
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of H2S/NO-donating artemisinin derivatives as potential antileukemic agents†
Xuemei Chena,
Pei Huang b,
Jing Wangcd,
Runmei Tianb,
Yan Chen*b,
Yongzheng Chencd,
Lei Zhang*cd and
Zhigui Ma*a
b,
Jing Wangcd,
Runmei Tianb,
Yan Chen*b,
Yongzheng Chencd,
Lei Zhang*cd and
Zhigui Ma*a
aDepartment of Pediatric Hematology, West China Second University Hospital, Sichuan University, Chengdu 610041, PR China. E-mail: zhiguima@scu.edu.cn
bDepartment of Pediatrics, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, PR China. E-mail: cyz600@163.com
cKey Laboratory of Biocatalysis & Chiral Drug Synthesis of Guizhou Province, School of Pharmacy, Zunyi Medical University, Zunyi 563003, PR China. E-mail: lzhang@zmu.edu.cn
dKey Laboratory of Basic Pharmacology of Ministry of Education, Joint International Research Laboratory of Ethnomedicine of Ministry of Education, Zunyi Medical University, Zunyi 563003, PR China
First published on 2nd January 2020
Abstract
Three H2S/NO-donating artemisinin derivatives were designed and synthesized. Their antiproliferative activities were evaluated against human acute myeloid leukemia (AML) cell lines of K562 and K562/ADR and human normal liver cells of LO2. Biological evaluation indicated that NO-donating compound 10c exhibited the most potent cytotoxicity against leukemia cells, similar to the bioactivity of clinical drug of homoharringtonine, but showed less toxicity than homoharringtonine against LO2 cells. Further mechanism studies revealed that 10c could enhance the levels of intracellular NO and ROS, induce apoptosis and S phase cell cycle arrest, and disturb the mitochondrial membrane potential in K562 and K562/ADR cells. Western blot results demonstrated that 10c noticeably promoted autophagy by up-regulating the levels of Beclin1 and L3-II expression, inhibited the AKT signaling, and stimulated the AMPK and JNK signaling in both leukemia cell lines. Overall, 10c exhibited the potential to be a promising candidate for the therapy of AML.
Introduction
Leukemia, a cancer of the blood or bone marrow, is the ninth most common cancer in the United States.1 Leukemia can be clinically classified into four major kinds: acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), acute myeloid leukemia (AML), and chronic myeloid leukemia (CML). AML is the most common type of acute leukemia in adults. There are several possible treatments for AML, such as allogeneic bone marrow transplantation, which involves high-dose chemotherapy and radiation, as well as a high risk of death. Chemotherapy is another countermeasure for newly diagnosed patients with AML, such as cytotoxic drugs (adriamycin and homoharringtonine). However, the resistance to chemotherapy drugs has developed in a significant portion of AML patients.2 Therefore, it is necessary to develop a novel strategy or drug for treatment of AML.3Artemisinin (1, Fig. 1), a naturally occurring sesquiterpene lactone isolated mainly from Artemisia annua L., had been used as a traditional Chinese medicine for treating malaria hundreds years ago.4 Nowadays, numerous derivatives of artemisinin have been synthesized, and several artemisinin derivatives with less toxicity, improved solubility and enhanced antimalarial activity, such as dihydroartemisinin (2, Fig. 1) and artesunate (3, Fig. 1), have been used as first-line antimalarial drugs widely. Besides, artemisinin and its derivatives have been shown to exhibit significantly antineoplastic activities in vitro and in vivo.5 Furthermore, they showed antiproliferative activities towards various human cancer cells, such as U937, NB-4, PC-3, MDA-MB-231 and MCF-7.6 Particularly, dihydroartemisinin and artesunate were promising candidates for leukemia treatment due to its potential anticancer activity in leukemic cell lines and murine models.7,8 Recently, our group also reported that artesunate and its derivative displayed anticancer activity against leukemia cells in vitro and in vivo.9,10 In addition, some studies have shown that artemisinin and its derivatives could be used as sensitizers to improve the cytotoxicity of other chemotherapeutic drugs.11,12 However, up to now, there were no artemisinin derivatives or artemisinin derivatives in combination chemotherapy had entered into clinical trials for the treatment of leukemia.
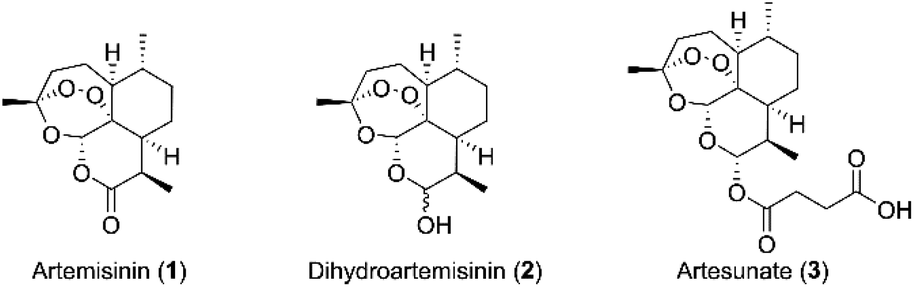 | ||
| Fig. 1 The structures of artemisinin and its derivatives. | ||
Hydrogen sulfide (H2S), nitric oxide (NO) and carbon monoxide (CO), are increasingly recognized as important endogenous mediators of diverse physiological processes, such as vasodilation and neuromodulation.13 H2S was reported to regulate protein kinases such as p38 mitogen-activated protein kinase and trigger cell apoptosis at concentration of 20 μM.14 There are several kinds of donor molecules which can release H2S under physiological conditions, such as inorganic salts (NaHS and Na2S). The drawback in using salts as H2S donors is the uncontrolled release profile. Therefore, many small organic compounds have been designed and synthetized for releasing H2S via endogenous enzymatic and/or non-enzymatic pathways, including 5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione (ADT-OH, 4, Fig. 2) and 4-hydroxy benzothiazamide (TBZ, 5, Fig. 2).15 Some reports had pointed out that H2S donors also displayed antitumor effects.16 Meanwhile, a number of synthetic H2S releasing molecules have been shown to exhibit anticancer activities in vitro and in vivo.17 Moreover, NO is a renowned gasotransmitter with a variety of physiological roles.18 Previous studies have reported that high levels of NO could inhibit proliferation and survival of tumor cells, and sensitize tumors to chemotherapy in vitro and in vivo, by inhibition of key transcription factors, DNA repair enzymes and drug efflux pumps.19,20 Like H2S donors, NO donors also have gained great attention for decades owing to the high reactivity and inconvenient handling, which contain organic nitrates, diazeniumdiolates, S-nitrosothiols and furoxans.21 For example, nitroglycerin (6), isosorbide 5-mononitrate (ISMN, 7) and isosorbide dinitrate (ISDN, 8) (Fig. 2) are organic nitrates, well-known vasodilators used in the therapy and prevention of angina pectoris by releasing NO,22 as well as exhibit antitumor and enhance chemosensitivity.23,24 Chen et al. demonstrated that the bioconversion of nitroglycerin to release NO occurred through mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2).25 Moreover, JS-K (9, Fig. 2), an NO-donating diazeniumdiolate, released high levels of NO when suitably activated by glutathione or GST, and exhibited anticancer activity against various human cancer cell lines.26,27 Particularly, Kodela and co-workers reported that NOSH-aspirin bearing both NO and H2S-releasing moieties suppressed eleven different human cancer cell lines, and NOSH-1 was the most potent hybrid, with an IC50 value of 48 nM in HT-29 cells.28 In 2017, Gu et al. synthesized a series of NO-releasing bifendate derivatives, and further biological evaluation showed that the most potent compound significantly inhibited the proliferation of resistant K562/ADR cells in vitro and in vivo by releasing high levels of NO.29
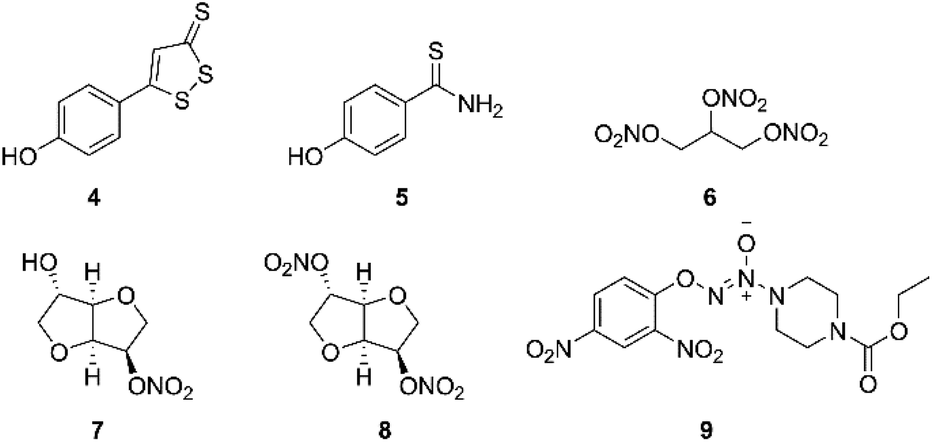 | ||
| Fig. 2 The structures of H2S/NO-donating agents. | ||
Given that both H2S/NO and artemisinin derivatives possess anticancer activity against various cancer cell lines, we hypothesized that the H2S/NO-donating artemisinin derivatives could exert potent cytotoxicity. In this study, to find valid candidates for the treatment of AML, we designed and synthesized three H2S/NO-donating artemisinin derivatives by coupling artesunate with H2S donors (ADT-OH and TBZ) and NO donor (ISMN) using the molecular hybridization strategy (Fig. 3). We further evaluated these H2S/NO-donating artemisinin derivatives for their antileukemic activities against AML cell lines, K562 and K562/ADR.
