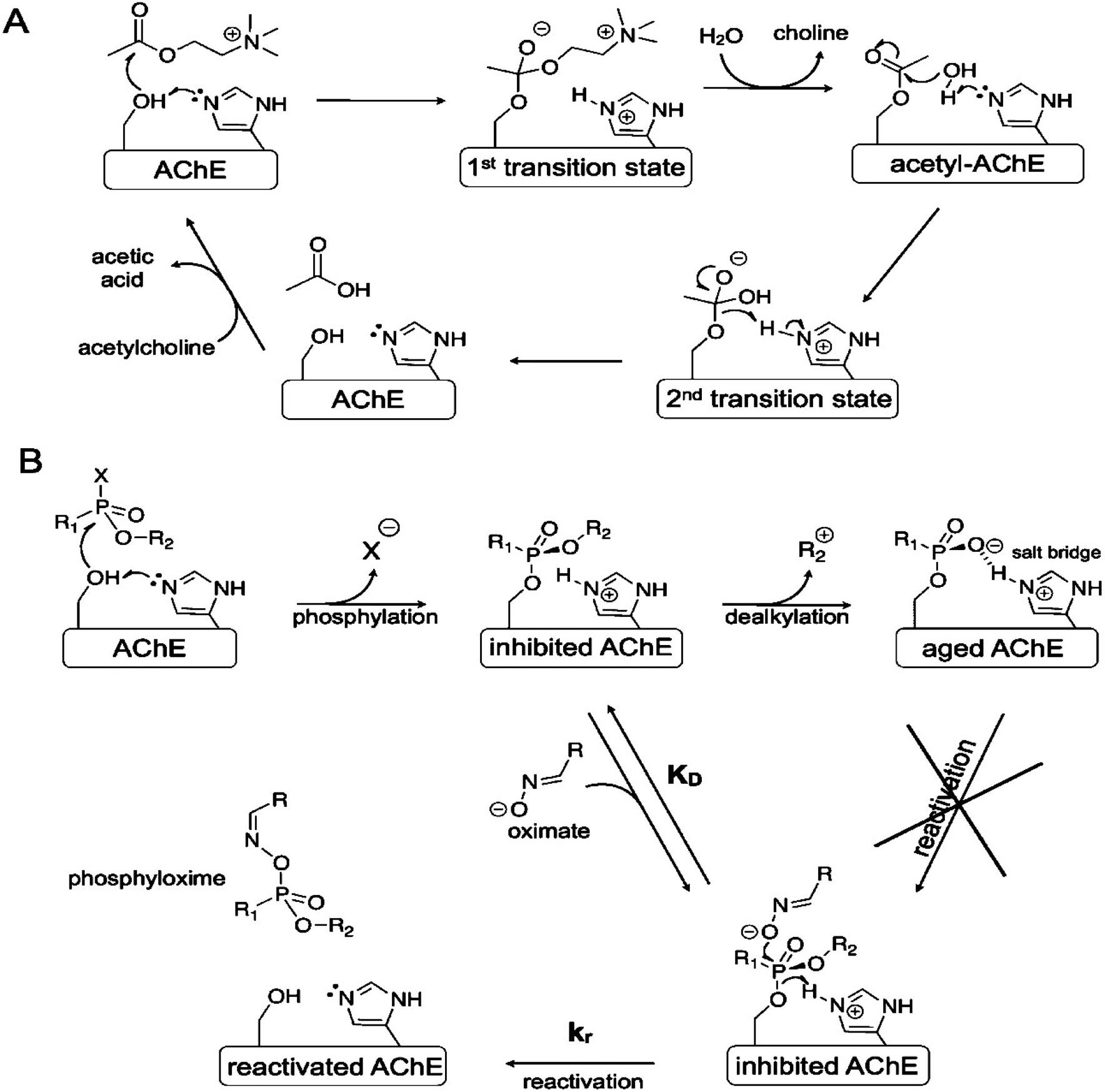 | ||
| Fig. 2 Inhibition of AChE. (A) Normal inhibition function of AChE; (B) inhibition of AChE by OP. This figure has been reproduced from ref. 37 with permission from Copyright (2012) American Chemical Society. | ||
The mechanism of inhibition of AChE by OP nerve agents is shown in Fig. 2(B). This inhibition action involves successive chemical events. The first is phosphorylation of the active site serine to produce a neutral adduct, which is a close structural analogue of the acylation transition state. This adduct is unreactive towards spontaneous hydrolysis. However, it can be reactivated by nucleophilic medicinal agents, such as oximes. Unfortunately, the initial neutral adduct is then converted to a monoanionic aged adduct via a dealkylation reaction, as shown in Fig. 2(B). This aged adduct is totally unreactive. Several efforts have been made to revert the function of the adduct but to no avail.36 Therefore, there is no medicinal agents available yet to reactivate the aged AChE.
Even when the OP-inhibited AChE can still be reactivated, unfortunately current available antidotal treatments are still not able to effectively counteract the acute toxic effects of OP. This is because of the limited ability of oximes to reactivate OP-inhibited AChE, especially in the case of acute poisoning with tabun, soman and cyclosarin.15 Tabun is one of the most resistant types of nerve agents due to its deleterious effects which are extraordinarily difficult to antagonise due to changes in hydrogen bonding and conformational changes of the AChE–tabun complex at the AChE active site.15,38,39
Oximes
Oximes are well known substances that have antidotal potency which is attributed to their ability to reactivate OP-inhibited AChE.40,41 In early studies by Wilson et al. (1955), they demonstrated that the site direction of nucleophilic oximes was quaternary cationic, which is found in the pyridinium aldoxime family.42 These are polar organic compounds with large negative lipophilicity values.35In the last five decades, five pyridinium oximes have been found to be worthy of use as antidotes against nerve agents in humans.43,44 Monopyridinium and bispyridinium aldoximes have been found to be effective in reactivating OP-inhibited AChE. However, the monopyridinium aldoximes are weak AChE reactivators compared to bispyridinium aldoximes.39 The bispyridinium oximes exhibit superior reactivation properties than monopyridinium oximes due to the fact that the second cation interacts with the peripheral anionic site of AChE, which increases the binding affinity.45
There are five common pyridinium aldoximes used to treat OP poisoning as shown in Table 1. Pralidoxime (2-PAM) is the most commonly used oxime whereas a number of other oximes are also available such as obidoxime (LüH-6), trimedoxime (TMB-4), HI-6, HLo-7, and methoxime (MMB-4). 2-PAM is currently the only approved oxime reactivator by the United States Food and Drug Administration (FDA).46
| Compound | Structure |
|---|---|
| Pralidoxime (2-PAM) | 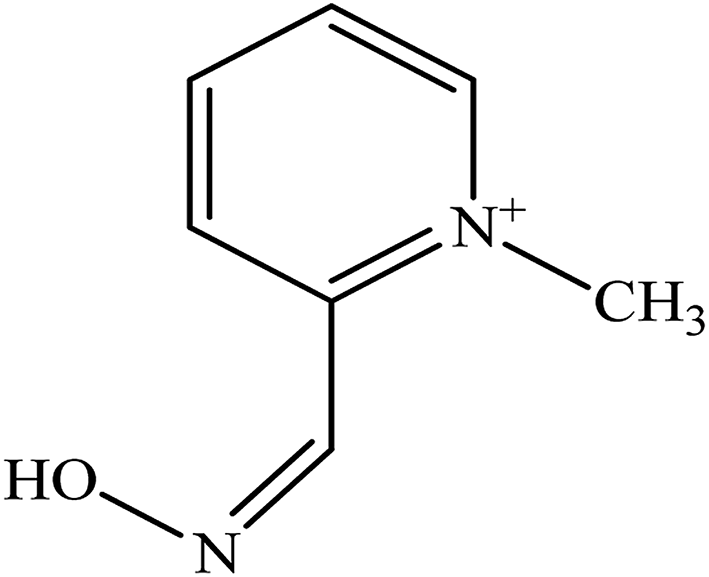 |
| HI-6 | 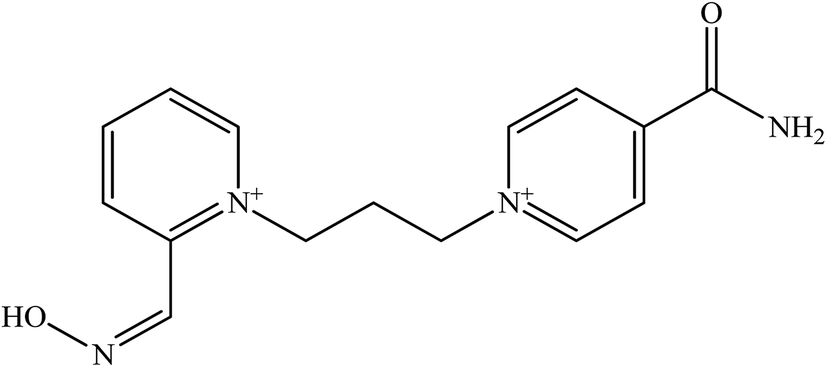 |
| Metoxime | 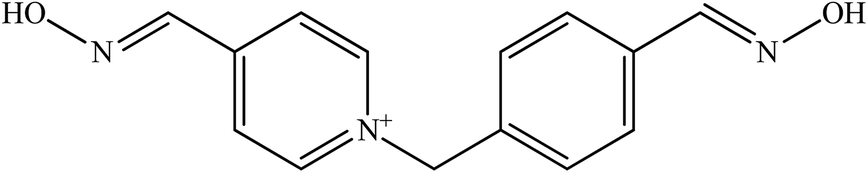 |
| Obidoxime | 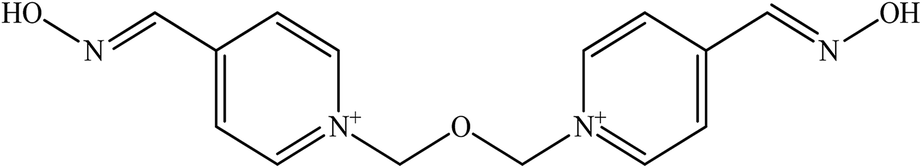 |
| Trimedoxime | 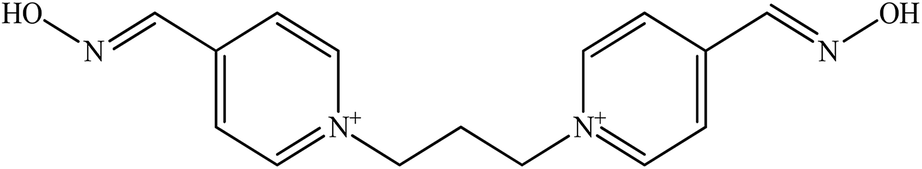 |
Currently, none of the available oximes are considered as real broad-spectrum reactivators for the several types of OP.47 Only two oximes, 2-PAM and obidoxime, are commercially available antidotes against CWAs.48 Most of these oximes are sufficiently effective to reactivate sarin- or VX-inhibited AChE, but their potency to reactivate soman- or tabun-inhibited AChE is generally low.49,50 2-PAM and obidoxime were also reported to be less effective against soman, cyclosarin, and Russian VX.51,52 Trimedoxime has been shown to have better efficacy in treating poisoning with tabun.53 HI-6 has since been considered as one of the better quaternary oximes for OP nerve agent poisoning so far54 as it was the first quaternary oxime that demonstrated reactivation efficiency for soman-inhibited AChE.19 However, it is not effective against tabun poisoning.52,53
The limited effectiveness of these established oximes has resulted in continuing research to develop several more novel oximes for the reactivation of OP-inhibited AChE.55 Table 2 shows examples of several newly developed oximes in attempt to improve the antidotes efficacy. For example, the oximes K 027 and K 048 are currently being tested in many countries because of their extraordinary potency and low toxicity.30 Oxime K 027 showed promising reactivation potency for tabun- and paraoxon-inhibited AChE. While the oxime K 048 has shown greater reactivation ability towards tabun-inhibited AChE compared to 2-PAM and its efficacy is comparable to obidoxime.
| Compound | Structure |
|---|---|
| K 027 |  |
| K 048 | 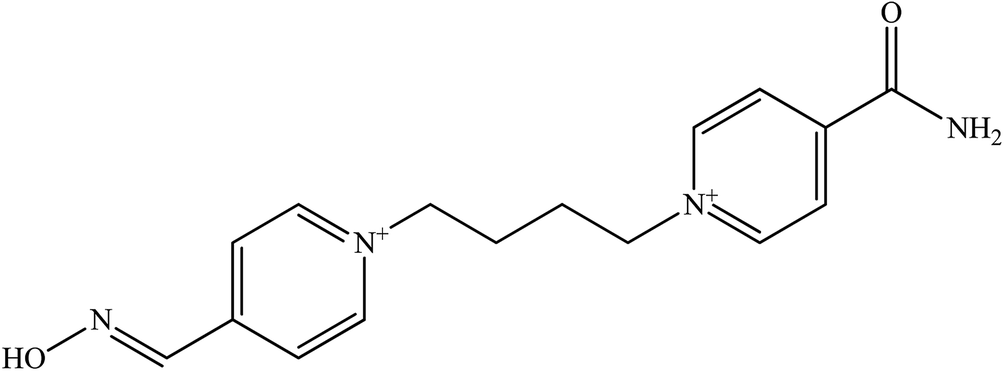 |
| K 074 | 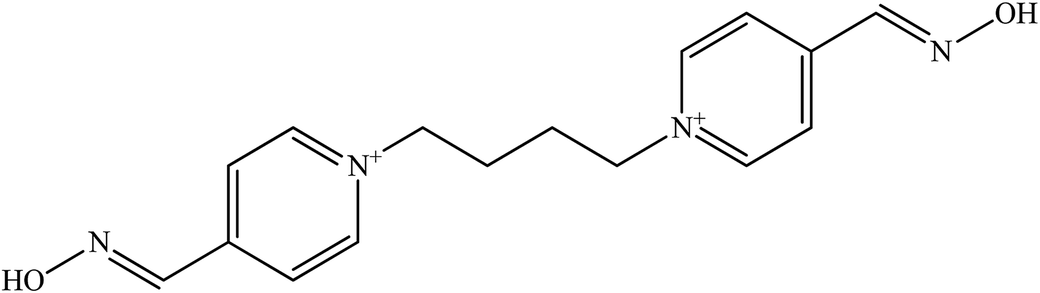 |
| K 075 | 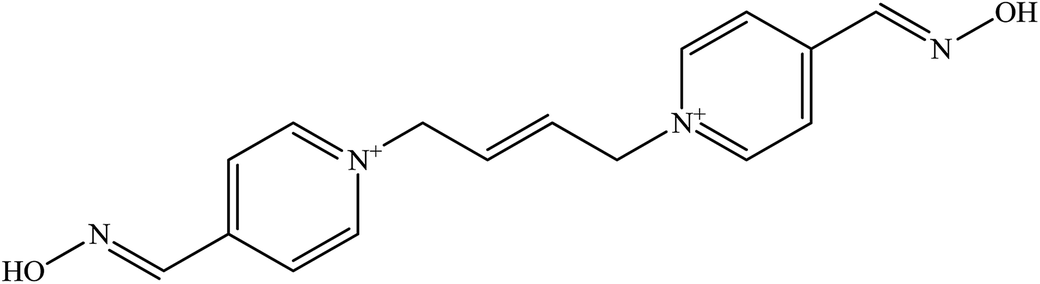 |
| K 156 | 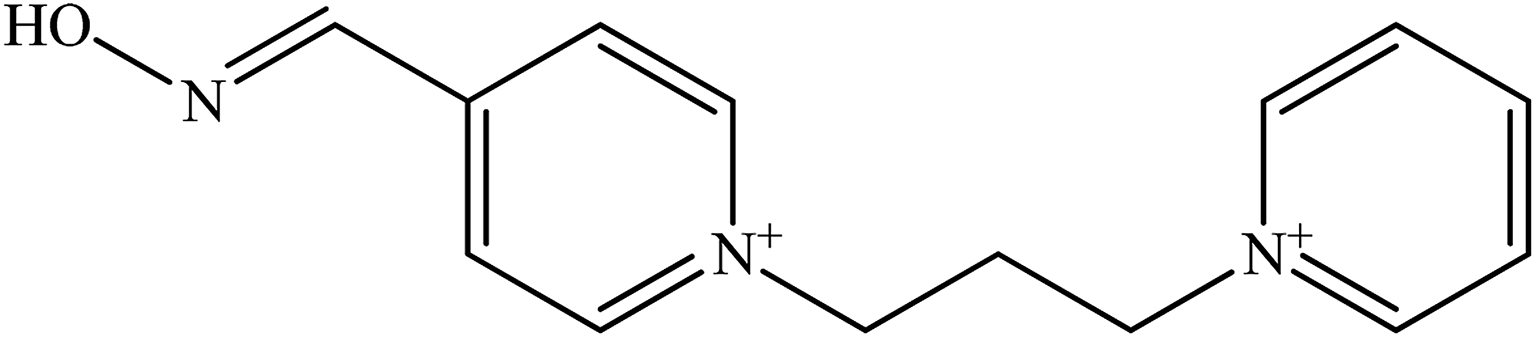 |
| K 203 | 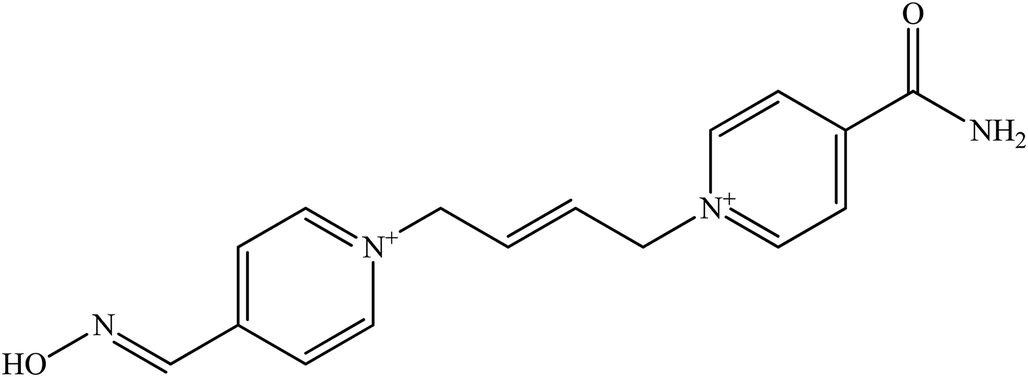 |
| 4-PAPE | 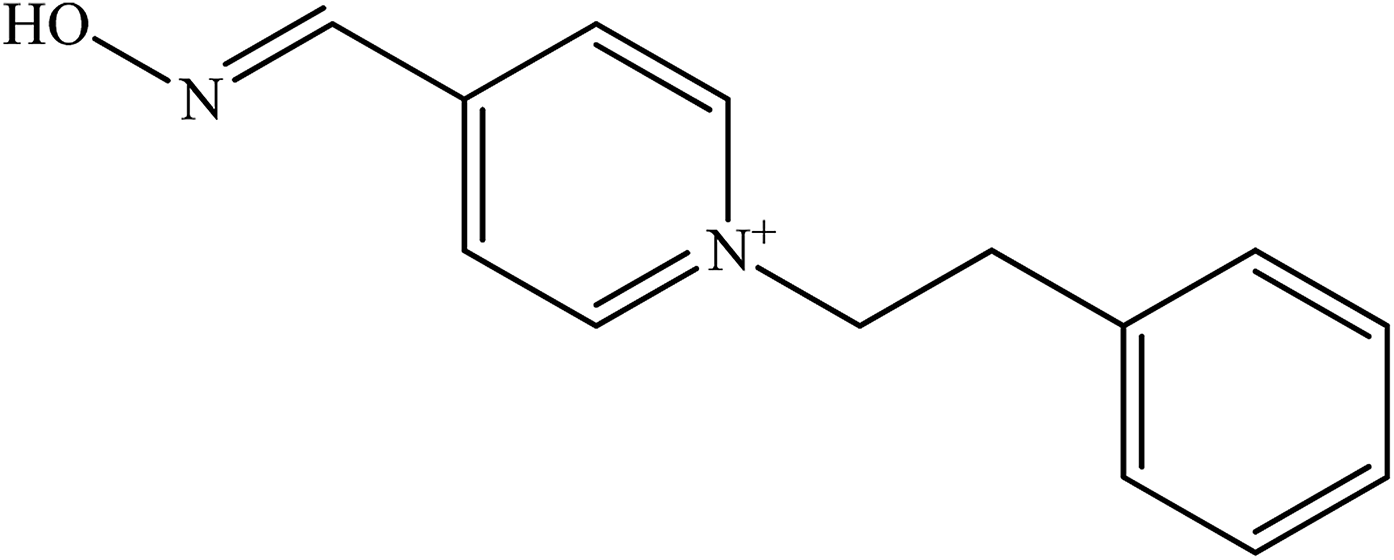 |
| 4-PAO | 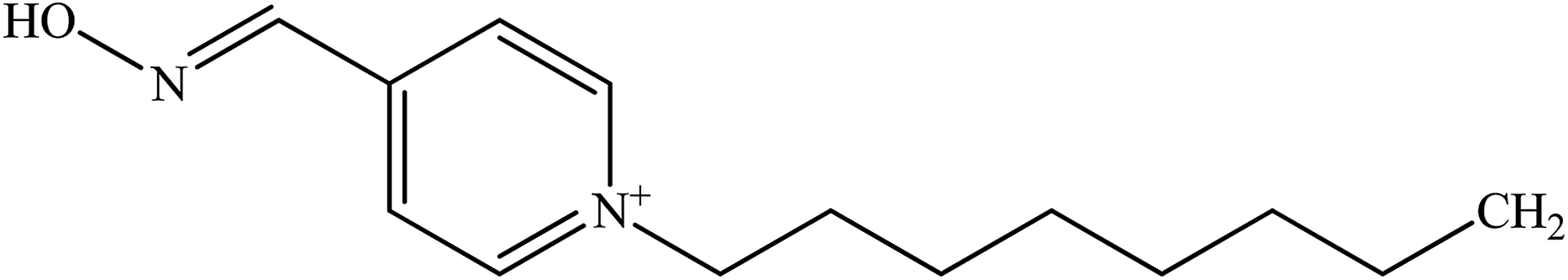 |
The oxime mechanism of action involves the reactivation of the phosphorylated AChE by displacing the phosphoryl moiety from the enzyme which has a very strong nucleophile as shown in Fig. 2. The reactivation rate is dependent on the structure of the phosphoryl moiety, source of enzyme, concentration of oximes, aging rate, and the stearic hindrance effects between oxime molecule and the phosphoryl moiety attached to the active site of AChE.4,11,36,43
Since the pyridinium oximes are polar organic compounds with large negative lipophilicity values, they are difficult to penetrate the BBB. Currently, several approaches are being considered in order to improve the potency of oximes to reactivate AChE. This includes the efficiency of oximes to penetrate the BBB which consequently results in the AChE reactivation in the CNS.30 Subsequently, we will discuss the issues related to BBB penetration by oximes.
