
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA magnetically separable and recyclable g-C3N4/Fe3O4/porous ruthenium nanocatalyst for the photocatalytic degradation of water-soluble aromatic amines and azo dyes†
Anupam Sahoo and
Srikanta Patra *
*
School of Basic Sciences, Indian Institute of Technology Bhubaneswar, Argul, Jatni, Khurda-752050, Odisha, India. E-mail: srikanta@iitbbs.ac.in
First published on 6th February 2020
Abstract
Herein, we present the development of a visible-light-driven magnetically retrievable nanophotocatalyst made of porous ruthenium nanoparticles supported on magnetic carbon nitride (g-C3N4/Fe3O4/p-RuNP) for the facile removal/degradation of aromatic amines and azo dyes from wastewater. Aromatic amines and azo-based dyes in water bodies are highly toxic and carcinogenic even at very low concentrations and are difficult to separate because of their high solubility. Our nanocatalyst can efficiently degrade/decompose the aromatic amines and azo dyes under visible light (LED/sunlight) at room temperature and in a wide pH range (pH 5.0–9.0) without using any external chemicals. The magnetic property of the nanocatalyst facilitates its efficient and facile separation from the reaction mixture for reuse in multiple photocatalytic cycles. The nanocatalyst-based degradation of azo dyes and aromatic amines presented here is simple and convenient in terms of efficiency, energy, reusability and cost. The process also does not require any external chemicals and forms gaseous/less harmful end products.
Introduction
Aromatic amines are disposed into the environment directly from industries manufacturing various products, such as resins, textile, plastic, photographic materials, and pharmaceuticals, and as the end products of pesticide degradation and bacterial/chemical conversion of azo compounds. These aromatic amines are highly toxic, carcinogenic or mutagenic and have been elaborated by various researchers.1–7 Owing to their toxic and carcinogenic nature, aromatic amines are considered as an important class of water pollutants.1–7 Aromatic amines can easily spread through soil and contaminate the groundwater due to their high polarity and high solubility in water. Azo dyes constitute more than 50% of the dyestuffs used for commercial purposes.5,8–13 Thus, it is essential to eliminate them from water bodies to avoid the environmental problems associated with them.Conventional processes, including extraction, steam distillation, sorption on activated carbon, UV-light irradiation, bacterial decomposition and chemical/electrochemical reaction (oxidation), have been extensively used for the removal of aromatic amines.7,8,14–21 Meanwhile, processes, such as catalytic degradation, precipitation and sedimentation, have been used for the removal of azo-based dyes.5,13,16,19,21–26 All these methods suffer from drawbacks, such as the use of harmful chemicals, generation of a large volume of sludge, high cost and poor efficiency (Table TS1†). This necessitates an improved method, which can address the aforesaid issues and achieve degradation in a greener way.
The photo-degradation of aromatic amines under visible light using a suitable photocatalyst is one of the green processes. This process does not require any external reagents, can convert the pollutants into their gaseous less-toxic forms and generate a small/negligible amount of solid waste. Despite this, the photo-degradation of organic pollutants is limited because of the poor efficiency and non-availability of suitable photocatalysts.26,27
Carbon nitride (g-C3N4) has emerged as a highly efficient photocatalyst in recent years and has exhibited potential in various applications due to its high thermal and chemical stabilities, electron-rich property, semiconducting property, propitious band gap and high affinity towards visible light.28–37 A major drawback of this system is the limited efficiency in separating photo-excited electron–hole pairs.38 Several modifications, such as chemical doping with metal and non-metals, have been attempted to improve the photocatalytic activity of pure g-C3N4.39–49
On the other hand, ruthenium nanoparticles are known for their potential in various catalysis-based applications.50–56 Because of their quantized nature, ruthenium nanoparticles can enhance the efficiency of photo-excited electron–hole pair separation and thereby, increase the photocatalytic activity of nanocomposites comprising g-C3N4.57,58 Recently, we have demonstrated that the trimetallic porous Au@Pd@RuNP and porous Si@p-RuNP systems exhibit excellent catalytic reactivity towards azo bond reduction.5,13 In addition, the porous Si@p-RuNP system demonstrated very good separation efficiency for water-soluble aromatic amines by oxidizing them to form polyamines. In both cases, external chemicals were used, and the separation of catalysts was not accomplished easily.
Nevertheless, heterogeneous catalysts are preferred over homogeneous ones due to their easy separability, recovery and reusability. Further, the separation of the catalyst can be made simpler by incorporating a magnetic support.46,59–68 A potentially efficient photocatalytic system would possess the combined advantages of g-C3N4, porous RuNP and magnetic Fe3O4NP. Considering this hypothesis, herein, we demonstrate the development of a ruthenium-based magnetic g-C3N4/Fe3O4/p-RuNP nanocomposite system and investigate its photocatalytic behaviour towards the decomposition of water-soluble aromatic amines and azo dyes from water bodies.
Results and discussion
Synthesis and characterisation
The g-C3N4/Fe3O4NP nanocomposite was synthesised by following a previously reported procedure.69,70 The g-C3N4 nanosheets were obtained by heating urea in a furnace at 520 °C.70,71 Fe3O4 nanoparticles were deposited on the g-C3N4 nanosheets by the deposition–precipitation method using a mixture of Fe(II)/Fe(III) salts (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and ammonia at 60 °C. CoNPs were deposited on g-C3N4/Fe3O4NP by reducing the Co2+ ions using NaBH4 under a nitrogen atmosphere. Porous ruthenium was deposited on g-C3N4/Fe3O4NP by the galvanic displacement of the in situ-formed CoNPs to yield the g-C3N4/Fe3O4/p-RuNP nanocomposite.5,13 A schematic representation of the synthesis of the g-C3N4/Fe3O4/p-RuNP nanocomposite is shown in Scheme 1.
:1) and ammonia at 60 °C. CoNPs were deposited on g-C3N4/Fe3O4NP by reducing the Co2+ ions using NaBH4 under a nitrogen atmosphere. Porous ruthenium was deposited on g-C3N4/Fe3O4NP by the galvanic displacement of the in situ-formed CoNPs to yield the g-C3N4/Fe3O4/p-RuNP nanocomposite.5,13 A schematic representation of the synthesis of the g-C3N4/Fe3O4/p-RuNP nanocomposite is shown in Scheme 1.
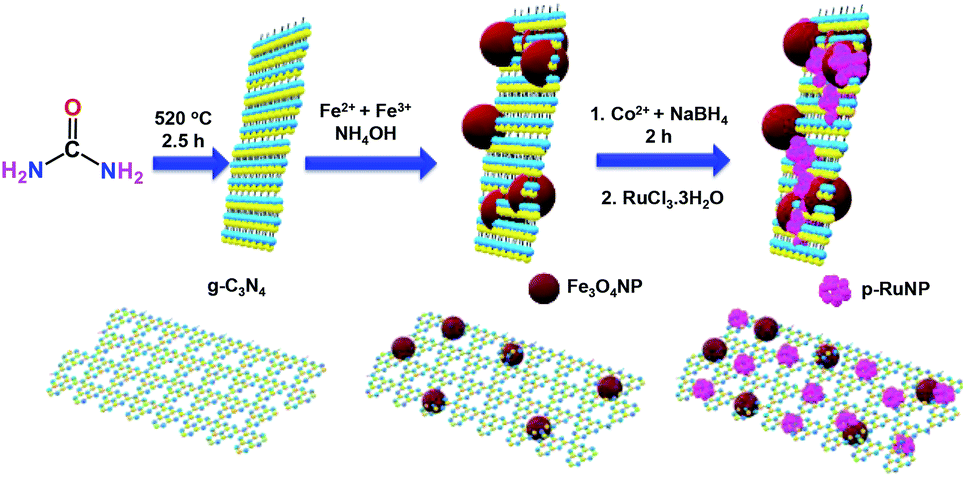 | ||
| Scheme 1 Schematic representation of the synthesis of the g-C3N4/Fe3O4/p-RuNP nanocomposite. | ||
The formation of the g-C3N4/Fe3O4/p-RuNP nanocomposite and its composition were analysed by FE-SEM and EDS spectroscopy (Fig. S1†). A clear distinction between g-C3N4, g-C3N4/Fe3O4 and g-C3N4/Fe3O4/p-RuNP nanocomposites was clearly visible in the SEM images. The surface morphology changed completely after the deposition of Fe3O4NPs and ruthenium on the g-C3N4 nanosheet (Fig. S1a–c†). The EDS analysis (Fig. S1d–f†) and atom mapping (Fig. S2†) of the aforesaid nanocomposites also supported the presence of iron and ruthenium in the respective nanocomposites.
The formation of the g-C3N4/Fe3O4/p-RuNP nanocomposite was further confirmed through high-resolution transmission electron microscopy (HRTEM) analysis. Fig. 1 represents the TEM and HRTEM images of the g-C3N4/Fe3O4/p-RuNP nanocomposite. The presence of ruthenium nanoparticles in the g-C3N4/Fe3O4/p-RuNP nanocomposite was clearly visible in the TEM images (Fig. 1a and b). The composition of elements in the g-C3N4/Fe3O4/p-RuNP nanocomposite was further confirmed through HRTEM analysis. The observed lattice fringes of 0.46 and 0.264 nm revealed the (020) and (311) planes of the Fe3O4 nanoparticles deposited on g-C3N4, respectively, (Fig. 1c–e).72,73 The lattice spacing of 0.246 nm confirmed the presence of Co in its oxidised form (CoO).74,75 The lattice spacing of 0.213 nm suggested that the deposited ruthenium nanoparticles had the (002) plane (Fig. 1e and f).76,77
 | ||
| Fig. 1 (a and b) TEM and (c and d) HRTEM images showing the lattice fringes of Fe3O4NP and (e and f) the lattice fringes of Ru in the g-C3N4/Fe3O4/p-RuNP nanocomposite. | ||
The surface composition and the oxidation state of elements on the surface of the g-C3N4/Fe3O4/p-RuNP nanocatalyst were examined by X-ray photoelectron spectroscopy (XPS) analysis (Fig. 2). Fig. 2a displays the XPS survey scan spectrum of g-C3N4/Fe3O4/p-RuNP, which shows the peaks for Ru, Fe, O, N and C on the surface. The high-resolution spectrum showed a peak at 280.4 eV corresponding to Ru 3d5/2, which confirmed the presence of ruthenium nanoparticles (metallic Ru) over the surface of the nanocatalyst (Fig. 2b).78–80 Further, the peak at 462.3 eV corresponding to Ru 3p3/2 confirmed that ruthenium was present at the zero oxidation state (Fig. 2c).79 The high-resolution XPS spectrum of Fe exhibited peaks at 710 eV and 724 eV corresponding to Fe 2p (Fig. 2d).69,81,82
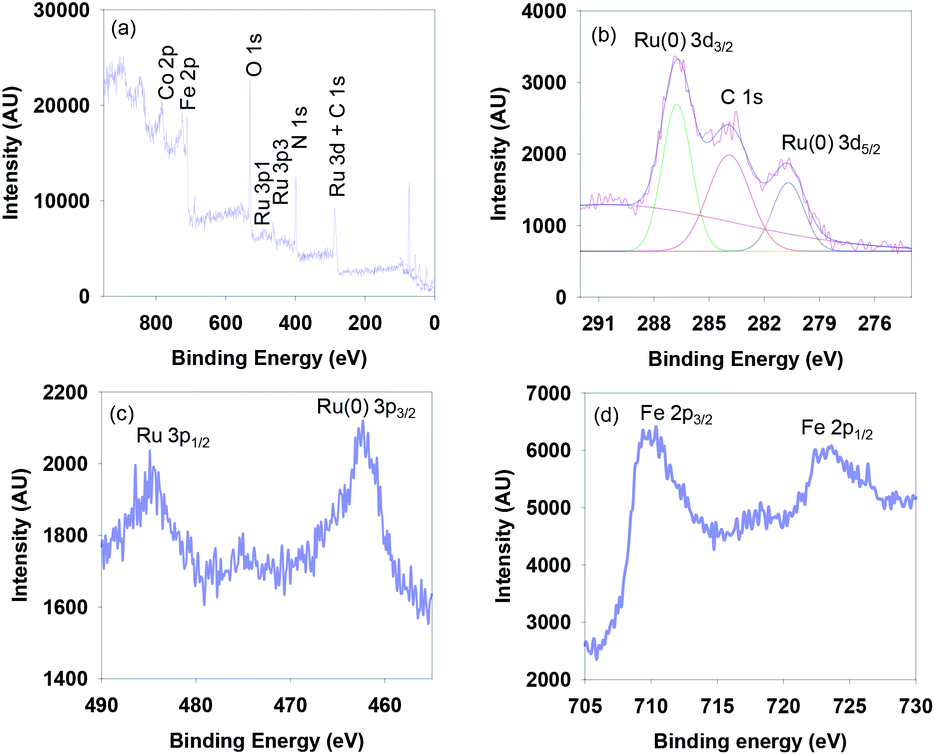 | ||
| Fig. 2 (a) XPS survey scan spectrum, and the high-resolution (b) Ru 3d (c) Ru 3p and (d) Fe 2p spectra of g-C3N4/Fe3O4/p-RuNP. | ||
The crystalline nature and formation of the g-C3N4/Fe3O4/p-RuNP nanocomposite were further authenticated by powder XRD analysis (Fig. S3†). A strong peak at 2θ = 27.5° corresponding to the characteristic interplanar stacking of the (002) plane of an aromatic system confirmed the formation of g-C3N4 nanosheets.83 The observed diffraction peaks of g-C3N4/Fe3O4NP at 2θ = 30.29°, 35.8°, 43.6°, 54°, 57.6°, 63.3° corresponded to the (200), (311), (400), (442), (511), and (440) planes of the fcc lattice structure of Fe3O4NPs, respectively.84,85 No additional peaks related to ruthenium nanoparticles were observed, which might be due to its smaller amount compared with the g-C3N4 nanosheet and Fe3O4NPs.
To confirm the porous nature of ruthenium in the g-C3N4/Fe3O4/p-RuNP nanocomposite, its N2 adsorption–desorption isotherms were studied at −196 °C (Fig. S4†). It was observed that the Brunauer–Emmett–Teller (BET) specific surface area of the g-C3N4/Fe3O4NP nanocomposite was 107.68 m2 g−1, which is larger than that of g-C3N4 (82.48 m2 g−1). After the deposition of porous ruthenium on g-C3N4/Fe3O4NP, there was a further increase in the surface area to 124.1 m2 g−1. Interestingly, with the increment in surface area, an increase in pore volume from 0.34 cm3 g−1 for g-C3N4/Fe3O4NP to 0.37 cm3 g−1 for the g-C3N4/Fe3O4/p-RuNP nanocomposite was observed, which supported the porous nature of the deposited ruthenium nanoparticles.
Absorption spectroscopy was conducted to understand the optical behaviour of the synthesised nanocomposites (Fig. 3a). The pure g-C3N4 nanosheet showed absorption bands in the UV-Vis region with absorption maxima at 387 nm and 337 nm in an aqueous solution. The intensity of the absorption maxima increased with a slight red-shift of the band to 390 nm when Fe3O4NP was deposited. A further red-shift of the absorption band in the visible region was observed when porous ruthenium was deposited on the g-C3N4/Fe3O4NP nanocomposite. This suggested that the g-C3N4/Fe3O4/p-RuNP nanocomposite can potentially act as a good photocatalyst in the visible region.