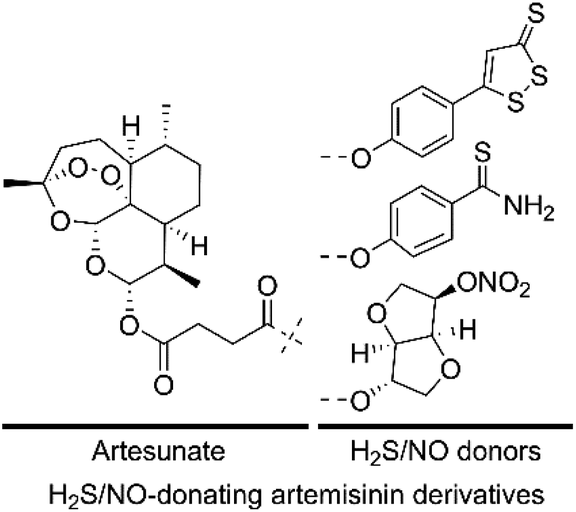 | ||
| Fig. 3 Design of H2S/NO-donating artemisinin derivatives. | ||
Results and discussion
The synthesis of hybrid molecules 10a–10c is outlined in Scheme 1. Firstly, intermediate 3 was prepared according to the method described previously30 via sodium borohydride (NaBH4) catalyzed reduction reaction of artemisinin (1) and 4-dimethylaminopyridine (DMAP) catalyzed esterification reaction. Then, the target molecules were obtained in good yields by reacting 3 and different H2S donors (4 and 5) or NO donor (7) via EDCI and DMAP catalyzed esterification reaction. The desired hybrid compounds were purified using column chromatography on silica gel and further characterized via 1H NMR, 13C NMR and high-resolution mass spectra (HRMS).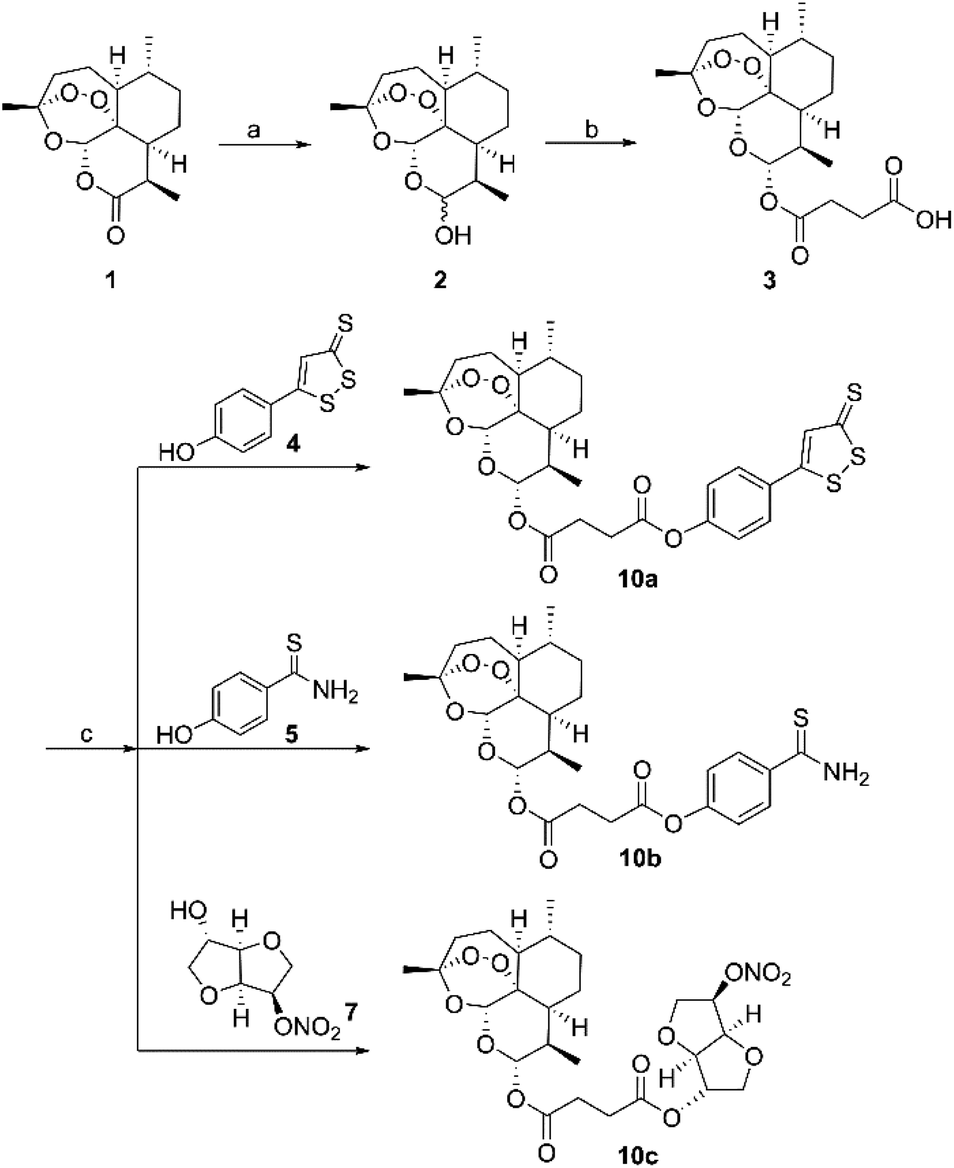 | ||
| Scheme 1 Synthesis of H2S/NO-donating artemisinin derivatives. Reagents and conditions: (a) NaBH4, MeOH, 0–5 °C; (b) succinic anhydride, DMAP, DCM, rt; (c) EDCI, DMAP, DMF, rt. | ||
The antiproliferative activities of target hybrids (10a–10c) were performed against AML cell lines of K562 and K562/ADR and human normal hepatic LO2 cells by the CCK-8 assay in vitro initially, compared with lead compounds artemisinin (1), dihydroartemisinin (2), artesunate (3), H2S releasing moieties (4 and 5), NO donor (7) and positive controls, homoharringtonine (HHT) and adriamycin (ADR). The IC50 values of test compounds were summarized in Table 1. All three H2S/NO-donating artemisinin derivatives were notably effective in inhibiting the growth of two leukemic cell lines, which displayed more potent antiproliferative activities than three lead compounds, artemisinin, dihydroartemisinin and artesunate. Nevertheless, all H2S/NO donors showed weak inhibitory activity with IC50 values more than 200 μM. Interestingly, hybrid 10c incorporated with ISMN showed stronger activity than 10a and 10b with H2S releasing moieties against both K562 and K562/ADR cells. In K562 cell line, 10c showed similar inhibitory activity (IC50 = 0.316 ± 0.021 μM) to clinical drug HHT (IC50 = 0.367 ± 0.011 μM) and exhibited superior cytotoxic activity to positive control ADR (IC50 = 0.672 ± 0.056 μM). Meanwhile, the inhibitory activity of hybrid 10c (IC50 = 6.634 ± 0.795 μM) against resistant K562/ADR cells was slightly stronger than that of ADR (IC50 = 9.161 ± 0.658 μM), but slightly weaker than that of HHT (IC50 = 3.256 ± 0.082 μM). Furthermore, in LO2 cells, NO releasing artemisinin derivative 10c also showed lower cytotoxicity (IC50 = 3.077 ± 0.281 μM) than two clinical drugs HHR (IC50 = 0.915 ± 0.149 μM) and ADR (IC50 = 2.736 ± 0.123 μM), suggesting that target compound 10c may be a promising candidate for further investigation.
| Compd | IC50a (μM) | ||
|---|---|---|---|
| K562 | K562/ADR | LO2 | |
| a The cells were treated with the indicated concentrations of each test compound for 72 h by the CCK-8 assay. The IC50 value (drug concentration inhibiting 50% of the cell proliferation) for each compound was calculated and expressed as mean IC50 (μM) ± SD from three independent experiments. | |||
| 1 | 11.085 ± 0.832 | >200 | 83.341 ± 1.988 |
| 2 | 8.666 ± 0.277 | 28.068 ± 1.095 | 19.893 ± 1.66 |
| 3 | 10.17 ± 0.135 | 36.433 ± 2.902 | 19.864 ± 0.331 |
| 4 | >200 | >200 | >200 |
| 5 | >200 | >200 | >200 |
| 7 | >200 | >200 | >200 |
| 10a | 2.981 ± 0.391 | 16.632 ± 2.058 | 9.862 ± 0.479 |
| 10b | 1.031 ± 0.116 | 8.479 ± 1.081 | 2.244 ± 0.359 |
| 10c | 0.316 ± 0.021 | 6.634 ± 0.795 | 3.077 ± 0.281 |
| HHT | 0.367 ± 0.011 | 3.256 ± 0.082 | 0.915 ± 0.149 |
| ADR | 0.672 ± 0.056 | 9.161 ± 0.658 | 2.736 ± 0.123 |
Compound 10c was composed of artesunate moiety (3) and ISMN moiety (7) (Scheme 1), and the IC50 values (shown in Table 1) of 10c for K562 (IC50 = 0.316 ± 0.021 μM) and K562/ADR cells (IC50 = 6.634 ± 0.795 μM) were significantly less than that of individual moieties, 3 (IC50 = 10.17 ± 0.135 and 36.433 ± 2.902 μM) and 7 (IC50 > 200 and 200 μM), respectively. Furthermore, in order to investigate the antiproliferative activity of the combination of 3 and 7, the growth inhibition of compounds 3, 7, and 3 + 7 against K562 and K562/ADR cells were determined by the CCK-8 assay. As seen in Fig. 4, it was notable that compounds 3 and 7 at 0.3 and 6 μM displayed less potent activity in both K562 and K562/ADR cell lines, respectively, however, conjugate 10c exhibited 2.98-fold and 2.67-fold increase in its antiproliferative activity on K562 and K562/ADR cells comparable to the combination of 3 and 7, respectively. These results suggested that the antitumor activity of hybrid 10c may be attributed to the synergic effects of artesunate and NO donor moiety in cancer cells.