Blood brain barrier
The BBB is formed by the brain's microvascular system which is a membrane barrier that separates blood from the extracellular fluid of brain within the CNS. The BBB is the reason for the high attrition rate of the treatments for many brain disorders, which then makes it one of the greater healthcare challenges in the twenty first century.56 Almost all macromolecule and small molecule drugs are unable to pass through the BBB.30,57 A schematic diagram of the BBB is shown in Fig. 3.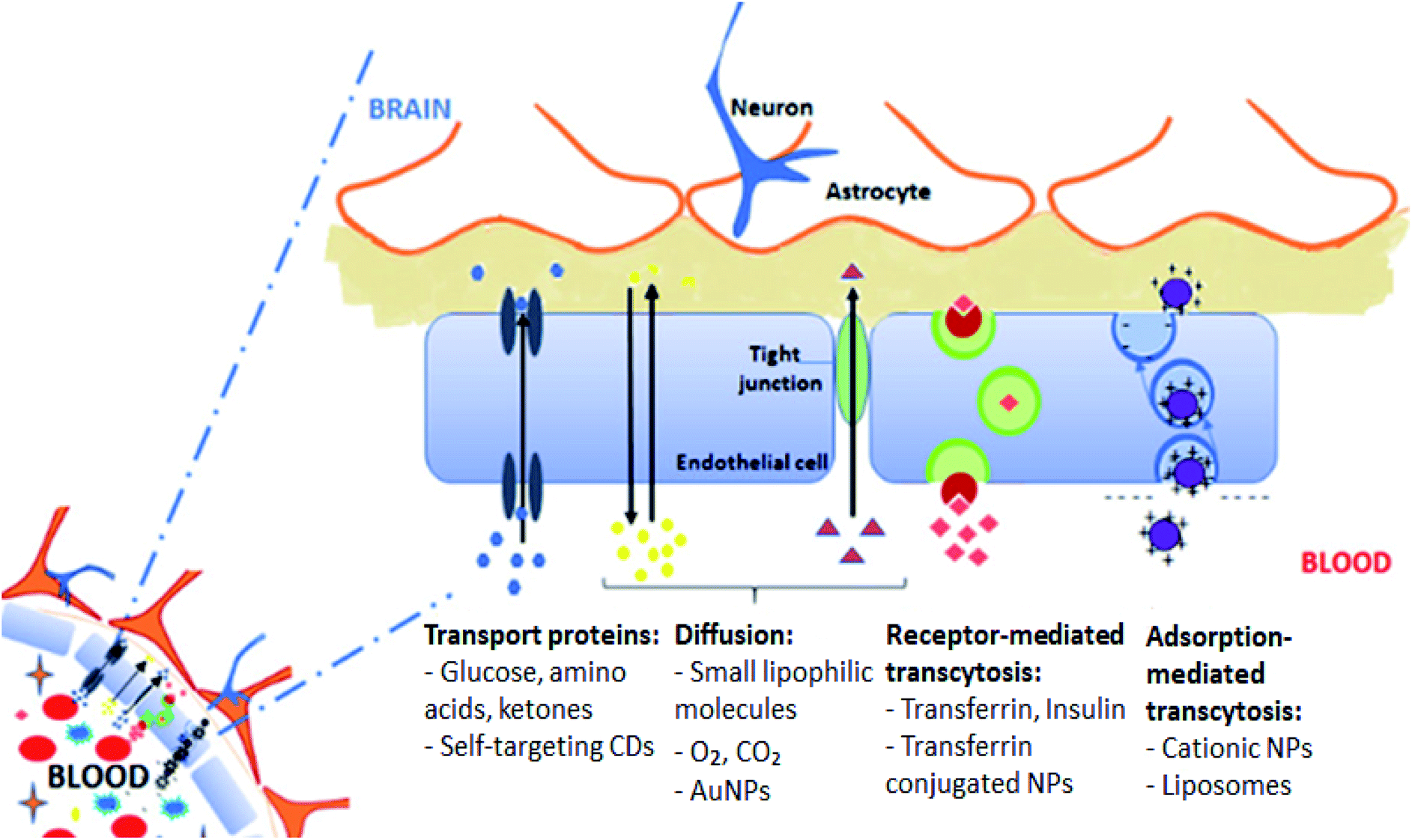 | ||
| Fig. 3 The blood brain barrier. This figure has been reproduced from ref. 61 with permission from Elsevier. | ||
Certain small molecule drugs may cross the BBB via lipid-mediated free diffusion, providing that drug has a molecular weight of less than 400 daltons, forms not more than 8 hydrogen bonds and is relatively uncharged molecule.11,26 Moreover, active drug transport is regulated by specific BBB transporters such as P-glycoprotein (Pgp).29 The transporter is overexpressed on the surface of the endothelial cells in the BBB and restricts cell entry. This transporter actively lowers the bioavailability of Pgp substrates in the brain. The Pgp helps in protecting susceptible organs from toxic compounds, preventing these compounds from entering the cytosol and extruding them to the exterior.58 Many charged drugs, including oximes and cytotoxic drugs for cancer treatment, are Pgp substrates and therefore unable to reach a sufficient availability at their site of action inside the brain.59,60
According to Korabecny et al. (2014), only 10% of mono-pyridinium 2-PAM was found to be able to cross the BBB, whereas bis-pyridinium oximes are able to penetrate at a lower concentration, approximately 1–3%.11 The concentration of obidoxime and HI-6 however were able to pass through the BBB at an estimated concentration of 3–5% and 10%, respectively.4 Monoisonitrosoacetone (MINA) is the only known BBB-penetrating oxime commercially available.62 Moreover, Karasova et al. (2011) evaluated the penetration of trimedoxime and oxime K 074 into different brain regions where it was found that the permeation of trimedoxime and oxime K 074 into the brain was very minimal, with only sporadic low reactivation activity expected to occur.63 The levels of these oximes in the brain were only found to be 0.58% for trimedoxime and 0.85% for oxime K 074. The concentration of oximes was highest in the frontal cortex region and lowest in the basal ganglia.
It is important to note that oximes are hardly penetrate the BBB, while OP nerve agents on the other hand can easily do so.19,62,64 This is because the OP nerve agents are small lipophilic molecules which can easily penetrate the BBB by free diffusion.
Joosen et al. (2011) and Shih et al. (2011) reported that the amount of oxime sufficient to reactivate inhibited AChE in order to provide survival in cases of exposure to OP nerve agents is uncertain.59,65 Thus, it may be postulated that even a slight increase in oxime concentration in the brain can contribute to significant enhancement of therapeutic efficacy and would improve medical countermeasure effectiveness.59 This could potentially reduce the actual dosage of oximes into the body which would decrease the toxicity side effects of oximes. It is known that when AChE reactivation was achieved in the CNS, good therapeutic effects of oximes and survival of poisoned organisms were observed.66
Therefore, several strategies were developed to increase the efficiency of BBB penetration by oximes such as through the development of new structure oximes, uncharged oximes, conjugation of uncharged oximes, and sugar oxime moieties.
Strategies to improve the blood brain barrier penetration of oximes
Previous researches had demonstrated the superiority of charged 2-PAM as a reactivator of inhibited AChE in the CNS but its penetration level across the BBB is still considered low. In this review, there are several approaches has been outlined in delivering the target oximes to the CNS. According to Patel et al. (2013), these approaches can be classified as follows:67(a) Direct delivery of the drug to the CNS. This is usually done by injection, convection enhanced delivery, or the use of biodegradable delivery systems (e.g., polymer wafers, nanoparticles, liposomes).
(b) Improving delivery across an intact BBB using non-specific mechanisms such as by manipulating the lipid solubility or through specific mechanisms (e.g., receptor-mediated and adsorptive transcytosis, modulation of transport systems such as Pgp).
(c) Using targeting technologies to increase local concentrations of drugs in the brain after systemic delivery, which include magnetic and immunological targeting, as well as ultrasound assisted drug release.
However, according to Hersh et al. (2016), direct injections into the brain, local exposure to high-intensity focused ultrasound, chemical disruption and osmotic opening by hypertonic mannitol have several disadvantages such as non-specific, low efficiency, costly and conflicting results in clinical trials.68 Hence, several other approaches are being investigated to find the most suitable method for drug penetration through the BBB into the CNS.
In this review, recent approaches to enhance oxime BBB penetration are classified into three strategies:
(a) Development of new structural oximes.
(b) Development of uncharged oximes.
(c) Oxime-lipophillic compound conjugations.
To the best of our knowledge, the development of uncharged oximes has received much attention among researchers when compared to the other approaches.
Development of new structural oximes
The molecular structure and molecular weight of a compound plays an important role in BBB penetration. It is expected that the BBB penetration of new developed structural oximes will be improved than the current available oximes as shown in Fig. 4.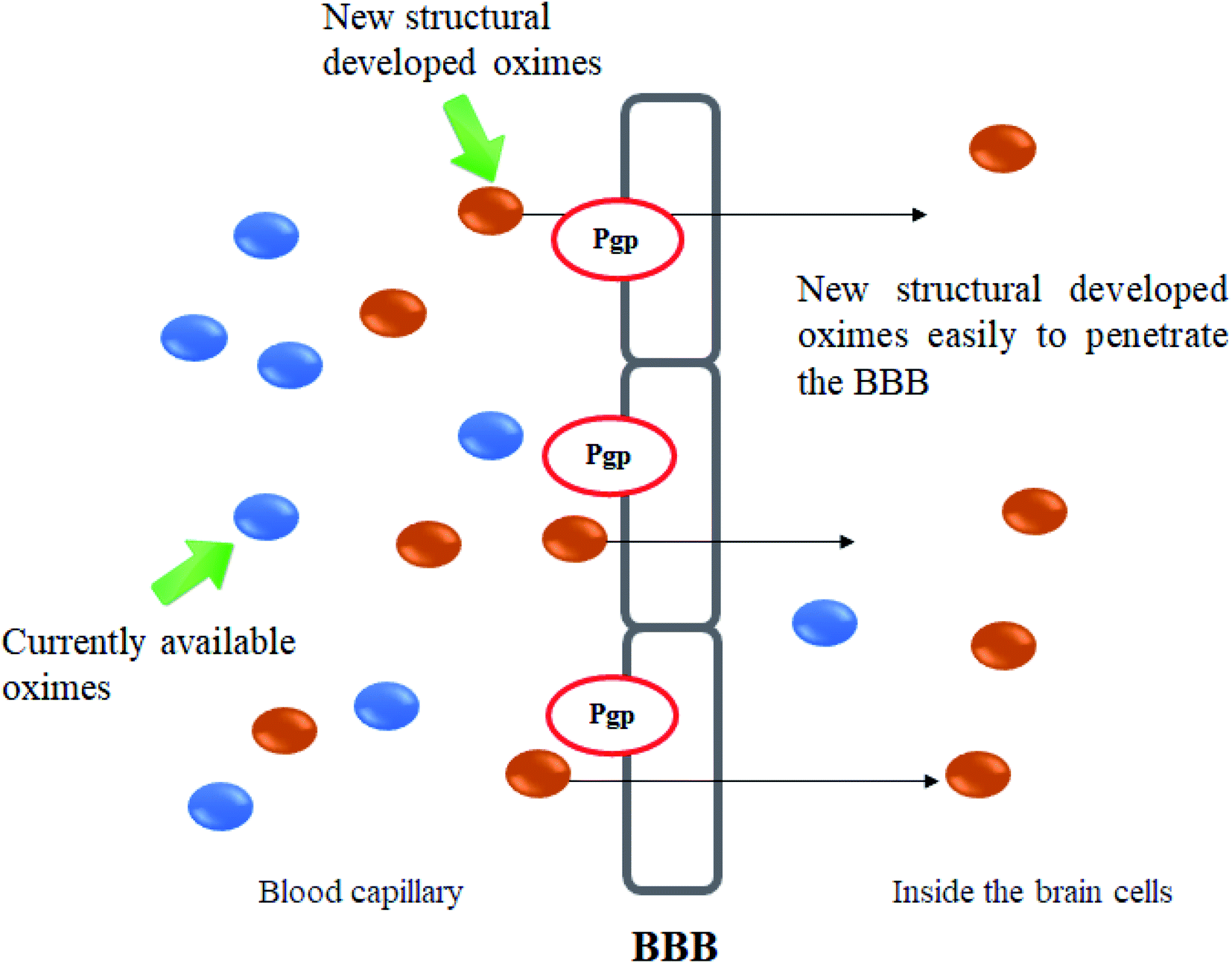 | ||
| Fig. 4 Schematic diagram of BBB penetration of new structural oximes. | ||
Previously, the alternative development was of the prodrug for 2-PAM that would undergo oxidation within the CNS to produce a functionally active quaternary oxime (see Fig. 5).69 It was reported that the pro-2-PAM was the first non-invasive means of administering a CNS therapeutic agent against OP nerve agent poisoning. The OP nerve agent exposed-guinea pigs showed a significant reduction in neurological damage when treated with pro-2-PAM. However, this approach has two apparent disadvantages; (1) the synthesis of the prodrug is inherently difficult, and (2) the pro-2-PAM is unstable and subjected to auto-oxidation.62
 | ||
| Fig. 5 Chemical structure of pro-2-PAM. | ||
Karasova et al. (2010) reported on the influence of the molecular structure and molecular weight of several different types of oximes upon BBB penetration (Fig. 6).63 Firstly, they found that the monoquaternary oximes had a ten to fifteen-fold higher BBB penetration compared to bisquaternary oximes. This is due to the presence of only one quaternary nitrogen in each molecule of the novel oxime and their lower molecular weight when compared to the bisquaternary and triquaternary oximes. Besides that, monoquaternary oximes are known to undergo active transport through the BBB. In contrast, there are no known transport systems available or suitable for bisquaternary oximes.
 | ||
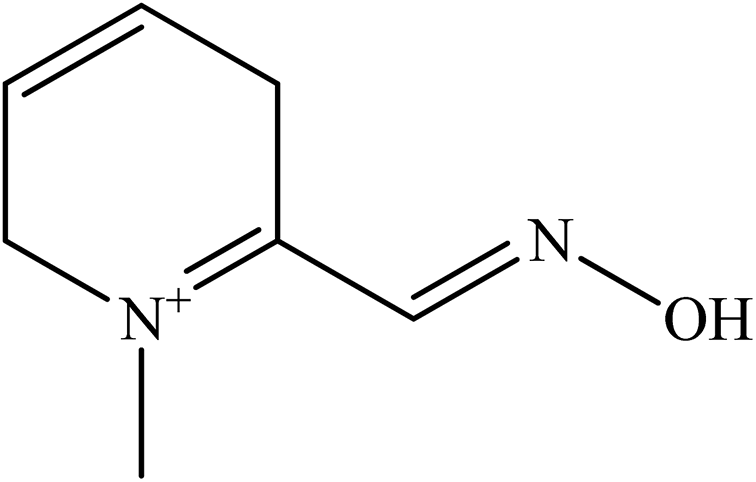
| Fig. 6 List of oximes as reported by Karasova et al. (2010).63 | ||
Secondly, the position of the oxime group on the pyridinium ring also influences the penetration of oximes. For monoquaternary structures (pralidoxime, oxime 18 and 19), as shown in Fig. 6, they have the best penetration with the para position (oxime 19) on the pyridinium ring. Similarly, some bispyridinium oximes are also found to behave in the same manner (oxime 10, 12, obidoxime or 15, 16, trimedoxime or 2, 7).
Thirdly, molecular weight also considered as an important factor for passive transport. The link connecting the two pyridinium rings also strongly influences the BBB. A simple, short link oxime (metoxime) tends to facilitate permeation better than other oximes. The extension of the carbon link is usually accompanied by a reduced ability to penetrate the BBB (compare oximes: 13, trimedoxime, metoxime or 8, obidoxime).
Moreover, substituent molecules such as xylene (oxime 7 and 8), double bond compounds (oxime 11 and 13) and oxygen (obidoxime and trimedoxime) all significantly reduce permeation through the BBB. If the cis/trans butane configuration is looked at, it is known that the cis isomer is able to penetrate into the CNS better than the trans isomer. Increasing the number of oxime groups has been found to have a reduced effect on predicted diffusivity (especially the HLö-7, HI-6, obidoxime and oxime 17). The type of substituents (as well as its position on the pyridinium ring) is also a factor that influences the predicted BBB diffusivity. For example, if the substitution of the oxime group is done using a carbamoyl group or amidoxime group, this markedly decreases the predicted penetration of AChE reactivators into the CNS (oxime 4, 6 and 11). This phenomenon is clearly seen when the oxime group is replaced by a carbamoyl group (oxime 3, 8 or 5, 14 or obidoxime, 9).
Oxime 1 has the worst ability to penetrate the BBB as it has three quaternary nitrogen, a branched link and three hydrophilic oxime groups. The authors propose that the design of a BBB penetrating oxime should consist of short carbon link (C1–C2) in the absence of additional functional groups and preferably with one or two oxime groups in the para position.
Chambers et al. (2016a and b), developed novel substituted phenoxyalkyl pyridinium oximes as shown in Fig. 7.18,46 These phenoxyalkyl pyridinium oximes have alkyl chains of 3–5 carbons, with various substitutions on the phenoxy group. Although it contains a quaternary ammonium, similar to other pyridinium oximes that do not cross the BBB, it is hypothesised that the phenoxyalkyl moiety could increase the lipophilicity of the oxime thus providing counterbalance to the positive charge from the quaternary ammonium, hence allowing BBB penetration. Based on in vitro studies, these five oximes have shown about 40–65% reactivation ability which is lower than that of 2-PAM (which is capable of 88–91% reactivation). Thus, all these oximes have some potential for reactivation, although with a lower potency than 2-PAM, with some approaching the efficacy of 2-PAM. The more effective novel oximes together with the 2-PAM were then screened in an in vivo study. Interestingly, all the oximes tested caused a reduction of brain AChE inhibition in rats of least 5–25% at 30 min after oxime administration. Meanwhile, treatment with 2-PAM did not cause any reduction in brain AChE inhibition. Oxime 20 exhibited a higher in vivo reactivation potency (25%) towards AChE inhibition. These oximes also showed an attenuation of the seizure-like behaviour among the rats when compared to the ones treated with 2-PAM. Therefore, this indicates that the presence of phenoxyalkyl moiety does seem to augment the anti-convulsive activity oxime, probably due to its increased lipophilicity and therefore results in enhanced BBB penetration.
 | ||
| Fig. 7 Structure of novel oximes as reported by Chambers et al. (2016a and b).18,46 | ||
Meanwhile, it has been described that most of the oximes often lack of reactivating potency towards AChE due to their low affinity towards AChE active site.17 Therefore, the development of a novel conceptual approach to the design of oximes through exploitation of existing knowledge of oxime structure is needed in order to increase the binding affinity between AChE and the oximes. An extra contribution towards affinity may be achieved by covalently connecting a peripheral site ligand (PSL) to an oxime, which may then result in a higher reactivation potency of the total construct (Fig. 8).17
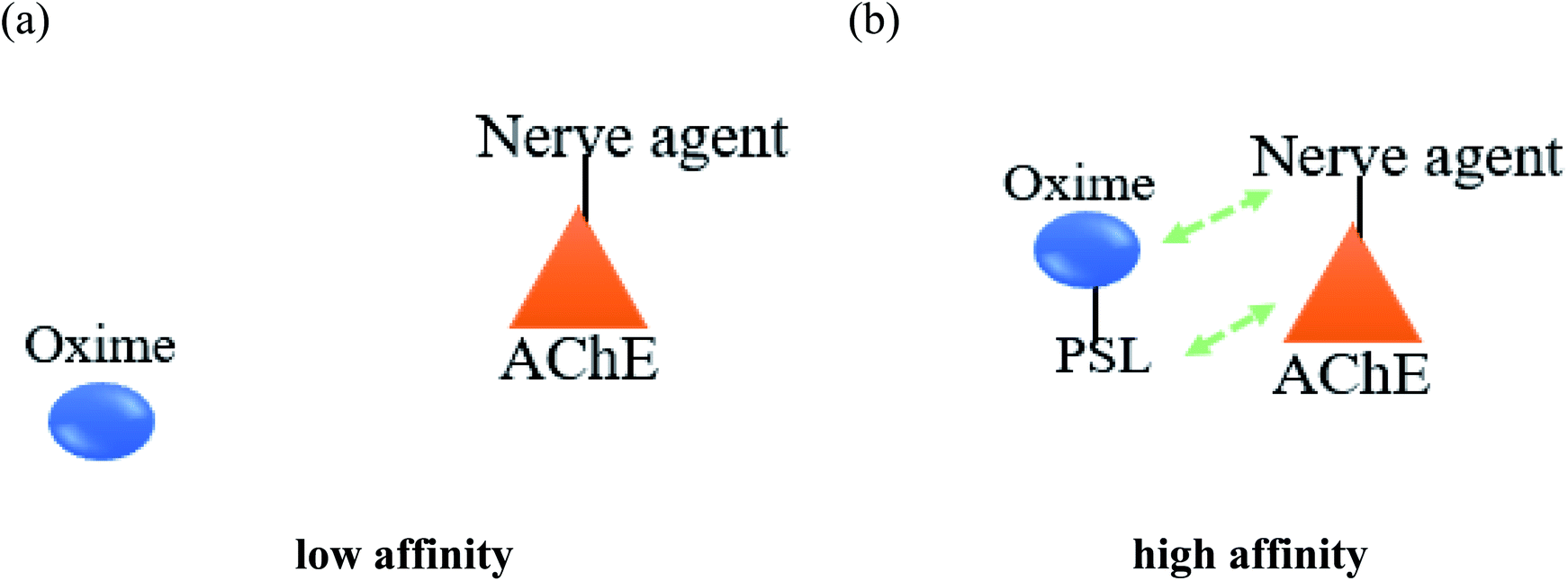 | ||
| Fig. 8 Schematic diagram of the interaction between inhibited AChE and a PSL conjugated oxime. | ||
Koning et al. (2011) developed the first steps towards the concept of PSL conjugation to pyridinium oximes in order to improve the reactivation potency of inhibited AChE and enhance BBB penetration.17 Three series of oximes containing PSL are shown in Fig. 9. Based on binding affinity analysis, oximes 21–23 show the highest affinity for AChE when compared to the control, 4-PAM. In the reactivation potency analysis, these oximes would be able to reactivate sarin- and VX-inhibited AChE and show higher reactivation potency than 4-PAM and HI-6 at pharmacologically relevant concentrations.
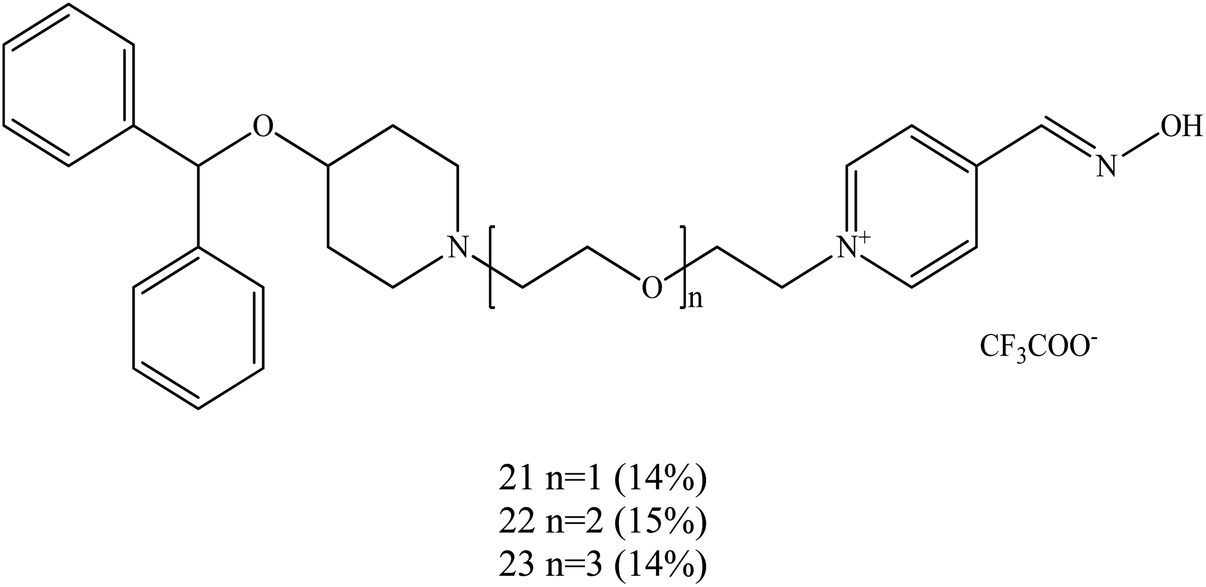 | ||
| Fig. 9 Structure of the oximes reported by Koning et al. (2011).17 | ||
Kalisiak et al. (2011) designed a new class of amidine-oximes.62 These compounds represent a novel group of oximes with enhanced BBB penetration capabilities. An amidine group is frequently used in drugs because of its non-toxic properties, pronounced water solubility, and its action as an efficient hydrogen-bond acceptor. In this study, the amidine group is represented as the PSL to the oxime structure. In the case of amidine oximes, which are shown in Fig. 10, the basic group readily undergoes protonation under physiological conditions to form a positively charged centre, similar to that of a pyridinium quaternary nitrogen. Protonated amidine groups presumably facilitate efficient binding to AChE through cation interactions with aromatic amino acid residues at the AChE active site.
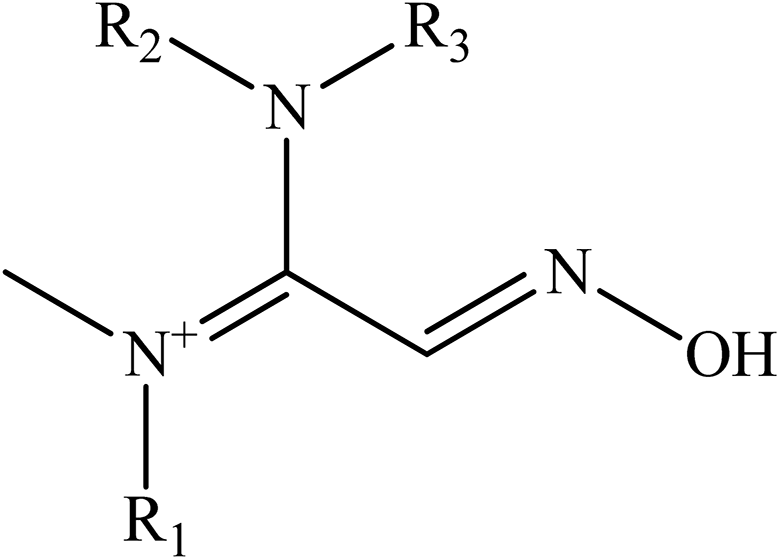 | ||
| Fig. 10 Basic amidine-oxime chemical structure. | ||
In this approach, the amidine moiety supplies a pseudopositive charge responsible for efficient binding to inhibited AChE, while the proximal oxime group is a nucleophile responsible for reactivation of the phosphylated enzyme. Amidine-oximes have several advantageous properties, including:
(i) Improved chemical stability as compared to pro-2-PAM.
(ii) Greatly improved lipophilicity as compared to quaternary oximes.
(iii) Increased in vitro reactivation efficacy of OP-inhibited AChE as compared to MINA.
(iv) Significant protection from the in vivo toxic effects of OP.
It was reported that the ability of oximes 24–28 to reactivate OP-inhibited AChE was attributed due to the superior functional activity compared to MINA (Fig. 11). The substituent on the amidine group of amidine-oximes moves from the hydrogen atom in oxime 24 to the N-butyl group in oxime 28 thus increasing its lipophilic character and therefore increasing reactivation potency. Amidine-oximes, 26, 27 and 28 were found to be the most lipophilic compounds. Despite the fact that the amidine-oximes examined did not afford better reactivation potency than 2-PAM for OP-inhibited AChE, two amidine-oximes were found to be efficacious in vivo. For example, pre-treatment with the amidine-oximes 26 and 27 was found to be able to protect mice from toxicity after their exposure to OP.
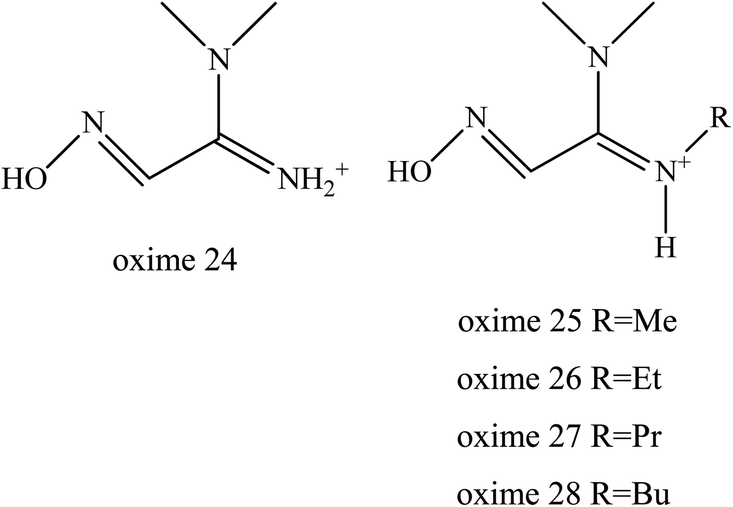 | ||
| Fig. 11 Amidine-oxime moieties. | ||
Moreover, in a post-treatment experiment, animals were exposed to the sarin nerve agent and then treated 5 minutes later with 27 or MINA. Subsequently it was found that all mice treated with the amidine-oxime survived and exhibited only minor signs of CNS toxicity, whereas in the case of animals treated with MINA, only 50% of these animals survived. Even though amidine-oximes are less potent than 2-PAM in the reactivation of OP-inhibited AChE in vitro, they are more lipophilic and achieve better penetration than 2-PAM into the CNS. These amidine-oximes appear to effectively protect against toxic OPs whether used as pre- or post-treatment.
Kalisiak et al. (2012) continued to design and synthesise a new class of cyclic amidine-oximes.62 The presence of the cyclic amidine-oximes is expected to have a higher interaction potency with AChE which was also noted earlier by Sauvaître et al. (2007).70 These recently developed amidine-oximes were tested for in vitro and in vivo for reactivation of sarin inhibited AChE. Fig. 12 shows the structure of the synthesised cyclic amidine-oximes 29, 30, 31, 32, and 33. Based on an in vitro reactivation study, all of the aforementioned amidine-oximes showed greater reactivation potency towards sarin-inhibited AChE compared to MINA, but were still less potent to 2-PAM. In the in vivo study, mice without amidine-oxime resulted in 6 out of 11 animals tested. Interestingly, post-treatment with all of these amidine-oximes successful protected all the mice that were poisoned with sarin. There were no seizure symptoms observed among the treated mice even after 24 hours. Thus, it was thus proven that the development of these uncharged amidine-oximes not only improved their BBB penetration, but also increased the binding affinity between the oxime and the inhibited AChE.
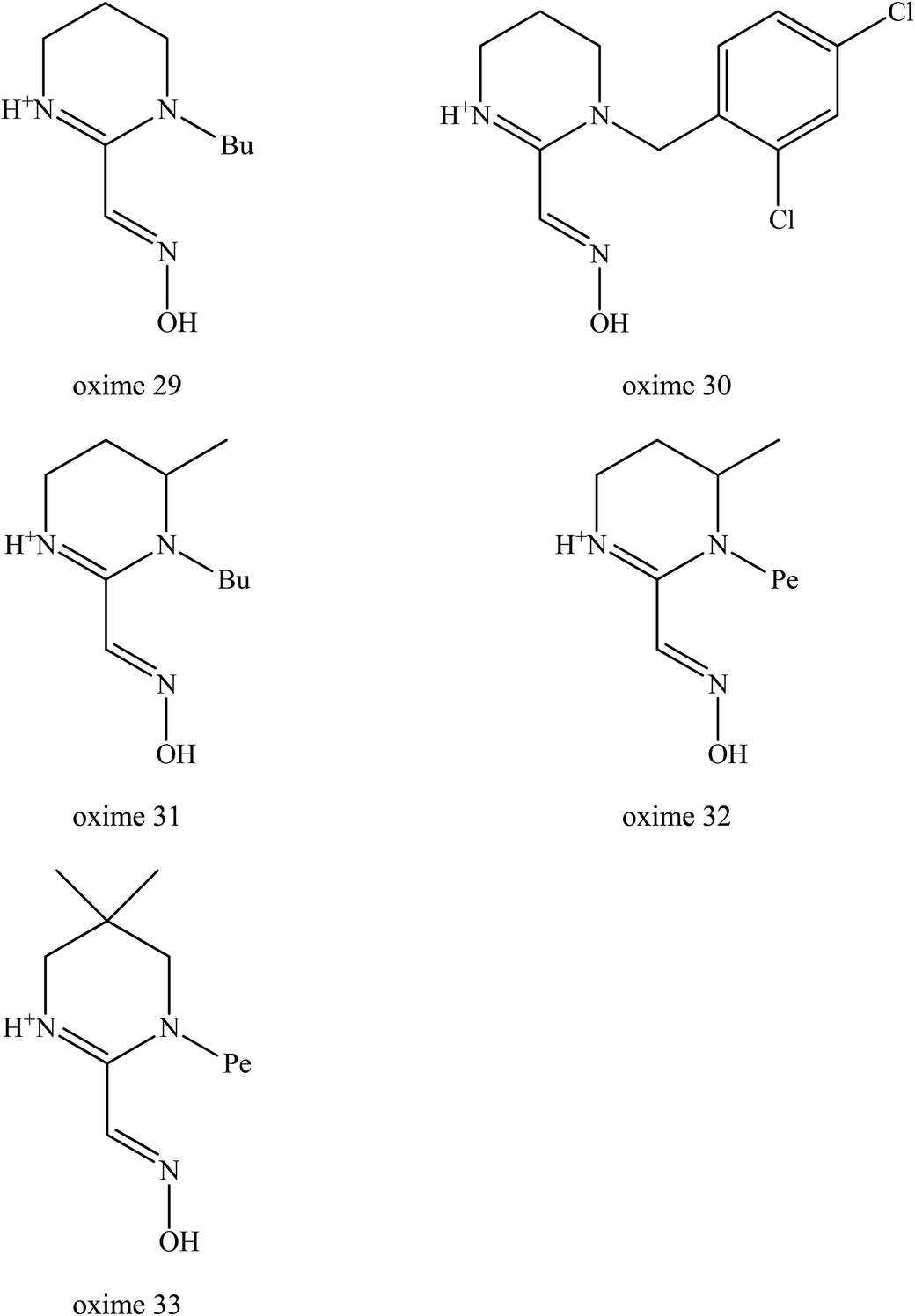 | ||
| Fig. 12 Chemical structure of amide-oximes developed by Kalisiak et al. (2012).62 | ||
Development of uncharged oximes
One of the major disadvantages of oximes is the presence of quaternary (charged) nitrogen that cannot pass through the BBB. Therefore, the development of novel uncharged oximes that are able to move efficiently across the BBB has received much attention by researchers as one of the more promising strategies to improve the effectiveness of oximes as reactivators of AChE. Korabecny et al. (2014) reported that the uncharged oximes represent a new hope for increasing bioavailability in the CNS and thus better therapeutic management of OP nerve agent poisonings.11 The uncharged oximes usually have better BBB penetration due to the increase in lipophilicity. Besides that, the absence of a charge in the oximes not only affects the reactivity of the nucleophilic oxime moiety, but also reduces the affinity of the oxime for the active site of AChE.71 The presence of PSL in the uncharged oximes has also been critically discussed in many reports. Fig. 13 shows the schematic diagram of BBB penetration for charged and uncharged oxime.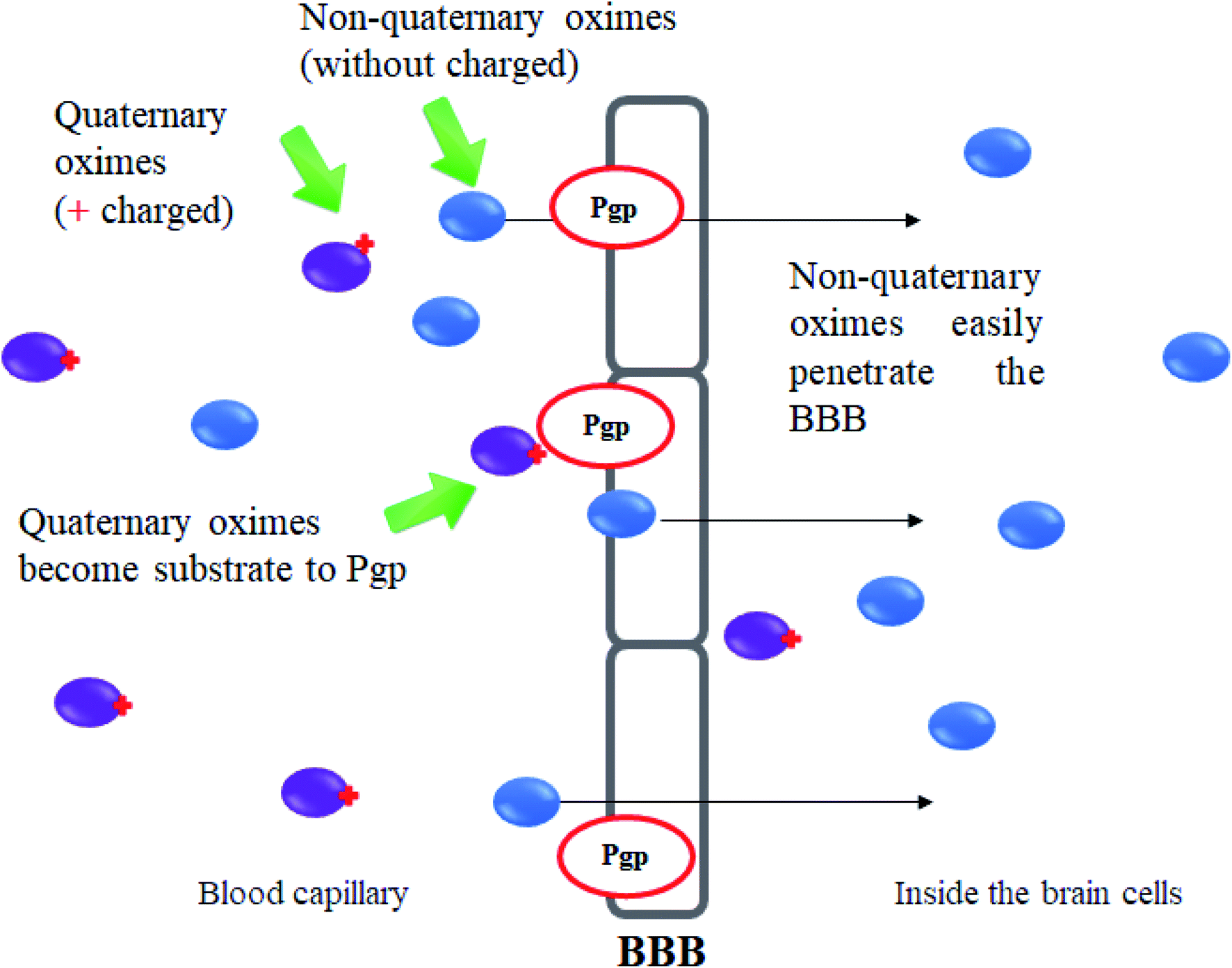 | ||
| Fig. 13 Schematic diagram of BBB penetration of charged and uncharged oximes. | ||
Similar to the earlier section, Koning et al. (2011) first developed the PSL conjugated to an uncharged pyridinium oximes as shown in Fig. 14.17 Their results spurred research in the field of uncharged oximes. The introduction of PSL into the uncharged oximes has not only increased affinity (Fig. 15) of the construct for AChE but also successfully enhanced the reactivation potency of AChE inhibited by sarin, VX and tabun. Although the developed uncharged oximes are still conventional oximes compared to the current pyridinium oximes, this approach holds promise for the future design and synthesis of uncharged oximes with improved BBB penetration and may also be suitable for non-oxime reactivators thus further widening the scope for ongoing research into broad-spectrum reactivators.
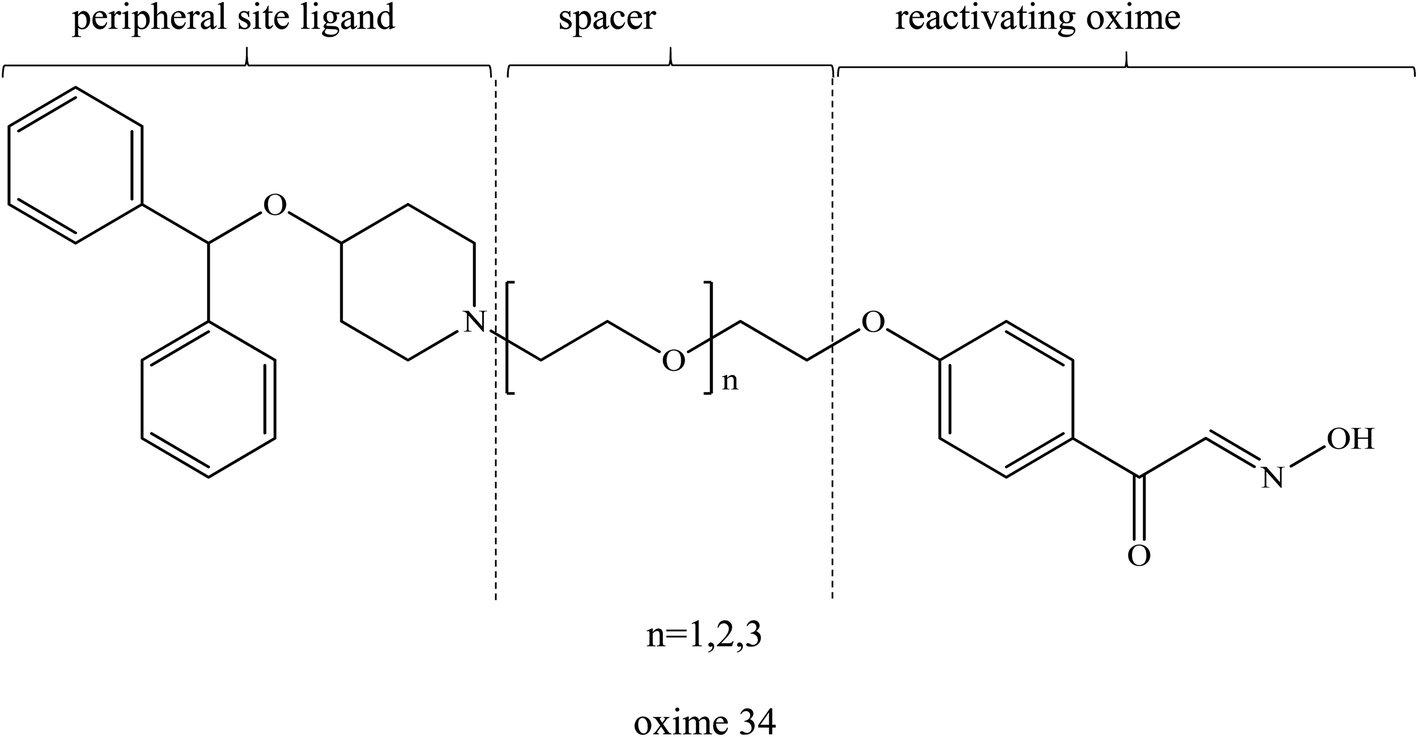 | ||
| Fig. 14 Chemical structure of uncharged pyridinium oxime containing PSL developed by Koning et al. (2011).17 | ||
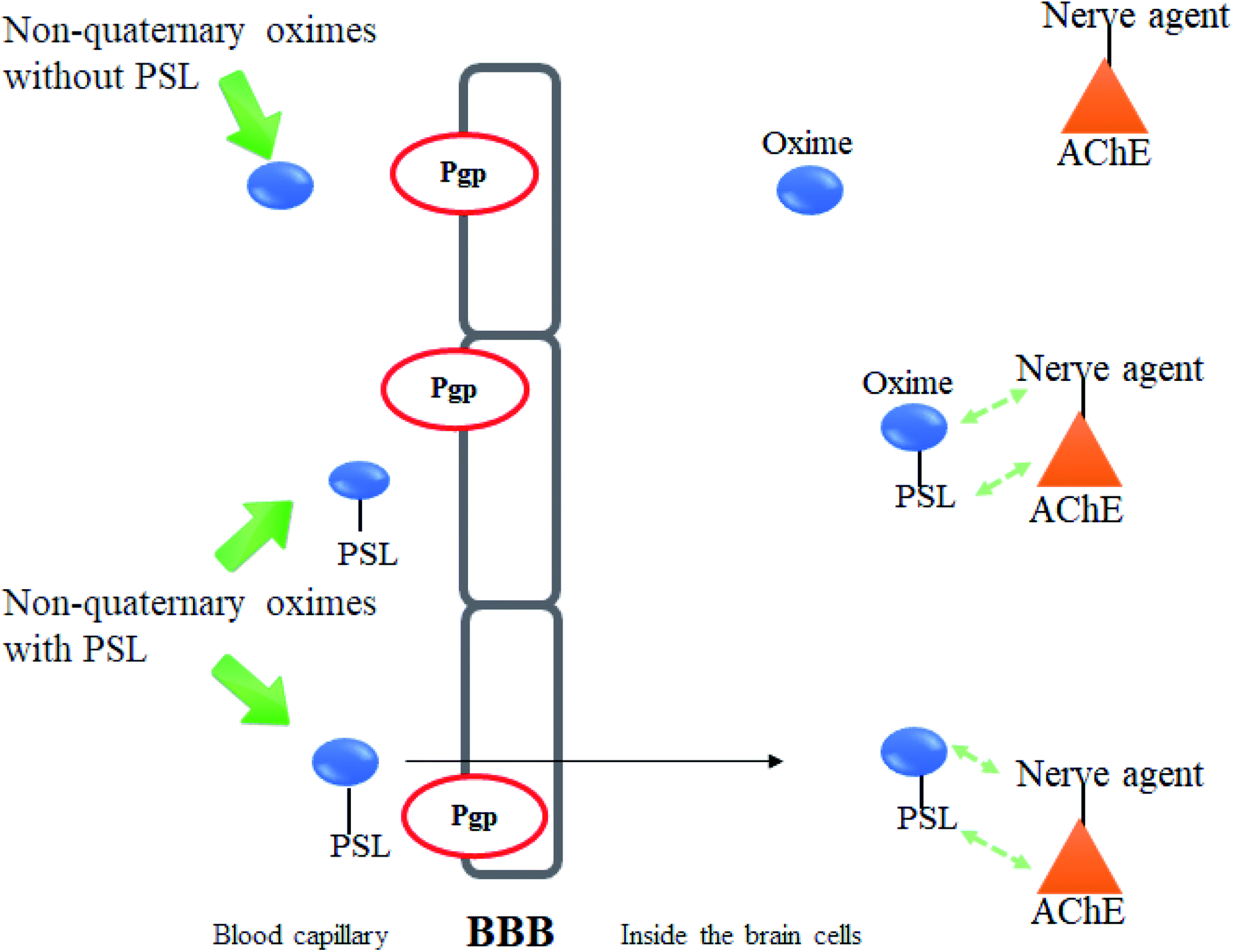 | ||
| Fig. 15 Schematic diagram of the interaction between inhibited AChE and PSL conjugated non-quaternary oxime. | ||
Mercey et al. (2011) designed the uncharged oximes that contain 3-hydroxy-2-pyridinealdoxime linked to 6,7-dimethoxy-1-phenyl-1,2,3,4-tetrahydroisoquinoline which represent PSL thus improving the binding affinity between the oximes and inhibited AChE.72 Based on the study by Saint-André et al. (2011), 3-hydroxy-2-pyridinealdoxime was found to be a very promising neutral α-nucleophile with a high reactivation rate constant.73 It is able to cleave the P–S bond of OP thereby yielding to the corresponding isoxazole analogue and non-toxic phosphonic adduct, thus through this preventing recapture phenomena.74 The two uncharged oximes (35 and 36) as shown in Fig. 16, had a superior or similar in vitro ability to reactivate AChE inhibited by VX and tabun as compared to HI-6, obidoxime, TMB-4 and HLö −7.
 | ||
| Fig. 16 Chemical structure of uncharged oximes developed by Mercey et al. (2011).72 | ||
The determination of reactivation potency of the VX- and tabun-inhibited AChE by oximes 35 and 36 under physiological conditions has shown that this new family of non-quaternary oximes is extremely promising. Their reactivation efficiencies exceed that of HI-6, obidoxime and HLö-7. Oxime 35 has been found to be two-fold more efficient than HI-6 for reactivating VX-inhibited AChE. Oxime 36 is about six-fold more efficient than HI-6 and obidoxime due to its better affinity and is at least as efficient as HLö-7.
