DOI:
10.1039/C9RA09203J
(Paper)
RSC Adv., 2020,
10, 3626-3635
Inhibition of human thrombin by the constituents of licorice: inhibition kinetics and mechanistic insights through in vitro and in silico studies†
Received
6th November 2019
, Accepted 13th January 2020
First published on 22nd January 2020
Abstract
Thrombin inhibition therapy is a practical strategy to reduce thrombotic and cardiovascular risks via blocking the formation of blood clots. This study aimed to identify naturally occurring thrombin inhibitors from licorice (one of the most popular edible herbs), as well as to investigate their inhibitory mechanisms. Among all tested licorice constituents, licochalcone A was found as the most efficacious agent against human thrombin (IC50 = 7.96 μM). Inhibition kinetic analyses demonstrated that licochalcone A was a mixed inhibitor against thrombin-mediated Z-Gly-Gly-Arg-AMC acetate hydrolysis, with a Ki value of 12.23 μM. Furthermore, mass spectrometry-based chemoproteomic assays and molecular docking simulations revealed that licochalcone A could bind to human thrombin at both exosite I and the catalytic site. In summary, our findings demonstrated that the chalcones isolated from licorice were a new class of direct thrombin inhibitors, also suggesting that licochalcone A was a promising lead compound for developing novel anti-thrombotic agents.
1. Introduction
Thrombus formation is one of the leading causes of cardiovascular disease (CVD), in which arterial thrombosis may result in myocardial infarction or stroke and phlebothrombosis may lead to deep venous thrombosis or pulmonary thromboembolism.1 Over the past sixty years, many therapeutic strategies have been proposed to modulate the formation of blood clots (thrombi) via targeting different targets. Among all reported therapeutic strategies, inhibition of thrombin (a crucial serine hydrolase distributed in circulation system, EC 3.4.21.5) has been recognized as a practical strategy to modulate the coagulation system and thereby block the formation of blood clots.2,3 Currently, several direct thrombin inhibitors (dabigatran, bivalirudin, argatroban, etc.) have been used in clinical settings to decrease the recrudesce rate of CVD events in patients.4–7 Although these direct thrombin inhibitors (DTIs) display outstanding anti-thrombin effects, the high-risk complications (such as major bleeding, dosage uncertainty and expensive price) of these DTIs greatly limit the widely applications of these agents in clinical settings.8 Thus, more efforts should be driven to find more thrombin inhibitors or modulators with high efficacy and good safety profiles.9
It is well-known that the natural compounds from herbs are the important sources for discovery of drug lead compounds, especially for the herbs which have been used in clinical settings for a long history with safety and efficacy.10,11 Licorice (the root of Glycyrrhiza glabra), one of the most frequently used herbs in both eastern and western countries, is quite popular in candies and sweets as a flavouring agent or health supplement around the world.12 In Asian countries, liquorice and its products have been frequently used for the adjuvant therapy of a variety of human disorders (such as digestive diseases, bronchitis, asthma, as well as circulatory disorders).13 Previous pharmacological studies have demonstrated that licorice and its major constituents have extensive biological activities, such as anti-obesity, anti-oxidative, anti-inflammatory, anti-cancer and enzyme regulatory activities.14,15 In addition, a previous study has demonstrated that glycyrrhizin is a weak thrombin inhibitor with an IC50 value around 150 μM.16 However, the anti-thrombotic effects of other constituents in licorice and their mechanisms of action are not well-investigated yet. In view of that some constituents isolated from licorice have been validated as strong inhibitors of a panel of serine hydrolases, including acetylcholinesterase, pancreatic lipase and phospholipase A2,17–21 we speculate that these constituents may also inhibit the hydrolytic activity of thrombin and then modulate the haemostatic-thrombotic system.
Herein, the anti-thrombin effects of the major constituents in licorice on human thrombin and their inhibitory mechanisms were investigated. Twenty-one constituents isolated from licorice were collected and their anti-thrombin effects were evaluated by a fluorescence-based thrombin inhibition assay. Preliminary screening found that licochalcone A and other chalcones exhibited a strong inhibition on human thrombin, while the flavonoids, coumarins, triterpenoids and others showed negligible inhibitory activity on human thrombin. Among all tested licorice constituents, licochalcone A was found as the most efficacious agent against human thrombin. These findings inspired us to further investigate the inhibitory mechanism of licochalcone A on human thrombin via a set of inhibition kinetic assays. Furthermore, the ligand-binding site(s) of licochalcone A on human thrombin were carefully investigated by mass spectrometry-based chemoproteomic assay and docking simulations. These investigations were extremely helpful for the deeply understanding of the inhibitory mechanism of licochalcone A against human thrombin, which may also facilitate the discovery of new anti-thrombotic agents.
2. Materials and methods
2.1. Chemical reagents and enzyme source
Twenty-one constituents (Fig. 1) isolated from licorice, including isoliquiritin (1), isoliquiritigenin (2), licochalcone A (3), licochalcone B (4), licochalcone C (5), echinatin (6), neoisoliquiritin (7), isoliquiritin apioside (8), liquiritin apioside (9), liquiritigenin (10), glabrol (11), liquiritin (12), neoliquiritin (13), licoflavonol (14), isolicoflavonol (15), glycycoumarin (16), isoglycyrol (17), glycyrrhetinic acid (18), glycyrrhizic acid (19), glabridin (20) and gancaomin I (21), with the purities over 98%, were collected from Chendu Pufeide (Sichuan, China). Thrombin (blood-coagulation factor IIa, Lot no. F1700752P2) isolated from human plasma was obtained from HYPHEN BioMed (France). Z-GGRAMC acetate, a fluorescence substrate for thrombin, was supplied by MCE CO., Ltd. (Shanghai, China) and its fluorescence product 7-amino-4-methylcoumarin (AMC) was obtained from Absin Biotechnology (Shanghai, China). LC grade dimethyl sulfoxide (DMSO), acetonitrile and formaldehyde were supplied by Tedia (Tedia, USA). NaBH3CN, chymotrypsin and other chemical reagents were purchased from Sigma-Aldrich (Shanghai). Tris–HCl buffer (pH = 8.3, 50 mM) and human thrombin (10 NIH per mL) were prepared using Millipore water. The substrate Z-GGRAMC acetate (50 mM) and each constituent (100 mM) in licorice, as well as the positive inhibitors were dissolved in DMSO. The stock solution of human thrombin was stored −80 °C and the stock solution of substrate or each tested compound stored at 4 °C until use.