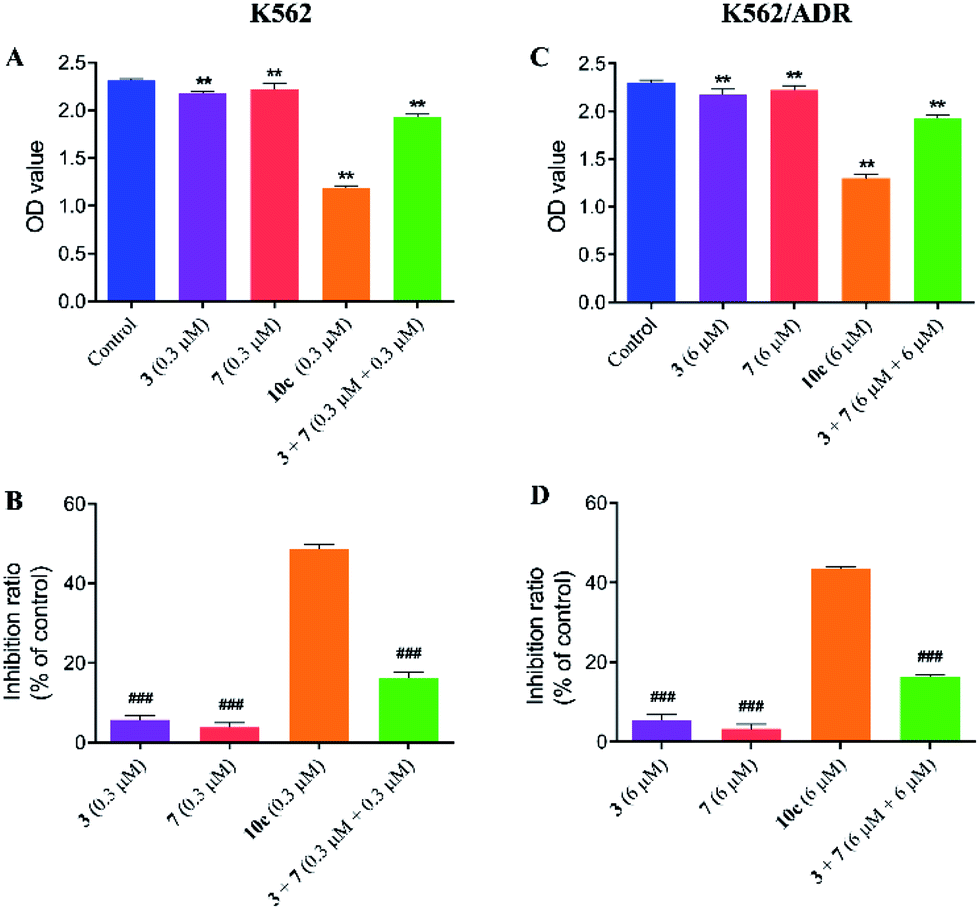 | ||
| Fig. 4 Compounds 3, 7, 10c and 3 + 7 inhibited the proliferation of K562 and K562/ADR cells. K562 cells were incubated with vehicle, 3 (0.3 μM), 7 (0.3 μM), 10c (0.3 μM) and 3 + 7 (0.3 μM + 0.3 μM) for 72 h, and K562/ADR cells were incubated with vehicle, 3 (6 μM), 7 (6 μM), 10c (6 μM) and 3 + 7 (6 μM + 6 μM) for 72 h, respectively. The cell proliferation was measured by CCK-8. Data were expressed as the means ± SD from three independent experiments. **P < 0.01 vs. the control group; ###P < 0.001 vs. the 10c group. | ||
It was reported that high levels of NO could result in nitrosative stress (NS) and induce tumor cell apoptosis.31 In order to verify the effect of NO on the anticancer activity of 10c, the intracellular NO levels were first determined using DAF-FM DA as a cell permeable fluorescent probe. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 10 μM and 100 μM), 3 (100 μM) and 7 (100 μM) for 3 h, respectively. And we measured NO generation by using fluorescence microscopy (Fig. 5A and C) and microplate reader (Fig. 5B and D), respectively. The results showed that hybrid 10c promoted the generation of NO in both K562 and K562/ADR cells in a dose-dependent manner compared with the control group. Notably, the parents 3 and 7 also increased the intracellular NO levels, however, the effects of 3 and 7 were significantly weaker than that of 10c in both K562 and K562/ADR cells, respectively, suggesting that the levels of intracellular NO induced by different compounds might be positively correlated with their antiproliferative activities. Furthermore, it was observed that treatment with test compounds caused lower levels of NO production in resistant K562/ADR cells, which might be associated with their anticancer activities in sensitive and resistant cancer cell lines. Interesting, parent 3 without NO donor also enhanced the intracellular NO levels, which may be related to previous study that the production of NO could be induced by inducible nitric oxide synthase (iNOS).32
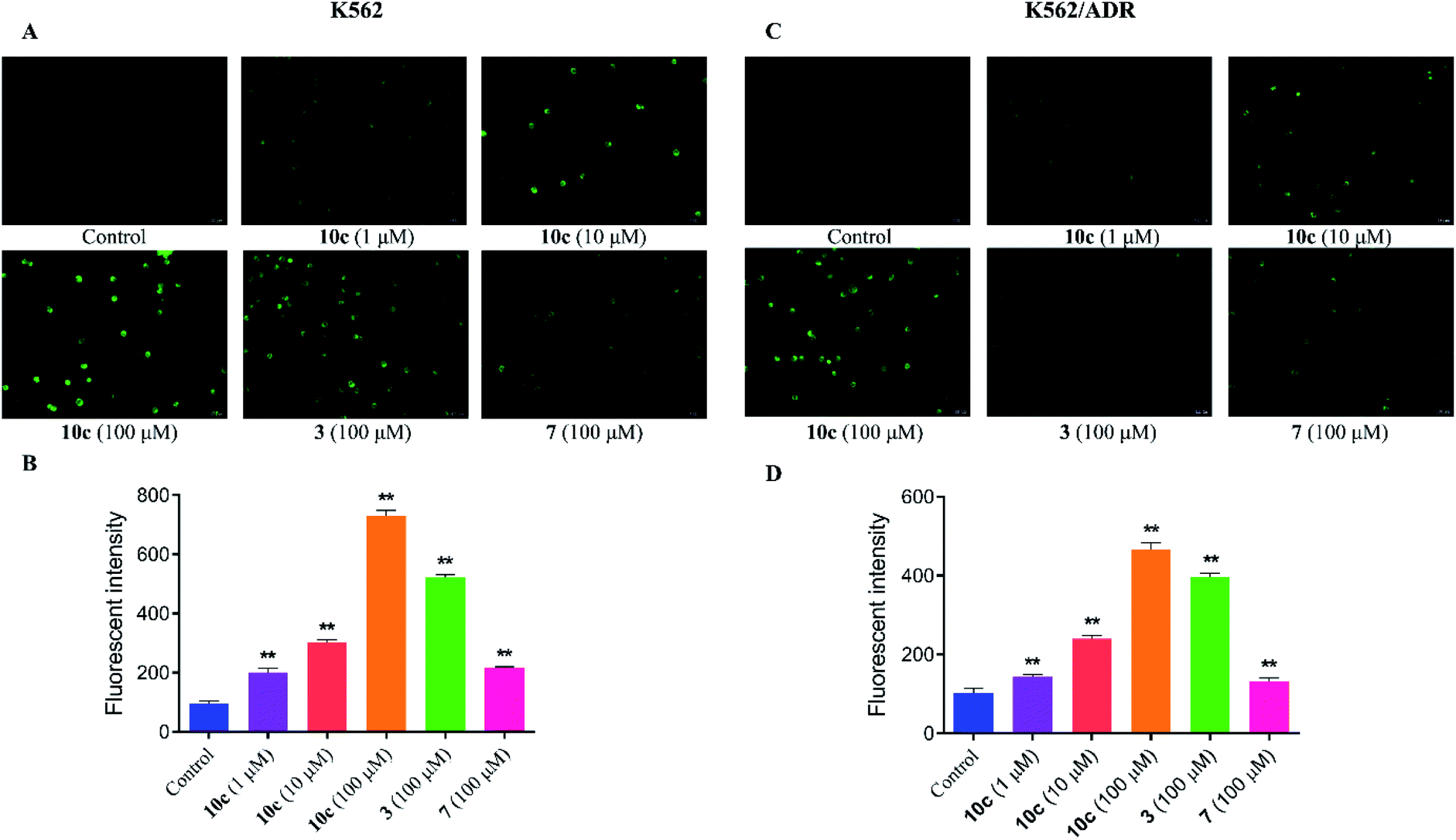 | ||
| Fig. 5 The generation of intracellular NO in K562 and K562/ADR cells. Cells were incubated with vehicle, 10c (1 μM, 10 μM and 100 μM), 3 (100 μM) and 7 (100 μM) for 3 h, respectively. (A and C) Intracellular NO was measured using DAF-FM DA staining by fluorescence microscopy. (B and D) NO production was assessed by microplate reader. The data were expressed as the mean ± SD from three independent experiments. **P < 0.01 vs. the control group. | ||
Reactive oxygen species (ROS) are cellular metabolites and play vital roles in cellular homeostasis. In addition, more evidences suggested that the excessive generation of ROS contributed to apoptosis and autophagy in various tumor cells.33 Extensive studies have showed that artemisinin and its derivatives induced apoptosis by ROS generation mediated by endoperoxide bridges.5 Therefore, in this study, the generation of intracellular ROS in K562 and K562/ADR cells was investigated by fluorescence microscopy and flow cytometer after DCFH-DA staining, respectively. Cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively. As shown in Fig. 6A and D, 10c-treated groups gave rise to increase in green fluorescence intensity in a dose-dependent manner, suggesting that 10c heightened the levels of intracellular ROS. Simultaneously, as the increased concentrations of 10c, the ROS production and percentage of ROS were gradually increased in K562 and K562/ADR cells (Fig. 6B, C, E and F). Moreover, incubation with 10c caused higher levels of ROS production in sensitive K562 cells compared with that in resistant K562/ADR cells. Nevertheless, NO and ROS were reported to react to form peroxynitrite (ONOO–).34,35 So, the correlation between NO and ROS induced by 10c needs our further studies.