The great improvements achieved in the reactivation of tabun-poisoned AChE where oxime 35 and oxime 36 were found to be ten-fold and twenty-fold more efficient than obidoxime as a result of their higher affinity. Additionally, oxime 36 is also much efficient compared to HLö-7. Despite a lower reactivation rate constant (two- and six-fold lower respectively), oximes 35 and 36 were two-fold and five-fold more efficient than trimedoxime due to their drastically improved affinity towards tabun-inhibited AChE.
Renou et al. (2013) also synthesised a series of seven new uncharged oximes (oximes 37 to 43) which are derived from 3-hydroxy-2-pyridinealdoxime and tested against VX- and tabun-inhibited AChE as shown in Fig. 17.10 The structures of these oximes were composed of two moieties: (1) an alpha-nucleophile, 3-hydroxy-2-pyridinealdoxime and (2) a PSL that could interact with AChE to increase the oxime's affinity towards OP-inhibited AChE.75,76 These two moieties are linked together through a carbon chain of various lengths. The reactivation potency of these oximes was determined and compared to HI-6, obidoxime and 2-PAM. Based on the in vitro reactivation potency of inhibited AChE, these oximes had a two-tenfold potential higher than 2-PAM. However, they were found to be less efficient than obidoxime and HI-6. This was due to the better affinity of these two oximes towards VX-inhibited AChE. For tabun-inhibited AChE, only oxime 42 exhibits the reactivation ability equivalent to 2-PAM. However, there has been no report for the in vivo reactivation ability of these oximes. Although these oximes have been shown to be less efficient than obidoxime and HI-6, this approach seem to be promising and has the potential in the future design and development of uncharged oximes.
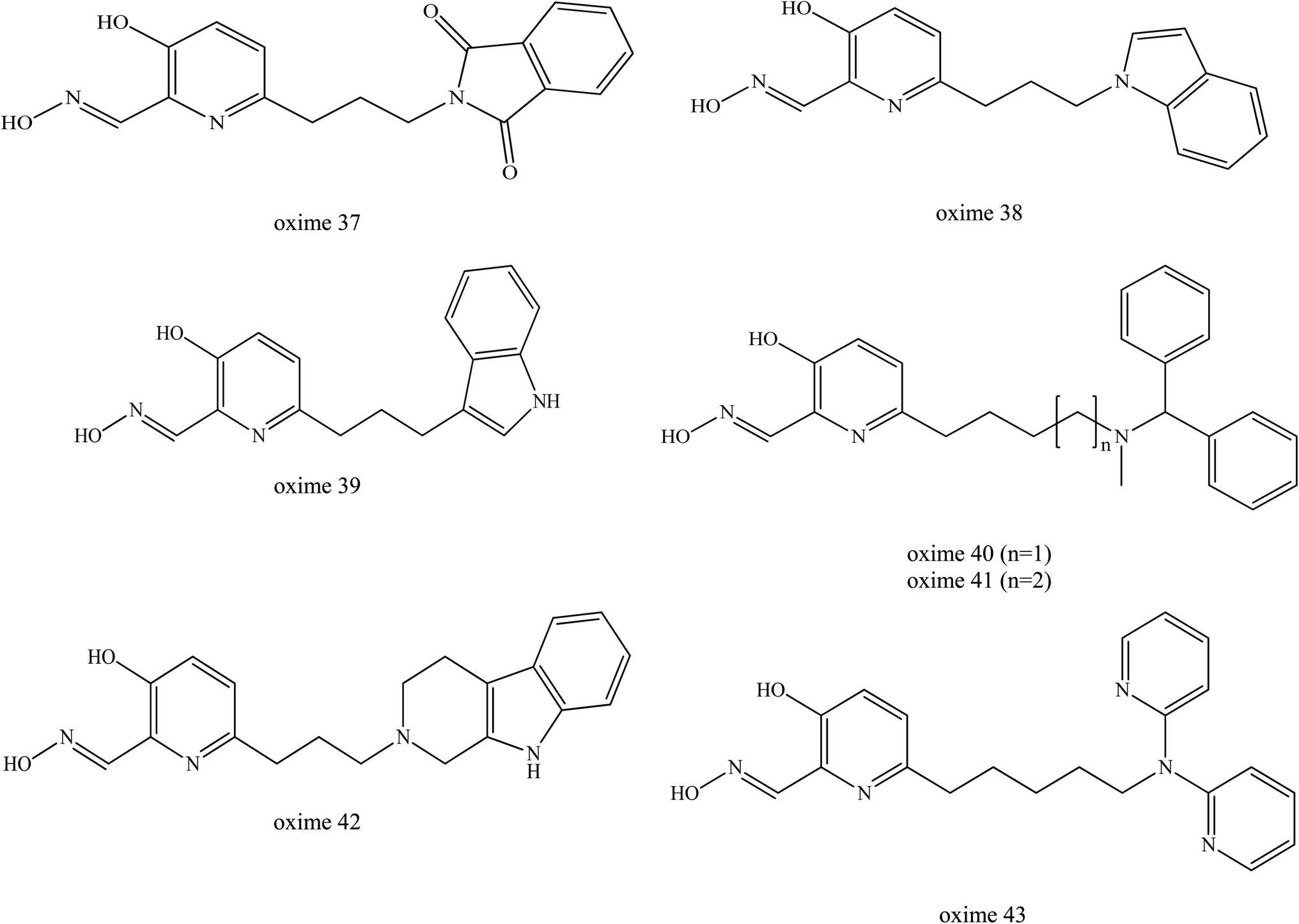 | ||
| Fig. 17 Chemical structure of oximes developed by Renou et al. (2013).9 | ||
The latest study on the enhancement of uncharged 3-hydroxy-2-pyridine aldoximes for the reactivation of AChE was reported by Zorbaz et al. (2018).9 A new series of pyridine aldoximes were designed as shown in Fig. 18. The five new designs of uncharged 3-hydroxy-2-pyridine aldoximes are connected via an aliphatic linker to simple protonatable tertiary amines (morpholine, piperidine) that can be fused with dimethoxybenzene (dimethoxy-tetrahydroisoquinoline) and substituted with N,N-dimethylaniline (1-(4-N,N dimethylaminophenyl)-1,2,3,4-tetrahydroisoquinoline) which represents PSL. These substituents could improve the binding affinity in the AChE and are capable of passive transport across membranes because they are ampholytes with a tertiary amine group, having also a fraction of non-ionised species. These novel oximes have been tested in terms of their inhibitory effect on VX-, tabun-, sarin-, cyclosarin-, and paraoxon-inihibited AChE. The reactivation of AChE was predicted for in vitro, in silico and ex vivo conditions. The reactivation rates for these three oximes (45, 46, 47) were found greater than 2-PAM and comparable to HI-6.
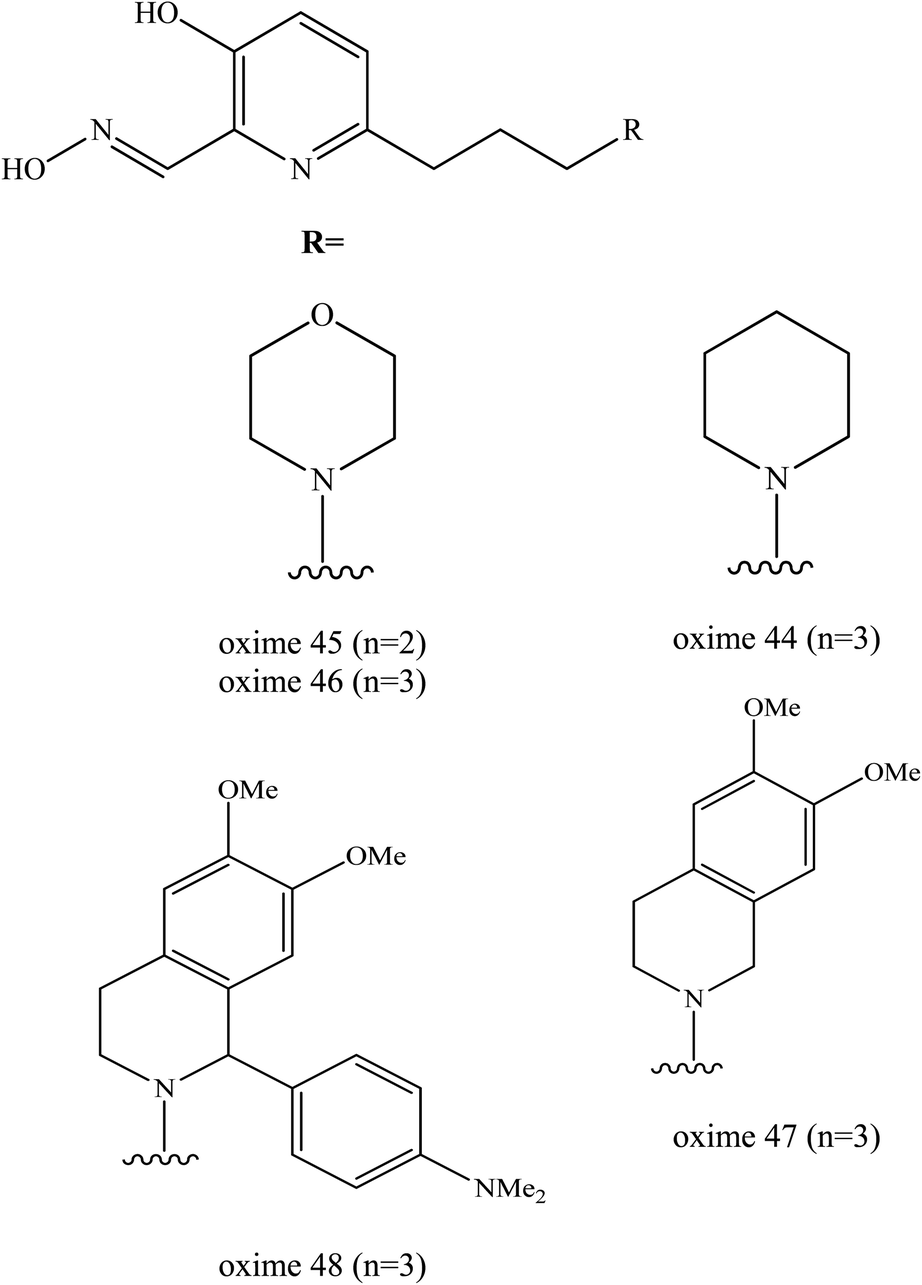 | ||
| Fig. 18 Chemical structure of oximes developed by Zorbaz et al. (2018).9 | ||
The majority of these oximes were highly capable in reactivating all of the tested inhibited AChE by up to 80–100% of the enzyme activity. Oximes 45 and 46 showed the most notable result in reactivating the VX-inhibited AChE as both had a two-fold higher reactivation rate compared to HI-6. In the case of sarin, the oximes 45 was the most potent reactivators followed by oximes and 47 and 48. The reactivation of cyclosarin-inhibited AChE by oximes 44–47 terminated at 100%, but this level was reached much more slowly compared to HI-6. Interestingly, in the case of tabun, all these 3-hydroxy-2-pyridine aldoximes reactivated 50–90% of the inhibited AChE which is unusual as tabun is known for its resistance to reactivation by standard oximes. These oximes were also evaluated with regards to the reactivation of paraoxon-inhibited AChE. All the developed oximes reactivated the paraoxon-inhibited AChE more potently than HI-6. The oxime 48 had the highest reactivation ability as a result of its high binding affinity.
Prediction of BBB penetration was carried out for oximes 44−47 and based on their physicochemical properties and in vitro brain membrane permeation ability. Among these evaluated oximes, two oximes 45 and 46 were shown to be promising AChE reactivators of inhibited AChE both in silico predictions and in vitro permeability tests. Moreover, based on the ex vivo study, efficient reactivation of VX-, sarin-, paraoxon- and cyclosarin-inhibited AChE was achieved by oximes 45 and 47.
Sit et al. (2011) synthesised a series of uncharged oximes (49–51) that contained a tertiary amine or imidazole protonable functional group.77 At first, the authors developed a series of oxime which consisted of primarily imidazole aldoximes and N-substituted 2-hydroxyiminoacetamides. They found that the oxime structures are shown in Fig. 19 exhibited a comparable reactivation efficacy with 2-PAM for cyclosarin- and VX-inhibited AChE. However, these oximes were not effective for paraoxon- and tabun-inhibited AChE. The oxime 49 had a maximal reactivation rate but only relatively weak binding to inhibited AChE indicating that the lead molecule was amenable to further structural refinement to gain a higher reactivation potency.
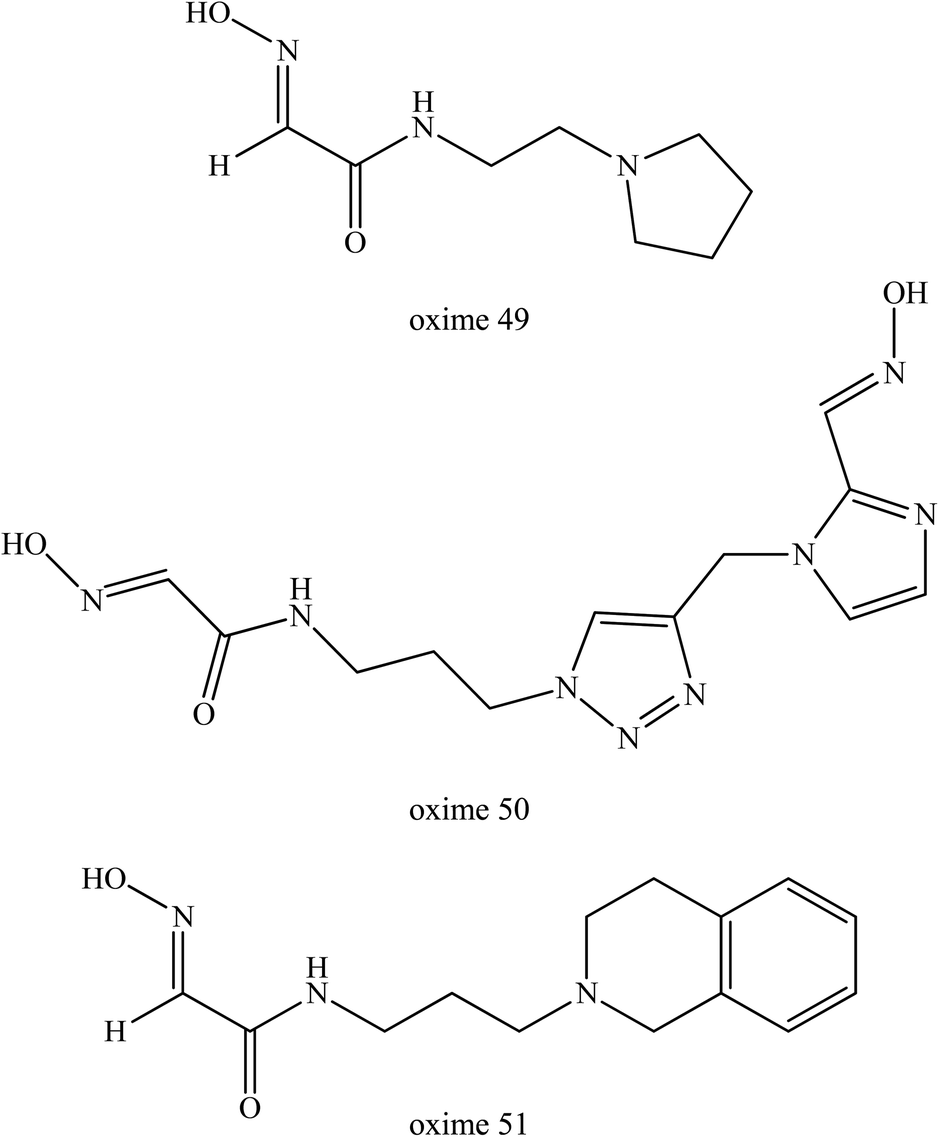 | ||
| Fig. 19 Chemical structure of oximes developed by Sit et al. (2011).77 | ||
With better understanding of the structure of hydroxyimino acetamido alkylamine oximes and through specifically improving oxime 49 reactivation properties, further research work was performed by Radić et al. (2011).78 They described systematic variations of the oxime 49 structure and made a detailed evaluation of the associated reactivating properties which then lead to several enhanced oxime developments. Through a better interaction potency with inhibited AChE, oxime 52 (Fig. 20) showed substantially improved in vitro reactivation kinetics compared to the initial lead oxime 49. This oxime was proven to be an efficient AChE reactivator compared to oxime 49 for sarin-, cyclosarin-, VX-, paraoxon- and tabun-inhibited AChE. It was found that this oxime was capable of reversible binding as a reactivator which caused progressive reactivation facilitated by a propyl linker and a bridge of azepane heterocycle. Based on the results obtained, this oxime appeared to have several fold higher reactivation potencies than alpha-ketoximes, 2,3-butanedione monoxime and MINA, primarily due to the enhanced molecular recognition of inhibited AChE. The in vivo study of this oxime in a mouse animal model resulted in great improvement of antidotal properties when compared to oxime 49 and 2-PAM.