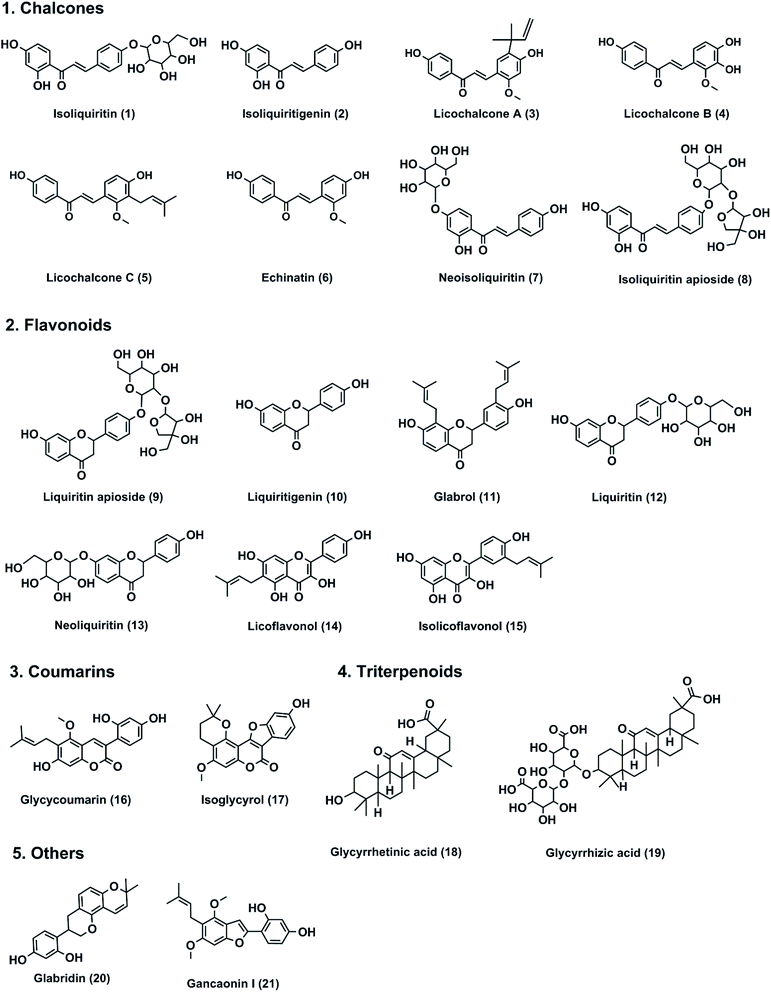 |
| | Fig. 1 The chemical structures of major constituents in licorice root. | |
2.2. Thrombin inhibition assay
Z-GGRAMC acetate was used as the substrate to assay the inhibition potentials of licorice constituents on human thrombin, while a multi-mode microplate reader (SpectraMaxs iD3, Molecular Devices, Austria) was used to determine the fluorescence intensity of the hydrolytic product (7-amino-4-methylcoumarin, AMC) in reaction mixture. The procedures and detail experimental for thrombin inhibition assay had been reported previously.22 In brief, 200 μL of incubation system containing Tris buffer (50 mM, pH 8.3), human thrombin (0.1 NIH per mL, final concentration), NaCl (100 mM), BSA (0.1 mg mL−1), and each tested compound were mixed. Following pre-incubation at 37 °C for 10 min, the reaction was initiated by adding Z-GGRAMC acetate (50 μM, final concentration). After 20 min incubation, the reaction mixture was recorded by a fluorescence microplate reader. To quantify the formation rate of the hydrolytic metabolite AMC, the excitation wavelength and the emission wavelength were set at 380 nm, and 440 nm, respectively. The standard curve of AMC was shown in Fig. S1.† Ginkgetin and baicalein were employed as positive inhibitors, while DMSO was used as a negative control.23,24 The florescence intensity of AMC in the negative control (DMSO only) was used as 100%.
2.3. Inhibition kinetic analyses
To investigate the inhibition kinetics of licochalcone A against human thrombin, a series of inhibition kinetic analyses was performed. In this study, varying concentrations of the substrate (Z-GGRAMC acetate) were employed to calculate the hydrolytic rates of human thrombin in the presence of various licochalcone A concentrations, which were then used to determine the inhibition modes (such as competitive, noncompetitive or mixed inhibition) and to get the inhibition constant (Ki). The Ki values were determined by using the mixed inhibition kinetic mode. The details for inhibition kinetic analyses, as well as the details for determination of the inhibition mode(s) and Ki value were described previously.25,26
2.4. Mass spectrometry-based chemoproteomic assay
A mass spectrometry-based chemoproteomic approach combined with direct lysine reactivity assay were used to characterize the ligand-binding site(s) of licochalcone A on human thrombin. The procedure of mass spectrometry-based chemoproteomic assay and the determination of labelled lysine residues using mass spectrometry-based chemoproteomic approach had been described recently.27–29 Briefly, a total of 200 μL HEPES buffer (pH 7.4, 20 mM), NaCl (200 mM) and human thrombin (20 μg) were pre-incubated under physiological conditions for 10 min. After then, the inhibitor licochalcone A (1.4 mg μL−1) was added and incubated for 30 min at 37 °C. Meanwhile, DMSO was used as the control group and also incubated under same conditions for 30 min. After 30 min incubation, human thrombin was labelled by adding NaBH3CN and CH2O at 37 °C for another 20 min and the lysine labelling reactivity was quenched by addition of NH4AC. The samples containing labelled protein were then digested by chymotrypsin at 37 °C for 24 h. Finally, the modified peptides were desalted and analysed by liquid chromatography-tandem mass spectrometry. The instrument information and analytical conditions of mass spectrometry detection were described previously.24 The ratio of labelled peptides was determined according to the following equation,
| The ratio of labelled peptides = (spectral counts of the peptides with labelled lysines)/(spectral counts of the peptides with labelled and unlabelled lysines) |
2.5. Molecular docking simulations
Herein, a crystal structure of human thrombin [PDB ID: 1PPB] at 1.92 Å resolution was used for docking simulations.30 To generate the proper protonation states, Epik 3.4 (Schrödinger, LLC, New York, NY, USA) of Ligprep 3.6 software (Schrödinger, NY, USA) was used to pre-process the ligand (licochalcone A). Docking simulations was performed by Glide 6.9 (Schrödinger, NY, USA) in SP (standard precision) mode, and the other parameters were set at default values.
3. Results
3.1. Inhibition of human thrombin by licorice constituents
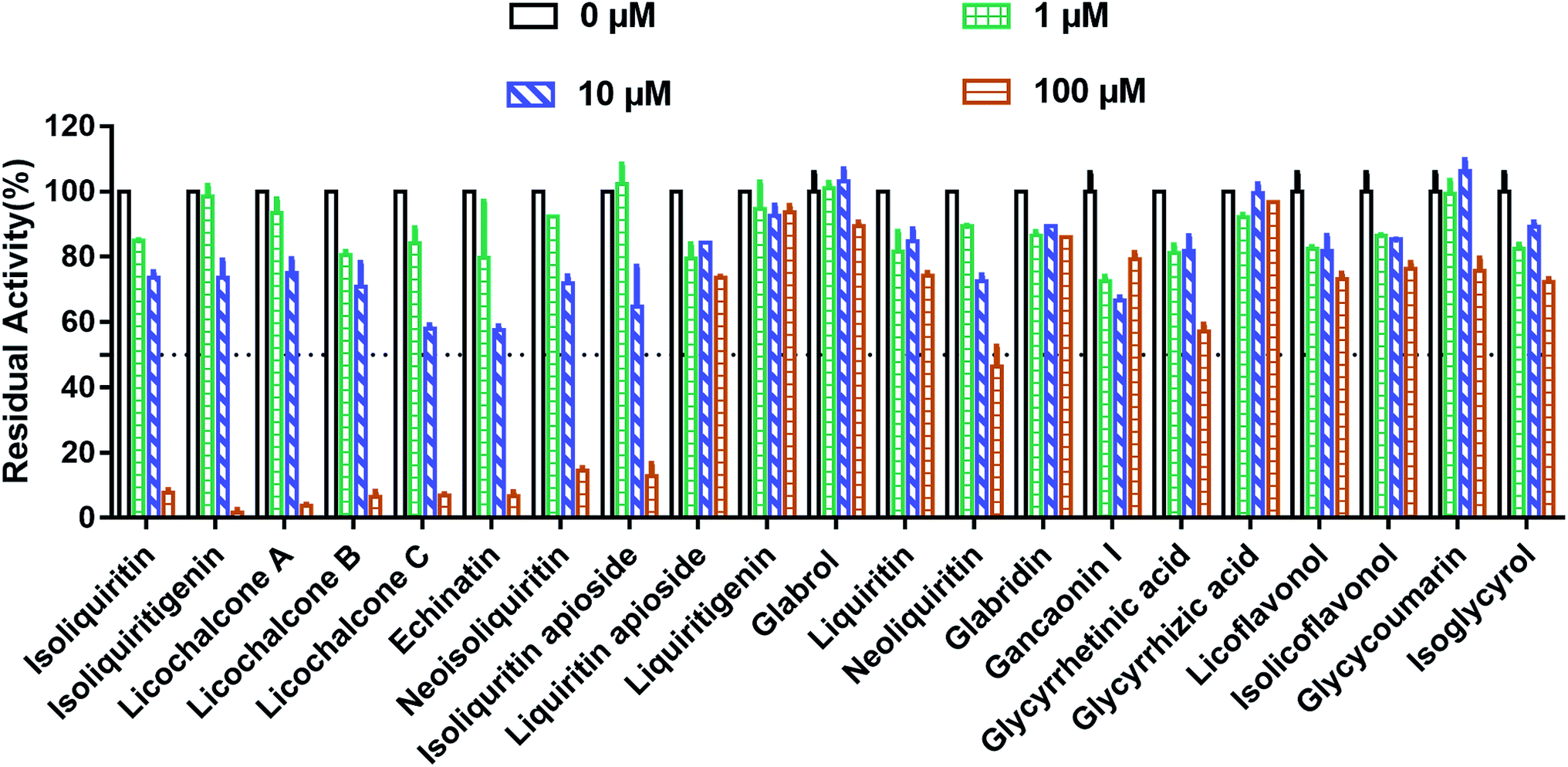
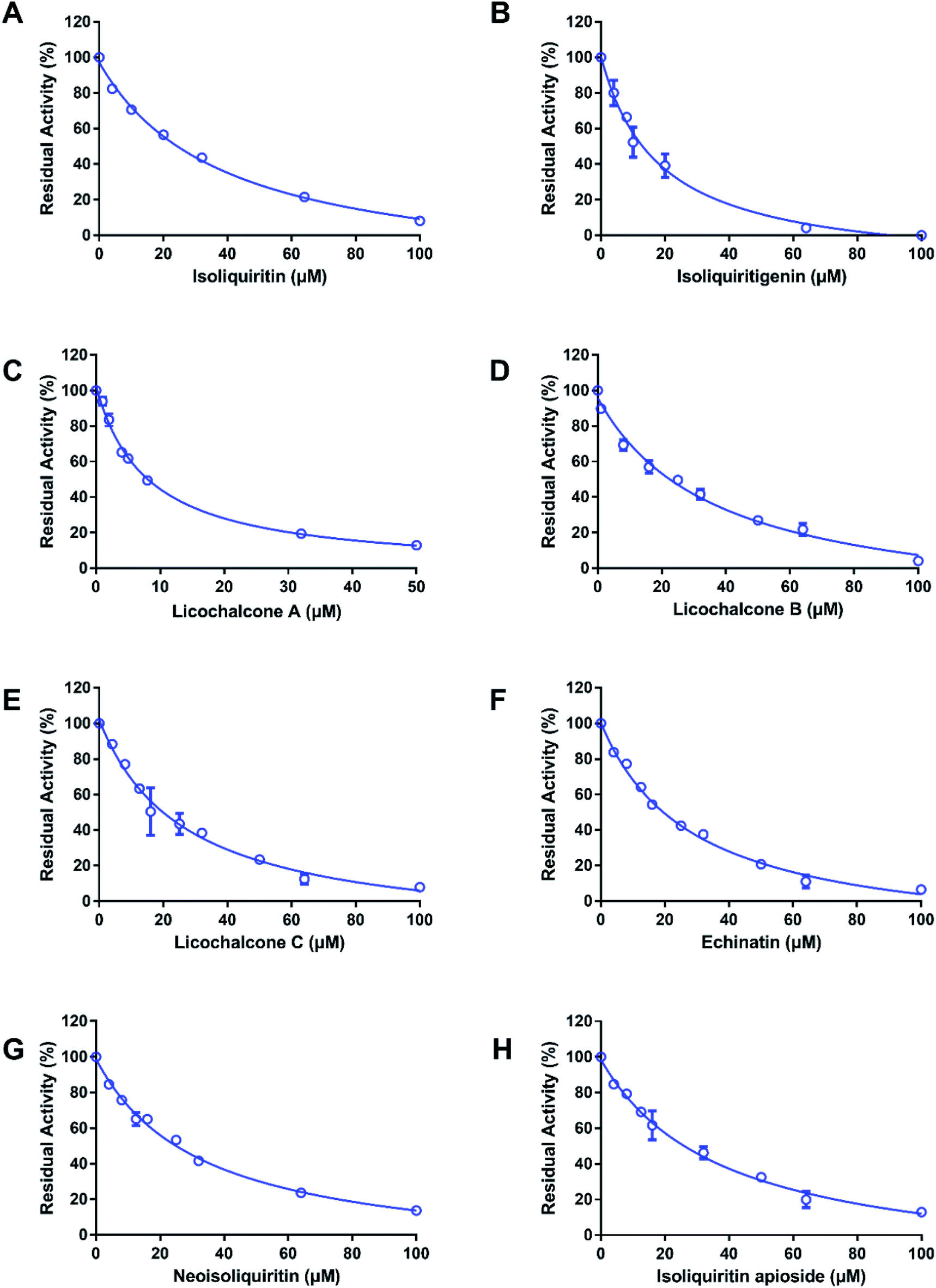
At first, the inhibition potentials of the twenty-one natural constituents from licorice on human thrombin were preliminarily screened. As shown in Fig. 2, preliminary screening results demonstrated eight licorice constituents could inhibit thrombin activity by 50% or more at the high dosage (100 μM). After that, the dose–response curves of these licorice constituents on thrombin were depicted (Fig. 3), while ginkgetin and baicalein were used as positive inhibitors (two naturally occurring thrombin inhibitors).23,24 The IC50 values of these eight licorice constituents against human thrombin were listed in Table 1. Among all tested licorice constituents, licochalcone A displayed the most potent inhibition on human thrombin (IC50 = 7.96 μM), while other chalcones isolated from licorice exhibited moderate inhibition on human thrombin, with the IC50 values ranging from 17.95 μM to 38.76 μM. By contrast, the flavonoids, coumarins, triterpenoids and others isolated from licorice showed negligible effects. Meanwhile, the dose–response curves of both ginkgetin and baicalein were also determined under same conditions, the calculated IC50 values of these two compounds were 8.05 μM, and 36.11 μM, respectively. These results demonstrated that the chalcones isolated from licorice were a new class of thrombin inhibitors, and licochalcone A exhibited the most potent anti-thrombin effect. These data inspired us to further examine the inhibitory mechanism of licochalcone A against human thrombin.
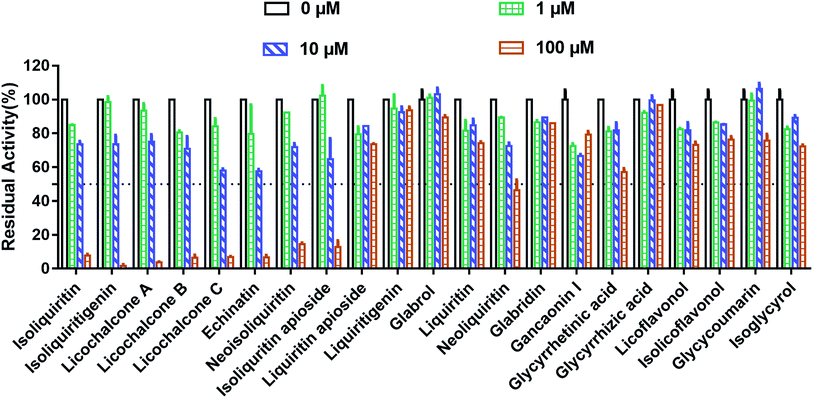 |
| | Fig. 2 Inhibitory effects of the major constituents in licorice on human thrombin at three different concentrations (0 μM, 1 μM, 10 μM, 100 μM). All inhibition experiments were performed in triplicate (n = 3) and the values are expressed as mean ± SD. | |
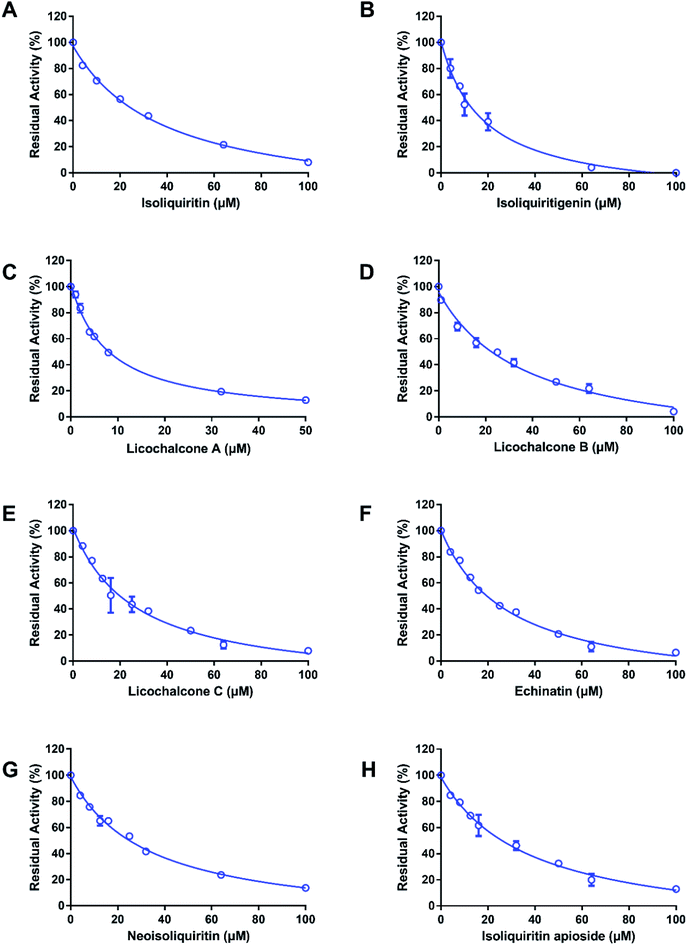 |
| | Fig. 3 The dose–response curves of components from licorice on human thrombin; isoliquiritin (A), isoliquiritigenin (B), licochalcone A (C), licochalcone B (D), licochalcone C (E), echinatin (F), neoisoliquiritin (G) and isoliquiritin apioside (H). All inhibition experiments were performed in triplicate (n = 3) and the values are expressed as mean ± SD. | |
Table 1 The IC50 values of the major constituents in licorice on human thrombin
| Class |
No. |
Constituent |
MW |
IC50 (μM) |
| Chalcones |
1 |
Isoliquiritin |
418.39 |
38.76 ± 4.04 |
| 2 |
Isoliquiritigenin |
256.30 |
17.95 ± 2.75 |
| 3 |
Licochalcone A |
338.30 |
7.96 ± 0.62 |
| 4 |
Licochalcone B |
286.28 |
36.90 ± 5.61 |
| 5 |
Licochalcone C |
338.40 |
26.64 ± 4.17 |
| 6 |
Echinatin |
270.28 |
28.46 ± 2.65 |
| 7 |
Neoisoliquiritin |
418.40 |
32.85 ± 3.38 |
| 8 |
Isoliquiritin apioside |
550.50 |
37.85 ± 5.78 |
| Flavonoids |
9 |
Liquiritin apioside |
550.50 |
>100 |
| 10 |
Liquiritigenin |
256.25 |
>100 |
| 11 |
Glabrol |
392.49 |
>100 |
| 12 |
Liquiritin |
418.39 |
>100 |
| 13 |
Neoliquiritin |
418.40 |
>100 |
| 14 |
Licoflavonol |
354.35 |
>100 |
| 15 |
Isolicoflavonol |
354.36 |
>100 |
| Coumarins |
16 |
Glycycoumarin |
368.37 |
>100 |
| 17 |
Isoglycyrol |
366.36 |
>100 |
| Triterpenoids |
18 |
Glycyrrhetinic acid |
470.64 |
>100 |
| 19 |
Glycyrrhizic acid |
822.92 |
>100 |
| Others |
20 |
Glabridin |
324.37 |
>100 |
| 21 |
Gancaonin I |
354.40 |
>100 |
| Positive inhibitors |
22 |
Ginkgetin |
552.49 |
8.05 ± 1.50 |
| 23 |
Baicalein |
270.20 |
36.11 ± 4.68 |
3.2. Inhibition kinetics of licochalcone A on human thrombin
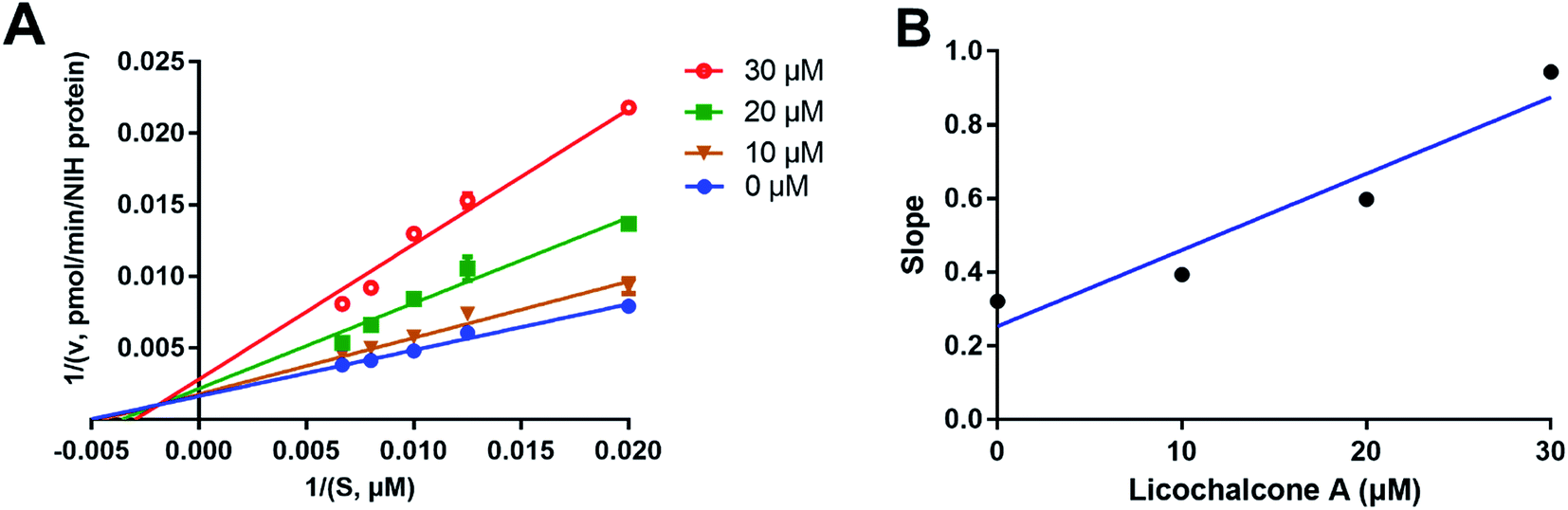
Considering that two classes of thrombin inhibitors (irreversible and reversible inhibitors) had been reported, it is necessary to identify the inhibition type of licochalcone A against human thrombin. To this end, time-dependent inhibition assays was conducted to depict the dose–inhibition curves with or without long pre-incubation time (30 min). As depicted in Fig. S2,† both the dose–inhibition curves and the IC50 values of licochalcone A against human thrombin were not changed along with 30 min pre-incubation, suggesting that this agent was a reversible inhibitor rather than an irreversible inactivator of human thrombin. After that, the inhibition mode and inhibition constant (Ki) of licochalcone A against human thrombin were carefully investigated by a panel of kinetic analyses. As shown in Fig. 4, the Lineweaver–Burk plots revealed that licochalcone A against thrombin-mediated Z-GGRAMC acetate hydrolysis via a mixed manner. Meanwhile, the inhibition constant (Ki) of licochalcone A against human thrombin was also determined as 12.23 μM (Table 2).
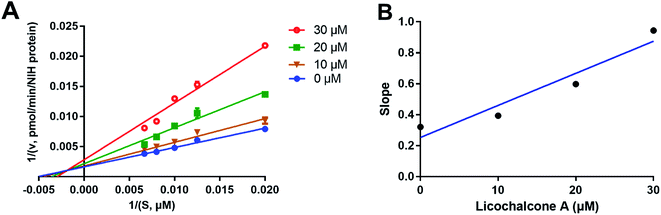 |
| | Fig. 4 The inhibitory behavior of licochalcone A on human thrombin. Left: the Lineweaver–Burk plot of licochalcone A (A) against thrombin-mediated Z-GGRAMC acetate hydrolysis. Right: the secondary plot from the Lineweaver–Burk plot for human thrombin inhibition by licochalcone A (B). All inhibition experiments were performed in triplicate (n = 3) and the values are expressed as mean ± SD. | |
Table 2 Inhibition constant and the inhibition mode of licochalcone A against thrombin-mediated Z-GGRAMC acetate hydrolysis
| Constituent |
IC50 (μM) |
Ki (μM) |
Inhibition mode |
Goodness of fit (R2) |
| Licochalcone A |
7.96 ± 0.62 |
12.23 |
Mixed |
0.92 |
3.3. Identification of the binding site(s) of licochalcone A on human thrombin
It had been reported that human thrombin had three major ligand-binding sites, including the catalytic cavity (for the substrates or the competitive inhibitors) and two positively charged sites on the surface of this key enzyme (exosite I and exosite II). Exosite I (fibrinogen-binding exosite) could recognize fibrinogen, thrombomodulin and protease-activated receptors, while exosite II (heparin-binding exosite) could interact with heparin, GPIbα, clotting factor V and VIII.31,32 Notably, the lysine residues surrounding at the exosite I (such as K107, K109 and K110) or exosite II (K236 and K240) were located on the surface of human thrombin and could be easily modified by NaBH3CN in the presence of CH2O. In these cases, mass spectrometry-based chemoproteomic assay combined with lysine reactivity assay were conducted to explore the binding site(s) of licochalcone A on human thrombin. The results clearly demonstrated that licochalcone A treatment could significantly decline the lysine labelling ratio from 80% to 0% at the exosite I (Tables 3 and S1†). In sharp contrast, the lysine labelling ratio at exosite II in both licochalcone A-treated group and the control group (DMSO only) were closed to each other. These findings clearly demonstrated that licochalcone A could directly bind on human thrombin at exosite I rather than exosite II, which could be used to explain why licochalcone A inhibited human thrombin via a non-competitive manner.
Table 3 The labelling ratios of key lysine residues of human thrombin (the amino acid numbers indicate human thrombin numbering)a
| Residues |
Sites |
Labelling ratios |
| Thrombin without licochalcone A |
Thrombin with licochalcone A |
| Other labelling ratios of lysine residues are provided in Table S1 (see ESI). |
| Lys-60F |
Active site |
Not labelled |
Not labelled |
| Lys-36 |
Exosite I |
93% |
100% |
| Lys-107, Lys-109, Lys-110 |
Exosite I |
80% |
0% |
| Lys-236 |
Exosite II |
Not labelled |
Not labelled |
| Lys-240 |
Exosite II |
95% |
70% |
Considering the lysine residue (K60F) on the catalytic cavity of human thrombin was deeply embedded in this enzyme and K60F was hardly labelled under physiological conditions, mass spectrometry-based chemoproteomic assay could not give us directly evidence to support whether licochalcone A could modulate human thrombin activity via directly occupying the catalytic cavity of this hydrolase. In these cases, molecular docking simulations were used to gain the deep insights into the key interactions between licochalcone A and human thrombin. The results demonstrated that licochalcone A could be well-docked into the catalytic cavity of human thrombin, and the binding area of licochalcone A on this enzyme was partially overlapped with that of the substrate (Z-GGRAME acetate). As shown in Fig. 5, licochalcone A could create strong interactions with Tyr-60A and Ser-195 via hydrogen bonding, as well as create strong interactions with Tyr-60A via Pi–Pi stacking. This finding suggested that licochalcone A might also inhibit human thrombin activity via a competitive inhibition manner by directly occupy the catalytic cavity of this enzyme. Collectively, both mass spectrometry-based chemo-proteomic assay and molecular docking simulations showed that licochalcone A could bind on human thrombin at both the catalytic cavity and exosite I, which agreed well with the mixed inhibition manner of licochalcone A (as shown in the inhibition kinetic analyses).