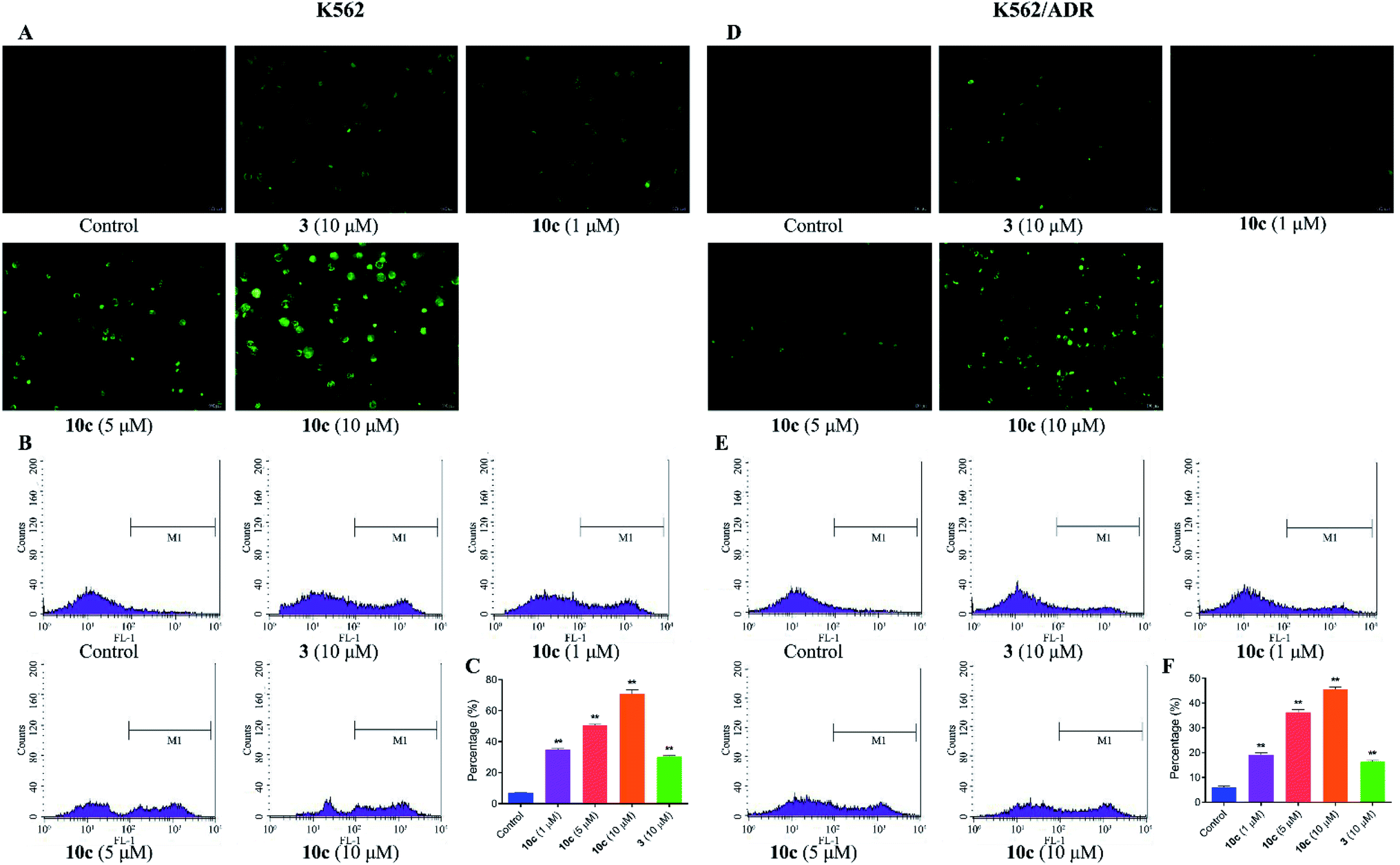 | ||
| Fig. 6 The generation of intracellular ROS in K562 and K562/ADR cells. Cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively. (A and D) Intracellular ROS was measured using DCFH-DA staining by fluorescence microscopy. (B, C, E and F) ROS production and percentage of ROS were measured by flow cytometer, respectively. The data were expressed as the mean ± SD from three independent experiments. **P < 0.01 vs. the control group. | ||
Cell cycle progression is vital to the cell processes, such as cell division and replication. Cell cycle arrest could inhibit cancer cell proliferation. Accumulating reports had reported that artemisinin derivatives inhibited cancer cells via cell cycle arrest.36 To examine the antineoplastic mechanism of 10c on cell cycle, cell cycle distribution was measured using flow cytometry after staining with PI. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively. As shown in Fig. 7, treatment of K562 and K562/ADR cells with 10c resulted in accumulation at the S phase a concentration-dependent manner, respectively, compared with the control group. Meanwhile, the levels of the accumulation of K562 cells in the S phase after treatment with 10c were slightly higher than the levels of the accumulation of K562/ADR cells.
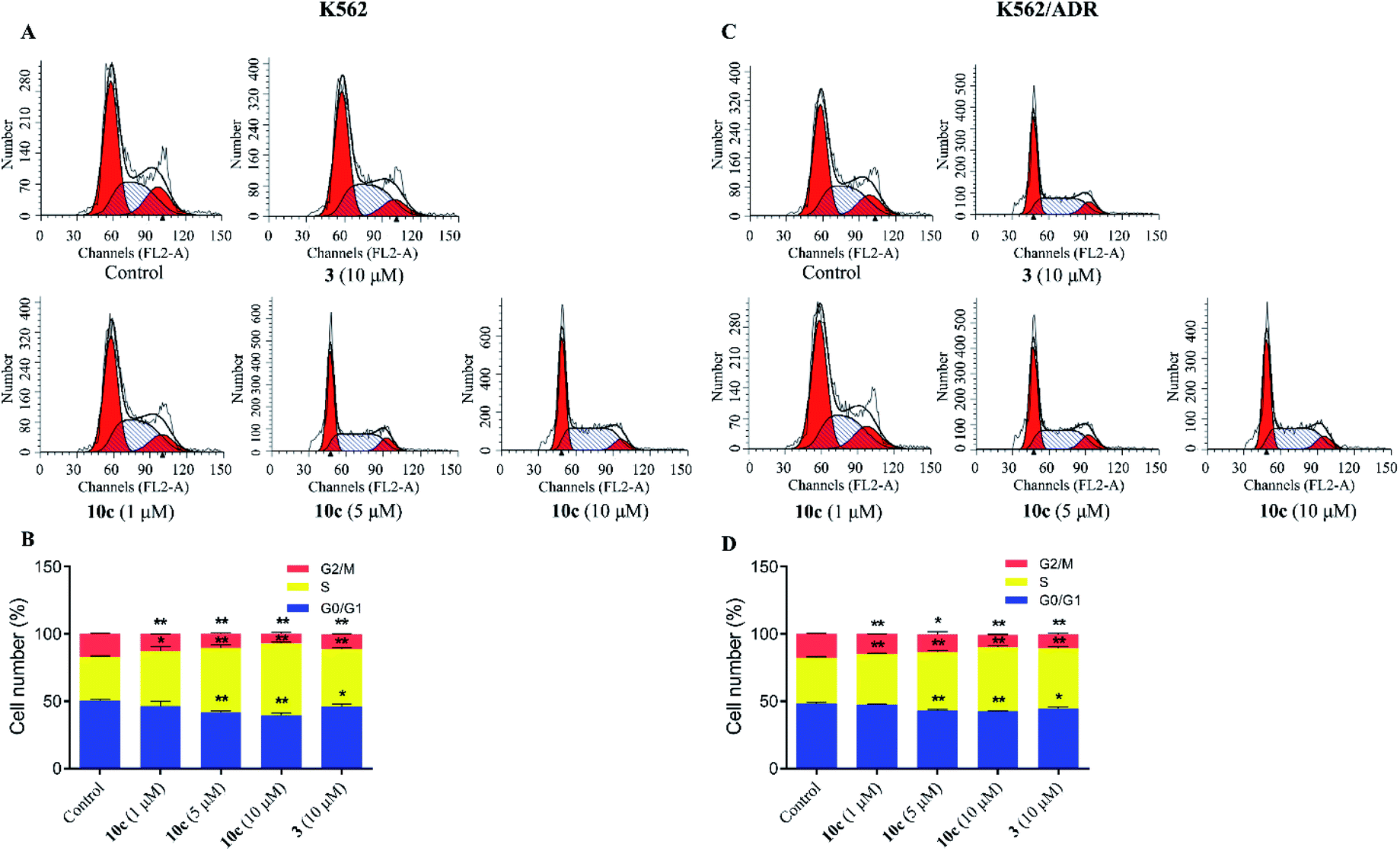 | ||
| Fig. 7 Cell cycle arrest effects of compound 10c. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and cell cycle distribution was measured using flow cytometry after staining with PI. The data were expressed as the mean ± SD from three independent experiments. (A and C) Flow cytometry analysis. (B and D) Quantitative analysis. *P < 0.05, **P < 0.01 vs. the control group. | ||
Previous studies have shown that artemisinins and its derivatives can induce cancer cell apoptosis by modulating the expression of apoptosis-related regulators and signal events. Accordingly, to further confirm the effect of 10c on induction of cancer cell apoptosis, K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and the percentages of apoptotic cells were measured by flow cytometry after staining with Annexin V-FITC and 7-AAD. As shown in Fig. 8, treatment with 10c induced apoptosis in both K562 and K562/ADR cells in a dose-dependent manner, and 10c at 10 μM showed significantly stronger effect than that of 3 in both K562 and K562/ADR cell lines.
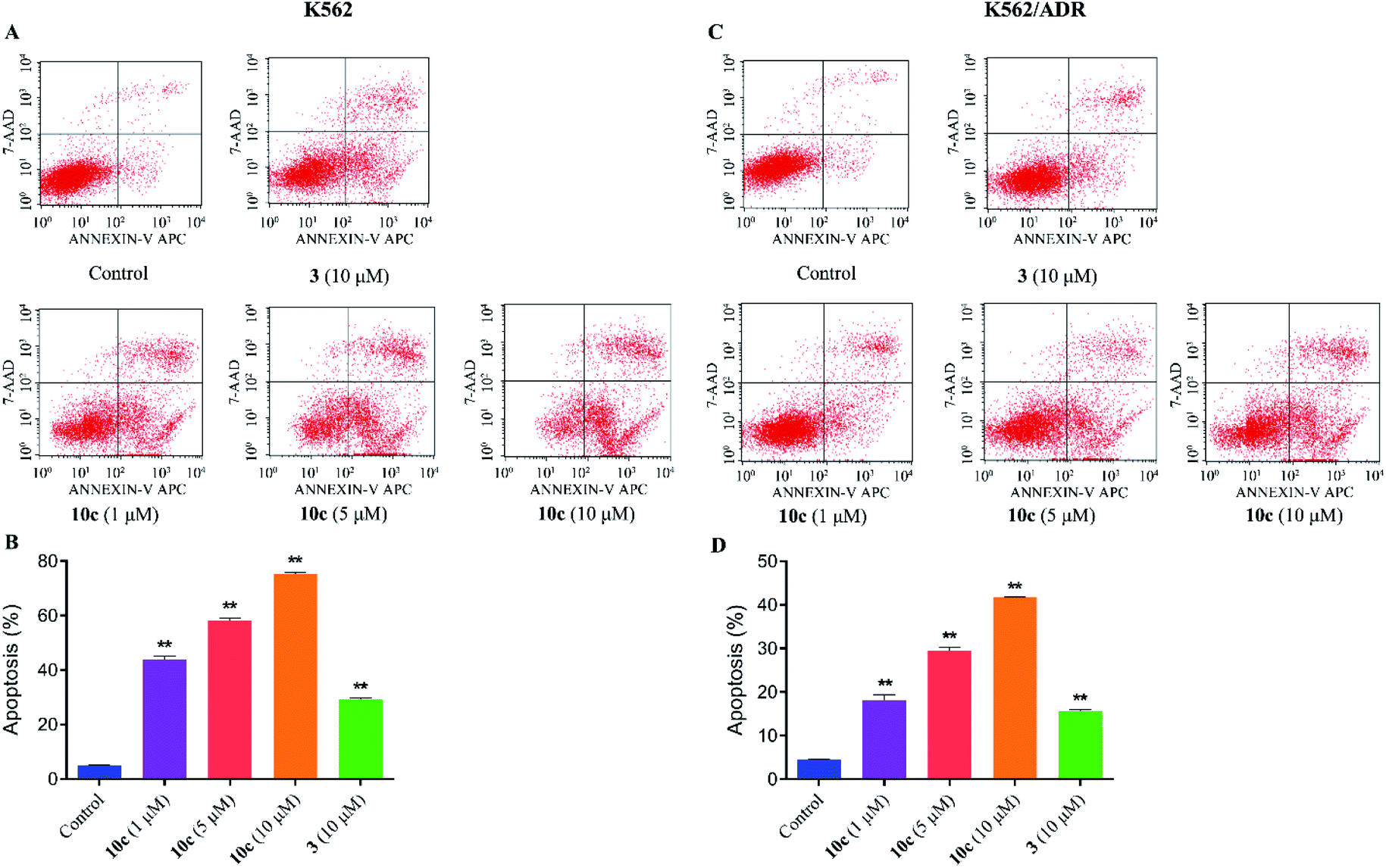 | ||
| Fig. 8 Compound 10c induced leukemia cells apoptosis. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and the percentages of apoptotic cells were measured by flow cytometry after staining with Annexin V-FITC and 7-AAD. The data were expressed as the mean ± SD from three independent experiments. (A and C) Flow cytometry analysis. (B and D) Quantitative analysis. **P < 0.01 vs. the control group. | ||
It was reported that the loss of mitochondrial membrane potential (MMP) was a significant marker of mitochondrial dysfunction and played a key role in the process of cell apoptosis.37 To uncover the involvement of mitochondria pathway in 10c induced apoptosis, the changes of mitochondrial membrane potentials were assessed in this paper. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and further stained with fluorochrome JC-1 dye followed by flow cytometry analysis. Fig. 9 showed that 10c obviously disturbed the mitochondrial membrane and decreased MMP in a dose-dependent manner in K562 and K562/ADR cells, which indicated that 10c could induce cell apoptosis through mitochondria pathway. At the same time, mitochondrial membrane potential in K562 cells was more sensitive to 10c than that in resistant K562/ADR cells.
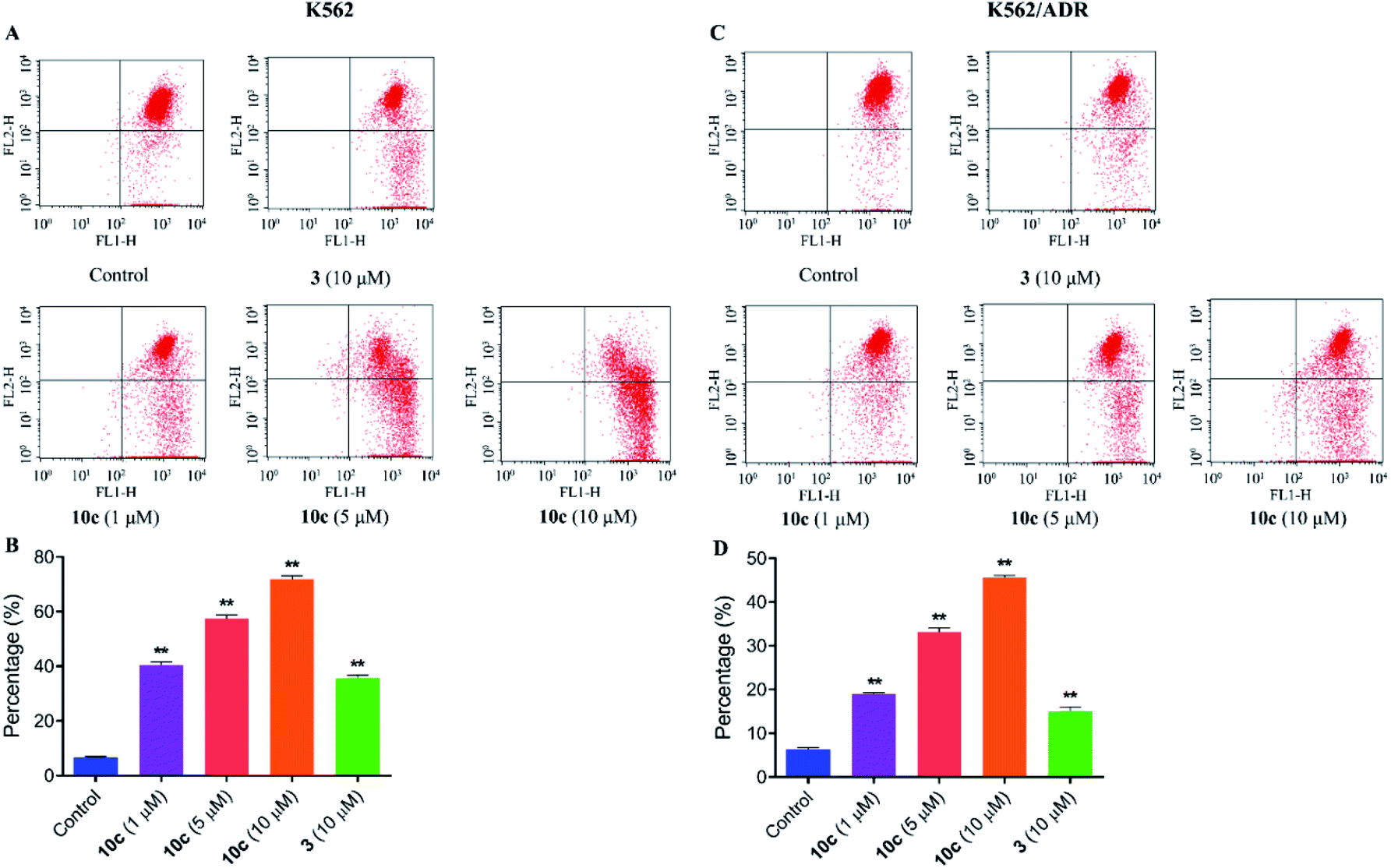 | ||
| Fig. 9 Effects of 10c on the mitochondrial membrane potential. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and stained with JC-1 dye followed by flow cytometry analysis. The data were expressed as the mean ± SD from three independent experiments. (A and C) Flow cytometry analysis. (B and D) Quantitative analysis. **P < 0.01 vs. the control group. | ||
In an effort to explore the molecular mechanisms underlying the 10c-induced cell cycle arrest and apoptosis, we determined the regulatory effects of 10c on the cycle- and apoptosis-related proteins in K562 and K562/ADR cells. Cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and the relative levels of CDK1, CDK2, cleaved caspase-3 and cleaved PARP expression were measured by western blot. After 24 h treatment with 10c, suppression of CDK1 and CDK2 proteins expression, and up-regulation of cleaved caspase-3 and cleaved PARP proteins expression were observed in a concentration-dependent manner compared with the control group (Fig. 10), and the effects of 10c were significantly stronger in K562 cells than those in K562/ADR cells.
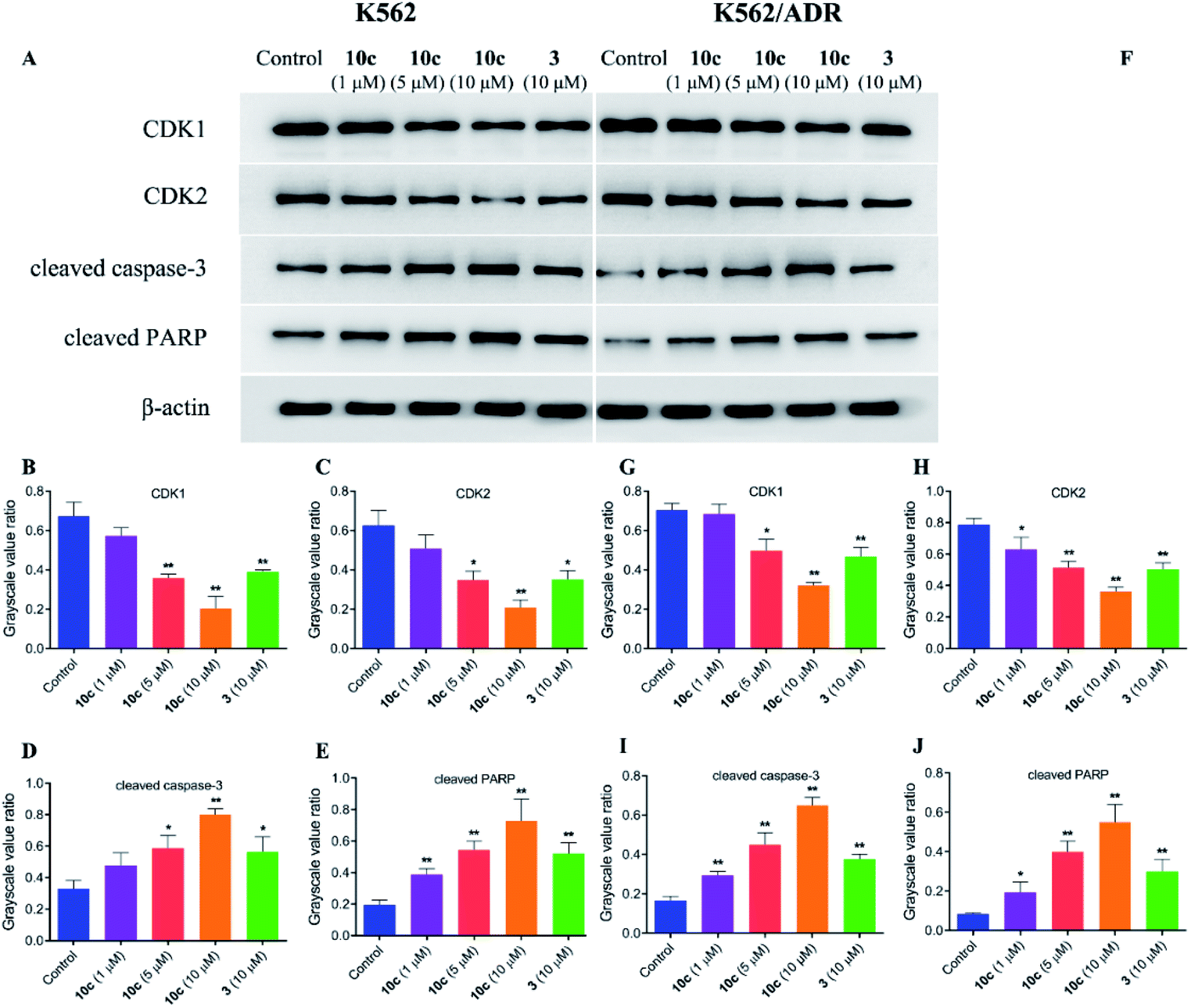 | ||
| Fig. 10 Effects of 10c on the cycle- and apoptosis-related proteins. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and the relative levels of CDK1, CDK2, cleaved caspase-3 and cleaved PARP expression were determined by western blot using β-actin as internal standard. The data were selected images and expressed as the mean ± SD from three independent experiments. (A and F) western blot analysis, (B–E, G–J) quantitative analysis. *P < 0.05, **P < 0.01 vs. the control group. | ||
Besides apoptosis, autophagy is another programmed cell death, which plays a dual role in the cancer therapy.38 Autophagy could induce cell death or protect cells depending upon the cell type and context.39 Therefore, to assess whether autophagy contributes to 10c-mediated inhibition of leukemia cell proliferation, we investigated the effects of 10c on levels of the autophagy-related proteins in K562 and K562/ADR cells. Cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and the relative levels of Beclin1 and LC3-II expression were determined by western blot. After treatment with 10c, the Beclin1 and cleavage of LC3 (LC3-II) levels were increased in a dose-dependent manner in K562 and K562/ADR cells (Fig. 11). We found that the level of autophagy in resistant K562/ADR cells was lower than that in sensitive K562 cells, and the 10c-induction of autophagy were weaker in K562/ADR cells than those in K562 cells.
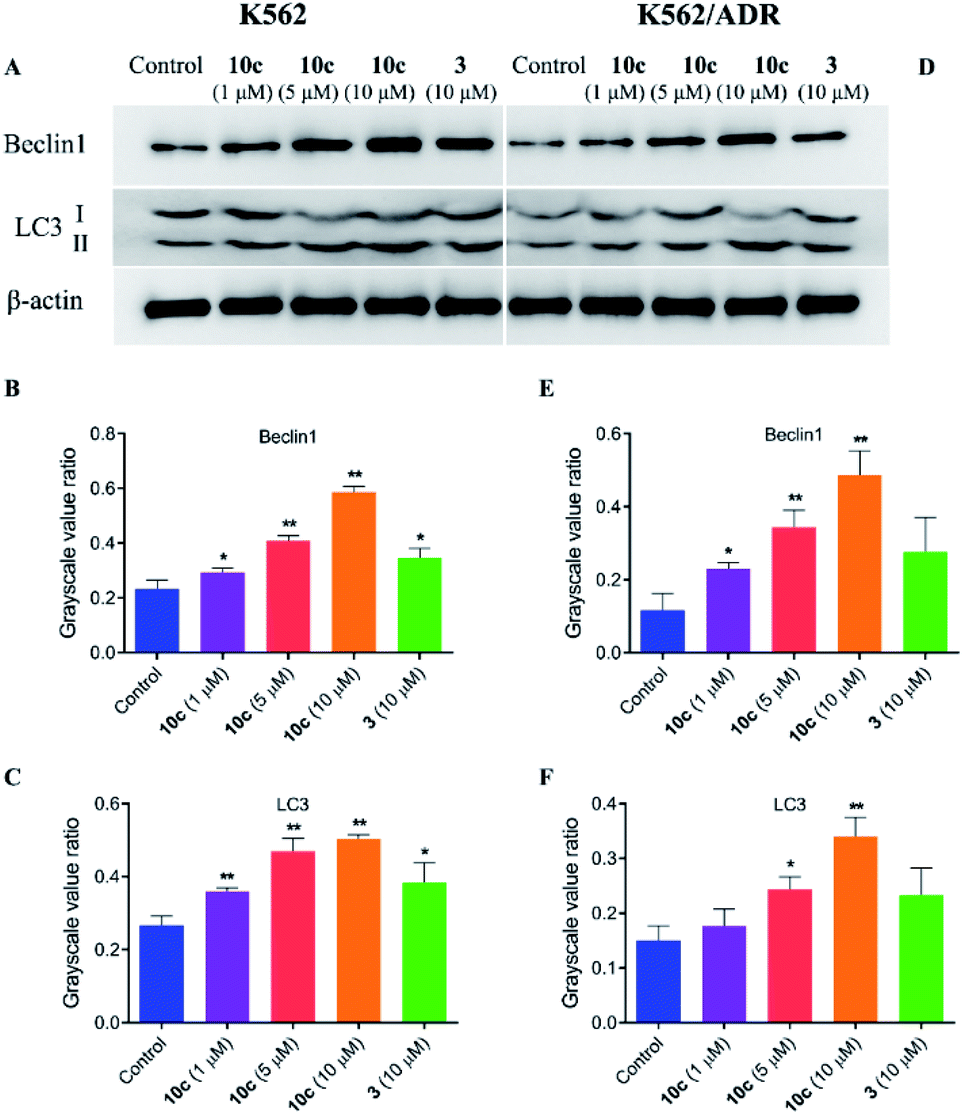 | ||
| Fig. 11 Effects of 10c on the autophagy-related proteins. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and the relative levels of Beclin1 and LC3 expression were determined by western blot using β-actin as internal standard. The data were selected images and expressed as the mean ± SD from three independent experiments. (A and D) western blot analysis, (B, C, E and F) quantitative analysis. *P < 0.05, **P < 0.01 vs. the control group. | ||
The crosstalk between apoptosis and autophagy is quite complex and remains controversial.40 Apoptosis and autophagy may be activated by the common upstream signals, giving rise to the activation of the combination of autophagy and apoptosis,41 such as AMPK, JNK and AKT signaling. Autophagy and apoptosis can be promoted by AMP activated protein kinase (AMPK), which is a key energy sensor and regulates cellular metabolism to maintain energy homeostasis,42 and c-Jun N-terminal kinase (JNK), which is a stress-activated protein kinase (SAPK) of the mitogen activated protein kinase (MAPK) family.43 Meanwhile, AKT is a serine/threonine protein kinase showing antiapoptotic activity, as well as inhibiting autophagy mediated by activation of mTOR, which is a negative regulatory axis of autophagy.44 To further explore the molecular mechanisms underlying the antiproliferative activity of 10c, we investigated the regulatory effects of treatment with 10c on the AMPK, JNK and AKT signaling in K562 and K562/ADR cells. Cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and the relative levels of AMPK, JNK and AKT phosphorylation were determined by western blot using β-actin as internal standard. As shown in Fig. 12, incubation with 10c significantly inhibited the AKT phosphorylation and stimulated the AMPK and JNK phosphorylation in a dose-dependent manner in both cancer cell lines. Similarly, treatment with 3 also clearly reduced the levels of AKT phosphorylation and up-regulated the levels of AMPK and JNK phosphorylation, while the regulatory effects of parent 3 (10 μM) in both cell lines were noticeably less than that of 10c (10 μM).