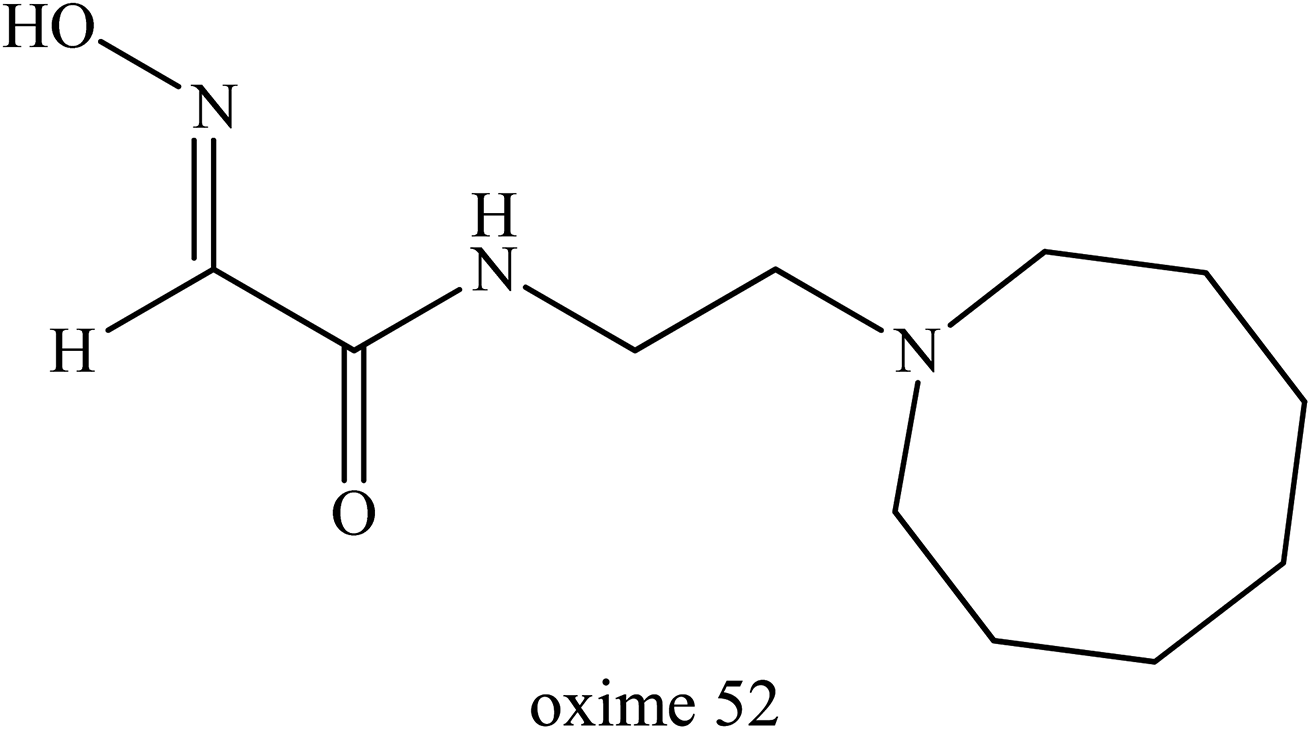 | ||
| Fig. 20 Chemical structure of oxime developed by Radić et al. (2011).78 | ||
Subsequently, Rosenberg et al. (2017) tested oxime 52 for post-exposure treatment on sarin- and paraoxon-inhibited AChE in macaques.79 The in vitro study showed that through administration of oxime 52 and atropine after continuous pulmonary exposure to sarin, it resulted in the rapid reactivation of the inhibited AChE in blood and reversed both early and advanced clinical symptoms of sarin-induced toxicity within one hour. The in vivo studies showed that post-exposure treatment with oxime 52 rapidly reversed both early and advanced classic clinical symptoms in macaques after 18 to 28 minutes exposure to sarin vapor. These data indicate that post-sarin exposure, oxime 52 resulted in the rapid reactivation of erythrocyte AChE activity in circulation, consistent with survival in the macaques, even for those exhibiting a panoply of symptoms including tremors and convulsions.
Meanwhile, Kovarik et al. (2013) developed and tested thirty uncharged oximes that also contain tertiary amine or imidazole protonable functional groups that could cross the BBB as unionised species.80 These oximes were tested on tabun-inhibited AChE along with three reference oximes, namely diacetylmonoxime, monoisonitrosoacetone, and 2-PAM. Interestingly, oxime 53 (Fig. 21) was highlighted as the most favourable oxime reactivator of tabun-inhibited AChE, even though it had lower reactivation activity the reference oximes. Oxime 53 contained a triazole ring as the PSL where this heterocycle was positioned at a certain distance from the oxime group and this structural position could enhance the reactivation of tabun-inhibited AChE. The triazole moiety was observed to improve affinity towards inhibited AChE.
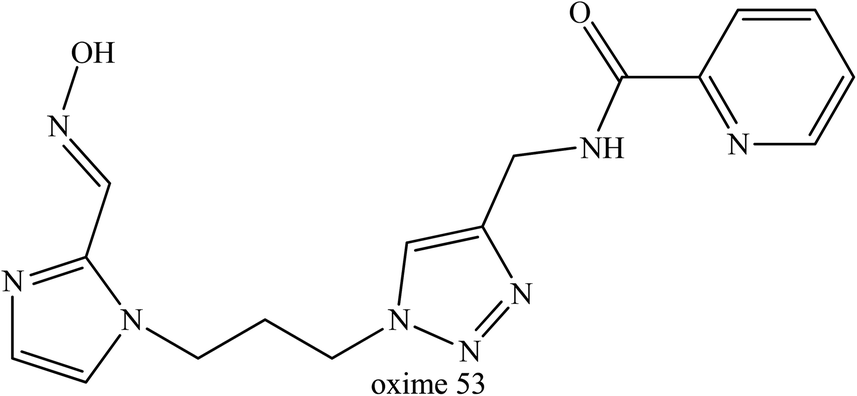 | ||
| Fig. 21 Chemical structure of oximes developed by Kovarik et al. (2013).80 | ||
McHardy et al. (2014) developed a series of uncharged oximes capable of reactivating cyclosarin-inhibited AChE.12 The structure activity relationships for AChE binding and reactivation of cyclosarin-inhibited AChE were identified. Two chemical series were highlighted, i.e. amide oxime analogs and heteroaryl keto-oxime analogs. Based on Fig. 22, the PSL replacement of the N-benzyl piperidine with the dihydroisoquinoline group (oxime 54) delivered oximes constructs with very good reactivation levels. Oxime 55 possessed a N-benzyl-4-piperidine moiety pattern with the ketoxime group on the pyridine ring and a 2,5-substitution pattern on the pyridine group providing 23% reactivation activity. Meanwhile, the 2,3-substitution pattern in oxime 56 displayed a marked improvement in reactivation activity (45%). Oxime 57 also produced a significant increase in reactivation activity (65%). However, future study is still needed to enhance their affinity toward the inhibited enzyme and also to investigate the in vivo behaviour of these uncharged oximes.
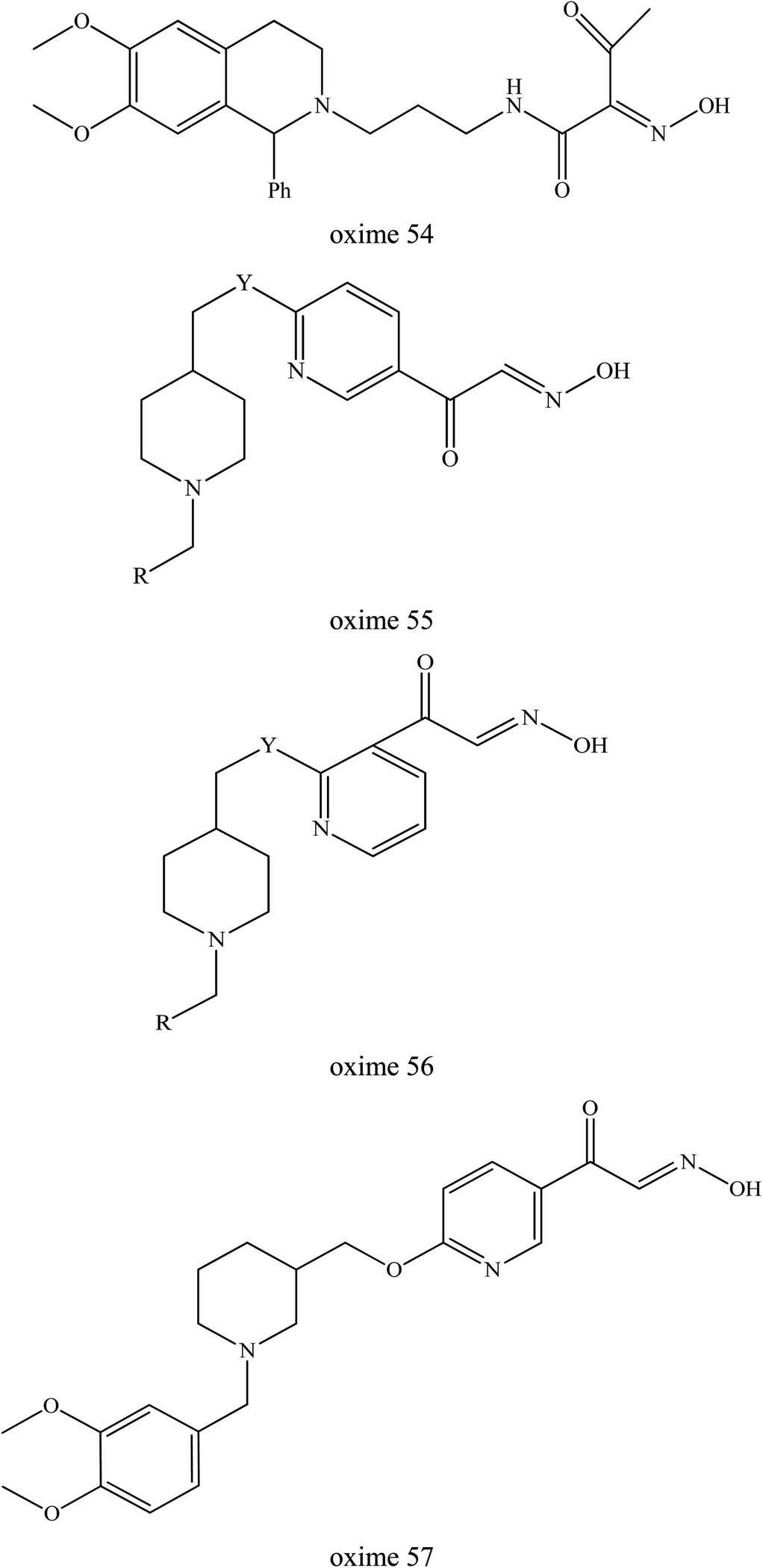 | ||
| Fig. 22 Chemical structure of oximes developed by McHardy et al. (2014).12 | ||
Kliachyna et al. (2014) reported on the design and synthesis of the novel tetrahydroacridine pyridine oxime hybrids as efficient uncharged reactivators of AChE.7 This oxime design was based on a 1,2,3,4-tetrahydroacridine (tacrine) substructure as the PSL which was able to bind to the AChE (Fig. 23). These oximes were coupled with various linkers to either 3-hydroxy-2-pyridine aldoxime or 3-hydroxy-2-pyridine amidoxime. The abilities of amidoximes 58–61, as well as of aldoximes 62–66 to reactivate in vitro VX-, tabun- and ethyl-paraoxon-inhibited AChE was investigated and compared to the standard oximes 2-PAM, obidoxime, HLö-7, TMB4 and HI-6. The 4-substituted tetrahydroacridine α-hydroxypyridine amidoximes 58, 60 and 61 were found to have improved affinity and higher reactivation efficiencies towards VX-inhibited AChE as compared to 2-PAM and TMB4. However, they remained less efficient than obidoxime, HLö-7, and HI-6 in reactivating VX-inhibited AChE. Interestingly, aldoximes 63 and 64 substituted in position 6 of the pyridine ring by the C5 and C4 alkyl linker were found to be more efficient in reactivating VX-inhibited AChE than 2-PAM, obidoxime, trimedoxime, and HI-6.
 | ||
| Fig. 23 Chemical structure of oximes developed by Kliachyna et al. (2014).7 | ||
Interestingly, the oxime 64 was able to reactivate tabun-inhibited AChE, where the OP complex is known to be difficult to reactivate due to its weak electrophilicity and to stearic hindrance imposed on the phosphorus atom in the tabun-inhibited AChE. Despite a lower reactivation rate constant compared to obidoxime and TMB4, oxime 64 was four-fold more efficient than TMB4, the best known pyridinium aldoxime of tabun-inhibited AChE. Oxime 64 also had better reactivity on both VX- and tabun-inhibited AChE, as compared to oxime 63. This suggested that a 4-carbon linker is optimum for both good binding to the inhibited enzyme and for accurate orientation of oxime function needed for displacementing of the phosphyl or phosphoramidyl moiety.
Oximes 63 and 64 were shown to be more efficient than obidoxime and HLö-7 towards paraoxon-inhibited AChE. However, these uncharged oximes were only tested in an in vitro study only and need to be evaluated in in vivo study. It is noteworthy that these oximes hold neither a permanent charge nor a tertiary amine which allow them to cross the BBB quite efficiently.
Wei et al. (2014) introduced a dual binding strategy to the uncharged non-pyridinium oximes.81 Pyridinium aldoximes are difficult to prepare due to their long synthetic process. Therefore, the development of alternatives to the pyridinium aldoximes by conjugating the imidazolium aldoxime to different PSL through alkyl chains was done. These were compared to several available oximes namely obidoxime, HI-6, MMB-4 and TMB-4. Several successful oxime conjugates were developed and the best oximes (67 and 68) are shown in Fig. 24. The resulting oxime conjugates were highlighted as the first efficient non-pyridinium oximes to reactivate sarin-, VX-, and tabun-inhibited AChE. It was proven that the conjugation of a proper PSL to the imidazolium aldoxime would lead to greatly enhanced reactivation potency. These two oximes emerged as the first efficient and broad-spectrum non-pyridinium oximes besides the quaternary oximes for use against sarin-, VX- and tabun-inhibited AChE. These oximes are potent and have the ability to reactivate sarin-inhibited AChE that is equal to and may even exceed the ability of MMB-4, HI-6 and obidoxime. Considering their simple synthesis, these novel imidazolium aldoximes show both economical superiority and hold promise for further design improvement for more efficient uncharged AChE reactivators.
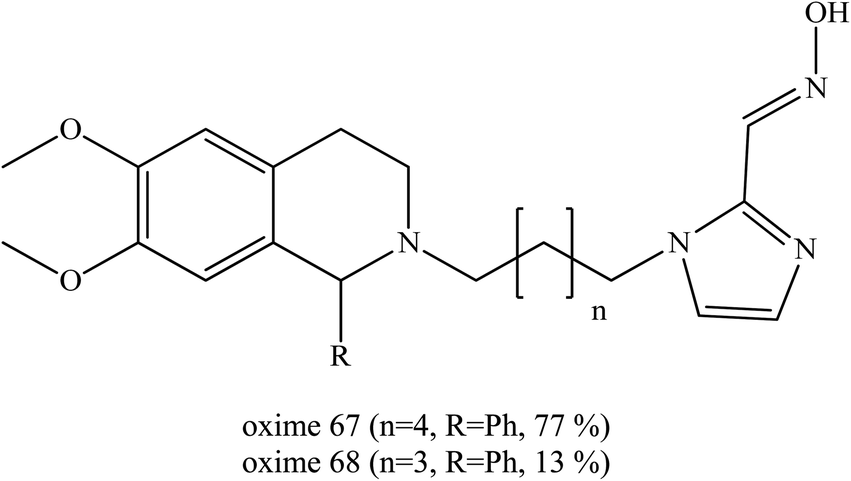 | ||
| Fig. 24 Chemical structure of oximes developed by Wei et al. (2014).81 | ||
In addition, Wei et al. (2016) developed another novel uncharged oxime to reactivate soman-inhibited AChE.19 Similar to their earlier work, they developed a dual site binding strategy. Simple aromatic groups were used as PSL, which was expected to interact with the aromatic group at the P-site and compensate for the ortho-hydroxybenzaldoximes's affinity to AChE. The ortho-hydroxybenzaldoximes which have a similar structure to the HI-6 were chosen as reactivation ligands of AChE to prevent the secondary poisoning of AChE and simple aromatic groups were used as PSL of AChE, which were linked to the oxime. A series of oximes were developed as shown in Fig. 25. The in vitro experiment demonstrated that some of the resulting conjugates have robust activity against soman-inhibited AChE. It can be concluded that oximes (69–71) with a 4-hydroxybenzyl PSL may produce higher reactivation activity against soman-inhibited AChE. At the same time, it was noticeable that oximes 69–71 were relatively heavier inhibitors of AChE. The oxime 70 was highlighted as the most efficient among them. However, the reactivation of these oximes was not as good as the HI-6. Although it is not as efficient as HI-6, they were expected to exhibit higher BBB penetration and demonstrate promising in vivo reactivation ability as a result of their non-quaternary structures. The author has not yet published the findings of the in vivo study of this developed oxime.
