
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTherapeutic potency of fermented field water-dropwort (Oenanthe javanica (Blume) DC.) in ethanol-induced liver injury†
Dong Hun Leea,
Jong Sung Leea,
Il Ho Leeb and
Jin Tae Hong *a
*a
aCollege of Pharmacy and Medical Research Center, Chungbuk National University, Osongsaengmyeong 1-ro 194-31, Osong-eup, Heungduk-gu, Cheongju, Chungbuk 28160, Republic of Korea. E-mail: jongjong135@naver.com; jinthong@chungbuk.ac.kr; leedonghun0154@gmail.com; Fax: +82-43-268-2732; Tel: +82-43-261-2813
bOSBio, Co. Ltd., CV Center Room 205, Osongsaengmyeong 1-ro 194-41, Osong-eup, Heungduk-gu, Cheongju, Chungbuk 28160, Republic of Korea. E-mail: oscarekr@hanmail.net; Fax: +82-43-238-2155; Tel: +82-43-238-2154
First published on 8th January 2020
Abstract
Alcohol overconsumption and abuse leads to alcoholic liver disease (ALD), which is a major chronic liver disease worldwide. Field water-dropwort (Oenanthe javanica (Blume) DC.) is a small perennial herb and has been cultivated in Asia for thousands of years and traditionally used to treat various diseases including hepatitis, jaundice, hypertension and polydipsia, as well as its therapeutic benefits have been recognized for centuries in Asia. Although several studies have reported that water-dropwort extracts have pharmacological effects on various diseases, the pharmacological ability of fermented field water-dropwort in ALD is not reported yet. Thus, we investigated the effect of fermented field water-dropwort extracts (FDE) on chronic plus binge ethanol-induced liver injury. C57BL/6 male mice (9 weeks old) were fed on a Lieber–DeCarli diet containing 6.6% ethanol for 10 days with parallel saline or FDE orally administered each day. Ethanol-induced hepatic triglyceride (TG) levels and the mRNA levels of TG synthesis-related genes such as sterol regulatory element binding protein 1 (SREBP1), acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) were decreased in the liver of mice with FDE administration. Moreover, FDE administered mice showed decreasing ethanol-induced oxidative stress such as increasing oxidised glutathione and lipid peroxidation in the liver. In primary hepatic cells, FDE treated cells exhibited decreased ethanol-induced lipid accumulation and the mRNA levels of TG synthesis-related genes, SREBP-1, ACC and FAS. In conclusions, FDE has the potential to be explored as a candidate treatment agent for ALD by inhibiting TG synthesis and blocking of oxidative stress.
Introduction
Alcoholic liver disease (ALD) caused by excessive alcohol intake is one of the chronic liver diseases, such as fatty liver, hepatic fibrosis, hepatocirrhosis and liver cancer. Three in 10 adults are heavy alcohol drinkers with a high risk for ALD and approximately 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths are related to alcohol abuse each year in the United States;1 ALD is also a major contributor to liver disease-related mortality in other countries.2
000 deaths are related to alcohol abuse each year in the United States;1 ALD is also a major contributor to liver disease-related mortality in other countries.2
The liver is the principle target of alcohol-induced damage because it is the primary organ of ethanol metabolism.3 Fatty liver, an early pathophysiological response of the liver to alcohol consumption, develops in most heavy drinkers, and a proportion of heavy drinkers develop more severe forms such as hepatic fibrosis, hepatocirrhosis, and liver cancer.4
During early stage of ALD, chronic alcohol exposure leads to upregulated fatty acid synthesis and accumulation of hepatic triglyceride (TG).5,6 Thus, TG, a major form of fat, excessively accumulates inside liver cells and represents an important indicator of ALD.7 Lipid accumulation in the liver leads to increased hepatic metabolic dysfunctions and hepatic sensitivity to toxins.8
Several studies have demonstrated that chronic alcohol ingestion elevates lipogenesis by activating sterol regulatory element binding protein 1 (SREBP1), which is a transcription factor regulating the expression genes involved in lipogenesis, including acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS).5 A significant amount of reactive oxygen species (ROS) are generated by the catalytic reaction of cytochrome P450 2E1 (CYP2E1), the expression of which increased during chronic alcohol abuse,9 and ROS are critically involved in TG accumulation via regulation of SREBP1.10 In addition, ROS leads to DNA damage and lipid peroxidation in ALD.11 Depletion of mitochondrial glutathione (GSH), which is an antioxidant, occurs preferentially in the liver in response to chronic ethanol ingestion.12 Moreover, treatment of human hepatic cells with hydrogen peroxide, a form of ROS, increased TG accumulation and SREBP1 activity.10 Lipid peroxidation is also involved in ethanol-induced liver injury by the formation of toxic aldehydes, including malondialdehyde (MDA), and the level of MDA was found to be significantly increased in the livers of patients with ALD.13 In addition, 4-hydroxynonenal (4-HNE), which is derived from lipid peroxidation, is a clinically relevant sensitizer of hepatotoxicity in ALD.14
Although there is a continuous increase in the number of patients with ALD, effective modern medications for treating or preventing ALD are still lacking. Thus, many researchers have been working to develop medications for ALD from numerous medicinal plants using various experimental models.15 Oenanthe javanica (Blume) DC. has been cultivated in the tropical and temperate regions of Asia for thousands of years and has long been widely used as a folk remedy for alleviating a wide spectrum of diseases.16 For example, this plant is commonly used in Asia for the treatment of various types of diseases17 including chronic and acute hepatitis,18,19 jaundice,20,21 fever,22,23 hypertension,21,24 abdominal pain, and urinary difficulties.25,26 Several studies reported that dropwort has various functional activities in the liver against lipid peroxidation by bromobenzene27 and hepatitis B virus activity.19 In addition, field water-dropwort extracts (FDE) which are naturally fermented by steeping with oligosaccharides, exhibited anti-proliferative effects on hepatocarcinoma cells and inhibits carbon tetrachloride (CCl4)-induced liver injury.20 However, the function of FDE in ALD has not been previously reported. Therefore, we investigated whether FDE has a hepatoprotective role in a mouse model of chronic ethanol-induced liver injury to confirm its potency as a medication for ALD.