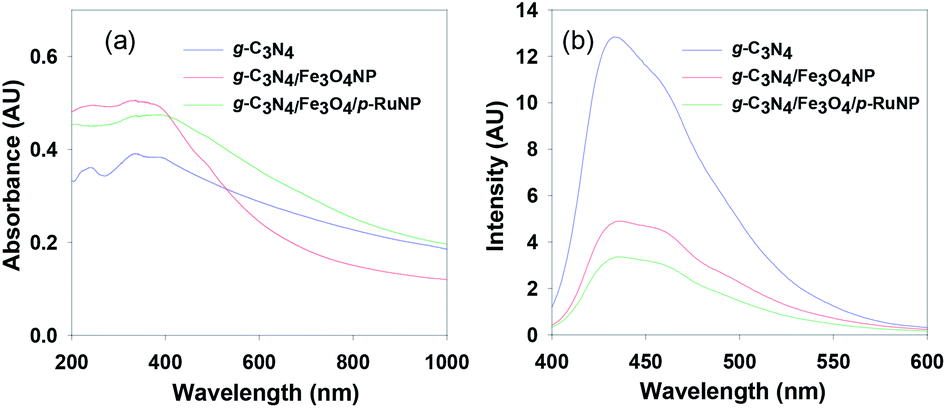 | ||
| Fig. 3 (a) Absorption and (b) emission spectra of the g-C3N4, g-C3N4/Fe3O4NP and g-C3N4/Fe3O4/p-RuNP nanocomposites recorded in water. | ||
In order to get more insights about the optical properties of the nanocomposites, a UV-Vis DRS study of the g-C3N4, g-C3N4/Fe3O4NP and g-C3N4/Fe3O4/p-RuNP nanocomposites was conducted, and all of them showed absorption maxima below 550 nm (Fig. S5†). The Tauc plot suggested that the band gaps for g-C3N4, g-C3N4/Fe3O4NP and g-C3N4/Fe3O4/p-RuNP were at ∼2.88 eV, ∼2.72 eV and ∼1.8 eV, respectively.86,87 This clearly verified the red-shifts of the band during the incorporation of Fe3O4 and RuNP and the better visible-light-harvesting capability of the g-C3N4/Fe3O4/p-RuNP nanocomposite for photocatalytic applications.
Photoluminescence (PL) spectral analysis was performed to probe the migration, transfer and recombination processes of the photo-induced electron–hole pairs in the nanocatalyst.31,39 As seen in Fig. 3b, the main emission peak was located at around 435 nm for pure g-C3N4 in water. The intensity of the photoluminescence spectrum had decreased remarkably for g-C3N4/Fe3O4NP. The addition of porous ruthenium further decreased the PL intensity, which indicated that the g-C3N4/Fe3O4/p-RuNP nanocomposite had a much lower recombination rate of the photo-induced electron–hole pairs compared to those in the pristine g-C3N4 nanosheet and g-C3N4/Fe3O4NP.41,49,61,88–92
Photocatalytic activity towards the removal of aromatic amines
We turned our attention to test the photocatalytic efficacy of the g-C3N4/Fe3O4/p-RuNP nanocomposite in the decomposition of aromatic amines as they are highly carcinogenic and toxic to humans and the environment. To check the visible-light-driven photocatalytic activity of the synthesised nanocomposites, aniline was used as the reference material. The photocatalytic decomposition of aniline (100 mg L−1) under visible-light (LED, 100 W, 8 klx) irradiation was monitored using UV-Vis spectroscopy by observing the change in absorbance at 230 nm at pH 7.0 (Fig. 4a). It was observed that all the nanocomposites (g-C3N4 nanosheet, g-C3N4/Fe3O4NP, and g-C3N4/Fe3O4/p-RuNP) were active towards photo-degradation of aniline; however, their reactivity differed significantly (Fig. 4b). The g-C3N4 nanosheet displayed very poor efficiency, whereas the g-C3N4/Fe3O4NP and g-C3N4/Fe3O4/p-RuNP nanocomposites exhibited moderate and good efficiency, respectively. This indicated the importance of the presence of ruthenium on the g-C3N4/Fe3O4NP nanocomposite, and further studies were carried out using the g-C3N4/Fe3O4/p-RuNP nanocomposite.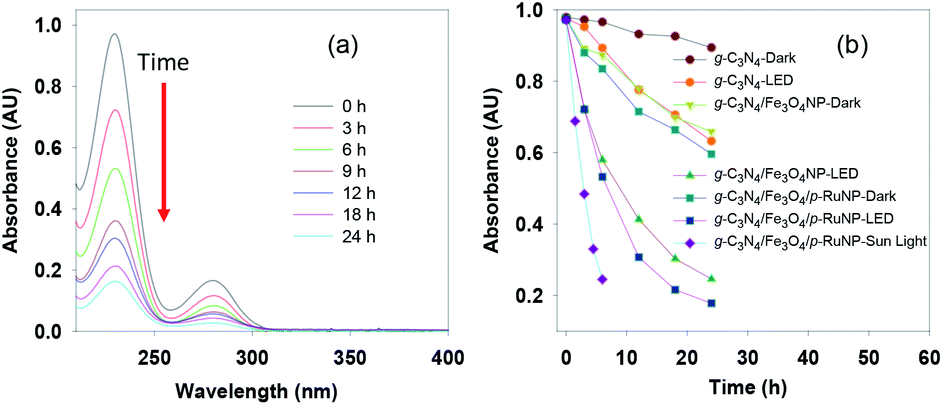 | ||
| Fig. 4 (a) Absorption spectra and (b) time course spectra of the photocatalytic degradation of aniline using the nanocomposites under LED, dark and sunlight. | ||
We then studied the effect of pH on the photo-degradation of aniline. The photo-degradation of aniline was carried out at different pH levels (5.0, 7.0 and 9.0) and was found to be the best at pH 7.0 (Fig. S6a†). It was also observed that the photodegradation of aniline depended on the amount of ruthenium present in the nanocomposite. The nanocomposite synthesised using 10 mM ruthenium (g-C3N4/Fe3O4/p-RuNP) displayed the best photocatalytic efficiency for the degradation of aniline (Fig. S6b†). The concentration of aniline was also varied (20–400 mg L−1) to check the maximum concentration of aniline that can be decomposed under light (Fig. S6c†). The optimised reaction condition for the photo-degradation of aniline (100 mg L−1) was found to be at pH 7.0 with a catalyst concentration of 80 mg L−1 (ruthenium concentration 3.44 ppm) at room temperature.
Thereafter, we explored the importance of light on the photo-degradation of aniline. The reactions were carried in the presence and absence of light (Fig. 4b). In the absence of light, the degradation of aniline was very poor, whereas, in the presence of light, it was significantly higher. This established that the nanocomposites were acting as photocatalysts. Interestingly, the photocatalytic activity of the g-C3N4/Fe3O4/p-RuNP nanocomposite was much higher than those of g-C3N4/Fe3O4NP and the g-C3N4 nanosheet, indicating the importance of the presence of ruthenium. Approximately, 30% reduction in aniline absorbance at 230 nm was observed under dark in the presence of both g-C3N4/Fe3O4NP and g-C3N4/Fe3O4/p-RuNP nanocomposites. This could be due to the sorption of aniline on the nanocomposite surface.
The photo-degradation of aniline was further conducted in the presence of sunlight, which was much faster than that under LED (Fig. 4b). This is expected as the intensity of sunlight (∼60 klx) is more than that of LED (8 klx).93,94 This further suggested that the g-C3N4/Fe3O4/p-RuNP nanocomposite is suitable for photo-degrading aromatic amines under normal environmental conditions.
Further, we extended our work to the degradation of other aromatic amines (Chart S1†). Aromatic amines, such as 2-bromoaniline, 3-chloro-4-fluoroaniline, 3-amino-4-hydroxy naphthalene-1-sulfonic acid, 2-aminophenol, and o-phenylenediamine, and the aromatic amines of reduced azo dyes (Chart S2†) were selected for photo-degradation in the presence of the g-C3N4/Fe3O4/p-RuNP nanocomposite under visible light (using LED light source). It was observed that the g-C3N4/Fe3O4/p-RuNP nanocomposite was able to efficiently photo-degrade all the aromatic amines (Chart S1†), which was confirmed by UV-Vis spectroscopy and HPLC analysis [Fig. S7† and Table 1 (entry 1–6)].
| Sr. no. | Name of the dye/amine | Concentration | Catalyst (per 10 mL) | Time of reaction | Separation efficiency%a |
|---|---|---|---|---|---|
| a The separation efficiency was calculated based on HPLC analyses. | |||||
| 1 | 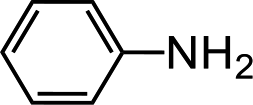 |
100 mg L−1 | 200 μL | 24 h | 76 ± 3.0 |
| 2 | 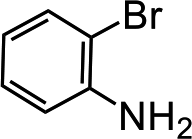 |
100 mg L−1 | 200 μL | 24 h | 96 ± 1.7 |
| 3 | 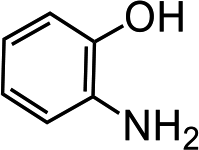 |
100 mg L−1 | 200 μL | 24 h | 99 ± 0.5 |
| 4 | 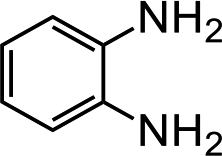 |
100 mg L−1 | 200 μL | 24 h | 99 ± 0.5 |
| 5 |  |
100 mg L−1 | 200 μL | 24 h | 83 ± 2.6 |
| 6 | 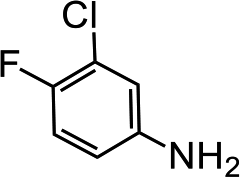 |
100 mg L−1 | 200 μL | 24 h | 87 ± 0.5 |
| 7 | Reduced CR | 200 mg L−1 | 200 μL | 24 h | 85 ± 1.9 |
| 8 | Reduced CB | 200 mg L−1 | 200 μL | 24 h | 73 ± 3.0 |
| 9 | Reduced EB | 200 mg L−1 | 200 μL | 24 h | 65 ± 1.9 |
| 10 | Reduced RR-120 | 200 mg L−1 | 200 μL | 24 h | 80 ± 1.0 |
Inspired by the above results, we extended our work to examine the photo-degradation of various coloured azo dyes as they are also known to be toxic and harmful to aquatic life and humans. Initially, the degradation efficiency of the g-C3N4/Fe3O4/p-RuNP nanocomposite was tested using the Congo red (CR) dye. The nanocomposite was tested with a 5 mg L−1 solution of CR under LED irradiation. Excitingly, within 2 h, the colour of the dye solution was bleached, indicating the photocatalytic performance of the nanocomposite (Fig. 5a). Higher concentrations of the dye (10–100 mg L−1) were then examined for photo-degradation (Fig. S8†). It was observed that up to 10 mg L−1 dye was degraded efficiently by the photocatalyst. However, at higher concentrations, the photocatalytic efficacy of the nanocomposite for the photo-degradation of CR reduced significantly (Fig. S8†). This was indeed expected due to the absorption of visible light by the dye molecules in solution, which reduces the photocatalytic activity of the g-C3N4/Fe3O4/p-RuNP nanocomposite. It is noteworthy that, although the nano-photocatalyst was inactive at higher concentrations, it could efficiently photo-degrade the repeatedly added CR dye (at least 5 times using 5 mg L−1 each time) (Fig. 5b). This suggested the excellent photocatalytic activity of the g-C3N4/Fe3O4/p-RuNP nanocomposite. We then extended this for the photo-degradation of other commercially used common azo dyes, such as Evans blue (EB), Chlorazol black (CB) and Reactive red 120 (RR-120). The g-C3N4/Fe3O4/p-RuNP nanocatalyst was found to efficiently photo-degrade the aforesaid azo dyes, but at different rates (Chart S2, Fig. 5a, S9 and Table T1†).
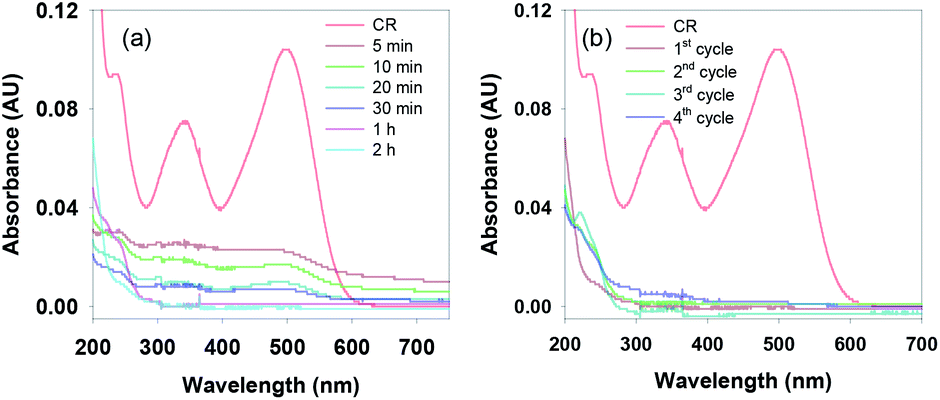 | ||
| Fig. 5 (a) UV-Vis spectra of the reduction of CR (5 mg L−1) in the presence of g-C3N4/Fe3O4/p-RuNP under LED with varying time, and (b) UV-Vis spectra of CR after the completion of each cycle. | ||
To check whether the nanocatalyst photo-degraded CR and its corresponding amines completely, HPLC analysis and FTIR spectral studies of the decomposed CR dye were carried out (Fig. S10 and S11†). No trace of azo dye and its corresponding amines were detected in HPLC (Fig. S10†). FTIR spectroscopy also revealed no peak for aromatic amine groups in the degraded solution of CR (Fig. S11†). The HPLC analyses of the photo-degraded products of other dyes also showed similar results to CR (Fig. S10†). These suggested that the nanocatalyst could eliminate azo dyes and its corresponding amines completely or convert them to less-toxic end products.
Although the nanocatalyst could not completely photo-degrade dyes above 20 mg L−1, it easily degraded and eliminated the reduced products (aromatic amines) of CR (200 mg L−1) under visible light (Table 1). The HPLC analyses of the azo dyes (200 mg L−1) before and after photo-degradation are displayed in Fig. S12,† and the removal efficiencies are is listed in Table 1 (entries 7–10). The photo-degradation of aromatic amines by the g-C3N4/Fe3O4/p-RuNP nanocomposite has already been observed. Thus, it may be envisioned that the present nanocatalytic system can provide a unique way of solving the problems associated with water-soluble aromatic amines and coloured azo-based dyes. The present nanocatalytic system does not require any external chemicals for eliminating the aromatic amines or azo dyes. This, in turn, reduces the cost, as well as the generation of solid waste and chemical oxygen demand, which is suitable for industrial applications.
Reusability
One of the major advantages of the g-C3N4/Fe3O4/p-RuNP nanocomposite is its ability to be separated and reused. Because of the incorporated magnetic property, the nanocomposite can be retrieved magnetically from the reaction mixture and reused. To study the stability and reusability, the synthesised g-C3N4/Fe3O4/p-RuNP nanocatalyst was separated magnetically after the photo-degradation of aromatic amines under visible light and reused for the next cycle. It was observed that the nanocomposite could photo-degrade aniline without losing its efficiency significantly (Fig. S13†). A ∼5% loss in efficiency was observed after 5 cycles. This observation verified that the hypothesis of combining the photocatalytic and magnetic behaviours of nanoparticles into the same framework to get an efficient separable catalytic system had worked out nicely.Several homogeneous and heterogeneous photocatalytic/catalytic systems are known. However, the facile separation aspect of the nanophotocatalyst after the photocatalytic reaction has been rarely explored. This report provides a simpler way for separating the nanophotocatalyst just by using an external magnet and at the same time, offers a greener way for the facile photo-degradation of aromatic amines and azo dyes at room temperature without using any additional chemicals at neutral pH. Thus, this nanocatalytic system has huge potential in eliminating the problems associated with azo dyes and aromatic amines.
Mechanism
Next, we studied the mechanism of the photodegradation of aromatic amines and azo dyes. The photodegradation process generally takes place through reactive oxygen species. To confirm the involvement of reactive oxygen species, the photo-degradation of CR was conducted in the presence and absence of dissolved oxygen. A clear difference in the reactivity was observed, which confirmed the major role of oxygen in the photo-degradation of CR (Fig. S14†). To investigate the type of reactive oxygen species involved in the photo-degradation of CR in the presence of the g-C3N4/Fe3O4/p-RuNP nanocomposite, different types of radical scavengers, namely sodium azide (NaN3) (for OH˙ and singlet oxygen),95,96 t-BuOH (for OH˙),69,97 ethylenediaminetetraacetate (EDTA) and formic acid (FA) (for h+),29,98 potassium iodide (KI) (for h+),98,99 ascorbic acid (AA) (for O2˙−),99 were used. The photocatalytic efficiency of the g-C3N4/Fe3O4/p-RuNP nanocomposite significantly reduced when t-BuOH and NaN3 were used, whereas it was not significant with EDTA, AA, FA and KI. This suggested that the g-C3N4/Fe3O4/p-RuNP nanocomposite induced the generation of OH˙ in the presence of photons. The holes (h+) or the oxygen radicals (O2˙−) did not have much impact on the photodegradation of CR. The moderate decrease in the photocatalytic efficiency in the presence of an N2 atmosphere suggested that the impact of oxygen radicals due to atmospheric O2 was less compared to that of the OH˙ radicals. Thus, combining the results a plausible mechanism could be drawn as follows. In the presence of light, the g-C3N4/Fe3O4/p-RuNP nanocomposite is photo-excited and generates electron–hole pairs. The excited electrons then react with dissolved/aerial oxygen and generate the reactive oxygen species (OH˙), which further reacts with the aromatic amines and decompose them into gaseous end products and intermediates (Fig. 6).100–104 The formation of polyamines from aromatic amines in the presence of photogenerated OH˙ radicals and the photocatalyst cannot be ruled out as well.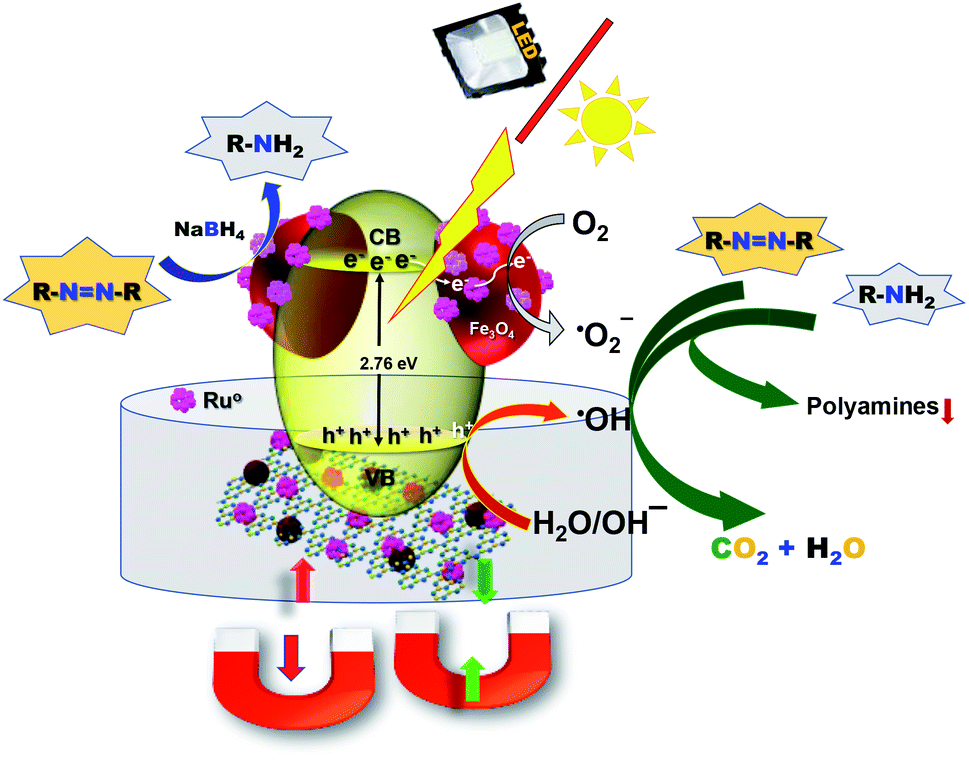 | ||
| Fig. 6 Schematic representation of the degradation mechanism of aromatic amines and azo dyes under visible light in the presence of the g-C3N4/Fe3O4/p-RuNP nanocomposite. | ||
Conclusions
In conclusion, we have developed a magnetically retrievable and reusable ruthenium-based g-C3N4/Fe3O4/p-RuNP nanocomposite. The nanocomposite efficiently photo-degrades aromatic amines and coloured azo dyes into gaseous/less toxic end-products in aqueous solutions without using any external chemicals or additives. The nanocomposite also offers a facile separation process by using a magnet, so that it can be reused after the completion of the reaction. Nanocatalytic systems that exhibit facile and greener ways for the elimination of toxic azo dyes and aromatic amines from aqueous solutions are rare. Thus, we envision that this nanocatalytic system presents the scope to eliminate toxic organics and offers a green and sustainable way for the treatment of industrial wastewater.Experimental methods
Materials
Urea (Spectrochem, 99.0%), ferric sulphate (Aldrich), ferrous sulphate (Aldrich), NH4OH solution (Finar, 30% v/v), aniline, 2-bromoaniline, 3-chloro-4-fluoroaniline, 3-amino-4-hydroxy naphthalene-1-sulfonic acid, 2-aminophenol, o-phenylenediamine, Congo red (CR), Reactive red 120 (RR-120) (Spectrochem), Chlorazol black (CB), and Evans blue (EB) (Aldrich) were used as received. All the glassware was washed with aqua regia and rinsed with triple distilled water before use.Instrumentation
Powder XRD studies were carried out on a Bruker D8 Advance diffractometer using Cu Kα radiation. The FTIR spectra were recorded in the range from 4000 to 500 cm−1 using a Bruker Alpha FTIR spectrometer with KBr pellets. The UV-Vis spectra were recorded on a PerkinElmer Lambda 35 UV-Vis spectrophotometer. UV-Vis DRS analyses were carried out using a Shimadzu UV-2600 spectrophotometer. FE-SEM images and EDS analyses were carried out using a Zeiss Merlin Compact Microscope and Oxford instruments, respectively. A Dynamica Velocity 18R refrigerated centrifuge was used for the separation of nanoparticles. An Agilent technologies Prostar HPLC instrument was used for the qualitative and quantitative estimation of aromatic amines. The HRTEM images were recorded using a JEOL JEM 2100 electron microscope. The nitrogen adsorption–desorption isotherms were obtained using a Quadrasorb SI (Quantachrome Instruments) BET surface area analyser. The fluorescence spectra of the photocatalysts were recorded on a Fluorimax fluorescence spectrophotometer at an excitation wavelength of 365 nm.HPLC analysis
The efficiency of the nanocatalyst in removing aromatic amines and azo-based dyes was monitored by HPLC analysis using a C-18 reverse-phase chromatographic column and methanol/water (1:1) as the mobile phase. The flow rate of the mobile phase was maintained at 0.2 mL min−1 at 25 °C and pH 7.0. The samples were filtered through 0.2 μm filter papers before being injected into the column. The injected volume of each sample was 20 μL. The wavelengths used for the different dyes and aromatic amines were 270 nm (for CR, CB, and EB), 258 nm (for RR-120), 230 nm (for aniline, 3-chloro-4-fluoroaniline, 2-bromoaniline, 2-aminophenol and o-phenylenediamine), and 235 nm (for 4-amino-3-hydroxy-1-naphthalenesulfonic acid).