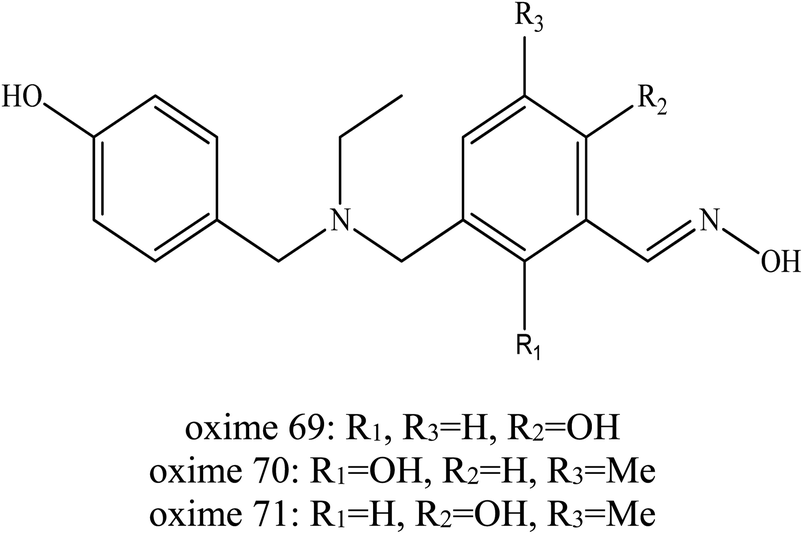 | ||
| Fig. 25 Chemical structure of oximes developed by Wei et al. (2016).19 | ||
Wei et al. (2017) continued their investigation by developing conjugates of salicylaldoximes and PSL.45 Salicylaldoximes are known to be able to quickly cleave the P–S bond of organophosphates and avoid the re-inhibition phenomenon in their reactivation process, but salicylaldoximes lack reactivation ability due to their poor affinity for.74 By using the dual binding site strategy, several PSLs were conjugated with salicylaldoximes as shown in Fig. 26 in order to increase their affinity. These developed oximes were reportedly to have advantageous properties including:
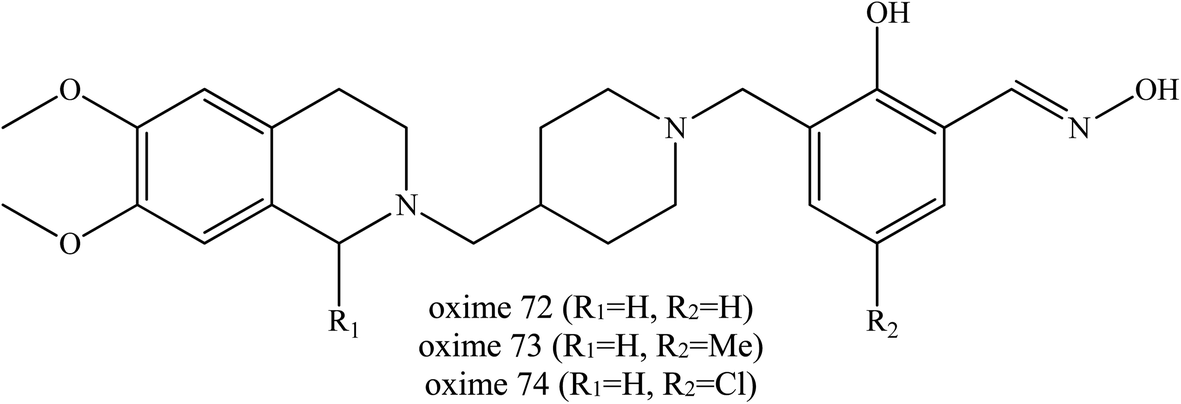 | ||
| Fig. 26 Chemical structure of oximes developed by Wei et al. (2017).45 | ||
(i) Improved chemical stability (compared to pro-2-PAM) and easier synthesis as compared to some pyridine aldoxime conjugates.
(ii) Greatly improved lipophilicity (compared to quaternary oximes).
(iii) Relatively increased in vitro reactivation efficacy of OP-inhibited AChE (compared to charged oximes).
Based on the in vitro reactivation experiments which demonstrated that some of the yielding conjugates exhibited similar or better ability to reactivate sarin-, VX- or tabun-inhibited AChE in comparison with the current pyridinium aldoximes used, it was found that, oximes (72–74) with a piperidine link gave the best reactivation activity for VX-, sarin-, and tabun-inhibited AChE. Moreover, due to their greatly improved lipophilicity, these uncharged oximes were significant constructs for the development of efficient centrally acting reactivators. These novel salicylaldoximes showed economical superiority due to their easier synthesis processes. The authors did mention that an in vivo study is in process and perhaps further modification would still be required in this development of centrally activating reactivators.
Wei et al. (2018) continued to develop other uncharged oximes of salicyaldoxime with isonicotinamide and its bioisostere.54 A similar dual site binding strategy was applied to construct a novel series of uncharged oximes against OP poisoning. The bioisostere 4-aminobenzamide and 3-aminobenzamide were introduced as PSL. Most of the new synthesised oximes were found to efficiently reactivate VX-inhibited AChE. Some of them even exceeded the reactivation activity of HI-6 and obidoxime, for example the oximes 75 to 85 as shown in Fig. 27. For VX-inhibited AChE, conjugates bearing R2 as the oxime part appeared to be relatively poor AChE reactivators. Whereas for soman-inhibited AChE, only oxime 83 was able to exhibit reactivation and even that only weakly, and it was lower than that of HI-6. For sarin and tabun poisoning, conjugates containing 4-aminobenzamide as a PSL exhibited promising reactivation potency, such as seen with oximes 76 to 80. Conjugates containing 3-aminobenzamide were reported to show reactivation efficiency for tabun-inhibited AChE. Moreover, conjugates with 4-aminobenzamide or 3-aminobenzamide linked to the ortho position and to the phenol function of salicylaldoxime even though piperidine also exhibited high efficiency reactivation. In addition, the introduction of methyl or halogen substitution in the para position of salicylaldoxime can increase reactivation ability. However, conjugates containing isonicotinamide have been reported to give unsatisfactory results against sarin- and tabun-inhibited AChE.
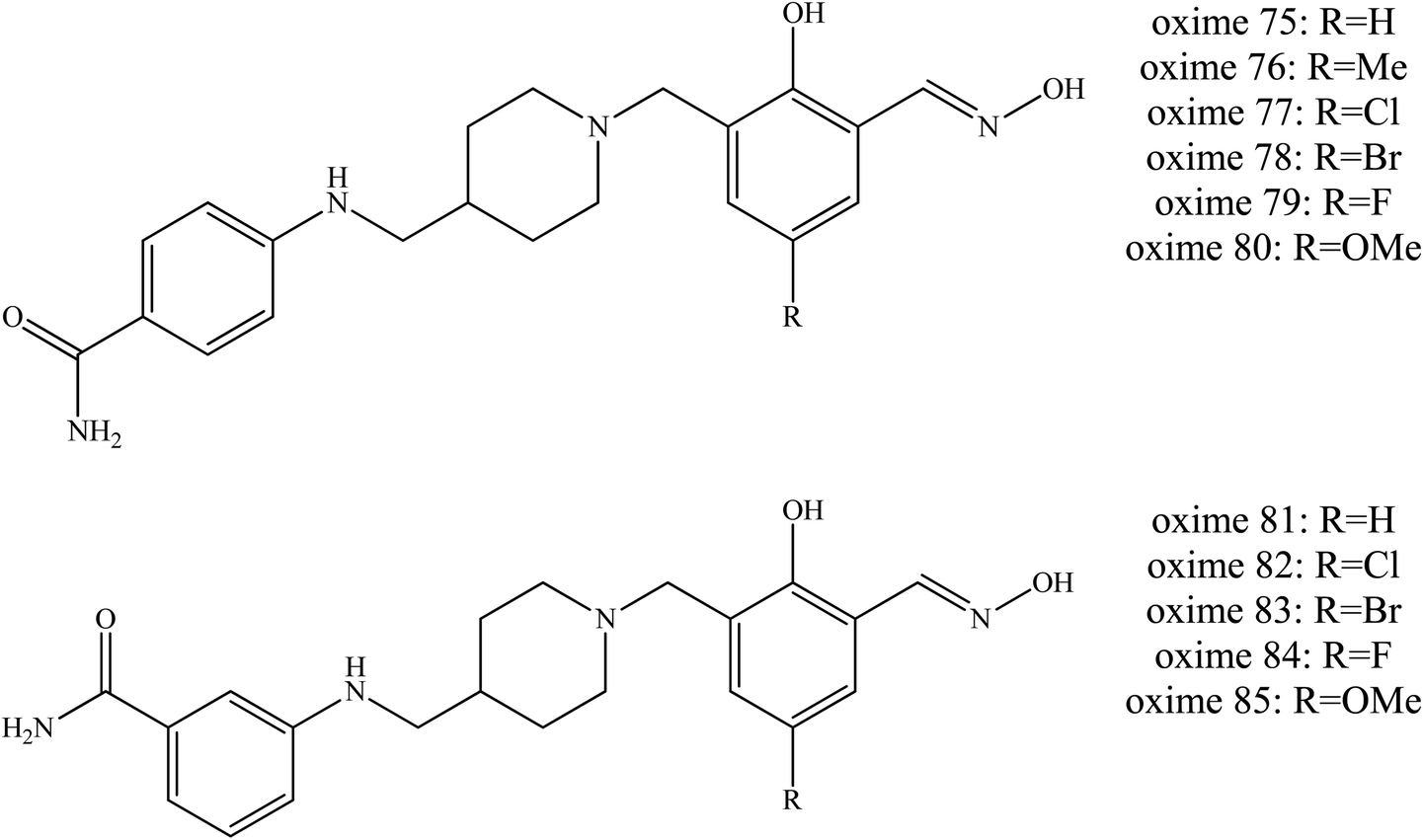 | ||
| Fig. 27 Chemical structure of oximes developed by Wei et al. (2018).54 | ||
Lima et al. (2017) too developed several uncharged oximes as AChE reactivators of parathion-inhibited AChE in eels.51 The presence of neutral ligands as PSL is expected to improve the affinity towards the inhibited AChE. It was reported that these developed uncharged oximes showed a lower affinity for AChE compared to 2-PAM. The oximes 86 and 87 as shown in Fig. 28 were able to reactivate parathion-inhibited eel AChE by 9% and 11% respectively which was lower as compared to the reactivation efficacy of 2-PAM (25%). However, the reactivation of OP-inhibited AChE by these oximes needs to be further analysed.
 | ||
| Fig. 28 Chemical structure of oximes developed by Lima et al. (2017).51 | ||
Strategy 3: oxime-lipophillic compounds conjugations
Oxime conjugations with lipophilic compounds have gained attention over the past decade and are being used in a number of applications, mainly in the field of bioconjugation.82 The BBB allows the passage of water, some gases, and lipid soluble molecules by passive diffusion, as well as the selective transport of molecules such as glucose and amino acids that are crucial for brain function. Glucose is the primary energy substrate of the brain. Glucose from blood can enter the brain by a glucose transport protein which is highly concentrated in BBB endothelial cells.83 This glucose transport protein also acts as therapeutic targets and provides routes for drugs to enter the brain for the treatment of neurological and neurovascular conditions and brain tumours. It is also one of the most extensively studied of all membrane transport proteins. The conjugation of glucose together with drugs including oximes could provide a very promising means for the reactivation of inhibited AChE in the CNS. Fig. 29 illustrates the mechanism of glucose conjugated oximes passing through the BBB. Glucose will be naturally degraded once past the BBB and will then release the oxime into the cerebrospinal fluid.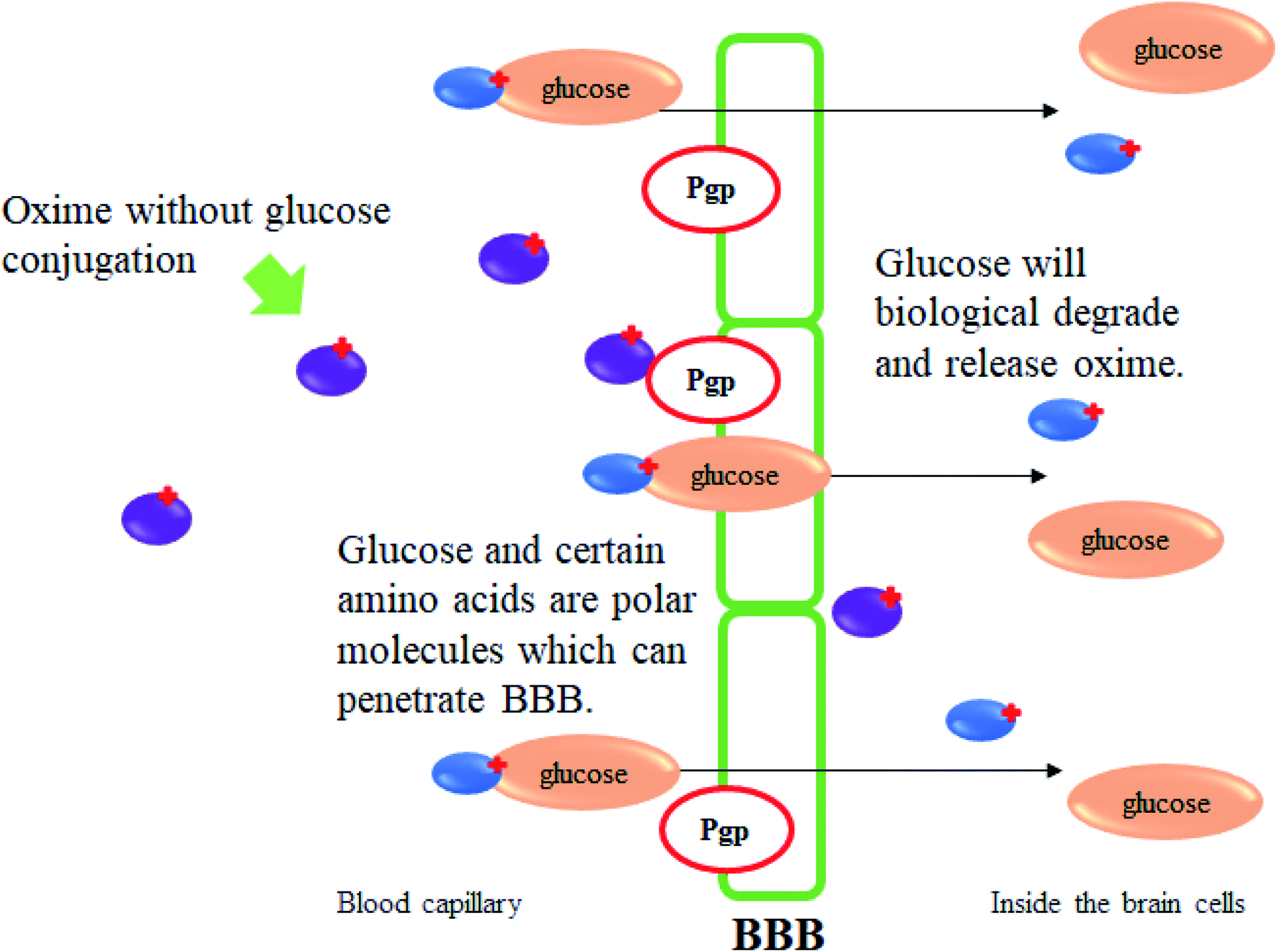 | ||
| Fig. 29 Schematic diagram of BBB penetration of glucose conjugated oxime. | ||
To the best of our knowledge, there is a lack of research focussing on oxime conjugation for BBB penetration in order to reactivate OP-inhibited AChE in cases of OP poisoning. Garcia et al. (2010) developed novel sugar-oxime conjugates whereby have the potential to be transported and cross the BBB.84 This is due to the presence of the sugar moiety which would be recognised by the facilitative glucose transporter. Several sugar-oximes have been synthesised and tested for their ability to reactivate AChE poisoned with sarin, VX, paraoxon, and DFP. Among the sugar-oximes developed, oxime 88 as shown in Fig. 30 was the most effective as compared to other oximes with its reactivation ability similar to that of pralidoxime. Besides that, oxime 89 had a low toxicity with a LD50 of 1590 mg kg−1 from a single IM dose tested in guinea pigs and >2000 mg kg−1 IP dose in the mice. Histopathological analysis showed that there were no apparent differences in the hippocampus, heart, liver, kidney sciatic nerve, or skeletal muscle between treated and untreated animals. These results suggested that the sugar-oximes developed in this study has the potential to be effective AChE reactivators even when administered in high doses.
 | ||
| Fig. 30 Chemical structure of sugar-oximes developed by Garcia et al. (2010).84 | ||
Other lipophilic compounds such as peptides, protein, caffeine and vitamins that can penetrate through the BBB can also be conjugated with oximes. According to Agten et al. (2016), the incorporation of ketones and aldehydes in proteins has received much attention over the past decade.82 Several methods are now available for the synthesis of ketone-containing proteins, thus making the starting point for oxime ligation available in many laboratories.