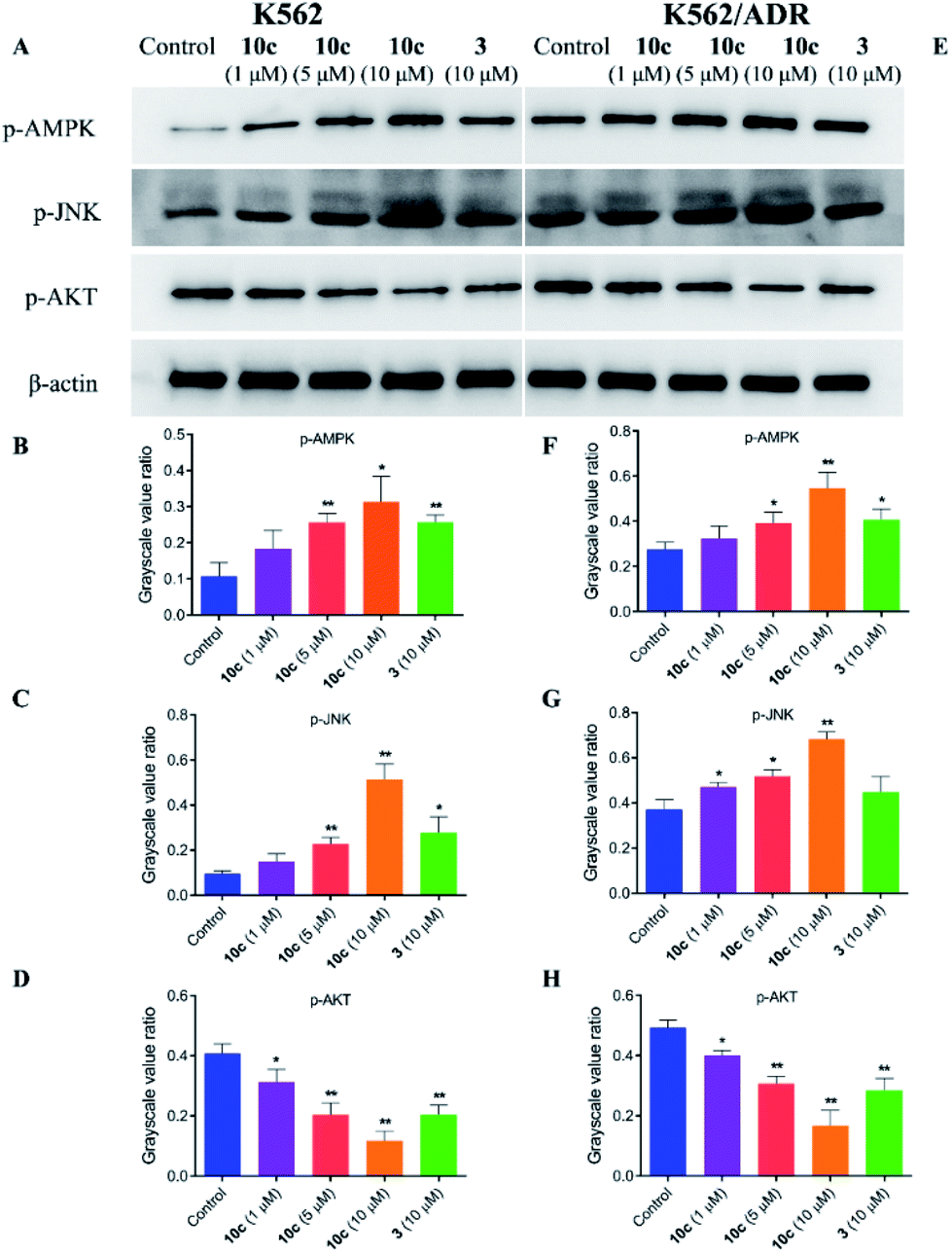 | ||
| Fig. 12 Effects of 10c on the AMPK, JNK and AKT signaling. K562 and K562/ADR cells were incubated with vehicle, 10c (1 μM, 5 μM and 10 μM) and 3 (10 μM) for 24 h, respectively, and the relative levels of AMPK, JNK and AKT phosphorylation were determined by western blot using β-actin as internal standard. The data were selected images and expressed as the mean ± SD from three independent experiments. (A and E) western blot analysis, (B–D and F–H) quantitative analysis. *P < 0.05, **P < 0.01 vs. the control group. | ||
Experimental
General
Commercially available reagents and solvents were analytical grade and used without further purification. 1H NMR and 13C NMR spectra were recorded with an Agilent 400 NMR spectrometer, using TMS as an internal standard. High resolution mass spectrometry (HRMS) were obtained on an Agilent 6520 Accurate-Mass Q-TOF. Column chromatography was carried out on silica gel (200–300 mesh).General procedure for the preparation of hybrid compounds 10a–10c
Intermediate 3 (0.26 mmol) was dissolved in dry N,N-dimethylformamide (3 mL), then 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride (EDCI) (2.0 eq.), 4-dimethylaminopyridine (DMAP) (2.4 eq.), and appropriate donor (0.9 eq. for 4, 0.95 eq. for 5 and 7) were added. The reaction mixture was stirred at room temperature for 3–5 h. The reaction solution was quenched by adding water (20 mL), and the crude was filtered and dried in vacuum oven overnight. The crude residue was purified by silica gel flash column chromatography (dichloromethane/methanol 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 10a, AcOEt/PE 1:4 for 10b and 1:8 for 10c) to afford the pure compound.
:1 for 10a, AcOEt/PE 1:4 for 10b and 1:8 for 10c) to afford the pure compound.
Hybrid compound 10a: red solid, yield 63%; 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 8.4 Hz, 2H), 7.40 (s, 1H), 7.28 (d, J = 8.4 Hz, 2H), 5.84 (d, J = 10 Hz, 1H), 5.46 (s, 1H), 2.99–2.84 (m, 4H), 2.61–2.56 (m, 1H), 2.43–2.35 (m, 1H), 2.06–2.02 (m, 1H), 1.93–1.88 (m, 1H), 1.80–1.64 (m, 4H), 1.43 (s, 3H), 1.40–1.30 (m, 3H), 1.25 (s, 1H), 0.98 (d, J = 6.0 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 215.45, 171.77, 170.81, 170.26, 153.49, 135.98, 129.19, 128.21, 122.96, 104.50, 92.42, 91.51, 80.09, 51.48, 45.15, 37.25, 36.15, 34.01, 31.77, 29.67, 29.14, 29.12, 25.93, 24.54, 21.96, 20.19, 12.07; HRMS-ESI (m/z): calcd for C28H33O8S3 [M + H]+ 593.1332, found 593.1335.
Hybrid compound 10b: yellow solid, yield 78%; 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 8.8 Hz, 2H), 7.75 (s, 1H), 7.38 (s, 1H), 7.15 (d, J = 8.8 Hz, 2H), 5.82 (d, J = 10.0 Hz, 1H), 5.46 (s, 1H), 2.95–2.81 (m, 4H), 2.60–2.54 (m, 1H), 2.42–2.34 (m, 1H), 2.06–2.02 (m, 1H), 1.90–1.87 (m, 1H), 1.80–1.70 (m, 3H), 1.65–1.60 (m, 1H), 1.54–1.46 (m, 1H), 1.42 (s, 3H), 1.39–1.26 (m, 3H), 0.97 (d, J = 5.6 Hz, 3H), 0.86 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ201.37, 170.96, 170.43, 153.36, 136.68, 128.48, 121.52, 104.53, 92.44, 91.51, 80.12, 51.48, 45.14, 37.23, 36.15, 34.00, 31.76, 29.09, 25.90, 24.53, 21.95, 20.18, 12.05; HRMS-ESI (m/z): calcd for C26H34NO8S [M + H]+ 520.2000, found 520.2001.
Hybrid compound 10c: white solid, yield 73%; 1H NMR (400 MHz, CDCl3) δ 5.80 (d, J = 9.6 Hz, 1H), 5.44 (s, 1H), 5.37–5.34 (m, 1H), 5.24 (d, J = 2.8 Hz, 1H), 5.00 (t, J = 5.2 Hz, 1H), 4.51 (d, J = 5.2 Hz, 1H), 4.06–3.97 (m, 3H), 3.93–3.89 (m, 1H), 2.73 (t, J = 6.4 Hz, 2H), 2.68–2.60 (m, 2H), 2.57–2.52 (m, 1H), 2.41–2.33 (m, 1H), 2.05–2.01 (m, 1H), 1.92–1.87 (m, 1H), 1.80–1.60 (m, 4H), 1.53–1.45 (m, 1H), 1.43 (m, 3H), 1.39–1.24 (m, 3H), 0.97 (d, J = 6.0 Hz, 3H), 0.86 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.08, 104.45, 92.27, 91.48, 86.43, 81.46, 81.30, 80.10, 77.60, 73.32, 69.22, 51.49, 45.17, 37.24, 36.16, 34.02, 31.79, 29.12, 28.88, 25.89, 24.53, 21.94, 20.19, 11.99; HRMS-ESI (m/z): calcd for C25H39N2O13 [M + NH4]+ 575.2447, found 575.2443.
Pharmacology
Conclusions
In summary, three H2S/NO-donating artemisinin derivatives were synthesized and evaluated. Among them, NO-donating artemisinin derivative 10c had potent inhibitory activity against AML cell lines of K562 and K562/ADR. In addition, compound 10c was observed to increase the levels of intracellular NO and ROS, induce apoptosis and S phase arrest, and lessen the mitochondrial membrane potential in a concentration-dependent manner in K562 and K562/ADR cells. Moreover, 10c could induce autophagy, activate AMPK and JNK signaling, and suppressed AKT signaling in both leukemia cell lines. We are also interested in further investigating the chemical mechanism of how NO improving activity of the parent. Overall, the data showed that 10c may be a potential antileukemic candidate for the therapy of AML. Subsequently, we will further evaluate its stability in vitro and efficiency in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (81860622); Joint Fund of the Department of Science and Technology of Zunyi City and Affiliated Hospital of Zunyi Medical University ([2018]56 and [2018]62).Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef.
- T. Arnason and T. Harkness, Cancers, 2015, 7, 2063–2082 CrossRef CAS PubMed.
- V. M. Rumjanek, R. S. Vidal and R. C. Maia, Biosci. Rep., 2013, 33, 875–888 CrossRef CAS PubMed.
- L. H. Miller and X. Su, Cell, 2011, 146, 855–858 CrossRef CAS PubMed.
- Y. K. Wong, C. Xu, K. A. Kalesh, Y. He, Q. Lin, W. S. F. Wong, H. M. Shen and J. Wang, Med. Res. Rev., 2017, 37, 1492–1517 CrossRef CAS.
- S. Luan, H. Zhong, X. Zhao, J. Yang, Y. Jing, D. Liu and L. Zhao, Eur. J. Med. Chem., 2017, 141, 584–595 CrossRef CAS PubMed.
- B. Kumar, A. Kalvala, S. Chu, S. Rosen, S. J. Forman, G. Marcucci, C. C. Chen and V. Pullarkat, Leuk. Res., 2017, 59, 124–135 CrossRef CAS PubMed.
- J. Lee, P. Shen, G. Zhang, X. Wu and X. Zhang, Biomed. Pharmacother., 2013, 67, 157–163 CrossRef CAS PubMed.
- L. Zhang, F. Chen, Z. Zhang, Y. Chen and J. Wang, Bioorg. Med. Chem. Lett., 2016, 26, 38–42 CrossRef CAS PubMed.
- M. Tan, Y. Rong, Q. Su and Y. Chen, Leuk. Res., 2017, 62, 98–103 CrossRef CAS.
- S. J. Wang, Y. Gao, H. Chen, R. Kong, H. C. Jiang, S. H. Pan, D. B. Xue, X. W. Bai and B. Sun, Cancer Lett., 2010, 293, 99–108 CrossRef CAS.
- L. Chen, C. Wang, N. Hu and H. Zhao, RSC Adv., 2019, 9, 1004–1014 RSC.
- L. Li, A. Hsu and P. K. Moore, Pharmacol. Ther., 2009, 123, 386–400 CrossRef CAS PubMed.
- G. Yang, X. Sun and R. Wang, FASEB J., 2004, 18, 1782–1784 CrossRef CAS.
- E. G. Mueller, Nat. Chem. Biol., 2006, 2, 185–194 CrossRef CAS PubMed.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- Z. J. Song, M. Y. Ng, Z. W. Lee, W. Dai, T. Hagen, P. K. Moore, D. Huang, L. W. Deng and C. H. Tan, RSC Med. Chem., 2014, 5, 557–570 RSC.
- L. J. Ignarro, J. Physiol. Pharmacol., 2002, 53, 503–514 CAS.
- A. J. Burke, F. J. Sullivan, F. J. Giles and S. A. Glynn, Carcinogenesis, 2013, 34, 503–512 CrossRef CAS PubMed.
- C. Riganti, E. Miraglia, D. Viarisio, C. Costamagna, G. Pescarmona, D. Ghigo and A. Bosia, Cancer Res., 2005, 65, 516–525 CAS.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- T. Münzel and T. Gori, Curr. Opin. Pharmacol., 2013, 13, 251–259 CrossRef PubMed.
- E. Pipili-Synetos, A. Papageorgiou, E. Sakkoula, G. Sotiropoulou, T. Fotsis, G. Karakiulakis and M. E. Maragoudakis, Br. J. Pharmacol., 1995, 116, 1829–1834 CrossRef CAS PubMed.
- H. Yasuda, K. Nakayama, M. Watanabe, S. Suzuki, H. Fuji, S. Okinaga, A. Kanda, K. Zayasu, T. Sasaki, M. Asada, T. Suzuki, M. Yoshida, S. Yamanda, D. Inoue, T. Kaneta, T. Kondo, Y. Takai, H. Sasaki, K. Yanagihara and M. Yamaya, Clin. Cancer Res., 2006, 12, 6748–6757 CrossRef CAS PubMed.
- Z. Chen, J. Zhang and J. S. Stamler, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8306–8311 CrossRef CAS PubMed.
- Z. Huang, L. Liu, J. Chen, M. Cao and J. Wang, Biomed. Pharmacother., 2018, 107, 1385–1392 CrossRef CAS PubMed.
- M. Z. Kaczmarek, R. J. Holland, S. A. Lavanier, J. A. Troxler, V. I. Fesenkova, C. A. Hanson, J. L. Cmarik, J. E. Saavedra, L. K. Keefer and S. K. Ruscetti, Leuk. Res., 2014, 38, 377–382 CrossRef CAS PubMed.
- R. Kodela, M. Chattopadhyay and K. Kashfi, ACS Med. Chem. Lett., 2012, 3, 257–262 CrossRef CAS PubMed.
- X. Gu, Z. Huang, Z. Ren, X. Tang, R. Xue, X. Luo, S. Peng, H. Peng, B. Lu, J. Tian and Y. Zhang, J. Med. Chem., 2017, 60, 928–940 CrossRef CAS PubMed.
- B. Xie, L. L. Meng, Y. J. Xiao, H. Y. Yang, B. L. Deng, S. Xiang and Y. Q. Kuang, Chin. J. Synth. Chem., 2011, 19, 62–65 CAS.
- G. C. Brown and V. Borutaite, Biochem. Soc. Trans., 2006, 34, 953–956 CrossRef CAS.
- H. A. Lee, B. R. Song, H. R. Kim, J. E. Kim, W. B. Yun, J. J. Park, M. L. Lee, J. Y. Choi, H. S. Lee and D. Y. Hwang, Exp. Ther. Med., 2017, 14, 4986–4994 CAS.
- J. Xu, Y. Wu, G. Lu, S. Xie, Z. Ma, Z. Chen, H. M. Shen and D. Xia, Redox Biol., 2017, 12, 198–207 CrossRef CAS PubMed.
- W. A. Pryor and G. L. Squadrito, Am. J. Physiol., 1995, 268, L699–L722 CAS.
- Z. Xie, M. Wei, T. E. Morgan, P. Fabrizio, D. Han, C. E. Finch and V. D. Longo, J. Neurosci., 2002, 22, 3484–3492 CrossRef CAS PubMed.
- C. Morrissey, B. Gallis, J. W. Solazzi, B. J. Kim, R. Gulati, F. Vakar-Lopez, D. R. Goodlett, R. L. Vessella and T. Sasaki, Anti-Cancer Drugs, 2010, 21, 423–432 CrossRef CAS PubMed.
- E. Gottlieb, S. M. Armour, M. H. Harris and C. B. Thompson, Cell Death Differ., 2003, 10, 709–717 CrossRef CAS PubMed.
- A. L. Edinger and C. B. Thompson, Curr. Opin. Cell Biol., 2004, 16, 663–669 CrossRef CAS PubMed.
- F. Nazio, F. Strappazzon, M. Antonioli, P. Bielli, V. Cianfanelli, M. Bordi, C. Gretzmeier, J. Dengjel, M. Piacentini, G. Fimia and F. Cecconi, Nat. Cell Biol., 2013, 15, 406–416 CrossRef CAS PubMed.
- G. Mariño, M. Niso-Santano, E. H. Baehrecke and G. Kroemer, Nat. Rev. Mol. Cell Biol., 2014, 15, 81–94 CrossRef PubMed.
- J. M. Gump and A. Thorburn, Trends Cell Biol., 2011, 21, 387–392 CrossRef CAS PubMed.
- C. S. Zhang and S. C. Lin, Cell Metab., 2016, 23, 399–401 CrossRef CAS PubMed.
- X. Sui, N. Kong, L. Ye, W. Han, J. Zhou, Q. Zhang, C. He and H. Pan, Cancer Lett., 2014, 344, 174–179 CrossRef CAS PubMed.
- E. A. Dunlop and A. R. Tee, Semin. Cell Dev. Biol., 2014, 36, 121–129 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08239e |
| This journal is © The Royal Society of Chemistry 2020 |