Results
FDE ameliorated ethanol-induced hepatosteatosis and liver damage in mice
To examine the effects of FDE in ethanol-induced liver injury, male mice (age 9 weeks) were orally administered FDE or saline daily in the chronic plus binge ethanol-feeding model. Firstly, to optimise the FDE concentration for administration, we gave an oral injection of FDE (50, 200, or 400 mg kg−1) or saline daily to mice (5 per group) during chronic ethanol-feeding. We observed that FDE 50 mg kg−1 administration did not attenuates ethanol-induced liver damage while FDE 400 mg kg−1 administration attenuated ethanol-induced liver injury, but it did not show a more dramatic therapeutic effect than 200 mg kg−1 administration in ethanol-induced liver injury as determined by measuring serum levels of aspartate transaminase (AST) and alanine transaminase (ALT) which are markers of liver injury (ESI Fig. 1†). Thus, we used 200 mg kg−1 of FDE for further experiment.Body weight and food intake were not significantly different among all groups (Fig. 1A). However, elevated serum AST and ALT levels induced by ethanol feeding were decreased in mice with FDE administration compared to saline administration (Fig. 1B).
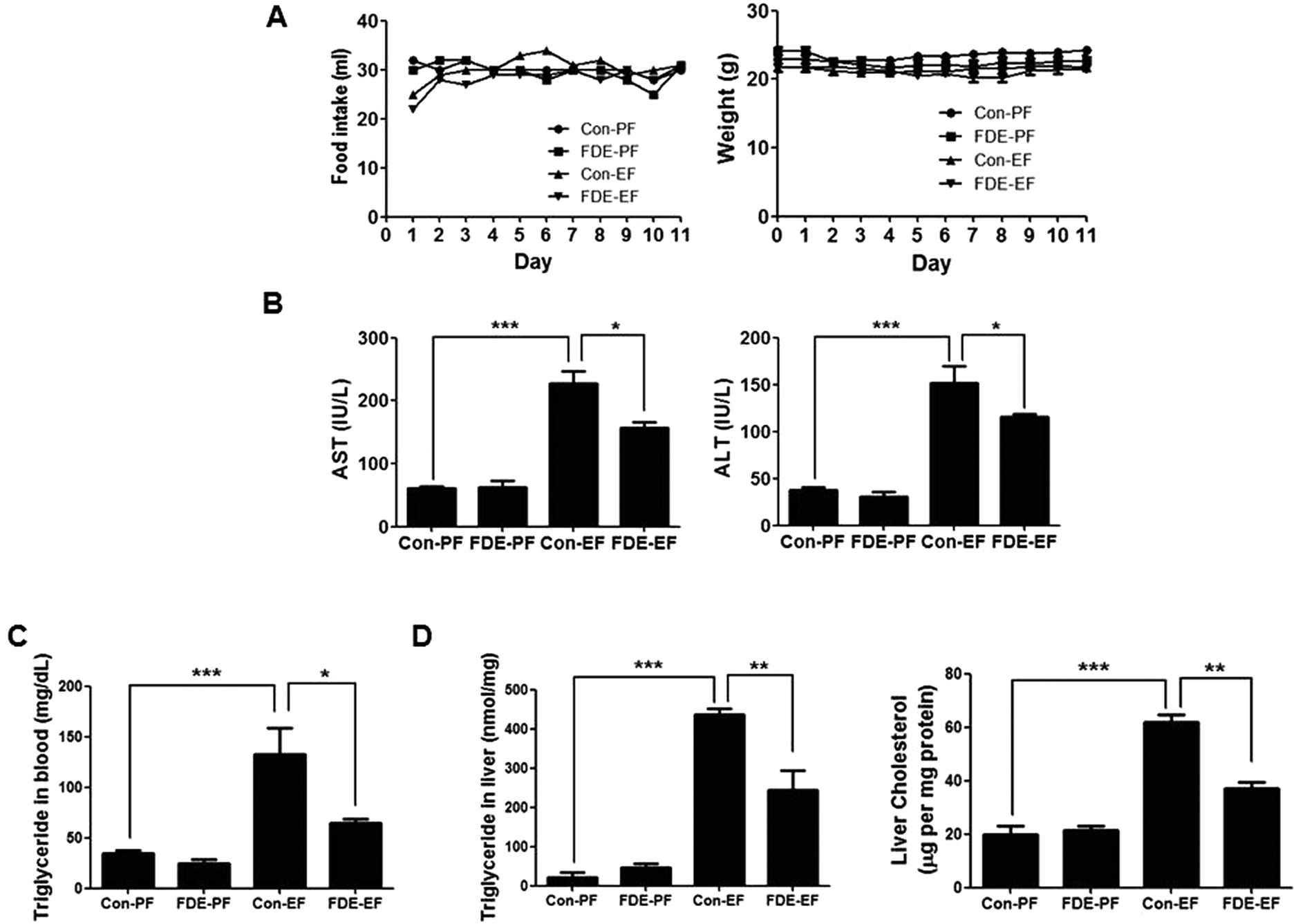 | ||
| Fig. 1 The food intake, body weight, serum biochemistry, and liver cholesterol and triglyceride in ethanol diet mice with fermented field water-dropwort (FDE) administration. (A) Food intake and bodyweight of pair-fed mice with saline administration (Con-PF), pair-fed mice with FDE administration (FDE-PF), ethanol-fed mice with saline administration (Con-EF) and ethanol-fed mice with FDE administration (FDE-EF) during food intake period. n = 20 per group; means ± SD of the mean (B) serum aspartate transaminase (AST) and alanine transaminase (ALT) levels of Con-PF mice, FDE-PF mice, Con-EF mice and FDE-EF mice. n = 20 per group; means ± SD of the mean. *p < 0.05 and ***p < 0.001 (C) triglyceride level in the blood and (D) triglyceride and cholesterol level in the liver of Con-PF mice, FDE-PF mice, Con-EF mice and FDE-EF mice. n = 20 per group; means ± SD of the mean. *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Hepatic steatosis is the most remarkable pathological feature of ALD, which is characterized by excessive hepatic lipid accumulation. To examine the degree of lipogenesis in the liver, we measured TG and total cholesterol level in the liver. The levels of hepatic TG and total cholesterol in the liver were increased by ethanol feeding and their increased levels were reduced by FDE administration (Fig. 1C). ALD caused by high alcohol intake is associated with elevated plasma TG.28 Like the TG level in the liver, the level of TG in the blood was increased by ethanol feeding and its increased level was reduced by FDE administration (Fig. 1D). Histopathological studies revealed vesicular hepatosteatosis in ethanol-fed mice, but its manifestation was attenuated by FDE administration (Fig. 2A). Similarly, oil red O staining for neutral lipids confirmed the decreased hepatosteatosis in the livers of ethanol-fed mice with FDE administration compared to ethanol-fed mice with saline administration (Fig. 2A). The mRNA level of SREBP1 and its target genes such as ACC and FAS involved in TG synthesis in the liver were increased by ethanol feeding but these increased levels were lowered in the liver of ethanol-fed mice with FDE administration (Fig. 2B).
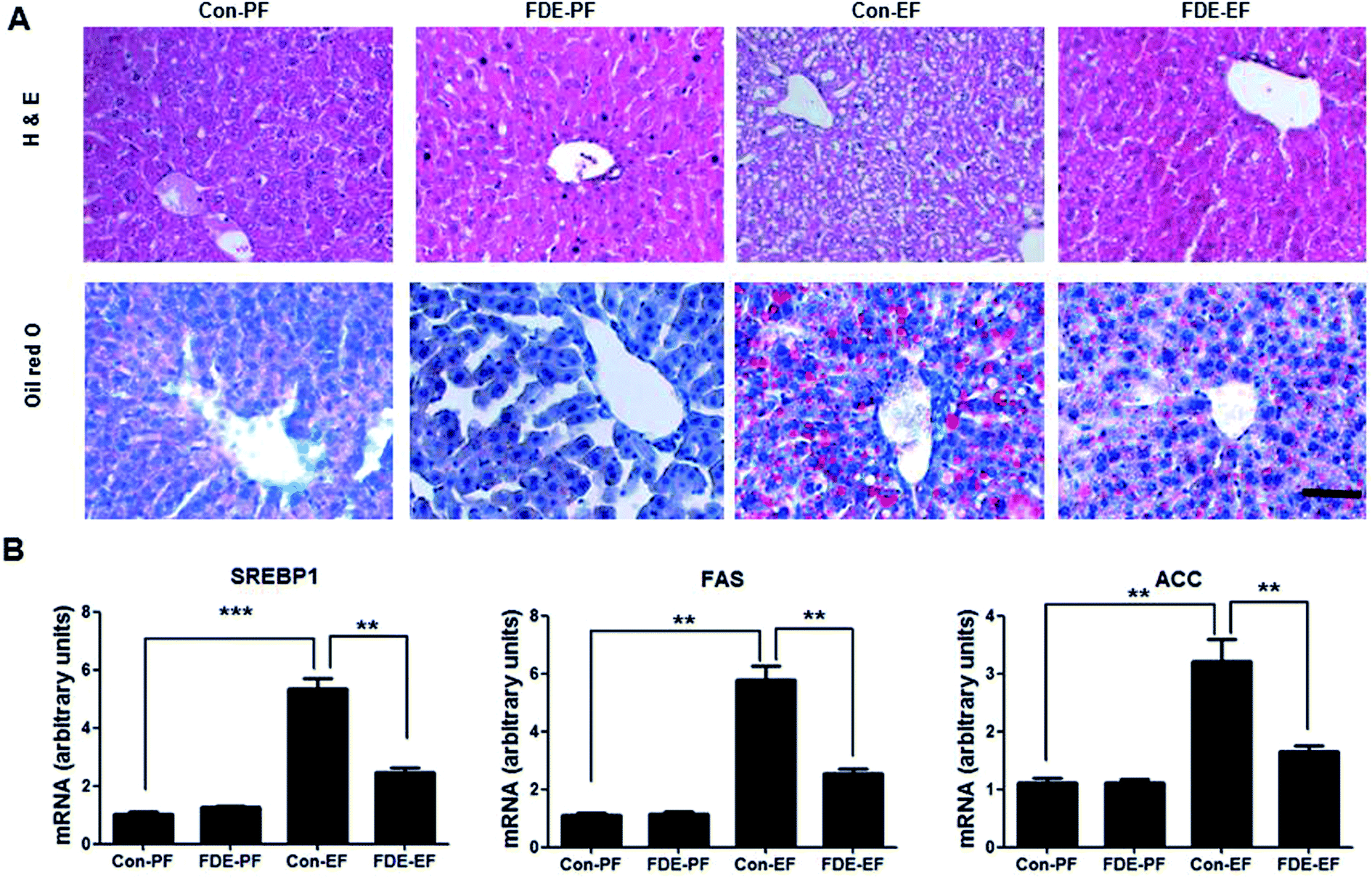 | ||
| Fig. 2 Effect of FDE on ethanol-induced fatty liver. (A) Liver sections of Con-PF mice, FDE-PF mice, Con-EF mice and FDE-EF mice were stained with hematoxylin and eosin (H&E) or oil red O (scale bars, 100 μm). (B) The mRNA level of triglyceride synthesis genes, SREPB1, ACC and FAS, in the liver of Con-PF mice, FDE-PF mice, Con-EF mice and FDE-EF mice. n = 20 per group; means ± SD of the mean. *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Ethanol-induced oxidative stress in the liver of mice was reduced by FDE administration
The liver is the organ most targeted by oxidative stress, and oxidative stress is critically involved in TG accumulation via regulation of SREBP1 activation.10 Ethanol-induced hepatic oxidative stress has been demonstrated by detecting ROS or by measuring the GSH/GSSG ratio and lipid peroxidation.29 When the level of oxidative stress is increased, GSH is decreased and GSSG (oxidised GSH) is accumulated; thus, the GSH/GSSG ratio is decreased. As shown Fig. 3A, the GSH/GSSG ratio in the liver of ethanol-fed mice with saline administration was lower than that in the liver of ethanol-fed mice with FDE administration.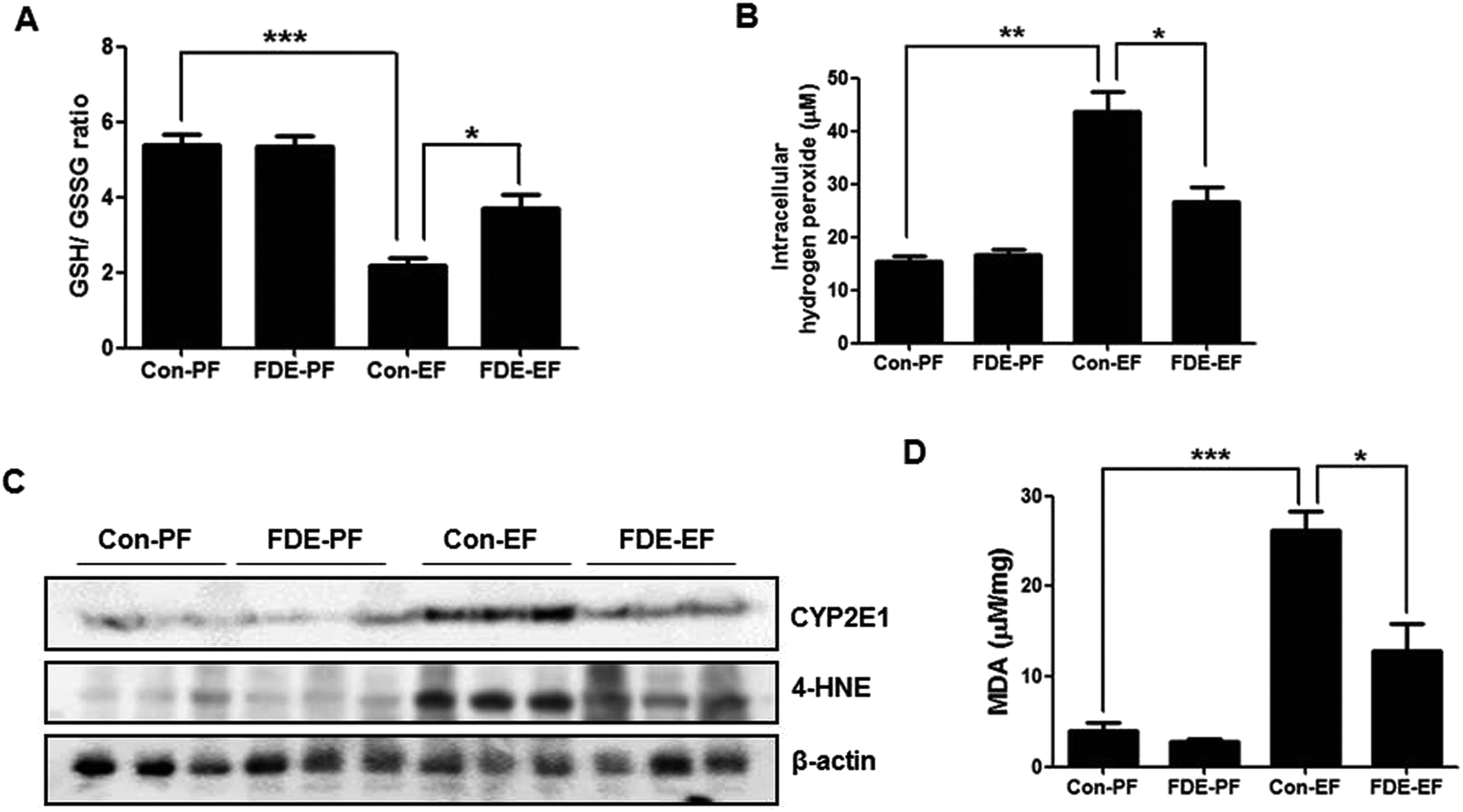 | ||
| Fig. 3 Effect of FDE on ethanol-induced oxidative stress. (A) The ratio of GSH/GSSG and (B) hydrogen peroxide level were measured in the liver of Con-PF mice, FDE-PF mice, Con-EF mice and FDE-EF mice. n = 20 per group; means ± SD of the mean. *p < 0.05, **p < 0.01 and ***p < 0.001 (C) the expression of CYP2E1 and 4-HNE were determined in the total protein extracts of mice liver tissues by western blot analysis. (D) MDA levels were measured in the liver of Con-PF mice, FDE-PF mice, Con-EF mice and FDE-EF mice. n = 20 per group; means ± SD of the mean. *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
The level of hydrogen peroxide, which is a form of ROS, was elevated in the liver of ethanol-fed mice whereas it was reduced in the liver of ethanol-fed mice with FDE administration (Fig. 3B). Ethanol can generate ROS through a CYP2E1-dependent mechanism.9 Therefore, we examined whether FDE attenuates TG synthesis by ethanol feeding through abrogating ROS generation during ethanol metabolism. CYP2E1 generates ROS during ethanol metabolism, and the degree of CYP2E1 induction is correlated with ROS production.30 Western blot analysis showed an induction of CYP2E1 in ethanol-fed mice. However, ethanol-induced CYP2E1 expression was reduced by FDE administration (Fig. 3C). In addition, the degree of lipid peroxidation, as assessed by the expression of 4-HNE using western blot analysis, was elevated in the liver of mice by ethanol feeding, but it was decreased in the liver by FDE administration (Fig. 3C). Accordingly, the level of MDA, a marker of lipid peroxidation, was also increased in the liver of ethanol-fed mice. However, the increase in the level of MDA caused by ethanol was suppressed in the liver of ethanol-fed mice with FDE administration (Fig. 3D).
FDE prevent ethanol-induced hepatic lipid accumulation in mouse primary hepatocytes
Because FDE administration resulted in a decrease of the lipid accumulation and oxidative stress in the liver induced by ethanol consumption, we investigated the effect of FDE on ethanol-treated mouse primary hepatic cells. To examine the effect of FDE on lipid accumulation induced by ethanol in hepatic cells, we evaluated the amount of lipid accumulation through oil red O staining in ethanol treated hepatic cells pre-treated with FDE or PBS. These experiments revealed that ethanol-treated hepatic cells co-treated with FDE exhibited greater reductions in lipid accumulation than ethanol-treated hepatic cells pre-treated with phosphate-buffered saline (PBS) (Fig. 4A). The mRNA expression levels of the principal factors related to lipid synthesis, namely SREBP-1 and its target genes such as ACC and FAS that are involved in TG synthesis, were downregulated by FDE administration to ethanol-treated hepatic cells (Fig. 4B).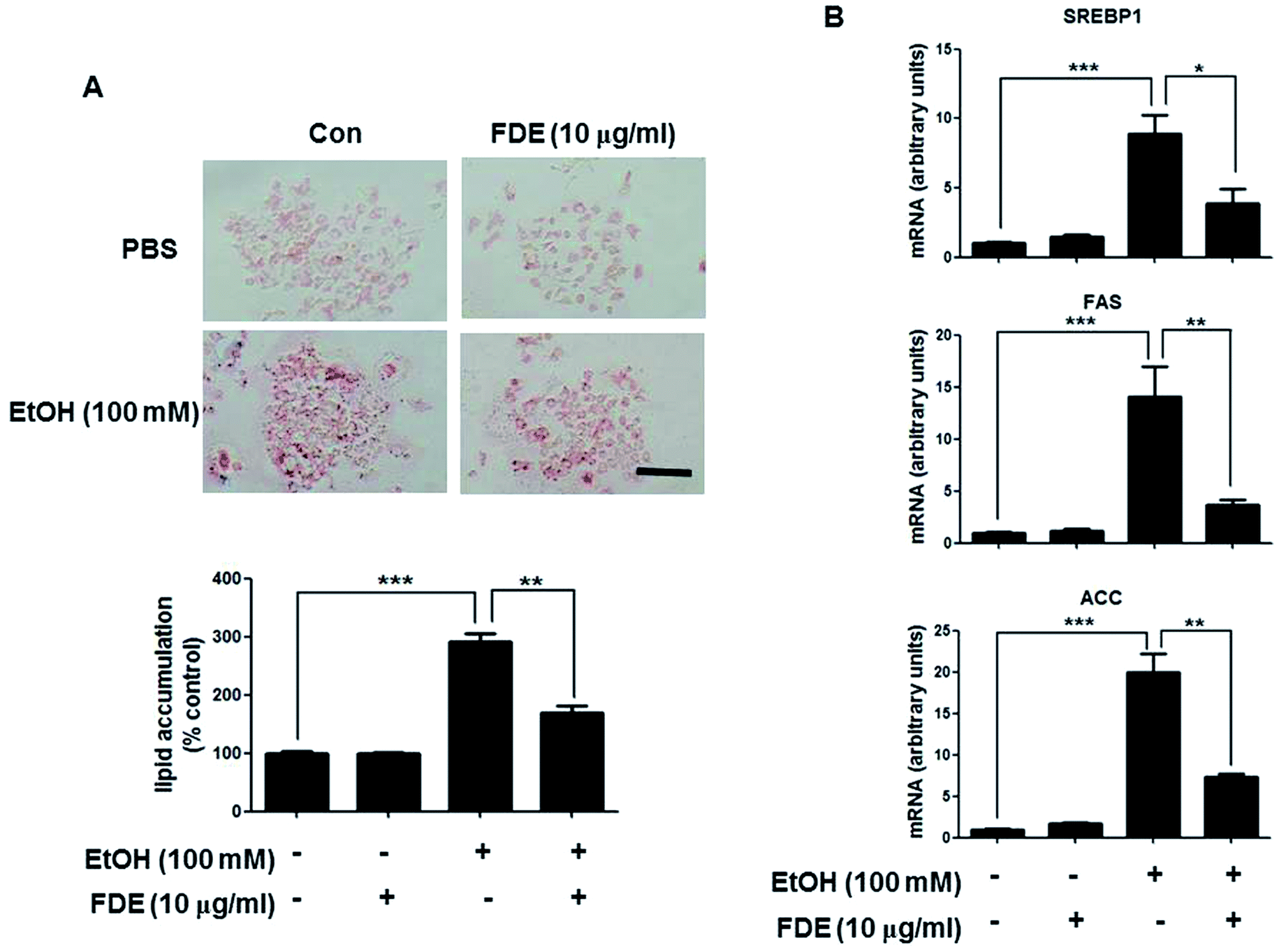 | ||
| Fig. 4 Effect of FDE on ethanol-induced lipid accumulation in primary hepatocyte from mice. (A) Hepatic lipid accumulation in FDE or PBS administered mouse primary hepatic cells treated with ethanol. Values are expressed as the mean ± SD of three different experiments conducted in triplicates. *p < 0.05, **p < 0.01 and ***p < 0.001. (scale bars, 100 μm). (B) The mRNA level of triglyceride synthesis genes, SREPB1, ACC and FAS, in FDE or PBS administered mouse primary hepatic cells treated with ethanol. Values are expressed as the mean ± SD of three different experiments conducted in duplicates. *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Discussion
A accumulating hepatic lipids play an important role in the pathogenesis of chronic liver disease. Increasing lipid synthesis in response to oxidative stress induced by continuous alcohol consumption is a noticeable feature of ALD.31 Several studies have reported that various plant extracts have been suggested to act as protectants against ethanol-induced liver damage.32 Like some other plant extracts, some studies have reported that dropwort extracts exhibit hepatoprotective effects. Ethanol extracts of dropwort have several antioxidants and antioxidant activities as demonstrated through a DPPH (1,1-diphenyl-2-picrylhydrazyl) and ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt) radical scavenging assay.33 Kim et al. reported that hot-water extracts of dropwort accelerate ethanol metabolism in ethanol-treated rabbits and mice.34 Recently a study reported that FDE showed hepatoprotective effects against CCl4-induced liver injury and that its major constituents are chlorogenic acid and caffeic acid which are known to exhibiting antioxidant activities.20 Thus, FDE may have therapeutic potential in various liver diseases with different pathogenesis. In this study, we have demonstrated a preventative effect of FDE against ethanol-induced fatty liver in the mouse by using a chronic plus binge ethanol-feeding model.Chronic alcohol consumption leads to fatty liver and liver injury. Hepatic steatosis due to excessive alcohol consumption includes excess fat accumulation, mainly TG, within cytosolic lipid droplets.35 We observed that the levels of ALT and AST, two major serum markers of hepatic damage, were increased by ethanol feeding, but those levels were decreased by FDE administration. In addition, hepatic steatosis, elevated blood and hepatic TG levels, and increased liver cholesterol induced by ethanol feeding were attenuated by FDE administration. Thus, we investigated how FDE alleviates ethanol-induced fatty liver disease.
SREBP1 has a major function in the development of ethanol-induced fatty liver injury.36 Induction of SREBP1 by ethanol exposure elevated the expression of several lipid synthesis genes as well as TG accumulation in the liver.36 In our results, the hepatic mRNA levels of SREBP1 and its target genes including ACC and FAS were increased by ethanol feeding, but they were suppressed by FDE administration in the mouse. Consistent with our in vivo data, ethanol-induced mRNA levels of SREBP1, ACC and FAS in mouse primary hepatic cells were also decreased by FDE treatment. These data suggest that FDE administration leads to down-regulation of TG synthesis related genes in hepatocytes and thus contributes to interfering with the process of ALD.
The importance of oxidative stress is emphasised in the pathogenesis of various degenerative disease, such as diabetes, cancer, cardiovascular disorders and neurodegenerative disease.13 It is well known that fatty liver ALD is associated with oxidative stress.11 Chronic alcohol exposure increases the production of ROS and lowers cellular antioxidant levels, leading to oxidative stress. Various metabolites produced by CYP2E1 and ROS, which are also generated during metabolism by CYP2E1, increase levels of polyunsaturated fatty acids to generate ω-hydroxylated fatty acids that then damaged hepatocytes through mitochondrial dysfunction.37 Therefore, we investigated ethanol-induced oxidative stress in the liver of mice with FDE or saline administration. Elevated CYP2E1 expression, induced by ethanol feeding, in the liver of mice was decreased by FDE administration.