Challenges and future directions
Reliable sources in the literature focussing on the importance of BBB penetration by oximes has been explored in this review. Three recent strategies discussed in this review have shown certain promising results in improving the performance of oximes in the reactivation of OP-inhibited AChE in the CNS. Several challenges were also described in this review. Firstly, there are several new developed oximes that show an excellent ability to reactivate OP poisoned AChE.85 However, most of these developed oximes have not been tested for BBB penetration. Therefore, it is necessary to further investigate these oximes for BBB penetration. Secondly, tabun is known as the most resistive OP nerve agent as compared to the other compounds. Very limited numbers of research as discussed above have been able to successfully reactivate this OP especially in the CNS. Focus must also be given to this OP in order to improve the reactivation potency of suitable oximes as well as their BBB penetration ability. Thirdly, there are several other approaches to improve BBB penetration such as the use of Pgp inhibitor and nanoparticles as drug carrier. However, there are limited reports focused on the use of these approaches for oximes. Therefore, research of the use of Pgp inhibitors and nanomaterial carriers for oximes delivery into CNS needs to be considered.Conflicts of interest
There are no conflicts to declare.References
- J. V. Peter, T. I. Sudarsan and J. L. Moran, Indian J. Crit. Care Med., 2014, 18, 735 CrossRef PubMed.
- M. Eddleston and M. R. Phillips, BMJ, 2004, 328, 42–44 CrossRef PubMed.
- E. Antonijevic, K. Musilek, K. Kuca, D. Djukic-Cosic, S. Vucinic and B. Antonijevic, Neurotoxicology, 2016, 55, 33–39 CrossRef CAS PubMed.
- M. Jokanović, in Handbook of Toxicology of Chemical Warfare Agents, Academic Press, 2015, pp. 1057–1070 Search PubMed.
- J. Kassa, J. Toxicol., Clin. Toxicol., 2002, 40, 803–816 CrossRef CAS PubMed.
- Handbook of toxicology of chemical warfare agents, ed. R. C.Gupta, Academic Press, 2015 Search PubMed.
- M. Kliachyna, G. Santoni, V. Nussbaum, J. Renou, B. Sanson, J. P. Colletier, M. Arboléas, M. Loiodice, M. Weik, L. Jean and P. Y. Renard, Eur. J. Med. Chem., 2014, 78, 455–467 CrossRef CAS PubMed.
- K. Kuca, J. Korabecny, R. Dolezal, E. Nepovimova, O. Soukup, L. Gorecki and K. Musilek, RSC Adv., 2017, 7, 7041–7045 RSC.
- T. Zorbaz, A. Braïki, N. Maraković, J. Renou, E. de La Mora, N. Maček Hrvat, M. Katalinić, I. Silman, J. L. Sussman, G. Mercey and C. Gomez, Chem.–Eur. J., 2018, 24, 9675–9691 CrossRef CAS PubMed.
- J. Renou, G. Mercey, T. Verdelet, E. Păunescu, E. Gillon, M. Arboléas, M. Loiodice, M. Kliachyna, R. Baati, F. Nachon and L. Jean, Chem.-Biol. Interact., 2013, 203, 81–84 CrossRef CAS PubMed.
- J. Korabecny, O. Soukup, R. Dolezal, K. Spilovska, E. Nepovimova, M. Andrs, T. Duong Nguyen, D. Jun, K. Musilek, M. Kucerova-Chlupacova and K. Kuca, Mini-Rev. Med. Chem., 2014, 14, 215–221 CrossRef CAS PubMed.
- S. F. McHardy, J. A. Bohmann, M. R. Corbett, B. Campos, M. W. Tidwell, P. M. Thompson, C. J. Bemben, T. A. Menchaca, T. E. Reeves, W. R. Cantrell Jr and W. E. Bauta, Bioorg. Med. Chem. Lett., 2014, 24, 1711–1714 CrossRef CAS PubMed.
- J. Kassa and V. Humlicek, Drug Chem. Toxicol., 2008, 31, 127–135 CrossRef CAS PubMed.
- M. Eddleston and F. R. Chowdhury, Br. J. Clin. Pharmacol., 2016, 81, 462–470 CrossRef CAS PubMed.
- J. Kassa, J. Hatlapatková and J. K. Žďárová, Acta Med., 2015, 58, 135–143 CAS.
- E. Antonijevic, K. Musilek, K. Kuca, D. Djukic-Cosic, M. Curcic, D. C. Miladinovic, Z. Bulat and B. Antonijevic, Food Chem. Toxicol., 2018, 121, 224–230 CrossRef CAS PubMed.
- M. C. de Koning, M. van Grol and D. Noort, Toxicol. lett., 2011, 206, 54–59 CrossRef CAS PubMed.
- J. E. Chambers, E. C. Meek and H. W. Chambers, Ann. N. Y. Acad. Sci., 2016, 1374, 52–58 CrossRef CAS PubMed.
- Z. Wei, Y. Q. Liu, Y. A. Wang, W. H. Li, X. B. Zhou, J. Zhao, C. Q. Huang, X. Z. Li, J. Liu, Z. B. Zheng and S. Li, Toxicol. Lett., 2016, 246, 1–6 CrossRef CAS PubMed.
- P. Taylor, Chapter 8: Anticholinesterase Agents, The Pharmacological Basis of Therapeutics, McGraw Hill, New York, 9th edn, 1996, pp. 161–176 Search PubMed.
- E. Gallagher, I. Minn, J. E. Chambers and P. C. Searson, Fluids Barriers CNS, 2016, 13, 10 CrossRef PubMed.
- M. Bonakdar, P. M. Graybill and R. V. Davalos, RSC Adv., 2017, 7, 42811–42818 RSC.
- K. Sakurada, K. Matsubara, K. Shimizu, H. Shiono, Y. Seto, K. Tsuge, M. Yoshino, I. Sakai, H. Mukoyama and T. Takatori, Neurochem. Res., 2003, 28, 1401–1407 CrossRef CAS PubMed.
- D. E. Lorke, M. Y. Hasan, S. M. Nurulain, R. Sheen, K. Kuča and G. A. Petroianu, J. Appl. Toxicol., 2007, 27, 482–490 CrossRef CAS PubMed.
- J. Z. Karasova, F. Zemek, K. Musilek and K. Kuca, Neurotoxic. Res., 2013, 23, 63–68 CrossRef CAS PubMed.
- L. Crawford, J. Rosch and D. Putnam, J. Controlled Release, 2016, 240, 251–266 CrossRef CAS PubMed.
- D. E. Lorke, H. Kalasz, G. A. Petroianu and K. Tekes, Curr. Med. Chem., 2008, 15, 743–753 CrossRef CAS PubMed.
- M. J. Joosen, S. M. Vester, J. Hamelink, S. D. Klaassen and R. M. van den Berg, Chem.-Biol. Interact., 2016, 259, 115–121 CrossRef CAS PubMed.
- G. F. Meerhoff, S. M. Vester, P. Hesseling, S. D. Klaassen, A. S. Cornelissen, P. J. Lucassen and M. J. Joosen, Neurotoxicology, 2018, 68, 167–176 CrossRef CAS PubMed.
- K. Musilek, J. Korabecny, D. Jun, J. Kassa, and K. Kuca, in Handbook of Toxicology of Chemical Warfare Agents, Academic Press, 2015, pp. 1071–1087 Search PubMed.
- E. Hilmas and C. J. Hilmas, in Handbook of Toxicology of Chemical Warfare Agents, Academic Press, 2015, pp. 1003–1034 Search PubMed.
- S. W. Wiener and R. S. Hoffman, J. Intensive Care Med., 2004, 19, 22–37 CrossRef PubMed.
- A. Radilov, V. Rembovskiy, I. Rybalchenko, E. Savelieva, E. Podolskaya, V. Babakov, E. Ermolaeva, S. Dulov, S. Kuznetsov, I. Mindukshev and A. Shpak, Russian VX, in Handbook of Toxicology of Chemical Warfare Agents, Academic Press, 2009, pp. 69–91 Search PubMed.
- S. Y. H. Tang and J. T. S. Chan, A review article on nerve agents, Hong Kong J. Emerg. Med., 2002, 9, 83–89 CrossRef.
- H. Kalász, S. M. Nurulain, G. Veress, S. Antus, F. Darvas, E. Adeghate, A. Adem, F. Hashemi and K. Tekes, J. Appl. Toxicol., 2015, 35, 116–123 CrossRef.
- D. Quinn, J. Topczewski, N. Yasapala and A. Lodge, Molecules, 2017, 22, 1464 CrossRef PubMed.
- G. Mercey, T. Verdelet, J. Renou, M. Kliachyna, R. Baati, F. Nachon, L. Jean and P. Y. Renard, Acc. Chem. Res., 2012, 45, 756–766 CrossRef CAS PubMed.
- M. Katalinić, G. Šinko, N. M. Hrvat, T. Zorbaz, A. Bosak and Z. Kovarik, Toxicology, 2018, 406, 104–113 CrossRef PubMed.
- J. Kassa, V. Sepsova, M. Tumova, A. Horova and K. Musilek, Basic Clin. Pharmacol. Toxicol., 2015, 116, 367–371 CrossRef CAS PubMed.
- J. Kim, Y. R. Malpani, J. Lee, J. S. Shin, S. B. Han and Y. S. Jung, Bioorg. Med. Chem. Lett., 2018, 28, 3784–3786 CrossRef CAS PubMed.
- B. Antonijevic and M. P. Stojiljkovic, Clin. Med. Res., 2007, 5, 71–82 CrossRef CAS PubMed.
- I. B. Wilson and S. Ginsburg, Arch. Biochem. Biophys., 1955, 54, 569 CrossRef CAS PubMed.
- M. Jokanovic and M. Prostran, Curr. Med. Chem., 2009, 16, 2177–2188 CrossRef CAS PubMed.
- M. P. Stojiljković and M. Jokanović, Arh. Hig. Rada Toksikol., 2006, 57, 435–443 Search PubMed.
- Z. Wei, Y. Q. Liu, S. Z. Wang, L. Yao, H. F. Nie, Y. A. Wang, X. Y. Liu, Z. B. Zheng and S. Li, Bioorg. Med. Chem., 2017, 25, 4497–4505 CrossRef CAS PubMed.
- J. E. Chambers, H. W. Chambers, K. E. Funck, E. C. Meek, R. B. Pringle and M. K. Ross, Chem.-Biol. Interact., 2016, 259, 154–159 CrossRef CAS PubMed.
- F. Worek, M. Koller, H. Thiermann and T. Wille, Toxicology, 2016, 350, 25–30 CrossRef PubMed.
- L. Nakab, I. Bardot, S. Bardot, S. Simar, D. Marzin and F. Nesslany, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2014, 762, 30–38 CrossRef CAS PubMed.
- K. Musilek, K. Kuca, D. Jun and M. Dolezal, J. Appl. Biomed., 2007, 5, 25–30 CrossRef CAS.
- J. Kassa, K. Kuca, L. Bartosova and G. Kunesova, Curr. Org. Chem., 2007, 11, 267–283 CrossRef CAS.
- J. A. Lima, L. P. Cavalcanti, A. P. Aguiar, C. M. Rezende, K. S. Lima and A. L. Lima, Def. Life Sci. J., 2017, 2, 363–369 CrossRef.
- P. M. Lundy, M. G. Hamilton, T. W. Sawyer and J. Mikler, Toxicology, 2011, 285, 90–96 CrossRef CAS PubMed.
- J. Kassa, K. Kuca and J. Cabal, Biomed. Pap. Med. Fac. Palacky Univ., Olomouc, 2005, 149, 419 CrossRef CAS PubMed.
- Z. Wei, H. Bi, Y. Q. Liu, H. F. Nie, L. Yao, S. Z. Wang, J. Yang, Y. A. Wang, X. Liu and Z. B. Zheng, Bioorg. Chem., 2018, 81, 681–688 CrossRef CAS PubMed.
- M. Winter, T. Wille, K. Musilek, K. Kuca, H. Thiermann and F. Worek, Toxicol. Lett., 2016, 244, 136–142 CrossRef CAS PubMed.
- J. Aparicio-Blanco, C. Martin-Sabroso and A. I. Torres-Suarez, Biomaterials, 2016, 103, 229–255 CrossRef CAS PubMed.
- A. Beloqui, M. Á. Solinís, A. Rodríguez-Gascón, A. J. Almeida and V. Préat, Nanomed. Nanotechnol. Biol. Med., 2016, 12, 143–161 CrossRef CAS PubMed.
- M. L. Amin, Drug Target Insights, 2013, 7, DTI–S12519 CrossRef PubMed.
- M. J. Joosen, M. J. van der Schans, C. G. van Dijk, W. C. Kuijpers, H. M. Wortelboer and H. P. van Helden, Toxicol. Lett., 2011, 206, 67–71 CrossRef CAS PubMed.
- J. Z. Karasova, M. Pohanka, K. Musilek, F. Zemek and K. Kuca, Toxicol. In Vitro, 2010, 24, 1838–1844 CrossRef CAS PubMed.
- Y. Zhou, Z. Peng, E. S. Seven and R. M. Leblanc, J. Controlled Release, 2018, 270, 290–303 CrossRef CAS PubMed.
- J. Kalisiak, E. C. Ralph, J. Zhang and J. R. Cashman, J. Med. Chem., 2011, 54, 3319–3330 CrossRef CAS PubMed.
- J. Z. Karasova, F. Zemek, J. Bajgar, M. Vasatova, P. Prochazka, L. Novotny and K. Kuca, J. Pharm. Biomed. Anal., 2011, 54, 1082–1087 CrossRef CAS PubMed.
- J. Kufleitner, S. Wagner, F. Worek, H. von Briesen and J. Kreuter, J. Microencapsulation, 2010, 27, 506–513 CrossRef CAS PubMed.
- T. M. Shih, J. A. Guarisco, T. M. Myers, R. K. Kan and J. H. McDonough, Toxicol. Mech. Methods, 2011, 21, 53–62 CrossRef CAS PubMed.
- J. Bajgar, J. Fusek, K. Kuca, L. Bartosova and D. Jun, Mini-Rev. Med. Chem., 2007, 7, 461–466 CrossRef CAS PubMed.
- M. Patel, E. B. Souto and K. K. Singh, Expert Opin. Drug Delivery, 2013, 10, 889–905 CrossRef CAS PubMed.
- D. S. Hersh, A. S. Wadajkar, N. B. Roberts, J. G. Perez, N. P. Connolly, V. Frenkel, J. A. Winkles, G. F. Woodworth and J. K. Anthony, Curr. Pharm. Des., 2016, 22, 1177–1193 CrossRef CAS PubMed.
- J. C. DeMar, E. D. Clarkson, R. H. Ratcliffe, A. J. Campbell, S. G. Thangavelu, C. A. Herdman, H. Leader, S. M. Schulz, E. Marek, M. A. Medynets and T. C. Ku, Chem.-Biol. Interact., 2010, 187, 191–198 CrossRef CAS PubMed.
- T. Sauvaître, M. Barlier, D. Herlem, N. Gresh, A. Chiaroni, D. Guenard and C. Guillou, J. Med. Chem., 2007, 50, 5311–5323 CrossRef PubMed.
- K. Kuca, D. Jun and K. Musilek, Mini-Rev. Med. Chem., 2006, 6, 269–277 CrossRef CAS PubMed.
- G. Mercey, T. Verdelet, G. Saint-André, E. Gillon, A. Wagner, R. Baati, L. Jean, F. Nachon and P. Y. Renard, Chem. Commun., 2011, 47, 5295–5297 RSC.
- G. Saint-André, M. Kliachyna, S. Kodepelly, L. Louise-Leriche, E. Gillon, P. Y. Renard, F. Nachon, R. Baati and A. Wagner, Tetrahedron, 2011, 67, 6352–6361 CrossRef.
- L. ise-Leriche, E. Pǎunescu, G. Saint-André, R. Baati, A. Romieu, A. Wagner and P. Y. Renard, Chem.–Eur. J., 2010, 16, 3510–3523 CrossRef PubMed.
- N. Khorana, K. Changwichit, K. Ingkaninan and M. Utsintong, Bioorg. Med. Chem. Lett., 2012, 22, 2885–2888 CrossRef CAS PubMed.
- A. Krasiński, Z. Radić, R. Manetsch, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, J. Am. Chem. Soc., 2005, 127, 6686–6692 CrossRef PubMed.
- R. K. Sit, Z. Radić, V. Gerardi, L. Zhang, E. Garcia, M. Katalinić, G. Amitai, Z. Kovarik, V. V. Fokin, K. B. Sharpless and P. Taylor, J. Biol. Chem., 2011, 286, 19422–19430 CrossRef CAS PubMed.
- Z. Radić, R. K. Sit, Z. Kovarik, S. Berend, E. Garcia, L. Zhang, G. Amitai, C. Green, B. Radić, V. V. Fokin and K. B. Sharpless, J. Biol. Chem., 2012, 287, 11798–11809 CrossRef PubMed.
- Y. J. Rosenberg, L. Mao, X. Jiang, J. Lees, L. Zhang, Z. Radic and P. Taylor, Chem.-Biol. Interact., 2017, 274, 50–57 CrossRef CAS PubMed.
- Z. Kovarik, N. Maček, R. K. Sit, Z. Radić, V. V. Fokin, K. B. Sharpless and P. Taylor, Chem.-Biol. Interact., 2013, 203, 77–80 CrossRef CAS PubMed.
- Z. Wei, Y. Q. Liu, X. B. Zhou, Y. Luo, C. Q. Huang, Y. A. Wang, Z. B. Zheng and S. Li, Bioorg. Med. Chem. Lett., 2014, 24, 5743–5748 CrossRef CAS PubMed.
- S. M. Agten, P. E. Dawson and T. M. Hackeng, J. Pept. Sci., 2016, 22, 271–279 CrossRef CAS PubMed.
- S. G. Patching, Mol. Neurobiol., 2017, 54, 1046–1077 CrossRef CAS PubMed.
- G. E. Garcia, A. J. Campbell, J. Olson, D. Moorad-Doctor and V. I. Morthole, Chem.-Biol. Interact., 2010, 187, 199–206 CrossRef CAS PubMed.
- D. E. Lorke and G. A. Petroianu, Front. Neurosci., 2019, 13, 427 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |