
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTeneligliptin protects against ischemia/reperfusion-induced endothelial permeability in vivo and in vitro
Lei Zhangab,
Weiqiong Yuanc,
Xiangli Kongab and
Bei Zhang *ab
*ab
aDepartment of Neurology, The First Affiliated Hospital of Xi'an Medical University, No. 48 Fenghao West Road, Lianhu District, Xi'an, Shaanxi province, China. E-mail: zhangbei5248@163.com; Fax: +86-29-84277356; Tel: +86-29-84277356
bShaanxi Key Laboratory of Brain Disorders, Xi'an Medical University, Xi'an, China
cDepartment of General Surgery, The Second Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi, China
First published on 22nd January 2020
Abstract
Ischemic stroke is a leading cause of disability and mortality worldwide, especially among the elderly population. Ischemia and reperfusion cause damage to cells and initiate an acute inflammatory response, which leads to cerebral endothelial dysfunction, increased endothelial permeability, and potentially permanent disability. Teneligliptin is a dipeptidyl peptidase-4 (DPP-4) inhibitor that has been used almost exclusively in the treatment of type 2 diabetes mellitus. However, it is still unknown whether teneligliptin possesses a protective effect in brain endothelial dysfunction in the context of ischemic stroke. In the present work, we demonstrate the potential of teneligliptin treatment to protect against ischemia/reperfusion-induced damage using a series of both in vivo and in vitro experiments. Our key findings are that administration of teneligliptin could reduce brain infarct volume, ameliorate neurological damage, and improve brain permeability by increasing the expression of the tight junction protein occludin in middle cerebral artery occlusion (MCAO) mice models. Importantly, teneligliptin displayed a robust protective effect against oxygen–glucose deprivation/reperfusion (OGD/R)-induced cell death of primary human brain microvascular endothelial cells (HBMVECs) in vitro. Notably, teneligliptin prevented OGD/R-induced increased endothelial monolayer permeability in HBMVECs by increasing the expression of occludin, which was mediated by the ERK5/KLF2 signaling pathway. These findings suggest that teneligliptin might serve as a potential therapeutic agent for the treatment of stroke
1. Introduction
Ischemic stroke is characterized by insufficient blood flow to the brain, usually resulting from arterial blockage or occlusion, and resulting in oxygen–glucose deprivation.1,2 Reperfusion of the affected area after an ischemic incident can cause significant damage due to cell death and the activation of an acute inflammatory response.3 Thus, timely treatment is of the utmost importance in patients suffering from ischemia/reperfusion injury.4,5 Brain blood vessels and their physiological function play a key role in maintaining normal brain function by ensuring that the tissues of the brain are supplied with oxygen and nutrients.6 The obstruction of these vessels leads to ischemia and significantly damages brain function.7 This disruption in healthy circulation leads to many of the hallmark symptoms of stroke including numbness, reduced motor control, and more severe side effects such as paralysis or blindness.8–10 The endothelium lines the blood vessels of the body and regulates numerous physiological processes, such as vascular permeability, vascular constriction and relaxation, and the expression of various signaling molecules.11 Endothelial dysfunction is responsible for a multitude of deleterious consequences related to stroke, including oxidative stress and the initiation of the inflammatory response.12,13 Dysregulation of the cerebral endothelium is considered to have a stark influence on the risk of stroke. Another antecedent in endothelial dysfunction and reperfusion injury is known as endothelial permeability. The endothelium acts as the blood–brain barrier (BBB), which ensures homeostasis through selective permeability. The development of brain endothelial dysfunction is a significant contributor to the process of ischemia/reperfusion injury as well as the failure of the BBB.14 A key aspect in the maintenance of healthy endothelial permeability is the function of tight junction proteins. Tight junction proteins form and maintain connections between endothelial cells and regulate the concentrations of solutes and water in the brain, which is necessary for cell function.15 In order to maintain its own function, the tight junction complex requires the presence of integral membrane proteins such as occludin. These proteins hold the tight junction in place and mediate its healthy function.16 Increasing the expression of occludin may be a novel treatment strategy to prevent reperfusion injury and infarction.Teneligliptin is part of the dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs, which is most commonly used in the treatment of type 2 diabetes mellitus. DPP-4 is an enzyme that rapidly degrades glucagon-like peptide-1 (GLP-1), which plays a vital role in insulin production, satiety, and other physiological processes.17 While a growing body of work has linked DPP-4 inhibition using teneligliptin with positive outcomes in other diseases, its main application remains as a treatment for type 2 diabetes mellitus.18 Teneligliptin possesses several pharmacological functions. For example, the administration of teneligliptin could prevent the development of non-alcoholic fatty liver disease (NAFLD) by reducing hepatic triglyceride (TG) and free fatty acid (FFA) levels.19 A recent study demonstrated that treatment with teneligliptin ameliorated the chemotherapy drug doxorubicin-induced damage in cardiomyocytes by reducing inflammation and mitigating apoptosis.20 Furthermore, teneligliptin has been shown to have vascular protective effects such as anti-inflammation.21 However, the pharmacological function of teneligliptin in ischemic stroke has not been reported before. In the present study, we endeavored to establish a basis of understanding of the mechanism of action of teneligliptin and to investigate the potential of this drug for use in the treatment of stroke and ischemia/reperfusion-related injury. Our findings demonstrate a promising role for teneligliptin in regulating endothelial permeability, rescuing occludin levels, and maintaining the function of the protective transcriptional factor Kruppel-like factor 2 (KLF2), as demonstrated both in vitro and in vivo.
2. Materials and methods
2.1. MCAO animal model and treatment method
C57/BL6 mice obtained from Jackson Laboratory were used in our series of in vivo experiments. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Xi'an Medical University and approved by the Animal Ethics Committee of the First Affiliated Hospital Of Xi'an Medical University. For animals in the MCAO + placebo and MCAO + teneligliptin groups, an acute cerebral ischemia MCAO stroke model was made via occlusion of the middle cerebral artery with a surgical filament followed by suture closure. At 24 h post-operation, the animals were allowed to undergo reperfusion by removing the closure suture for another 24 h period. The area of brain infarction was evaluated using the 2,3,5-triphenyl tetrazolium (TTC) staining method (Sigma-Aldrich, USA). Briefly, the brain tissues were sliced into sections with a thickness of 2 mm and stained with a 2% solution of 2,3,5-triphenyltetrazolium (TTC) at 37 °C for 10 min in darkness. Then, the sections were fixed using 4% paraformaldehyde (Beyotime Biotech. Shanghai, China) at 37 °C for 30 min. Infarct areas were measured blinded using the National Institutes of Health (NIH) Image J software and summed to determine stroke volume. For the administration of teneligliptin, the drug was delivered to mice via intraperitoneal (IP) injection for 14 consecutive days before the stroke procedure as well as during the cerebral ischemia and reperfusion process. The approximate intake of teneligliptin was 30 mg per kg body weight per day.2.2. Primary human brain microvascular endothelial cells (HBMVECs) culture and oxygen–glucose deprivation/reperfusion (OGD/R)
HBMVECs obtained from Cell Systems were used in all in vitro experiments. The cells were maintained using a Complete Medium Kit with 10% FBS and CultureBoost-R™ from Cell Systems. Briefly, HBMVECs were exposed to hypoxic conditions (1% O2, 5% CO2, 94% N2) in an air-tight incubator with deoxygenated media for 6 h, followed by exposure to reperfusion media (21% O2, 5% CO2) at 37 °C for different time points (24 and 48 h) in the presence or absence of teneligliptin (1, 3 μM).2.3. Neurological deficit scoring method
Neurological deficit scores were evaluated by an observer who was blinded to the surgical procedure to compare neurological dysfunction in all experimental mice once they had fully recovered from anesthesia. The severity of the neurological deficit in MCAO mice was assessed using a five-point grading scale. The scores used are as follows: grade 0 = no observable sign of neurological deficit; grade 1 = failure to fully extend the contralateral forepaw when held by the tail; grade 2 = animals move in ipsilateral circles; more severe: grade 3 = animals fall to one side contralateral to brain damage; grade 4 = animals cease movement and display minimal signs of consciousness.2.4. Blood–brain barrier vascular leakage assay
To assess BBB permeability in vivo, the Evans blue staining method was used. Directly following stereotactic injection, mice in all groups were injected with 2% Evans blue dye (EBD; 4 ml kg−1) through the tail vein. Mice in the sham group had only burr holes and were used as a control. After the indicated treatment, mice in all groups were collected and sacrificed. The brains of the mice were collected as quickly as possible immediately following saline perfusion. The brains were then weighed and homogenized in 50% trichloroacetic acid (TCA), and then visualized using a fluorescence microscope. The concentration of EBD was indexed by measuring the fluorescence intensity with excitation at 620 nm and emission at 680 nm.2.5. Immunostaining of tight junctions in vitro
Next, we performed a tight junction protein immunostaining assay to determine the integrity of the BBB using antibodies against occludin. After the indicated treatments, the mice in each group were collected and sacrificed. The brains were extracted and perfused with phosphate buffer saline (PBS), and the whole-brain tissues were sliced into 8 μm thick sections. OCT compound was used to embed the tissue sections. The embedded sections were then stained with primary antibodies against occludin (#ab31721, Abcam, USA), followed by the corresponding Alexa Fluor 488-labeled anti-rabbit secondary antibody (#ab150077, Abcam, USA). Fluorescence microscopy was used to visualize the images (Zeiss) with an excitation wavelength at 495 nm and an emission wavelength at 519 nm. Fluorescence intensity was analyzed using the National Institutes of Health (NIH) Image J software.2.6. Real-time polymerase chain reaction (PCR) analysis
The expression levels of occludin and KLF2 were measured using real-time PCR analysis. Briefly, the total RNA was extracted from HBMVECs using an RNeasy Micro kit (Qiagen, Cat. 74004), and then quantified using a Nanodrop spectrophotometer. To synthesize cDNA, iScript™ Reverse Transcription Supermix for RT-qPCR (Invitrogen (Cat. 1708840)) was employed using 1 μg total RNA. The synthesized cDNA were used for SYBR-based real-time PCR. The analysis was performed on the ABI 7500 platform. The following primers were used in this study: mouse occludin: F, 5′-CCAGGCAGCGTGTTCCT-3′ and R, 5′-TTCTAAATAACAGTCACCTGAGGGC-3′; mouse GAPDH: F, 5′-AGAAGGCTGGG-GCTCATTTG-3′ and R, 5′-AGGGGCCATCCACAGTCTTC-3′(reverse); human occludin: F, 5′-AGTGTGATAATAGTGAGTGCTATCC-3′ and R, 5′- TGTCATACCTGTCCATCTTTCTTC-3’; human KLF2, F, 5′-TGCCATCTGTGCGATCGT-3′ and R, 5′-GGCTACATGTGCCGTTTCATG-3′; human GAPDH: F, 5′-ACCACAGTCCACGCCATCAC-3′ and R, 5′-TCCACCACCCTGTTGCTGTA-3′.2.7. Western blot analysis
To determine the protein expression levels of occludin and KLF2, HBMVECs were lysed using radio-immunoprecipitation assay (RIPA) buffer supplemented with protease inhibitor cocktail after undergoing the indicated treatment. The proteins were then separated by size by loading 20 μg total cell lysates onto 4–20% precasted polyacrylamide gel electrophoresis (PAGE) gels, and then transferring the separated protein mixture onto polyvinylidene fluoride (PVDF) membranes. Specific antibodies were used to determine the protein expression levels. The following antibodies were used in this study: p-ERK5 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500, #3371, Cell Signaling Technology, USA); ERK5 (1:1000, #12950, Cell Signaling Technology, USA); KLF2 (1:1000, #ab194486, Abcam, USA); occludin (1:500, #ab31721, Abcam, USA); tubulin (1:10000, #ab210797, Abcam, USA); anti-rabbit IgG, HRP-linked antibody (1:2000, #7074, Cell Signaling Technology, USA); anti-mouse IgG, HRP-linked Antibody (1:2000, #7076, Cell Signaling Technology, USA).
:500, #3371, Cell Signaling Technology, USA); ERK5 (1:1000, #12950, Cell Signaling Technology, USA); KLF2 (1:1000, #ab194486, Abcam, USA); occludin (1:500, #ab31721, Abcam, USA); tubulin (1:10000, #ab210797, Abcam, USA); anti-rabbit IgG, HRP-linked antibody (1:2000, #7074, Cell Signaling Technology, USA); anti-mouse IgG, HRP-linked Antibody (1:2000, #7076, Cell Signaling Technology, USA).
2.8. Endothelial cell permeability in vitro assay (Transwell method, FITC–dextran)
The Transwell method was used to determine the permeability of the endothelial layer in vitro. Briefly, a 24-well receiver plate with 24 individual hanging cell culture inserts was used for the assay. HBMVECs were seeded onto collagen-coated inserts, and when an endothelial monolayer had formed, the inserts were exposed to OGD for 6 h. Reperfusion media was then introduced for 24 h. FITC–Dextran was added on top of the cell culture media. The rate at which the fluorescent molecules passed through the endothelial cell monolayer was considered to be proportional to the permeability of the monolayer. The fluorescent signal observed in the receiver plate well solution was used to measure the extent of permeability. The resulting fluorescence intensity was normalized to the baseline values and comparisons were made between various time points.2.9. LDH release
To determine cell death, a commercial LDH kit (#ab65391, Abcam, USA) was used to measure the release of LDH. Briefly, 50 μL of cell culture medium was collected from each well and mixed with an equal volume of reaction buffer and incubated for 30 min at room temperature in darkness. The OD value was measured at 490 nm using a fluorescence microscope.2.10. MTT assay
After the indicated treatment, the cell viability of HBMVECs was determined using the 3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-di- phenytetrazoliumromide (MTT) assay method. Briefly, the cells were washed 3 times with fresh PBS, followed by the addition of 5 mg L−1 MTT and incubation at 37 °C in darkness in a 5% CO2 incubator for 4 h. Once the cell culture medium was removed, the resulting formazan crystals were dissolved in dimethyl sulfoxide (DMSO). A fluorescent microscope was used to record the absorbance at 590 nm.2.11. Statistical analysis
The results of all in vivo and in vitro experiments are expressed as means ± standard deviation (SD). To assess the significance of the differences between groups, we used the SPSS statistical software package (v15.0). Statistical significance was determined using the analysis of variance (ANOVA) method, followed by Bonferroni's post-hoc test. A P value of less than 0.05 was determined to be statistically significant.3. Results
Teneligliptin, classified as a class III DPP-4 inhibitor, has a unique structural feature that provides strong binding to DPP-4 enzymes. The molecular structure of teneligliptin is shown in Fig. 1. In the present study, we explored the reperfusion damage-mitigating effects of teneligliptin in both in vivo and in vitro experiments. In our in vivo tests, we utilized MCAO mice in three groups; sham, MCAO + placebo, and MCAO + teneligliptin. MCAO mice were administered 30 mg kg−1 of teneligliptin a day for a period of two weeks before inducing ischemic conditions. We began our research by organizing brain infarction images and neurological score graphs. Brain infarction images demonstrated that while hypoxia upregulated ischemia conditions by approximately 33%, teneligliptin rescued this number by about half, as shown in Fig. 2A. Neurological score graphs also demonstrated significant damage due to ischemia/reperfusion with an initial upregulation to 3.4 from 0. However, teneligliptin reduced neurological damage to only 1.9 (Fig. 2B). | ||
| Fig. 1 Molecular structure of teneligliptin. | ||
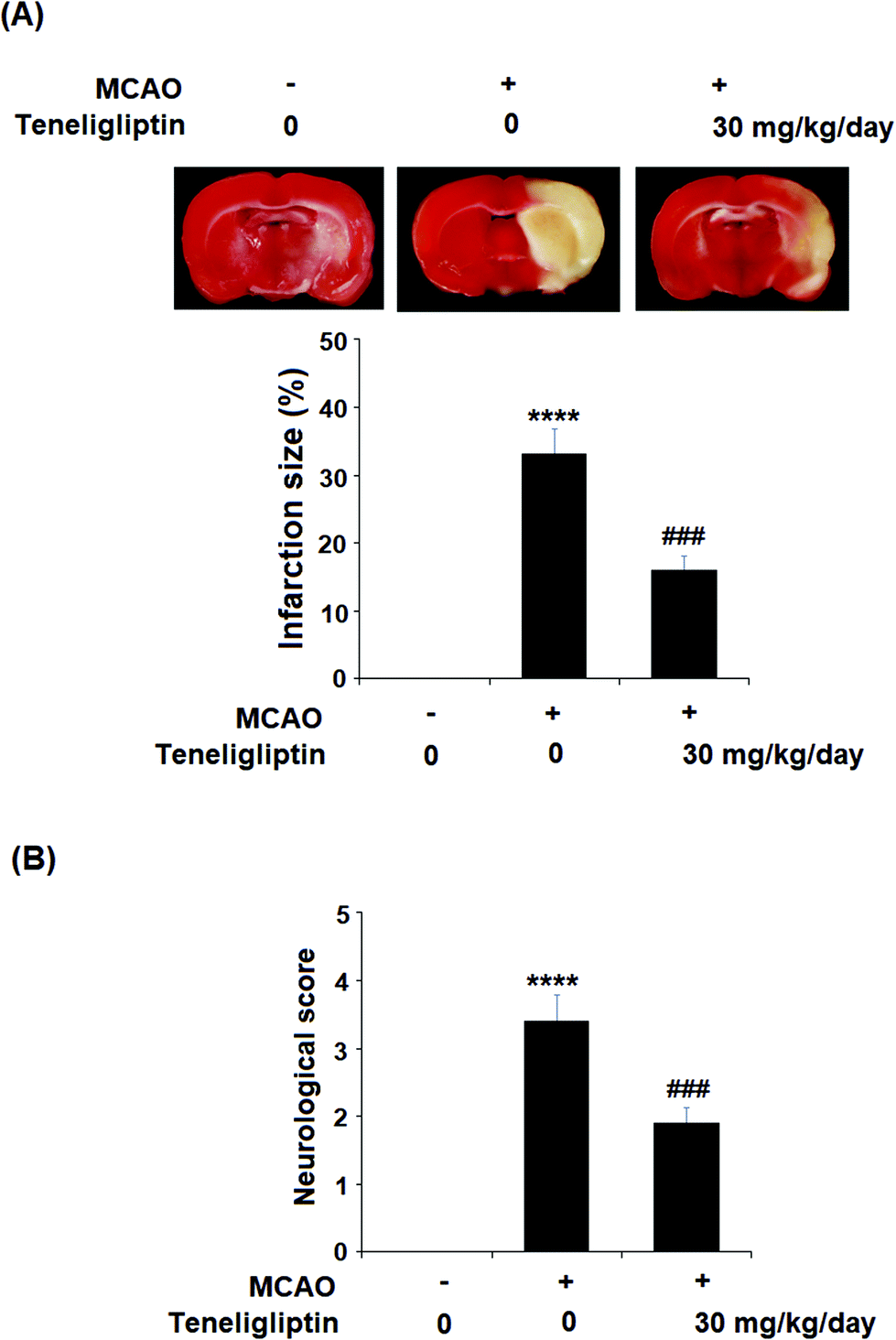 | ||
| Fig. 2 The protective effects of teneligliptin in MCAO mice. Mice were divided into 3 groups: sham group, MCAO + placebo, and MCAO + teneligliptin (30 mg kg−1 day−1) for 2 weeks before the MCAO experiment and during the MCAO experiment. (A) Representative brain infarction images and quantitative analysis; (B) neurological score graph of the three experimental groups (****, P < 0.0001 vs. sham group; ###, P < 0.001 vs. MCAO group). | ||
We then went on to explore the effect of teneligliptin on BBB permeability. We found that in mice in the MCAO + placebo group, permeability increased to nearly twice the basal value. However, this effect was almost completely ameliorated in mice in the MCAO + teneligliptin group, as demonstrated in Fig. 3. Tight junction proteins including occludin play an important role in regulating the integrity of BBB permeability. Teneligliptin was further shown to possess protective actions on the expression levels of the tight junction protein occludin. Using the previous parameters, PCR analysis demonstrated that while MCAO mice without teneligliptin treatment experienced a drop in occludin of nearly 60% basal expression, while teneligliptin rescued this number to near-basal levels, as shown in Fig. 4A. We went on to use immunostaining to determine occludin expression at the protein levels and found that the initial drop in expression of roughly 50% was reduced to only approximately 10% by teneligliptin, as shown in Fig. 4B.
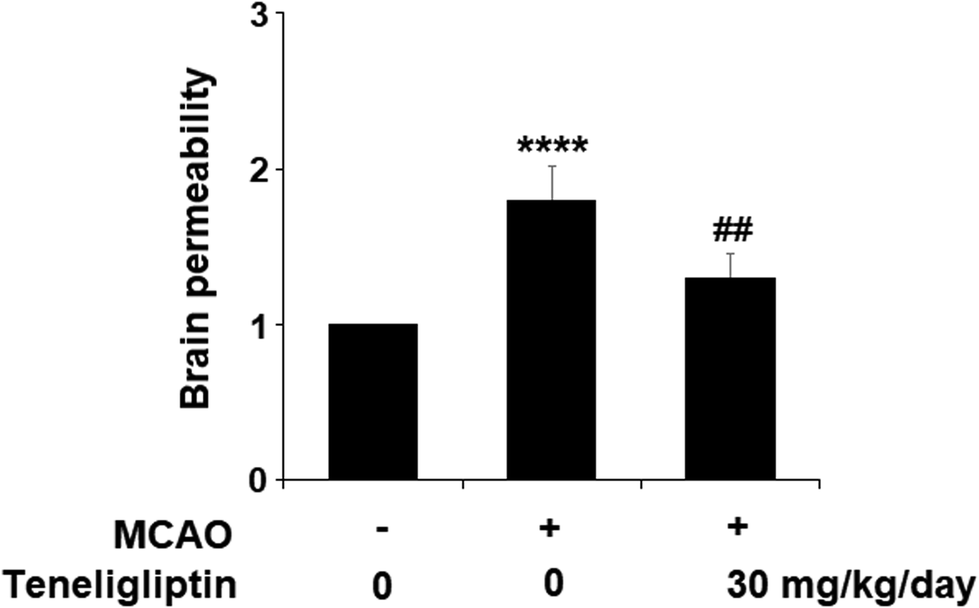 | ||
| Fig. 3 The protective effects of teneligliptin against ischemia/reperfusion-induced impairment of brain permeability in MCAO mice. MCAO + placebo, and MCAO + teneligliptin (30 mg kg−1 day−1) for 2 weeks before the MCAO experiment and during the MCAO experiment. Brain permeability of MCAO mice was measured by Evans blue staining assay (****, P < 0.0001 vs. sham group; ##, P < 0.01 vs. MCAO group). | ||
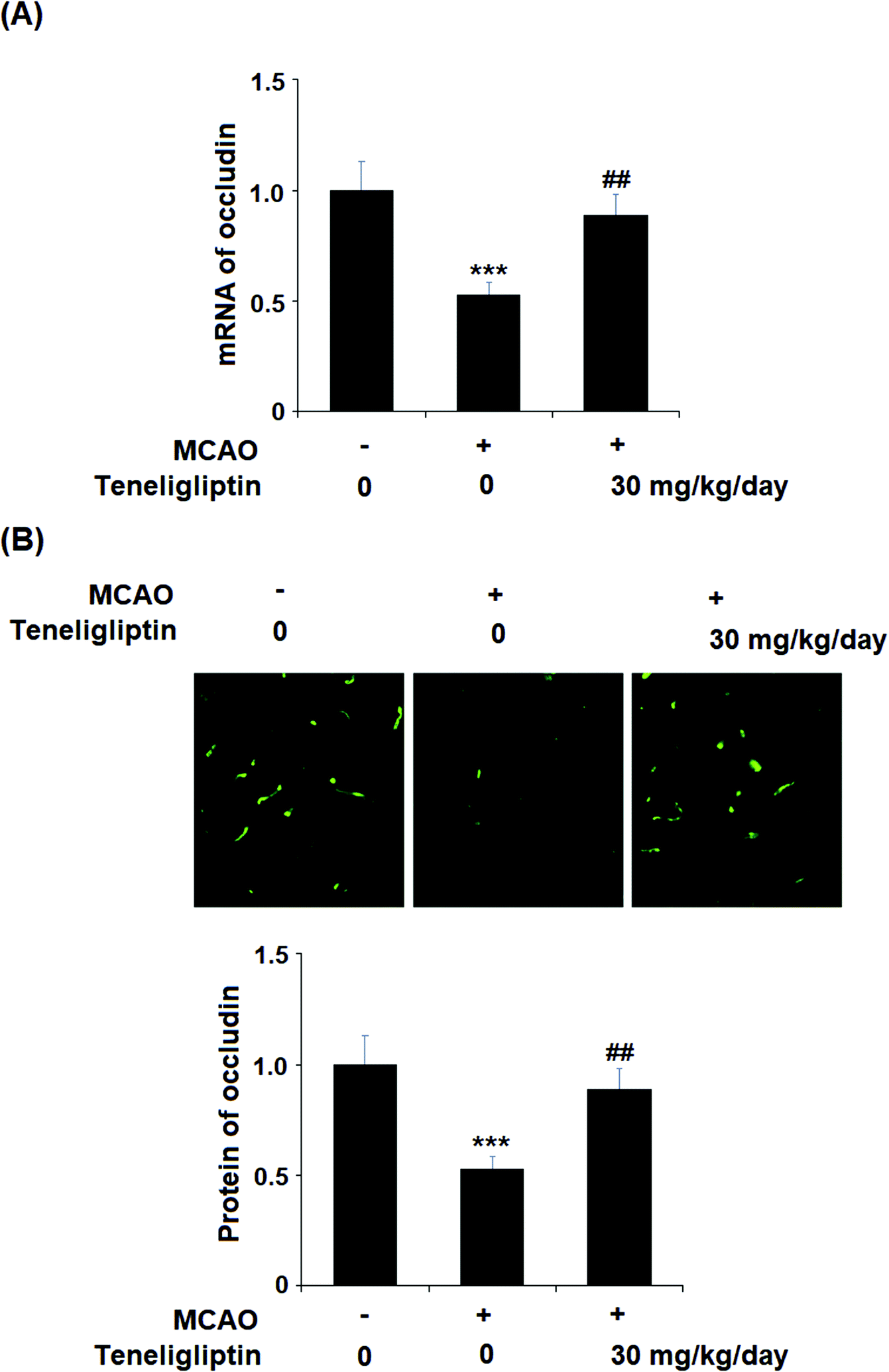 | ||
| Fig. 4 The protective effects of teneligliptin against ischemia/reperfusion-induced reduction of the tight junction protein occludin in MCAO mice. MCAO + placebo, and MCAO + teneligliptin (30 mg kg−1 day−1) for 2 weeks before the MCAO experiment and during the MCAO experiment. (A) Real-time PCR analysis of occludin; (B) Immunostaining of occludin (***, P < 0.001 vs. sham group; ##, P < 0.01 vs. MCAO group). | ||
To further explore the underlying mechanism whereby teneligliptin protects ischemia/reperfusion-induced brain endothelial cells, an in vitro study using human brain microvascular endothelial cells (HBMVECs) subjected to OGD/R was performed. As shown in Fig. 5A, teneligliptin was capable of significantly attenuating the ischemia-induced reduction in cell viability in a dose-dependent manner. This finding was further corroborated by studying the levels of lactate dehydrogenase (LDH) release from HBMVECs, which is widely used as an indicator of cell death. The release of LDH was increased in OGD/R conditions, while treatment with teneligliptin significantly ameliorated this effect, as shown in Fig. 5B. Remarkably, both cell viability and LDH release were maintained at near-basal levels by teneligliptin treatment.
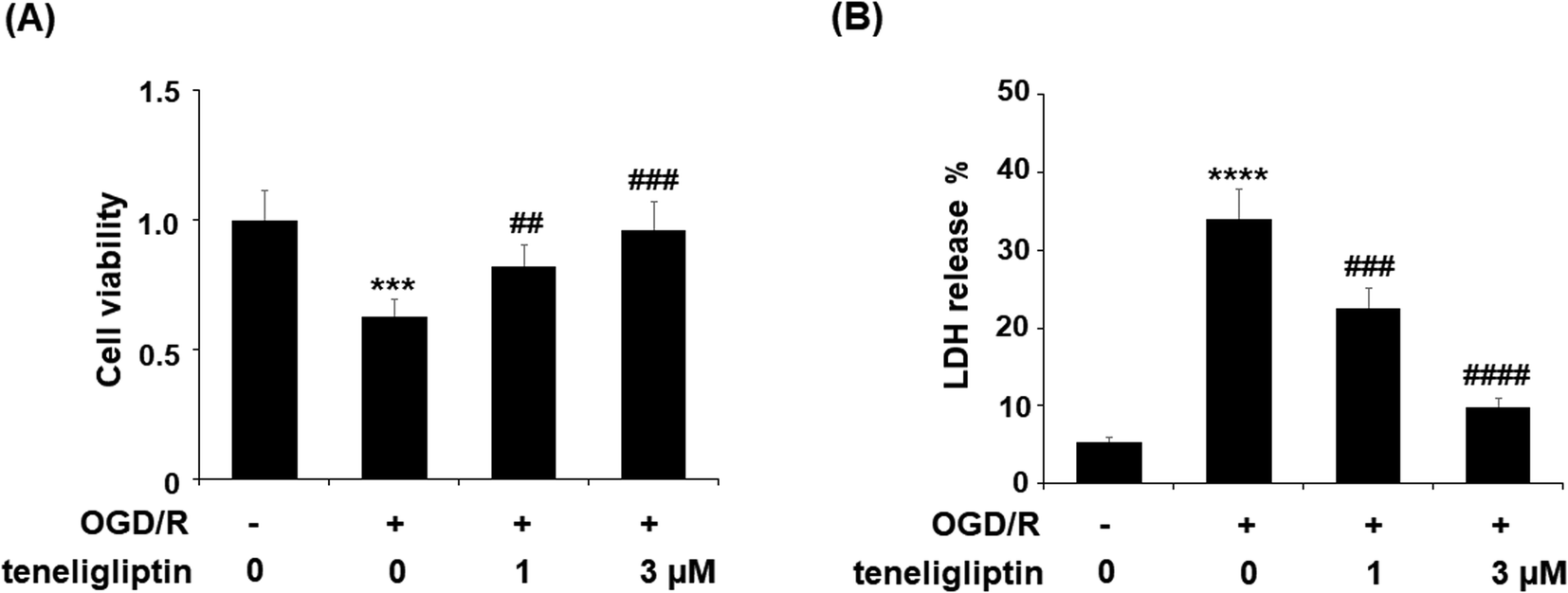 | ||
| Fig. 5 The protective effects of teneligliptin against OGD/R-induced cell death of HBMVECs. HBMVECs were exposed to hypoxic conditions for 6 h, followed by exposure to reperfusion media for different time points (48 h) in the presence or absence of teneligliptin (1, 3 μM). (A) Cell viability was determined by MTT assay; (B) LDH release was determined by a commercial kit (***, ****, P < 0.001, 0.0001 vs. control group; ##, ###, ####, P < 0.01, 0.001, 0.0001 vs. OGD/R group). | ||
Endothelial permeability is another important treatment target that plays a role in ischemia/reperfusion injury. The results in Fig. 6 show that upon exposure to OGD/R, endothelial permeability was significantly increased. However, teneligliptin reduced the level of endothelial permeability to just under 36% in a dose-dependent manner. Occludin levels were also significantly affected by OGD/R. As shown in Fig. 7, real-time PCR and western blot analyses demonstrated that mRNA and protein expression levels of occludin were reduced by roughly half upon exposure to OGD/R. However, both the mRNA and protein expression of occludin were significantly restored to approximately 95% in a dose-dependent manner. KLF2 has been identified as a central regulator of endothelial permeability by increasing the transcription of occludin and endothelial function. It is unknown whether teneligliptin has an influence on the expression of KLF2. Here, we found that the expression of KLF2 followed a similar pattern to occludin. OGD/R caused an initial reduction of KLF2 by roughly half at both the mRNA and protein levels. As shown in Fig. 8, real-time PCR and western blot analysis revealed that KLF2 expression was almost completely restored by teneligliptin treatment in a dose-dependent manner.
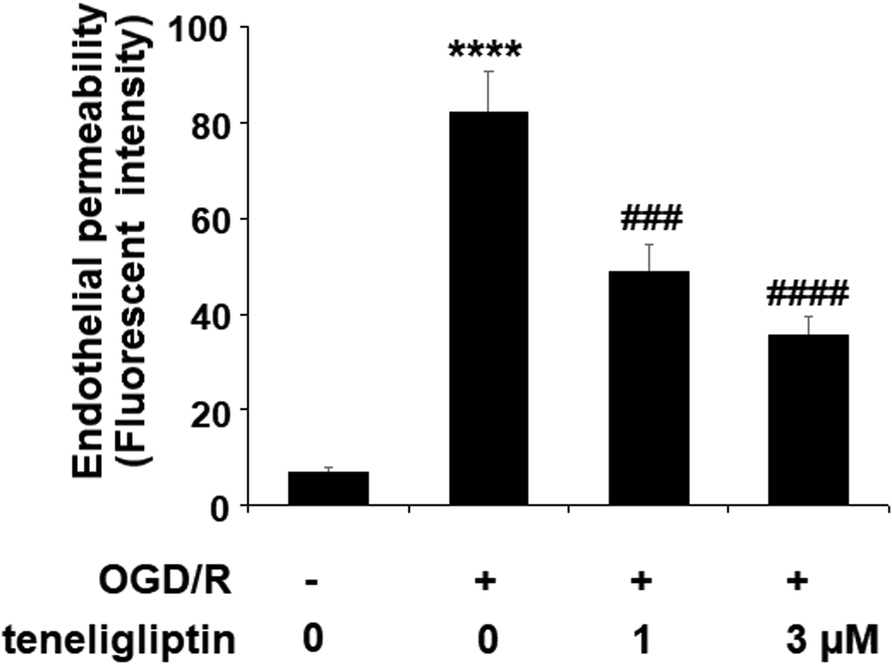 | ||
| Fig. 6 The protective effects of teneligliptin against OGD/R-induced increase in endothelial monolayer permeability in HBMVECs. HBMVECs were exposed to hypoxic conditions for 6 h, followed by exposure to reperfusion media for different time points (24 h) in the presence or absence of teneligliptin (1, 3 μM). Endothelial permeability was assessed by FITC-dextran permeation (****, P < 0.0001 vs. control group; ###, ####, P < 0.001, 0.0001 vs. OGD/R group). | ||
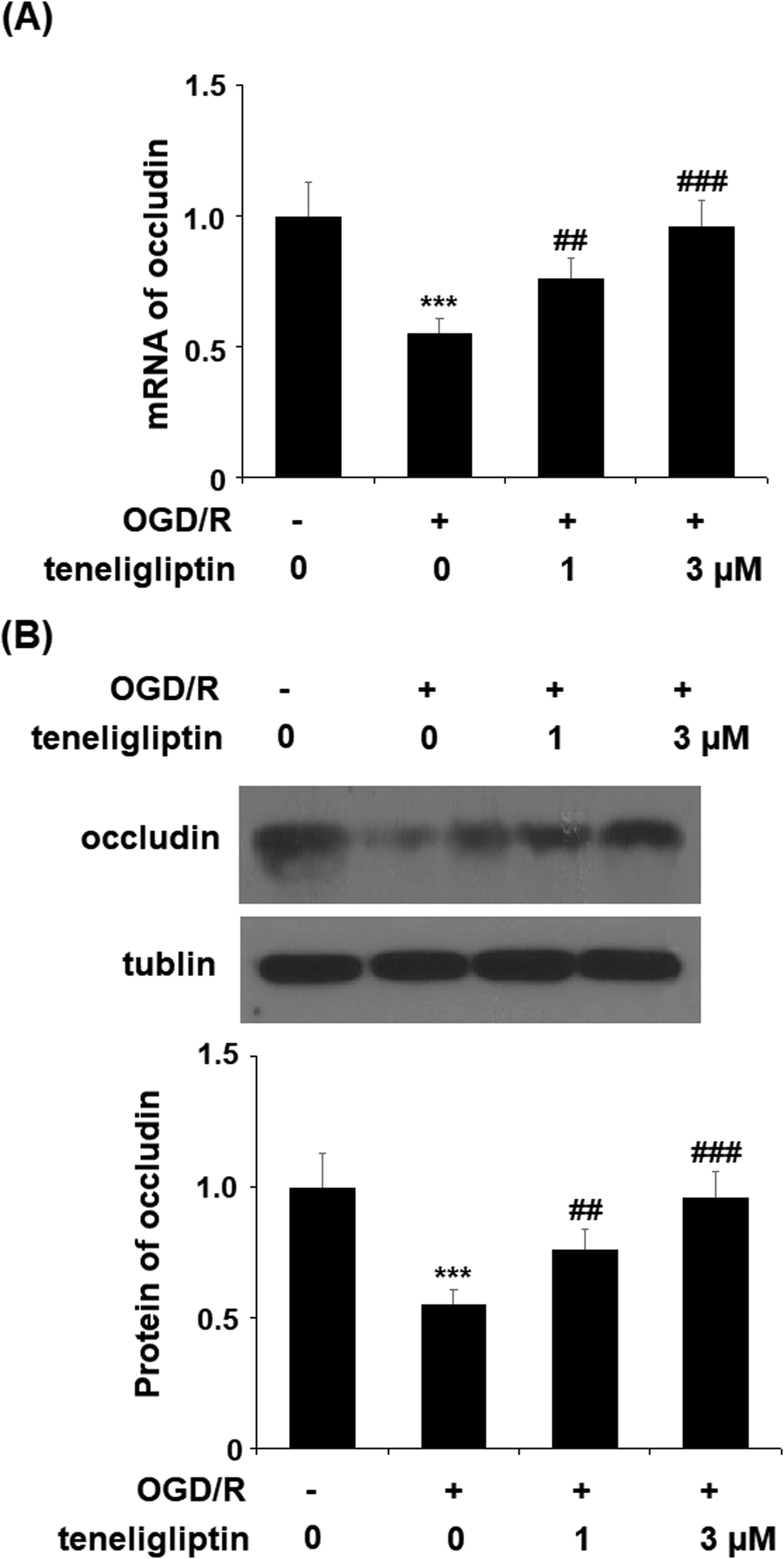 | ||
| Fig. 7 The protective effects of teneligliptin against OGD/R-induced reduction of occludin in HBMVECs. HBMVECs were exposed to hypoxic conditions for 6 h, followed by exposure to reperfusion media for different time points (24 h) in the presence or absence of teneligliptin (1, 3 μM). (A) Real-time PCR of occludin; (B) western blot analysis of occluding (***, P < 0.001 vs. control group; ##, ###, P < 0.01, 0.001 vs. OGD/R group). | ||
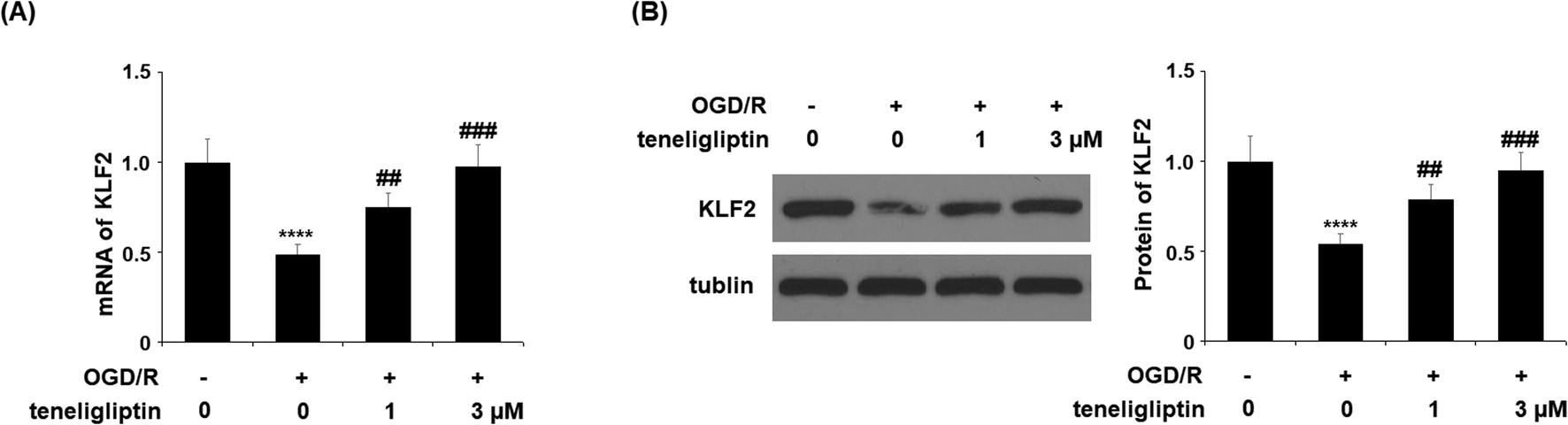 | ||
| Fig. 8 The protective effects of teneligliptin against OGD/R-induced reduction of KLF2 in HBMVECs. HBMVECs were exposed to hypoxic conditions for 6 h, followed by exposure to reperfusion media for different time points (24 h) in the presence or absence of teneligliptin (1, 3 μM). (A) Real-time PCR analysis of KLF2; (B) western blot analysis of KLF2 (****, P < 0.0001 vs. control group; ##, ###, P < 0.01, 0.001 vs. OGD/R group). | ||
The expression of KLF2 has been shown to be regulated by ERK5. To further clarify the underlying mechanism, we studied the effect of teneligliptin on ERK5 phosphorylation to determine whether this signaling pathway mediates the effects of teneligliptin on KLF2 expression. We subjected HBMVECs to OGD/R in the presence or absence of teneligliptin and measured the total and phosphorylated levels of ERK5. In cells exposed to OGD/R, the expression of ERK5 was reduced by more than half, while the addition of teneligliptin significantly ameliorated this reduction to roughly 89%, as shown in Fig. 9A. To confirm the involvement of ERK5 in the teneligliptin-mediated rescue of KLF2, we performed an ERK5 inhibition experiment using the ERK5 inhibitor XMD8-92. As shown in Fig. 9B and C, knockdown of ERK5 abolished the effects of teneligliptin on OGD/R-induced reduced KLF2 expression. Interestingly, ERK5 inhibition yielded similar results in terms of the effects of teneligliptin on occludin expression. The results in Fig. 9D and E demonstrate that ERK5 signaling is required for teneligliptin treatment to rescue OGD/R-induced reduced expression of occludin. This suggests that the effects of teneligliptin in the expression of KLF2 and occludin are mediated by ERK5.
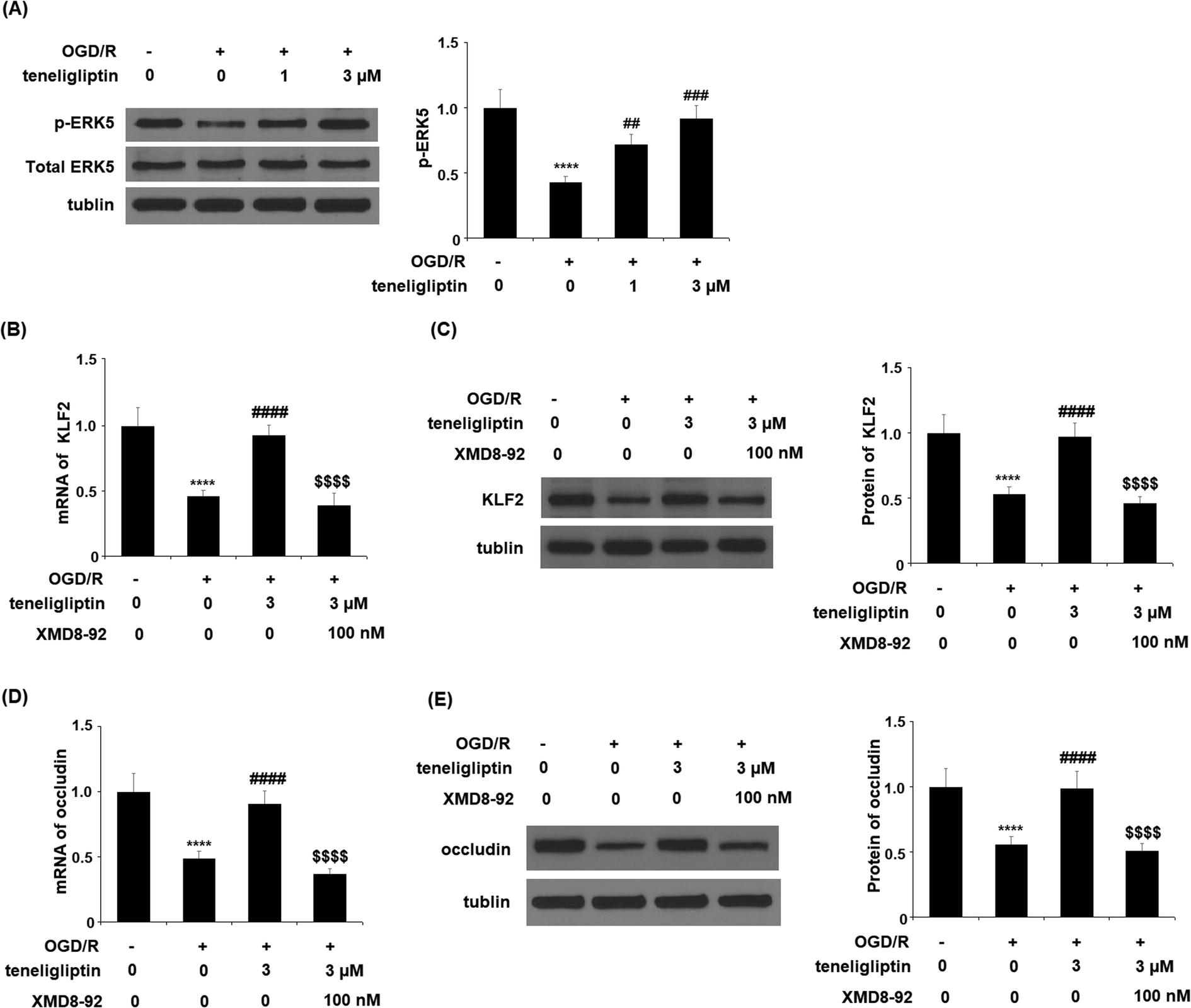 | ||
| Fig. 9 The protective effects of teneligliptin against OGD/R-induced reduction of ERK5 phosphorylation in HBMVECs. HBMVECs were exposed to hypoxic conditions for 6 h, followed by exposure to reperfusion media for different time points (24 h) in the presence or absence of teneligliptin (1, 3 μM). (A) Western blot analysis of phosphorylated and total ERK5 (****, P < 0.0001 vs. control group; ##, ###, P < 0.01, 0.001 vs. OGD/R group); (B and C). HBMVECs were exposed to hypoxic conditions for 6 h, followed by exposure to reperfusion media for different time points (24 h) in the presence or absence of teneligliptin (3 μM) or the ERK5 inhibitor XMD8-92 (100 nM). mRNA and Protein of KLF2 were measured; (D and E). HBMVECs were exposed to hypoxic conditions for 6 h, followed by exposure to reperfusion media for different time points (24 h) in the presence or absence of teneligliptin (3 μM) or the ERK5 inhibitor XMD8-92 (100 nM). mRNA and Protein of occludin was measured (****, P < 0.0001 vs. vehicle group; ####, P < 0.0001 vs. OGD/R group; $$$$, OGD/R+ teneligliptin group). | ||
4. Discussion
Ischemia/reperfusion injury is a key event in the pathogenesis of stroke. The rapid reperfusion following ischemic injury triggers the release of a myriad of signaling molecules that induce an acute inflammatory response, oxidative stress, cell death, and excessive endothelial permeability, thereby allowing blood flow to saturate the tissues of the brain. Previous research has provided ample evidence that DPP-4 inhibitors may confer neuroprotective effects during and immediately after the occurrence of stroke, including reduced oxidative stress, inflammation, and infarct volume, among other things.22 Here, we investigated the effects of teneligliptin on infarct size and neurological score in vivo using an MCAO mouse model. We found that teneligliptin significantly improved these parameters by reducing cerebral endothelial permeability. The results of our in vitro endothelial permeability assay corroborated these findings.Several lines of evidence have shown that brain vascular endothelial cells are an important cell type in the pathogenesis of stroke. A main function of brain endothelial cells is to regulate the integrity of the BBB. Occludin, a 65 kDa tetraspan integral membrane protein, contributes to tight junction stabilization and optimal barrier function.23 Tight junction dysregulation has been associated with the pathogenesis of various diseases, including stroke.24 We investigated the role of the DPP-4 inhibitor teneligliptin in the preservation of occludin expression both in vivo and in vitro. Indeed, we found that teneligliptin could significantly attenuate OGD/R-induced reduced occludin levels both in vivo and in vitro. Consistently, it has been recently reported that inhibition of DPP-4 reduced bacterial translocation in short bowel syndrome (SBS) by reinforcing the tight junction proteins E-cadherin and occludin.25 The preservation of cell viability following ischemia is vital to mitigating reperfusion damage. A recent study found that teneligliptin could rescue endothelial cell viability in rat cardiac microvascular endothelial cells exposed to hypoxia/reoxygenation injury.26 While there is considerable evidence that DPP-4 inhibition can improve endothelial cell viability and function after ischemia/reperfusion, the role of teneligliptin in protecting brain endothelial cells from OGD/R-induced injury is not clear.27 Our experiments demonstrate that teneligliptin significantly rescued this OGD/R-induced decreased cell viability in a dose-dependent manner, and remarkably, the higher dose almost completely restored cell viability.
KLF2 is expressed in both the brain macrovasculature and microvascular endothelial cells. Its expression is known to play a vital role in the maintenance of endothelial function. Long-proven in this regard, KLF2 has been shown to exert numerous endothelial-protective effects, such as aiding in c-Jun N-terminal kinase signaling and mitogen-activated protein kinase signaling.28 KLF2 is induced by laminar shear stress and inhibited by pro-inflammatory cytokines. KLF2 strongly attenuates the expression of cellular adhesion molecules including VCAM-1 and E-selectin in endothelial cells.29 Importantly, endothelial KLF2 has been identified as a key regulator of the integrity of the BBB and a novel neuroprotective factor in ischemic stroke.30 Therefore, we assessed the ability of teneligliptin to rescue KLF2 and its endothelial-protective downstream actions in our study. Following OGD/R conditions in our in vitro testing, KLF2 expression was found to be significantly reduced at both the mRNA and protein levels. Teneligliptin treatment rescued KLF2 levels in a dose-dependent manner, thereby indicating a distinct protective effect of teneligliptin against OGD/R-induced injury.
In addition to acting as a kinase, ERK5 is a transcriptional co-activator with a unique C-terminus transactivation domain. ERK5 activation increases KLF2 expression in endothelial cells, with consequent anti-inflammatory and athero-protective effects.31 The deficiency of ERK5 in endothelial cells has been shown to accelerate atherosclerosis formation in LDL receptor deficient (LDLR−/−) mice.32 Consistently, endothelial apoptosis and inflammation are accelerated in endothelial-specific ERK5 knockout mice.33 The ERK5 pathway has been shown to play an important role in protection against stroke as well as neuroprotection against ischemic injury.34,35 Our results show that teneligliptin almost completely rescued ERK5 expression, which was significantly reduced by exposure to OGD/R. This finding was further verified by the results of our ERK5 knockdown experiment using the specific ERK5 inhibitor XMD8-92. Indeed, inhibition of ERK5 abolished the effects of teneligliptin on the expression of KLF2 at both the mRNA and protein levels. These findings demonstrate a significant role of DPP-4 inhibition by teneligliptin as a treatment against ischemia/reperfusion damage.
The protective effects of teneligliptin against post-ischemic injury, such as reduced infarct volume, improved cell viability, reduced endothelial permeability, and the preservation of occludin expression, have been clearly demonstrated in the present work. Additionally, we elucidated the importance of the ERK5 pathway in mediating the ability of teneligliptin to rescue the expression of the protective transcriptional factor KLF2. Thus, teneligliptin may serve as a novel therapy to prevent or treat neurological injury due to stroke or ischemia/reperfusion.
Conflicts of interest
There is no conflict interest among the authors of the present study.Acknowledgements
This study was supported by the Key Research and Development Project of the Shan-xi Science and Technology Department (2018SF-087).References
- I. S. Party, National clinical guideline for stroke, London, Royal College of Physicians, 2012 Sep Search PubMed.
- A. Ciccone, L. Valvassori, M. Nichelatti, A. Sgoifo, M. Ponzio, R. Sterzi and E. Boccardi, Endovascular treatment for acute ischemic stroke, N. Engl. J. Med., 2013, 368(10), 904–913 CrossRef CAS PubMed.
- R. Khatri, A. M. McKinney, B. Swenson and V. Janardhan, Blood–brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke, Neurology, 2012, 79(13 Supplement 1), S52–S57 CrossRef PubMed.
- S. M. Lai, M. Alter, G. Friday and E. Sobel, A multifactorial analysis of risk factors for recurrence of ischemic stroke, Stroke, 1994, 25(5), 958–962 CrossRef CAS.
- G. A. Donnan, M. Fisher, M. Macleod and S. M. Davis, Stroke., Lancet, 2008, 371, 1612–1623 CrossRef CAS.
- A. Shuaib, K. Butcher, A. A. Mohammad, M. Saqqur and D. S. Liebeskind, Collateral blood vessels in acute ischaemic stroke: a potential therapeutic target, Lancet Neurol., 2011, 10(10), 909–921 CrossRef.
- G. A. Rosenberg, Ischemic brain edema, Prog. Cardiovasc. Dis., 1999, 42(3), 209–216 CrossRef CAS.
- T. S. Harwell, L. L. Blades, C. S. Oser, C. C. Fogle, S. D. Helgerson, D. Gohdes, D. W. Dietrich, A. M. Burnett, N. J. Okon, M. J. Allen and D. V. Rodriguez, Peer Reviewed: Rural Community Knowledge of Stroke Warning Signs and Risk Factors, Prev. Chronic Dis., 2005, 2(2), A14 Search PubMed.