ROS include the free radicals superoxide, hydroxyl radicals, hydrogen peroxide and reactive nitrogen species.37 Of these, hydrogen peroxide is produced by CYP2E1 during ethanol metabolism and dismutation of superoxide, one of the ROS generated, is produced by superoxide dismutase (SOD). Because hydrogen peroxide is more stable compared with other ROS, it may play an important role in intracellular signalling. In the present study, hydrogen peroxide levels were significantly elevated in the liver of ethanol-fed mice with saline administration but not in the liver of ethanol-fed mice with FDE administration. Oxidative stress depends on the balance between oxidant and antioxidant particles.13 For example, the level of hydrogen peroxide is significantly elevated in glutathione peroxidase knock-out mice compared to wild-type mice.38 Therefore, we examined ratio of GSH and GSSG (oxidised GSH). The GSH/GSSG ratio was increased in the liver of ethanol-fed mice but it was inhibited by FDE administration.
Mouse models and patients with ALD showed lipid peroxidation levels were increased compared to normal liver.39 Increased expression of 4-HNE and the level of MDA, a marker of lipid peroxidation, were suppressed in the liver of ethanol-fed mice by FDE administration. These data suggested that FDE might suppress SREBP1 and thus TG synthesis via decreasing oxidative stress through inhibition of CYP2E1 expression.
Experimental
Plant materials and preparation of FDE
FDE was kindly provided by Dr Lee (Keimyung University, South Korea) and a HPLC chromatogram of the FDE exhibited two major peaks (chlorogenic acid and caffeic acid) and many minor peaks.20 In brief, to obtain the FDE, field water dropwort (O. javanica (Blume) DC) was cultivated in the dry land region of Bissell Chunglog Farm (Daegu, South Korea) and naturally fermented by steeping with oligosaccharide (1:1, v/v) at room temperature for one year, then the fermented extract was stored at 4 °C for two years for maturation. For the animal experiment and cell treatment, the FDE was concentrated and freeze dried (<5% of water content remaining).
Animals experiment and housing condition
Male C57BL/6 mice (9 weeks age) were purchased from DBL Co., Ltd (South Korea). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Chungbuk National University and approved by the Animal Ethics Committee of Chungbuk National University (CBNUA-1272-19-01). The mice were acclimatised to the laboratory environment, maintained throughout the experiment at 22 ± 1 °C and a relative humidity of 55 ± 10%, with 12 h light–dark cycles. The FDE was prepared by dissolving it in saline (10 mg ml−1) for administration. For the chronic ethanol consumption experiment, we used the short-term NIAAA model.40,41 Briefly, all mice were fed for 5 days with ad libitum Lieber/DeCarli regular liquid diet-control, which was dissolved in tap water following the manufacture's protocol, (Dyets, Cat # 710027). Following acclimation, the mice were randomly divided into four groups (n = 20/group): (1) paired-fed standard diet and saline administration (Con-PF); (2) paired-fed standard diet and FDE administration (200 mg kg−1) (FDE-PF); (3) ethanol diet and saline administration (Con-EF); (4) ethanol diet and FDE administration (200 mg kg−1) (FDE-EF), feeding ad libitum with a Lieber/DeCarli regular liquid diet-ethanol (Dyets, Cat # 710260) containing 5% (vol/vol) ethanol or pair-fed a Lieber/DeCarli regular liquid diet-control for 10 days. During feeding of the liquid diets for 10 days, FDE or saline was orally administered each day. On the morning of the 11th day, the mice were gavaged with a single dose of ethanol (5 g kg−1 bodyweight) or maltodextrin and were sacrificed 9 h later.41Measurements of serum aspartate transaminase and alanine transaminase
Blood was collected at 9 h after ethanol administration. Serum was separated by centrifugation at 3000 rpm for 8 min at 21 °C. Serum AST and ALT levels were measured using a biochemical analyzer (AU480, Beckman Coulter, CA, USA).Histopathological analysis
For histological processing, liver tissues were fixed in 4% formalin solution. Then, the liver tissues were embedded in paraffin. Specimens were sectioned at 4 μm and stained with hematoxylin and eosin (H&E), then observed with a light microscope (Nikon, Tokyo, Japan).Mouse primary hepatocyte isolation and cell culture
The primary mouse hepatic cells were obtained from the liver tissue of 9 weeks-old male C57BL/6 mice as described previously.42 Briefly, we anaesthetise the mice by intraperitoneally injecting a mixture of ketamine (80 mg kg−1) and xylazine (5 mg kg−1) in 200 μl of saline. After perfusion with Hank's Balanced Salt Solution (HBSS)-EGTA solution (0.5 mM EGTA in HBSS, Gibco, Grand Island, NY, USA) (pH = 8), we collected the liver and then dissected it to release hepatocytes. The resulting cells were gently pressed through a 100 μm cell strainer (BD, Franklin Lakes, New Jersey, USA). The filtered cells were washed with Dulbecco's modified Eagle (DMEM) medium and plated into 100 mm2 dishes. The cells were grown at 37 °C in 5% CO2-humidified air in DMEM medium that contained 10% foetal bovine serum (FBS), 100 U ml−1 penicillin, and 100 mg ml−1 streptomycin. DMEM, penicillin, streptomycin, and FBS were purchased from Gibco Life Technologies (Grand Island, NY, USA). To confirm the role of FDE in ethanol-treated mouse primary hepatic cells, the cells from the livers of the mice were pre-treated with FDE (10 μg ml−1) or PBS for 2 h then treated with ethanol (100 mM) for 24 h.Oil red O staining
Liver tissues were fixed in 10% formalin in PBS and cut by the frozen section procedure to 10 mm thick sections. Next, these sections were rinsed with propylene glycol and stained with a 0.2% oil red O solution in propylene glycol for 30 min at room temperature, and subsequently washed with tap water. Primary mouse hepatic cells were washed twice with PBS, fixed with 0.5% glutaraldehyde for 3 h at room temperature, washed again with PBS, and allowed to dry completely. Next, the fixed cells were stained with a 0.2% oil red O solution in isopropanol diluted in distilled water (6:4) for 1 h at room temperature, and subsequently washed twice with PBS. Stained lipid droplets were observed under a light microscope (Nikon, Tokyo, Japan).
Western blot analysis
Homogenised liver tissues were lysed by protein extraction solution (PRO-PREP, iNtRON, Sungnam, Korea) and the total protein concentration was determined using the Bradford reagent (Bio-Rad, Hercules, CA, USA). Then, 100 μg of extracted total protein was separated by SDS/PAGE and transferred to Immobilon® PVDF membranes (Millipore, Bedford, MA, USA). The membrane was blocked with 5% dried skimmed milk for 1 h at room temperature, followed by incubation with specific primary antibodies overnight at 4 °C. The membranes were washed with Tris-buffered saline containing 0.05% Tween-20 (TBST) and incubated with diluted horse radish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After washing, binding of antibodies to the PVDF membrane was detected using the Immobilon Western Chemilum HRP substrate (Millipore, Bedford, MA, USA). The band intensities were measured using the Fusion FX 7 image acquisition system (Vilber Lourmat, Eberhardzell, Germany). Specific primary antibodies were purchased from Abcam (CYP2E1 and 4-HNE; Cambridge, MA, USA). Secondary antibodies were purchased from Santo Cruz Biotechnology (anti-mouse and anti-rabbit; Dallas, TX, USA).RNA isolation and quantitative real-time RT-PCR
Total RNA from the liver tissues was extracted by RiboEx™ Total RNA isolation solution (GeneAll Biotechnology, Seoul, Korea) and cDNA was synthesised using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time RT-PCR was performed on a 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA) using custom-designed primers with β-actin used as a house-keeping control with the HiPi Real-Time PCR 2X Master Mix (ELPIS, Daejeon, Korea). Cycling conditions consisted of a denaturation of 15 s at 94 °C, a combined annealing for 30 s at 55 °C and extension of 60 s at 72 °C for 40 cycles. The values obtained for the target gene expression were normalized to β-actin and quantified relative to the expression in the control samples.Measurement of triglycerides and liver cholesterol
The blood and hepatic level of triglycerides (TG) were measured as described in the manufacturer's protocol using triglycerides assay kit (Abcam, Cambridge, MA, USA). Liver cholesterol was measured using a biochemical analyser (AU480, Beckman Coulter, CA, USA).Oxidative stress assay
The hydrogen peroxides assay was performed as described in the manufacturer's protocol (Cell Biolabs, San Diego, CA, USA). Hepatic levels of reduced glutathione (GSH) and oxidised glutathione (GSSG) were measured using a GSH/GSSG Ratio Detection Assay Kit (Abcam, Cambridge, MA, USA). Lipid peroxidation was measured by determining the generation of malondialdehyde (MDA; TBARS Assay kit, Cayman, Ann Arbor, MI, USA).Statistical analysis
The data were analyzed using the GraphPad Prism 4 version 4.03 software (Graph-Pad Software, La Jolla, CA). Data are presented as mean ± SD. The differences were assessed by one-way analysis of variance. When the P value in the analysis of variance test indicated statistical significance, the differences were further assessed by Tukey's test. A value of p ≤ 0.05 was considered to be statistically significant.Conclusion
Currently, there is no effective treatment or potential agents for the treatment of ALD. Therefore, we should seek novel and effective interventions for ALD, and we focused on water-dropwort, which has potential therapeutic effects for various diseases. Our study demonstrated that FDE ameliorates ethanol-induced fatty liver, and our results suggest that FDE can be a potential medicinal agent for the treatment early stage ALD.Abbreviation
| ALD | Alcoholic liver disease |
| FDE | Naturally fermented field water-dropwort (Oenanthe javanica (Blume) DC.) extracts by steeping with oligosaccharides |
| CCl4 | Carbon tetrachloride |
| TG | Triglyceride |
| SREBP1 | Sterol regulatory element binding protein 1 |
| ACC | Acetyl-CoA carboxylase |
| FAS | Fatty acid synthase |
| ROS | Reactive oxygen species |
| CYP2E1 | Cytochrome P450 2E1 |
| GSH | Glutathione |
| GSSG | Oxidized GSH |
| MDA | Malondialdehyde |
| 4-HNE | 4-Hydroxynonenal |
| Con-PF | Paired-fed standard diet with saline administration |
| FDE-PF | Paired-fed standard diet with FDE administration |
| Con-EF | Ethanol diet with saline administration |
| FDE-EF | Ethanol diet with FDE administration |
| AST | Aspartate transaminase |
| ALT | Alanine transaminase |
| H&E | Hematoxylin and eosin stain |
| PBS | Phosphate-buffered saline |
| DPPH | 1,1-Diphenyl-2-picrylhydrazyl |
| ABTS | 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt |
Funding information
This work was supported by the National Research Foundation of Korea [NRF] grant funded by the Korea government (MSIP) [Grant Number MRC2017R1A5A2015541].Conflicts of interest
There are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.Acknowledgements
We would like to thank Editage (www.editage.co.kr) for English language editing.References
- S. M. Penny, Radiologic technology, 2013, 84, 577–592 Search PubMed , quiz 593–575.
- J. Rehm, C. Mathers, S. Popova, M. Thavorncharoensap, Y. Teerawattananon and J. Patra, Lancet, 2009, 373, 2223–2233 CrossRef.
- M. Galicia-Moreno and G. Gutierrez-Reyes, Rev. Gastroenterol. Mex., 2014, 79, 135–144 CAS.
- B. Gao and R. Bataller, Gastroenterology, 2011, 141, 1572–1585 CrossRef CAS.
- L. E. Nagy, Annu. Rev. Nutr., 2004, 24, 55–78 CrossRef CAS.
- M. You, Q. Cao, X. Liang, J. M. Ajmo and G. C. Ness, J. Nutr., 2008, 138, 497–501 CrossRef CAS.
- R. Guo, X. Xu, S. A. Babcock, Y. Zhang and J. Ren, J. Hepatol., 2015, 62, 647–656 CrossRef CAS.
- E. Brandon-Warner, L. W. Schrum, C. M. Schmidt and I. H. McKillop, Alcohol, 2012, 46, 715–725 CrossRef.
- Y. Lu and A. I. Cederbaum, Free Radical Biol. Med., 2008, 44, 723–738 CrossRef CAS.
- M. Sekiya, A. Hiraishi, M. Touyama and K. Sakamoto, Biochem. Biophys. Res. Commun., 2008, 375, 602–607 CrossRef CAS PubMed.
- T. M. Leung and N. Nieto, J. Hepatol., 2013, 58, 395–398 CrossRef CAS PubMed.
- A. I. Cederbaum, Y. Lu and D. Wu, Arch. Toxicol., 2009, 83, 519–548 CrossRef CAS PubMed.
- H. Cichoz-Lach and A. Michalak, World J. Gastroenterol., 2014, 20, 8082–8091 CrossRef PubMed.
- X. Dou, S. Li, Z. Wang, D. Gu, C. Shen, T. Yao and Z. Song, Am. J. Pathol., 2012, 181, 1702–1710 CrossRef CAS PubMed.
- N. Ghosh, R. Ghosh, V. Mandal and S. C. Mandal, Pharm. Biol., 2011, 49, 970–988 CrossRef CAS PubMed.
- C. L. Lu and X. F. Li, Evid.-Based Complementary Altern. Med., 2019, 2019, 6495819 Search PubMed.
- S. Li and X. Luo, Compendium of materia medica : Bencao gangmu, Foreign Languages Press, Beijing, 1st edn, 2003 Search PubMed.
- Z. M. Huang, X. B. Yang and W. B. Cao, Chin. Tradit. Herb. Drugs, 2001, 32, 59–61 CAS.
- Y. Q. Han, Z. M. Huang, X. B. Yang, H. Z. Liu and G. X. Wu, J. Ethnopharmacol., 2008, 118, 148–153 CrossRef CAS PubMed.
- S. A. Yang, Y. S. Jung, S. J. Lee, S. C. Park, M. J. Kim, E. J. Lee, H. J. Byun, K. H. Jhee and S. P. Lee, Food Chem. Toxicol., 2014, 67, 154–160 CrossRef CAS PubMed.
- W. N. Wang, X. B. Yang, H. Z. Liu, Z. M. Huang and G. X. Wu, Acta Pharmacol. Sin., 2005, 26, 587–592 CrossRef CAS.
- S. K. Ku, M. S. Han and J. S. Bae, Thromb. Res., 2013, 132, e58–63 CrossRef CAS.
- J. H. Park, J. H. Cho, I. H. Kim, J. H. Ahn, J. C. Lee, B. H. Chen, B. N. Shin, H. J. Tae, K. Y. Yoo, S. Hong, I. J. Kang, M. H. Won and J. D. Kim, Chin. Med. J., 2015, 128, 2932–2937 CrossRef CAS PubMed.
- J. C. Park, H. S. Young, Y. B. Yu and J. H. Lee, Planta Med., 1995, 61, 377–378 CrossRef CAS.
- B. H. Chen, J. H. Park, J. H. Cho, I. H. Kim, B. N. Shin, J. H. Ahn, S. J. Hwang, B. C. Yan, H. J. Tae, J. C. Lee, E. J. Bae, Y. L. Lee, J. D. Kim, M. H. Won and I. J. Kang, Neural Regener. Res., 2015, 10, 271–276 CrossRef CAS PubMed.
- S. K. Ku, T. H. Kim, S. Lee, S. M. Kim and J. S. Bae, Food Chem. Toxicol., 2013, 53, 197–204 CrossRef CAS PubMed.
- J. C. Park, Y. B. Yu, J. H. Lee, M. Hattori, C. K. Lee and J. W. Choi, Planta Med., 1996, 62, 488–490 CrossRef CAS PubMed.
- B. Klop, A. T. do Rego and M. C. Cabezas, Curr. Opin. Lipidol., 2013, 24, 321–326 CrossRef CAS PubMed.
- Z. Zhou, L. Wang, Z. Song, J. C. Lambert, C. J. McClain and Y. J. Kang, Am. J. Pathol., 2003, 163, 1137–1146 CrossRef CAS.
- K. Sugimoto and Y. Takei, Hepatol. Res., 2017, 47, 70–79 CrossRef PubMed.
- P. Mandrekar, A. Ambade, A. Lim, G. Szabo and D. Catalano, Hepatology, 2011, 54, 2185–2197 CrossRef CAS.
- E. Madrigal-Santillan, E. Madrigal-Bujaidar, I. Alvarez-Gonzalez, M. T. Sumaya-Martinez, J. Gutierrez-Salinas, M. Bautista, A. Morales-Gonzalez, M. Garcia-Luna y Gonzalez-Rubio, J. L. Aguilar-Faisal and J. A. Morales-Gonzalez, World J. Gastroenterol., 2014, 20, 14787–14804 CrossRef CAS.
- C. R. Hwang, I. G. Hwang, H. Y. Kim, T. S. Kang, Y. B. Kim, S. S. Joo, I. S. Lee and H. S. Jeong, J. Korean Soc. Food Sci. Nutr., 2011, 40, 316–320 CrossRef CAS.
- J. Y. Kim, K. H. Kim, Y. J. Lee, S. H. Lee, J. C. Park and D. H. Nam, BMB Rep., 2009, 42, 482–485 CrossRef CAS PubMed.
- X. Tong, P. Li, D. Zhang, K. VanDommelen, N. Gupta, L. Rui, M. B. Omary and L. Yin, J. Lipid Res., 2016, 57, 1219–1230 CrossRef CAS PubMed.
- M. You, M. Fischer, M. A. Deeg and D. W. Crabb, J. Biol. Chem., 2002, 277, 29342–29347 CrossRef CAS PubMed.
- J. Aubert, K. Begriche, L. Knockaert, M. A. Robin and B. Fromenty, Clin. Res. Hepatol. Gastroenterol., 2011, 35, 630–637 CrossRef CAS PubMed.
- D. D. Harrison-Findik and S. Lu, Biomolecules, 2015, 5, 793–807 CrossRef CAS PubMed.
- A. Kasdallah-Grissa, B. Mornagui, E. Aouani, M. Hammami, N. Gharbi, A. Kamoun and S. El-Fazaa, Alcohol Alcohol., 2006, 41, 236–239 CrossRef CAS.
- S. H. Ki, O. Park, M. Zheng, O. Morales-Ibanez, J. K. Kolls, R. Bataller and B. Gao, Hepatology, 2010, 52, 1291–1300 CrossRef CAS.
- A. Bertola, S. Mathews, S. H. Ki, H. Wang and B. Gao, Nat. Protoc., 2013, 8, 627–637 CrossRef.
- M. Severgnini, J. Sherman, A. Sehgal, N. K. Jayaprakash, J. Aubin, G. Wang, L. Zhang, C. G. Peng, K. Yucius, J. Butler and K. Fitzgerald, Cytotechnology, 2012, 64, 187–195 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08976d |
| This journal is © The Royal Society of Chemistry 2020 |