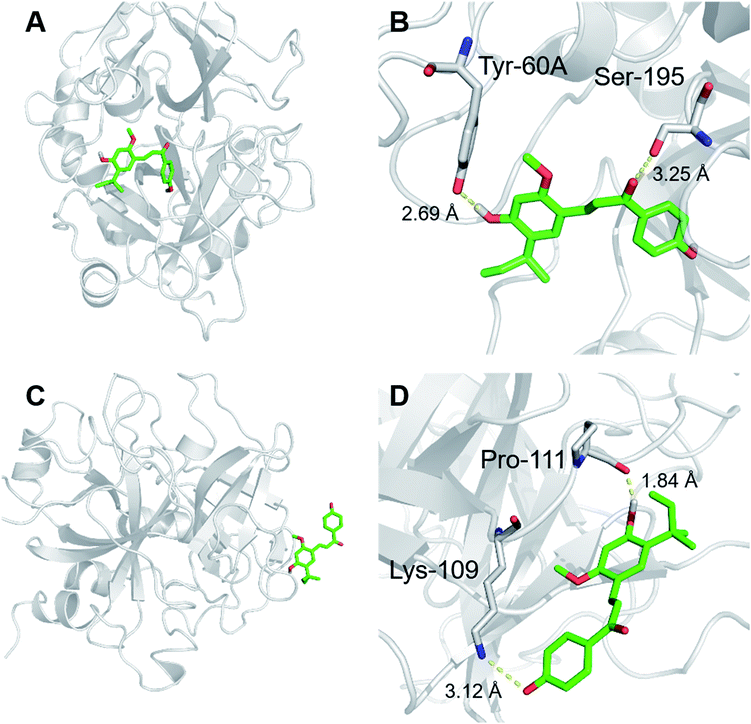 |
| | Fig. 5 A stereo view of the crystal structure of thrombin and a stereodiagram of Z-GGRAMC acetate docked with licochalcone A. Inhibitor could bind to the active site (A), exosite I (C) of human thrombin. The detailed view represents the interactions between licochalcone A and the amino acids in the active site (B) and exosite I (D) of human thrombin. | |
4. Discussion
Although more and more attention has been paid to the prevention of thrombus formation and thrombosis-related cardiovascular diseases, cardiovascular and thrombotic diseases are still one of the leading causes of morbidity and mortality around the world.33 Nowadays, inhibition of thrombin activity has become a pivotal strategy to modulate the processes of both venous and arterial thrombosis.34 In fact, several synthetic DTIs have been used to prevent and treat cardiovascular and thrombotic diseases in the clinical settings, but the severe side effects of these DTIs have motivated the medicinal chemists to find more DTIs with good safety profiles. In view of that natural compounds are one of the leading sources of the drug lead compounds, many groups are devoted to find active ingredients with strong anti-thrombotic activity and good safety from herbal medicines. In fact, a variety of natural compounds with strong thrombin inhibition activity have been reported by several groups over the past two decades. For instance, Liu et al. have reported that some natural flavonoids are thrombin inhibitors.23 Wang et al. have identified isoquinoline alkaloid (such as berberine) as a novel structural scaffold of thrombin inhibitors.35 Lu et al. have found three tanshinones with a good anti-thrombin activity.36 Ge et al. have demonstrated that naphthodianthrone (such as hypericin) and anthraquinone (such as obtusifolin) are new classes of thrombin inhibitor.37,38 Guo et al. have characterized four biflavones as mix thrombin inhibitors.24 All these findings have indicated that herbal constituents hold great promise for the discovery of thrombin inhibitors and inspire us to find more thrombin inhibitors with new structurally diverse scaffolds.
Licorice, one of the oldest and most popular herbs used in the world, also the major ingredient for preparing a variety of Chinese herbal prescriptions have been used to prevent and treat cardiovascular and circulation disorders in Asian countries.13 However, the anti-thrombin effects of licorice constituents and the underlying molecular mechanisms have not been identified yet. In the present study, our findings clearly demonstrate that licochalcone A, a bioactive compound in licorice, exhibit strong inhibition on human thrombin, with the IC50 value of 7.96 μM. In addition to licochalcone A, other chalcones isolated from licorice, such as isoliquiritin, isoliquiritigenin, licochalcone B, licochalcone C, echinatin, neoisoliquiritin and isoliquiritin apioside also show moderate anti-thrombin activities. By contrast, other constituents in licorice, such as flavonoids, coumarins, triterpenoids and others, display negligible inhibition on human thrombin. These date indicate that the chalcones are major bioactive constituents in licorice with anti-thrombin effects, while licochalcone A could serve as a leading compound for the development of chalcone-type thrombin inhibitors.
Although licochalcone A has been found with relatively strong anti-thrombin effect, its potency is not as strong as that of the marketed synthetic thrombin inhibitors (the IC50 values at the nanomolar level).39,40 Hence, it is essential to develop more efficacious thrombin inhibitors using licochalcone A as a lead compound or using chalcone as a basic skeleton.41,42 It is well-known that the chalcones (such as licochalcone A) can be easily obtained from licorice, a widely distributed and cultivated herb in the northern hemisphere.43 From the views of chemical modifications, these naturally occurring chalcones can generate more chalcone derivatives via chemical modification due to the presence of multiple functional groups.44 It is noteworthy that the chalcones can be totally synthesized and easily to obtain,42,45 which strongly facilitates the medicinal chemists to design and develop more chalcone derivatives for further investigations on structure–activity relationships of chalcones as thrombin inhibitors.46–48 In this study, some key information on structure–activity relationships of chalcones as thrombin inhibitors have been revealed. For instance, the IC50 values of isoliquiritigenin (2), and its mono-glycoside isoliquiritin (1) and its di-glycoside isoliquiritin apioside (8) against human thrombin are 17.95 μM, 38.76 μM and 37.85 μM, respectively. These results demonstrate that the introduction of one or two glycoside group will reduce the inhibition activity of these chalcones on human thrombin. Meanwhile, the IC50 values of licochalcone A (3), isoliquiritigenin (2), and licochalcone B (4) against human thrombin are 7.96 μM, 17.95 μM and 36.90 μM, respectively. These results suggest that introduction of hydrophobic substituents on the right ring of the chalcones may be beneficial for thrombin inhibition. These findings will be very helpful for the design and development of the next generation of chalcone-type thrombin inhibitors.
In addition to improve the inhibition potency of the chalcones on thrombin, the specificity of the chalcones on thrombin over other human hydrolases should also been improved. It has been reported that the chalcones can inhibit a panel of human hydrolases (such as lipase and carboxylesterases), the specificity of chalcone derivatives against human thrombin over other human hydrolases should be carefully investigated.26,49 In near future, more efficacious and highly specific thrombin inhibitors can be designed via computer-aided drug design, on the basis of the crystal structures of human thrombin and other concerned serine hydrolases. In addition, considering that licochalcone A can be rapidly metabolized by hepatic drug metabolizing enzymes,50–52 the metabolic stability of licochalcone A analogous should be improved in the future, which will be beneficial for the development of chalcone derivatives with outstanding in vivo anti-thrombin effects.
In view of licorice and its products have been frequently used as dietary supplements or herbal medicines, the potential risks of drug–drug interactions (DDI) between licorice and other anticoagulant or antiplatelet agents should be noted in the clinical settings, especially for the cases of the licorice-containing products used in combination with those agents with narrow therapeutic windows (such as warfarin and clopidogrel). The occurrence of DDIs can reduce the safety of a drug.53,54 Previous studies have demonstrated that licorice constituents may increase the bleeding risk when anticoagulant or antiplatelet drugs are co-administrated with this herb.55 It should be noted that some previous investigations have revealed that licorice constituents can affect the pharmacokinetic behaviour of some therapeutic drugs via inhibiting the hepatic drug metabolizing enzymes (cytochrome P450 enzymes and UDP-glucuronosyltransferases).56–58 In this study, our results demonstrate that thrombin activity can be inhibited by the chalcones in licorice. Therefore, it is conceivable that both pharmacokinetic and pharmacodynamics interactions may occur when anticoagulant or antiplatelet drugs are used in combination with licorice-containing products. It also has been reported that licochalcone A can block collagen-induced platelet aggregation59,60 and function as a inhibitor of clotting factor Xa,61 suggesting that licorice constituents may regulate the blood coagulation cascade via multiple mechanisms. Therefore, the clinical consequences of licorice in combination with anticoagulant or antiplatelet agents should be carefully investigated from the views of both pharmacokinetic and pharmacodynamics interactions. In future, more efforts should be made to predict the possible risks of DDI between licorice and the clinically used anticoagulant or antiplatelet drugs.
5. Conclusion
In summary, the inhibitory effects of twenty-one constituents in licorice on human thrombin were investigated. The results clearly demonstrated that the licochalcone A, one of the abundant chalcones in licorice, exhibited strong inhibition on human thrombin. Further investigation on inhibition kinetic analyses demonstrated that licochalcone A acted as a mixed inhibitor of human thrombin, with the inhibitory constant of 12.23 μM. Finally, the mass spectrometry-based chemoproteomic assay and molecular docking simulations revealed that licochalcone A could bind on human thrombin at both the active site and the exosite I. Collectively, these findings provided solid evidence to support that licorice and licorice-related products might bring beneficial effects on cardiovascular disease via inhibiting human thrombin. In future, licochalcone A could serve as a leading compound for the development of novel anti-thrombotic agents bearing the chalcone scaffold.
Author contributions
Ge G., Guo W., and Zhang L. designed the whole experiment. Shi C., Chen T., Zhang Q., Huang C., Zhu Y. performed the experiment. Shi C., Chen T., Ge G. and Guo W. written the paper. Wei L., Liu H., Bai Y. and Wang F. were supportive during the experiment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFC1700200, 2017YFC1702000), the National Natural Science Foundation of China (81973286, 81803489, U1604282, 81973393, 81922070, 81773687), the Program of Shanghai Academic/Technology Research Leader (18XD1403600), the Basic and Frontier Technology Research Program of Henan Science and Technology Commission Foundation (142300410039) and TaiShan Industrial Experts Program [tscy20180234].
References
- S. Cheng, M. Tu, H. Chen, Z. Xu, Z. Wang, H. Liu, G. Zhao, B. Zhu and M. Du, Food Funct., 2018, 9, 6391–6400 RSC.
- N. Mackman, Nature, 2008, 451, 914–918 CrossRef CAS PubMed.
- M. Di Nisio, S. Middeldorp and H. R. Buller, N. Engl. J. Med., 2005, 353, 1028–1040 CrossRef CAS.
- P. J. O'Brien and L. Mureebe, J. Cardiovasc. Pharmacol. Ther., 2012, 17, 5–11 CrossRef.
- S. Bielecki, D. Lee and B. Hamad, Nat. Rev. Drug Discovery, 2018, 17, 617–618 CrossRef CAS PubMed.
- H. A. Blair and G. M. Keating, Drugs, 2017, 77, 331–344 CrossRef.
- R. W. Yeh and I. K. Jang, Am. Heart J., 2006, 151, 1131–1138 CrossRef CAS PubMed.
- P. C. Kam, N. Kaur and C. L. Thong, Anaesthesia, 2005, 60, 565–574 CrossRef CAS PubMed.
- D. J. P. Pinto, M. J. Orwat, L. M. Smith, 2nd, M. L. Quan, P. Y. S. Lam, K. A. Rossi, A. Apedo, J. M. Bozarth, Y. Wu, J. J. Zheng, B. Xin, N. Toussaint, P. Stetsko, O. Gudmundsson, B. Maxwell, E. J. Crain, P. C. Wong, Z. Lou, T. W. Harper, S. A. Chacko, J. E. Myers, Jr., S. Sheriff, H. Zhang, X. Hou, A. Mathur, D. A. Seiffert, R. R. Wexler, J. M. Luettgen and W. R. Ewing, J. Med. Chem., 2017, 60, 9703–9723 CrossRef CAS PubMed.
- Y. Zhang, C. Wang, L. Wang, G. S. Parks, X. Zhang, Z. Guo, Y. Ke, K. W. Li, M. K. Kim, B. Vo, E. Borrelli, G. Ge, L. Yang, Z. Wang, M. J. Garcia-Fuster, Z. D. Luo, X. Liang and O. Civelli, Curr. Biol., 2014, 24, 117–123 CrossRef CAS PubMed.
- D. D. Wang, L. W. Zou, Q. Jin, J. Hou, G. B. Ge and L. Yang, Fitoterapia, 2017, 117, 84–95 CrossRef CAS PubMed.
- W. He, J. J. Wu, J. Ning, J. Hou, H. Xin, Y. Q. He, G. B. Ge and W. Xu, Toxicol. In Vitro, 2015, 29, 1569–1576 CrossRef CAS PubMed.
- X. Wang, H. Zhang, L. Chen, L. Shan, G. Fan and X. Gao, J. Ethnopharmacol., 2013, 150, 781–790 CrossRef CAS PubMed.
- H. Hosseinzadeh and M. Nassiri-Asl, Phytother. Res., 2015, 29, 1868–1886 CrossRef.
- G. Pastorino, L. Cornara, S. Soares, F. Rodrigues and M. Oliveira, Phytother. Res., 2018, 32, 2323–2339 CrossRef CAS PubMed.
- I. M. Francischetti, R. Q. Monteiro and J. A. Guimaraes, Biochem. Biophys. Res. Commun., 1997, 235, 259–263 CrossRef CAS PubMed.
- R. B. Birari, S. Gupta, C. G. Mohan and K. K. Bhutani, Phytomedicine, 2011, 18, 795–801 CrossRef CAS PubMed.
- L. W. Zou, Y. G. Li, P. Wang, K. Zhou, J. Hou, Q. Jin, D. C. Hao, G. B. Ge and L. Yang, Eur. J. Med. Chem., 2016, 112, 280–288 CrossRef CAS.
- S. Ji, Z. Li, W. Song, Y. Wang, W. Liang, K. Li, S. Tang, Q. Wang, X. Qiao, D. Zhou, S. Yu and M. Ye, J. Nat. Prod., 2016, 79, 281–292 CrossRef CAS PubMed.
- C. Xie, X. Li, J. Wu, Z. Liang, F. Deng, W. Xie, M. Zhu, J. Zhu, W. Zhu, S. Geng and C. Zhong, Inflammation, 2015, 38, 1639–1648 CrossRef CAS PubMed.
- A. Kusano, T. Nikaido, T. Kuge, T. Ohmoto, G. Delle Monache, B. Botta, M. Botta and T. Saitoh, Chem. Pharm. Bull., 1991, 39, 930–933 CrossRef CAS PubMed.
- P. K. Liu, Z. M. Weng, G. B. Ge, H. L. Li, L. L. Ding, Z. R. Dai, X. D. Hou, Y. H. Leng, Y. Yu and J. Hou, Int. J. Biol. Macromol., 2018, 118, 2216–2223 CrossRef CAS.
- L. Liu, H. Ma, N. Yang, Y. Tang, J. Guo, W. Tao and J. Duan, Thromb. Res., 2010, 126, e365–e378 CrossRef CAS PubMed.
- T.-R. Chen, L.-H. Wei, X.-Q. Guan, C. Huang, Z.-Y. Liu, F.-J. Wang, J. Hou, Q. Jin, Y.-F. Liu, P.-H. Wen, S.-J. Zhang, G.-B. Ge and W.-Z. Guo, Bioorg. Chem., 2019, 92 Search PubMed.
- Y. Q. Song, Z. M. Weng, T. Y. Dou, M. Finel, Y. Q. Wang, L. L. Ding, Q. Jin, D. D. Wang, S. Q. Fang, Y. F. Cao, J. Hou and G. B. Ge, Chem.-Biol. Interact., 2019, 308, 339–349 CrossRef CAS PubMed.
- Y. Q. Song, X. Q. Guan, Z. M. Weng, Y. Q. Wang, J. Chen, Q. Jin, S. Q. Fang, B. Fan, Y. F. Cao, J. Hou and G. B. Ge, Int. J. Biol. Macromol., 2019, 137, 261–269 CrossRef CAS PubMed.
- Y. Zhou, Z. Liu, J. Zhang, T. Dou, J. Chen, G. Ge, S. Zhu and F. Wang, Chem. Commun., 2019, 55, 4311–4314 RSC.
- Z. Liu, Y. Zhou, J. Liu, J. Chen, A. J. R. Heck and F. Wang, TrAC, Trends Anal. Chem., 2019, 118, 771–778 CrossRef CAS.
- Y. Zhou, Y. Wu, M. Yao, Z. Liu, J. Chen, J. Chen, L. Tian, G. Han, J. R. Shen and F. J. A. C. Wang, Anal. Chem., 2016, 88, 12060 CrossRef CAS PubMed.
- R. C. Pereira, A. L. Lourenco, L. Terra, P. A. Abreu, V. Laneuville Teixeira and H. C. Castro, Mar. Drugs, 2017, 15, pii: E79 CrossRef PubMed.
- E. W. Davie and J. D. Kulman, Semin. Thromb. Hemostasis, 2006, 32(suppl 1), 3–15 CrossRef CAS PubMed.
- J. A. Huntington, Biochim. Biophys. Acta, 2012, 1824, 246–252 CrossRef CAS PubMed.
- G. A. Roth, C. Johnson, A. Abajobir, F. Abd-Allah, S. F. Abera, G. Abyu, M. Ahmed, B. Aksut, T. Alam and C. Murray, J. Am. Coll. Cardiol., 2017, 70, 1–25 CrossRef.
- J. I. Weitz and J. Harenberg, Thromb. Haemostasis, 2017, 117, 1283–1288 CrossRef PubMed.
- X. Wang, Y. Zhang, Y. Yang, X. Wu, H. Fan and Y. Qiao, Sci. Rep., 2017, 7, 44040 CrossRef PubMed.
- J. Lu, H. P. Song, P. Li, P. Zhou, X. Dong and J. Chen, J. Pharm. Biomed. Anal., 2015, 109, 85–90 CrossRef CAS PubMed.
- X. Yu, L. H. Wei, J. K. Zhang, T. R. Chen, Q. Jin, Y. N. Wang, S. J. Zhang, T. Y. Dou, Y. F. Cao, W. Z. Guo, G. B. Ge and L. Yang, Phytochemistry, 2019, 165, 112025 CrossRef CAS PubMed.
- L. H. Wei, T. R. Chen, H. B. Fang, Q. Jin, S. J. Zhang, J. Hou, Y. Yu, T. Y. Dou, Y. F. Cao, W. Z. Guo and G. B. Ge, Int. J. Biol. Macromol., 2019, 134, 622–630 CrossRef CAS PubMed.
- N. H. Hauel, H. Nar, H. Priepke, U. Ries, J. M. Stassen and W. Wienen, J. Med. Chem., 2002, 45, 1757–1766 CrossRef CAS PubMed.
- T. E. Warkentin, A. Greinacher and A. Koster, Thromb. Haemostasis, 2008, 99, 830–839 CrossRef CAS PubMed.
- Y. Zhong, Y. T. Lu, Y. Sun, N. G. Li, T. Gu, W. Y. Wu, S. P. Yu and Z. H. Shi, Med. Chem., 2018, 14, 478–484 CrossRef CAS PubMed.
- M. F. A. Mohamed, M. S. A. Shaykoon, M. H. Abdelrahman, B. E. M. Elsadek, A. S. Aboraia and G. Abuo-Rahma, Bioorg. Chem., 2017, 72, 32–41 CrossRef CAS PubMed.
- S.-S. Wei, M. Yang, X. Chen, Q.-R. Wang and Y.-J. Cui, Chin. J. Nat. Med., 2015, 13, 232–240 CAS.
- M. N. Gomes, E. N. Muratov, M. Pereira, J. C. Peixoto, L. P. Rosseto, P. V. L. Cravo, C. H. Andrade and B. J. Neves, Molecules, 2017, 22, pii: E1210 CrossRef PubMed.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS.
- Z. Sang, K. Wang, P. Zhang, J. Shi, W. Liu and Z. Tan, Eur. J. Med. Chem., 2019, 180, 238–252 CrossRef CAS.
- D. Kar Mahapatra, V. Asati and S. K. Bharti, Expert Opin. Ther. Pat., 2019, 29, 385–406 CrossRef CAS PubMed.
- J. Suresh, S. C. Baek, S. P. Ramakrishnan, H. Kim and B. Mathew, Int. J. Biol. Macromol., 2018, 108, 660–664 CrossRef CAS.
- Y. G. Li, J. Hou, S. Y. Li, X. Lv, J. Ning, P. Wang, Z. M. Liu, G. B. Ge, J. Y. Ren and L. Yang, Fitoterapia, 2015, 101, 99–106 CrossRef CAS.
- Y. K. Lee, Y. W. Chin, J. K. Bae, J. S. Seo and Y. H. Choi, Planta Med., 2013, 79, 1656–1665 CrossRef CAS PubMed.
- L. Huang, D. Nikolic and R. B. van Breemen, Anal. Bioanal. Chem., 2017, 409, 6937–6948 CrossRef CAS.
- J. Guo, D. Liu, D. Nikolic, D. Zhu, J. M. Pezzuto and R. B. van Breemen, Drug Metab. Dispos., 2008, 36, 461–468 CrossRef CAS PubMed.
- Q. H. Zhou, Y. D. Zhu, F. Zhang, Y. Q. Song, S. N. Jia, L. Zhu, S. Q. Fang and G. B. Ge, Chin. J. Nat. Med., 2019, 17, 858–870 Search PubMed.
- G. B. Ge, Chin. J. Nat. Med., 2019, 17, 801–802 Search PubMed.
- H. H. Tsai, H. W. Lin, Y. H. Lu, Y. L. Chen and G. B. Mahady, PLoS One, 2013, 8, e64255 CrossRef CAS PubMed.
- H. Xin, X. Y. Qi, J. J. Wu, X. X. Wang, Y. Li, J. Y. Hong, W. He, W. Xu, G. B. Ge and L. Yang, Food Chem. Toxicol., 2016, 90, 112–122 CrossRef CAS PubMed.
- U. M. Kent, M. Aviram, M. Rosenblat and P. F. Hollenberg, Drug Metab. Dispos., 2002, 30, 709–715 CrossRef CAS.
- G. Li, C. Simmler, L. Chen, D. Nikolic, S. N. Chen, G. F. Pauli and R. B. van Breemen, Eur. J. Pharm. Sci., 2017, 109, 182–190 CrossRef CAS.
- L. M. Lien, K. H. Lin, L. T. Huang, M. F. Tseng, H. C. Chiu, R. J. Chen and W. J. Lu, Int. J. Mol. Sci., 2017, 18 Search PubMed.
- A. Okuda-Tanino, D. Sugawara, T. Tashiro, M. Iwashita, Y. Obara, T. Moriya, C. Tsushima, D. Saigusa, Y. Tomioka, K. Ishii and N. Nakahata, PLoS One, 2017, 12, e0173628 CrossRef PubMed.
- M. Tawata, K. Aida, T. Noguchi, Y. Ozaki, S. Kume, H. Sasaki, M. Chin and T. Onaya, Eur. J. Pharmacol., 1992, 212, 87–92 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09203j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence g,
Wen-Zhi Guo*f,
Li-Rong Zhang*a and
Guang-Bo Ge
g,
Wen-Zhi Guo*f,
Li-Rong Zhang*a and
Guang-Bo Ge