Preparation of samples for ICP AES analysis
The sample preparation was carried out by following a procedure reported elsewhere.105 Briefly, 100 μL of g-C3N4/Fe3O4/p-RuNP was dissolved in a 900 μL solution of HCl, HNO3, and HF in the ratio of 6:1:1. After complete dissolution, the solution was diluted to 10 mL. The ICP AES analysis was carried out with this sample using an ARCOS Simultaneous ICP spectrometer.
Synthesis of catalysts
g-C3N4 was synthesised by heating urea in a furnace at 520 °C for 2.5 h. g-C3N4/Fe3O4NP was prepared by the deposition precipitation method.61 Briefly, g-C3N4 nanosheets (125 mg) were added to 500 mL of ethanol/water (1:2) mixture and dispersed well by sonicating for 2 h at room temperature. FeSO4·7H2O (1.84 g, 0.022 mol) and Fe2(SO4)3·xH2O (0.70 g, 0.011 mol) were dissolved in 20 mL of distilled water and added to the dispersed g-C3N4 nanosheet solution. The reaction mixture was stirred at 60 °C for 30 min, and then, 10 mL of NH4OH was added quickly to it. The resulting solution was stirred for an additional 1 h, after which it was cooled to room temperature and magnetically separated. The nanocomposite was washed several times with triple-distilled water. Porous ruthenium was deposited on g-C3N4/Fe3O4NP by the galvanic displacement of CoNPs to obtain g-C3N4/Fe3O4/p-RuNP (Scheme 1).5,13 To 20 mL of the stirred solution of g-C3N4/Fe3O4NP, 15 mM CoCl2·6H2O was added and deaerated. N2 was purged throughout the reaction. NaBH4 was added to the solution for the reduction of Co2+ to Co0 on g-C3N4/Fe3O4NP. After complete decomposition of NaBH4, 10 mM of RuCl3·xH2O was added to the solution for ruthenium to be deposited on g-C3N4/Fe3O4NP in a porous manner. The solution was stirred for an additional 6 h and centrifuged to remove the oxidized Co2+ ions and redispersed in 20 mL distilled water. This solution was used for further studies.
Reduction of azo dyes into aromatic amines
The azo dyes (CR, RR-120, CB, and EB) were reduced using NaBH4 by following the previously reported procedure using g-C3N4/Fe3O4/p-RuNP to obtain the corresponding aromatic amines, which were used for further photodegradation.5Photocatalytic activity towards aromatic amines
The photocatalytic activity of the nanocomposites was evaluated by the degradation of an aqueous solution of aniline under visible light. The visible light source was a 100 W LED light (OREVA Led lighting, ORFLD-100W-SLIM, 10 klx). To an aqueous solution of aniline (10 mL, 100 mg L−1), 200 μL of the photocatalyst (80 mg L−1; ruthenium loading 3.44 ppm) was added and kept under visible light irradiation at room temperature. During this process, the sample was collected periodically, and the catalyst was magnetically separated for recording the absorbance of the solution. The absorbance measurements were recorded using a UV-Vis spectrophotometer over a period of 24 h. A similar procedure was adopted for the other aromatic amines (Chart S1†), such as 2-bromoaniline, 3-chloro-4-fluoroaniline, 3-amino-4-hydroxynaphthalene-1-sulfonic acid, 2-aminophenol, and o-phenylenediamine, and the aromatic amines of reduced azo dyes (Chart S2†) to study their photo-degradation in the presence of the g-C3N4/Fe3O4/p-RuNP nanocomposite.Photocatalytic activity towards azo dyes
The photocatalytic activity of the catalyst towards azo dyes was examined by taking 5 mg L−1 of the dye solution under LED irradiation in the presence of 80 mg L−1 of the g-C3N4/Fe3O4/p-RuNP nanocomposite. The decrease in the absorbance of the degraded azo dye solutions was monitored by UV-Vis spectroscopy. The samples were collected periodically for absorbance measurements. After the complete degradation of the dyes, the solutions were filtered using 2 μm filter papers, and HPLC analysis of the degraded solutions was performed.Mechanism studies
To investigate the role of radicals involved in the photocatalytic degradation of dyes, reactions were carried out under light in the presence of g-C3N4/Fe3O4/p-RuNP (80 mg L−1) with different types of radical scavengers (1% w/v solution), including NaN3 (for 1O2), t-BuOH (for OH˙), EDTA (for h+), potassium iodide (KI) (for h+), ascorbic acid (AA) (for O2˙−), and formic acid (FA) (for O2˙−). The concentration of the dye (CR) was 5 mg L−1. A 10 mL solution containing both the dye and radical scavengers was kept under LED light, and the nanocatalyst (80 mg L−1) was added. The change in the absorbance of the dye solution was monitored by recording UV-Vis spectra after 30 min, 60 min, and 120 min at 498 nm.Reusability studies
To check the reusability, the g-C3N4/Fe3O4/p-RuNP nanocatalyst was magnetically separated from the reaction mixture after 24 h using an external magnet. The solution was removed by decantation. The catalyst was washed with fresh distilled water and again magnetically separated. A fresh solution of aromatic amine was added and monitored under a light with stirring. This process was repeated after each catalytic cycle.Conflicts of interest
This work is submitted for the Indian Patent Application (File Number: TEMP/E-1/951/2019-KOL; Reference No. Application Number: 201931000965).Acknowledgements
The authors would like to acknowledge IIT Bhubaneswar for financial supports. AS is grateful to DST, India for providing Inspire Research Fellowship.Notes and references
- P. Vineis and R. Pirastu, Cancer Causes Control, 1997, 8, 346–355 CrossRef CAS PubMed.
- E. Kriek, Biochim. Biophys. Acta, Rev. Cancer, 1974, 355, 177–203 CrossRef CAS.
- P. L. Skipper, M. Y. Kim, H. L. Patty Sun, G. N. Wogan and S. R. Tannenbaum, Carcinogenesis, 2009, 31, 50–58 CrossRef PubMed.
- D. W. Hein, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2002, 506–507, 65–77 CrossRef CAS.
- A. Sahoo and S. Patra, ACS Appl. Nano Mater., 2018, 1, 5169–5178 CrossRef CAS.
- I. Casero, D. Sicilia, S. Rubio and D. Pérez-Bendito, Water Res., 1997, 31, 1985–1995 CrossRef CAS.
- H. M. Pinheiro, E. Touraud and O. Thomas, Dyes Pigm., 2004, 61, 121–139 CrossRef CAS.
- K. Singh and S. Arora, Crit. Rev. Environ. Sci. Technol., 2011, 41, 807–878 CrossRef CAS.
- J.-S. Bae, H. S. Freeman and S. D. Kim, Fibers Polym., 2006, 7, 30–35 CrossRef CAS.
- H. U. Kaefferlein, A. Slowicki and T. Bruening, Gefahrstoffe–Reinhalt. Luft, 2009, 69, 423–430 CAS.
- T. M. Fonovich, Drug Chem. Toxicol., 2013, 36, 343–352 CrossRef CAS PubMed.
- J. L. Parrott, A. J. Bartlett and V. K. Balakrishnan, Environ. Pollut., 2016, 210, 40–47 CrossRef CAS PubMed.
- A. Sahoo, S. K. Tripathy, N. Dehury and S. Patra, J. Mater. Chem. A, 2015, 3, 19376–19383 RSC.
- Z. Karim and Q. Husain, Int. Biodeterior. Biodegrad., 2009, 63, 587–593 CrossRef CAS.
- S. Wada, H. Ichikawa and K. Tastsumi, Biotechnol. Bioeng., 1995, 45, 304–309 CrossRef CAS PubMed.
- V. López-Grimau, M. Riera-Torres, M. López-Mesas and C. Gutiérrez-Bouzán, Color. Technol., 2013, 129, 267–273 Search PubMed.
- E. González-Pradas, A. Valverde-garcía and M. Villafranca-sánchez, J. Chem. Technol. Biotechnol., 2007, 47, 15–22 CrossRef.
- Q. Husain and U. Jan, J. Sci. Ind. Res., 2000, 59, 286–293 CAS.
- E. Yilmaz, S. Memon and M. Yilmaz, J. Hazard. Mater., 2010, 174, 592–597 CrossRef CAS PubMed.
- A. Volkov, G. Tourillon, P. C. Lacaze and J. E. Dubois, J. Electroanal. Chem., 1980, 115, 279–291 CrossRef CAS.
- E. Akceylan, M. Bahadir and M. Yılmaz, J. Hazard. Mater., 2009, 162, 960–966 CrossRef CAS PubMed.
- A. Pirkarami and M. E. Olya, J. Saudi Chem. Soc., 2017, 21, S179–S186 CrossRef CAS.
- R. G. Saratale, G. D. Saratale, J. S. Chang and S. P. Govindwar, J. Taiwan Inst. Chem. Eng., 2011, 42, 138–157 CrossRef CAS.
- E. Forgacs, T. Cserháti and G. Oros, Environ. Int., 2004, 30, 953–971 CrossRef CAS.
- E. Hosseini Koupaie, M. R. Alavi Moghaddam and S. H. Hashemi, J. Hazard. Mater., 2011, 195, 147–154 CrossRef CAS.
- A. Dolbecq, P. Mialane, B. Keita and L. Nadjo, J. Mater. Chem., 2012, 22, 24509 RSC.
- A. Anshuman, S. Saremi-Yarahmadi and B. Vaidhyanathan, RSC Adv., 2018, 8, 7709–7715 RSC.
- J. Yu, S. Wang, J. Low and W. Xiao, PCCP Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- X. Bai, L. Wang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2013, 117, 9952–9961 CrossRef CAS.
- T. Ma, J. Bai, Q. Wang and C. Li, Dalton Trans., 2018, 47, 10240–10248 RSC.
- A. Nikokavoura and C. Trapalis, Appl. Surf. Sci., 2018, 430, 18–52 CrossRef CAS.
- M. Aleksandrzak, W. Kukulka and E. Mijowska, Appl. Surf. Sci., 2017, 398, 56–62 CrossRef CAS.
- N. Ullah, S. Chen and R. Zhang, Appl. Surf. Sci., 2019, 487, 151 CrossRef CAS.
- R. Wang, H. Li, L. Zhang, Y.-J. Zeng, Z. Lv, J.-Q. Yang, J.-Y. Mao, Z. Wang, Y. Zhou and S.-T. Han, J. Mater. Chem. C, 2019, 7, 10203–10210 RSC.
- D. Yuan, W. Huang, X. Chen, Z. Li, J. Ding, L. Wang, H. Wan, W.-L. Dai and G. Guan, Appl. Surf. Sci., 2019, 489, 658–667 CrossRef CAS.
- J. Xu, Z. Wang and Y. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 27727–27735 CrossRef CAS PubMed.
- X. Yuan, C. Zhou, Y. Jin, Q. Jing, Y. Yang, X. Shen, Q. Tang, Y. Mu and A.-K. Du, J. Colloid Interface Sci., 2016, 468, 211–219 CrossRef CAS PubMed.
- H. Yan, Chem. Commun., 2012, 48, 3430 RSC.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- S. Kumar, S. Karthikeyan and A. Lee, Catalysts, 2018, 8, 74 CrossRef.
- S. Patnaik, S. Martha and K. M. Parida, RSC Adv., 2016, 6, 46929–46951 RSC.
- L. Wang, Y. Zhu, D. Yang, L. Zhao, H. Ding and Z. Wang, Appl. Surf. Sci., 2019, 488, 728 CrossRef CAS.
- D. Masih, Y. Ma and S. Rohani, Can. J. Chem. Eng., 2019, 97, 2632–2641 CrossRef CAS.
- R. Ma, S. Zhang, L. Li, P. Gu, T. Wen, A. Khan, S. Li, B. Li, S. Wang and X. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9699–9708 CrossRef CAS.
- R. Dadigala, R. Bandi, B. R. Gangapuram and V. Guttena, Nanoscale Adv., 2019, 1, 322–333 RSC.
- C. G. Liu, X. T. Wu, X. F. Li and X. G. Zhang, RSC Adv., 2014, 4, 62492–62498 RSC.
- S. L. Prabavathi, K. Govindan, K. Saravanakumar, A. Jang and V. Muthuraj, J. Ind. Eng. Chem., 2019, 80, 558 CrossRef CAS.
- H. Wang, Z. Lei, L. Li and X. Wang, Sol. Energy, 2019, 191, 70–77 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Dalton Trans., 2015, 44, 1249–1257 RSC.
- S. Iqbal, S. A. Kondrat, D. R. Jones, D. C. Schoenmakers, J. K. Edwards, L. Lu, B. R. Yeo, P. P. Wells, E. K. Gibson, D. J. Morgan, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 5047–5059 CrossRef CAS.
- Q. Yao, Z.-H. Lu, K. Yang, X. Chen and M. Zhu, Sci. Rep., 2015, 5, 15186 CrossRef CAS PubMed.
- J. Wang, Z. Wei, S. Mao, H. Li and Y. Wang, Energy Environ. Sci., 2018, 11, 800–806 RSC.
- J. Mondal, S. K. Kundu, W. K. Hung Ng, R. Singuru, P. Borah, H. Hirao, Y. Zhao and A. Bhaumik, Chem.–Eur. J., 2015, 21, 19016–19027 CrossRef CAS PubMed.
- R. B. Nasir Baig and R. S. Varma, ACS Sustainable Chem. Eng., 2013, 1, 805–809 CrossRef CAS.
- T. Umegaki, Y. Enomoto and Y. Kojima, Catal. Sci. Technol., 2016, 6, 409–412 RSC.
- S. Mondal, R. Singuru, S. Chandra Shit, T. Hayashi, S. Irle, Y. Hijikata, J. Mondal and A. Bhaumik, ACS Sustainable Chem. Eng., 2018, 6, 1610–1619 CrossRef CAS.
- X. Liu, K. X. Yao, C. Meng and Y. Han, Dalton Trans., 2012, 41, 1289–1296 RSC.
- Y. Wang, Y. Zhang, H. Sha, X. Xiong and N. Jia, ACS Appl. Mater. Interfaces, 2019, 11(40), 36299 CrossRef CAS PubMed.
- S. P. Gubin, Y. A. Koksharov, G. B. Khomutov and G. Y. Yurkov, Russ. Chem. Rev., 2005, 74, 489–520 CrossRef CAS.
- S. Chi, C. Ji, S. Sun, H. Jiang, R. Qu and C. Sun, Ind. Eng. Chem. Res., 2016, 55, 12060–12067 CrossRef CAS.
- S. Kumar, T. Surendar, B. Kumar, A. Baruah and V. Shanker, J. Phys. Chem. C, 2013, 117, 26135–26143 CrossRef CAS.
- Y. Yang, W. Zhang, F. Yang, B. Zhou, D. Zeng, N. Zhang, G. Zhao, S. Hao and X. Zhang, Nanoscale, 2018, 10, 2199–2206 RSC.
- J. Gao, H. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097–1107 CrossRef CAS PubMed.
- C. Prasad, H. Tang and W. Liu, J. Nanostruct. Chem., 2018, 8, 393–412 CrossRef CAS.
- F. Gao, ChemistrySelect, 2019, 4, 6805–6811 CrossRef CAS.
- A. Moslehi and M. Zarei, New J. Chem., 2019, 43, 12690–12697 RSC.
- P. Zhang, Y. Wang, Y. Zhou, H. Zhang, X. Wei, W. Sun, S. Meng and L. Han, Mol. Catal., 2019, 465, 24–32 CrossRef CAS.
- I. Cano, C. Martin, J. A. Fernandes, R. W. Lodge, J. Dupont, F. A. Casado-Carmona, R. Lucena, S. Cardenas, V. Sans and I. de Pedro, Appl. Catal., B, 2020, 260, 118110 CrossRef CAS.
- S. Kumar, T. Surendar, B. Kumar, A. Baruah and V. Shanker, J. Phys. Chem. C, 2013, 117, 26135–26143 CrossRef CAS.
- D. Xiao, K. Dai, Y. Qu, Y. Yin and H. Chen, Appl. Surf. Sci., 2015, 358, 181–187 CrossRef CAS.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- H. Hu, H. Yang, P. Huang, D. Cui, Y. Peng, J. Zhang, F. Lu, J. Lian and D. Shi, Chem. Commun., 2010, 46, 3866 RSC.
- B. Andrzejewski, W. Bednarski, M. Kaźmierczak, A. Łapiński, K. Pogorzelec-Glaser, B. Hilczer, S. Jurga, G. Nowaczyk, K. Załęski, M. Matczak, B. Łęska, R. Pankiewicz and L. Kępiński, Composites, Part B, 2014, 64, 147–154 CrossRef CAS.
- W. Qian, Z. Chen, S. Cottingham, W. A. Merrill, N. A. Swartz, A. M. Goforth, T. L. Clare and J. Jiao, Green Chem., 2012, 14, 371–377 RSC.
- F. D. Wu and Y. Wang, J. Mater. Chem., 2011, 21, 6636 RSC.
- G. Li, H. Nagasawa, M. Kanezashi, T. Yoshioka and T. Tsuru, J. Mater. Chem. A, 2014, 2, 9185–9192 RSC.
- S. Agarwal and J. N. Ganguli, RSC Adv., 2014, 4, 11893 RSC.
- A. Lewera, W. P. Zhou, C. Vericat, J. H. Chung, R. Haasch, A. Wieckowski and P. S. Bagus, Electrochim. Acta, 2006, 51, 3950–3956 CrossRef CAS.
- M. Zahmakiran and S. Zkar, Langmuir, 2009, 25, 2667–2678 CrossRef CAS PubMed.
- Q. He, B. Shyam, M. Nishijima, X. Yang, B. Koel, F. Ernst, D. Ramaker and S. Mukerjee, J. Phys. Chem. C, 2013, 117, 1457–1467 CrossRef CAS.
- O. V Manaenkov, J. J. Mann, O. V Kislitza, Y. Losovyj, B. D. Stein, D. Gene Morgan, M. Pink, O. L. Lependina, Z. B. Shifrina, V. G. Matveeva, E. M. Sulman and L. M. Bronstein, ACS Appl. Mater. Interfaces, 2016, 8, 21285 CrossRef PubMed.
- L. Wang, J. Luo, Q. Fan, M. Suzuki, I. S. Suzuki, M. H. Engelhard, Y. Lin, N. Kim, J. Q. Wang and C. J. Zhong, J. Phys. Chem. B, 2005, 109, 21593–21601 CrossRef CAS.
- Y. Li, F. Li, X. Wang, J. Zhao, J. Wei, Y. Hao and Y. Liu, Int. J. Hydrogen Energy, 2017, 42, 28327–28336 CrossRef CAS.
- Y. Wang, Y. Shen, A. Xie, S. Li, X. Wang and Y. Cai, J. Phys. Chem. C, 2010, 114, 4297–4301 CrossRef CAS.
- F. Chen, Q. Chen, S. Fang, Y. Sun, Z. Chen, G. Xie and Y. Du, Dalton Trans., 2011, 40, 10857–10864 RSC.
- M. Mousavi and A. Habibi-Yangjeh, J. Colloid Interface Sci., 2016, 465, 83–92 CrossRef CAS PubMed.
- P. Sharma and Y. Sasson, Green Chem., 2017, 19, 844–852 RSC.
- X. Yue, S. Yi, R. Wang, Z. Zhang and S. Qiu, Sci. Rep., 2016, 6, 22268 CrossRef CAS PubMed.
- Z. Wu, X. Chen, X. Liu, X. Yang and Y. Yang, Nanoscale Res. Lett., 2019, 14, 147 CrossRef PubMed.
- G. Jiang, K. Geng, Y. Wu, Y. Han and X. Shen, Appl. Catal., B, 2018, 227, 366–375 CrossRef CAS.
- A. Kumar, M. Khan, X. Zeng and I. M. C. Lo, Chem. Eng. J., 2018, 353, 645–656 CrossRef CAS.
- X. N. Wei and H. L. Wang, J. Alloys Compd., 2018, 763, 844–853 CrossRef CAS.
- P. L. Turner and M. A. Mainster, Br. J. Ophthalmol., 2008, 92, 1439–1444 CrossRef CAS PubMed.
- M. Spitschan, G. K. Aguirre, D. H. Brainard and A. M. Sweeney, Sci. Rep., 2016, 6, 26756 CrossRef CAS PubMed.
- S. Leichnitz, J. Heinrich and N. Kulak, Chem. Commun., 2018, 54, 13411–13414 RSC.
- A. Sreedhara, A. J. D. Freed and J. A. Cowan, J. Am. Chem. Soc., 2000, 122, 8814–8824 CrossRef CAS.
- M. Von Piechowski, M.-A. Thelen, J. Hoigné and R. E. Bühler, Bunsen-Ges. Phys. Chem., Ber., 1992, 96, 1448–1454 CrossRef CAS.
- A. A.-W. Wei Deng, H. Zhao, F. Pan, X. Feng, B. Jung and Y. L. Bill Batchelor, Environ. Sci. Technol., 2017, 51, 13372–13379 CrossRef PubMed.
- R. Palominos, J. Freer, M. A. Mondaca and H. D. Mansilla, J. Photochem. Photobiol., A, 2008, 193, 139–145 CrossRef CAS.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- M. L. Canle, J. A. Santaballa and E. Vulliet, J. Photochem. Photobiol., A, 2005, 175, 192–200 CrossRef.
- G. Liu, S. Liao, D. Zhu, L. Liu, D. Cheng and H. Zhou, Mater. Res. Bull., 2011, 46, 1290–1295 CrossRef CAS.
- X. Li, D. Shao, H. Xu, W. Lv and W. Yan, Chem. Eng. J., 2016, 285, 1–10 CrossRef CAS.
- A. Alshehri, M. A. Malik, Z. Khan, S. A. Al-Thabaiti and N. Hasan, RSC Adv., 2017, 7, 25149–25159 RSC.
- A. V. Shaverina, A. R. Tsygankova and A. I. Saprykin, J. Anal. Chem., 2015, 70, 28–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08631e |
| This journal is © The Royal Society of Chemistry 2020 |