- E. M. Khedr, O. El Shinawy, T. Khedr, Y. A. Aziz Ali and E. M. Awad, Assessment of corticodiaphragmatic pathway and pulmonary function in acute ischemic stroke patients, Eur. J. Neurol., 2000, 7(3), 323–330 CrossRef CAS PubMed.
- D. Herderschee, A. Hijdra, A. Algra, P. J. Koudstaal, L. J. Kappelle and J. Van Gijn, Silent stroke in patients with transient ischemic attack or minor ischemic stroke. The Dutch TIA Trial Study Group, Stroke, 1992, 23(9), 1220–1224 CrossRef CAS.
- R. F. Keep, A. V. Andjelkovic, S. M. Stamatovic, P. Shakui and S. R. Ennis. Ischemia-induced endothelial cell dysfunction, in Intracranial Pressure and Brain Monitoring XII 2005, vol. 95, pp. 399–402 Search PubMed.
- A. Ceriello, R. Assaloni, R. Da Ros, A. Maier, L. Piconi, L. Quagliaro, K. Esposito and D. Giugliano, Effect of atorvastatin and irbesartan, alone and in combination, on postprandial endothelial dysfunction, oxidative stress, and inflammation in type 2 diabetic patients, Circulation, 2005, 111(19), 2518–2524 CrossRef CAS PubMed.
- I. A. Van den Oever, H. G. Raterman, M. T. Nurmohamed and S. Simsek, Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus, Mediators Inflammation, 2010, 2010, 792393 Search PubMed.
- G. J. del Zoppo and J. M. Hallenbeck, Advances in the vascular pathophysiology of ischemic stroke, Thromb. Res., 2000, 98(3), 73–81 CrossRef CAS.
- K. E. Sandoval and K. A. Witt, Blood-brain barrier tight junction permeability and ischemic stroke, Neurobiol. Dis., 2008, 32(2), 200–219 CrossRef CAS.
- K. M. McCarthy, I. B. Skare, M. C. Stankewich, M. Furuse, S. Tsukita, R. A. Rogers, R. D. Lynch and E. E. Schneeberger, Occludin is a functional component of the tight junction, J. Cell Sci., 1996, 109(9), 2287–2298 CAS.
- M. Kishimoto, Teneligliptin: a DPP-4 inhibitor for the treatment of type 2 diabetes, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2013, 6, 187 CrossRef CAS.
- S. K. Sharma, A. Panneerselvam, K. P. Singh, G. Parmar, P. Gadge and O. C. Swami, Teneligliptin in management of type 2 diabetes mellitus, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2016, 9, 251 CrossRef CAS PubMed.
- K. Nakamura, S. Fukunishi, K. Yokohama, H. Ohama, A. Asai, Y. Tsuda and K. Higuchi, Su1886 Long-Lasting Dipeptidyl Peptidase-4 Inhibitor, Teneligliptin, Can Be a Preventative Drug Not a Treatment Drug for the Development of NAFLD, Gastroenterology, 2015, 148(4), S-5–43 Search PubMed.
- W. Peng, D. Rao, M. Zhang, Y. Shi, J. Wu, G. Nie and Q. Xia, Teneligliptin prevents doxorubicin-induced inflammation and apoptosis in H9c2 cells, Arch. Biochem. Biophys., 2019, 108238 CrossRef PubMed.
- T. Iwakura, Z. Zhao, J. A. Marschner, S. K. Devarapu, H. Yasuda and H. J. Anders, Dipeptidyl peptidase-4 inhibitor teneligliptin accelerates recovery from cisplatin-induced acute kidney injury by attenuating inflammation and promoting tubular regeneration, Nephrol., Dial., Transplant., 2019, 34(10), 1669–1680 CrossRef PubMed.
- V. Darsalia, M. Larsson, D. Nathanson, T. Klein, T. Nyström and C. Patrone, Glucagon-like receptor 1 agonists and DPP-4 inhibitors: potential therapies for the treatment of stroke, J. Cereb. Blood Flow Metab., 2015, 35(5), 718–723 CrossRef CAS.
- P. M. Cummins, Occludin: one protein, many forms, Mol. Cell. Biol., 2012, 32(2), 242–250 CrossRef CAS PubMed.
- C. Förster, Tight junctions and the modulation of barrier function in disease, Histochem. Cell Biol., 2008, 130(1), 55–70 CrossRef PubMed.
- R. Sueyoshi, K. Miyahara, N. Nakazawa-Tanaka, N. Fujiwara, T. Ochi and A. Yamataka, DPP4 inhibitor reinforces cell junction proteins in mouse model of short bowel syndrome, Pediatr. Surg. Int., 2019, 1–7 Search PubMed.
- Z. Zhang, X. Jin, C. Yang and Y. Li, Teneligliptin protects against hypoxia/reoxygenation-induced endothelial cell injury, Biomed. Pharmacother., 2019, 109, 468–474 CrossRef CAS PubMed.
- M. S. Rajput, P. D. Sarkar and N. P. Nirmal, Inhibition of DPP-4 activity and neuronal atrophy with genistein attenuates neurological deficits induced by transient global cerebral ischemia and reperfusion in streptozotocin-induced diabetic mice, Inflammation, 2017, 40(2), 623–635 CrossRef CAS PubMed.
- R. A. Boon, T. A. Leyen, R. D. Fontijn, J. O. Fledderus, J. M. Baggen, O. L. Volger, G. P. van Nieuw Amerongen and A. J. Horrevoets, KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium, Blood, 2010, 115(12), 2533–2542 CrossRef CAS PubMed.
- M. R. DiChiara, J. M. Kiely, M. A. Gimbrone Jr, M. E. Lee, M. A. Perrella and J. N. Topper, Inhibition of E-selectin gene expression by transforming growth factor β in endothelial cells involves coactivator integration of Smad and nuclear factor κB-mediated signals, J. Exp. Med., 2000, 192, 695–704 CrossRef CAS PubMed.
- H. Shi, B. Sheng, F. Zhang, C. Wu, R. Zhang, J. Zhu, K. Xu, Y. Kuang, S. C. Jameson, Z. Lin, Y. Wang, J. Chen, M. K. Jain and G. B. Atkins, Kruppel-like factor 2 protects against ischemic stroke by regulating endothelial blood brain barrier function, Am. J. Physiol.: Heart Circ. Physiol., 2013, 304(6), H796–H805 CrossRef CAS PubMed.
- K. M. Parmar, H. B. Larman, G. Dai, Y. Zhang, E. T. Wang, S. N. Moorthy, J. R. Kratz, Z. Lin, M. K. Jain and M. A. Gimbrone Jr, Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by kruppel-like factor 2, J. Clin. Invest., 2006, 116, 49–58 CrossRef CAS PubMed.
- N. T. Le, K. S. Heo, Y. Takei, H. Lee, C. H. Woo, E. Chang, C. McClain, C. Hurley, X. Wang, F. Li, H. Xu, C. Morrell, M. A. Sullivan, M. S. Cohen, I. M. Serafimova, J. Taunton, K. Fujiwara and J. Abe, A crucial role for p90rsk-mediated reduction of erk5 transcriptional activity in endothelial dysfunction and atherosclerosis, Circulation, 2013, 127, 486–499 CrossRef CAS PubMed.
- S. J. Sohn, B. K. Sarvis, D. Cado and A. Winoto, Erk5 mapk regulates embryonic angiogenesis and acts as a hypoxia- sensitive repressor of vascular endothelial growth factor expression, J. Biol. Chem., 2002, 277, 43344–43351 CrossRef CAS PubMed.
- J. Sun and G. Nan, The mitogen-activated protein kinase (MAPK) signaling pathway as a discovery target in stroke, J. Mol. Neurosci., 2016, 59(1), 90–98 CrossRef CAS PubMed.
- R. Jin, L. Liu, S. Zhang, A. Nanda and G. Li, Role of inflammation and its mediators in acute ischemic stroke, J Cardiovasc Transl Res., 2013, 6(5), 834–851 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |