DOI:
10.1039/C9RA08208E
(Paper)
RSC Adv., 2020,
10, 86-94
Boronate sol–gel method for one-step fabrication of polyvinyl alcohol hydrogel coatings by simple cast- and dip-coating techniques†
Received
9th October 2019
, Accepted 16th December 2019
First published on 23rd December 2019
Abstract
The self-assembly of polyvinyl alcohol (PVA) and benzene-1,4-diboronic acid (DBA) is employed as a sol–gel method for one-step fabrication of hydrogel coatings with versatile functionalities. A mixture of PVA and DBA in aqueous ethanol is prepared as a coating agent. The long pot life of the mixture allows for the coating of a wide range of materials with hydrogel films by simple cast- and dip-coating techniques. The resultant films show negligible dissolution in water and the intrinsic hydrophilicity of PVA provides the films with functional properties, such as improved antifogging property and resistance to protein and cell fouling. The self-assembling process shows adaptive inclusion properties toward nanoscale materials, such as metal–organic coordination polymers and inorganic nanoparticles, affording composite films. Furthermore, the coating film exhibits a unique secondary functionalization reactivity toward boronic acid-appended fluorescent dyes, through which a variety of materials are converted into fluorescent materials.
Introduction
The self-assembly of molecules on solid surfaces offers rich opportunities to build functional structures as coating films. For the control of the self-assembling processes, supramolecular chemistry serves as a valuable tool through the design of molecular structures and their intermolecular interactions in equilibrium and even nonequilibrium systems.1–3 Among the known supramolecular strategies, harnessing specific interactions between component molecules and material surfaces leads to the formation of well-defined monolayers and multilayers on the materials in a simple manner, and therefore, Langmuir–Blodgett methods,4–9 self-assembled monolayer methods,10–15 and layer-by-layer adsorption methods16–20 have been employed widely for practical applications. More recently, mussel-inspired methods21–24 and metal–phenolic assembly methods25–27 have emerged as alternative coating methods. These methods are based on the self-assembly of network structures from component molecules through oxidative polymerization of dopamine and coordination complexation of polyphenol tannic acid with metal ions, respectively, instead of being driven by specific interactions between component molecules and substrates, and thus allow for the fabrication of stable molecular networks as universal coatings for a wide range of material surfaces.28,29 However, such self-assembling processes often cause spontaneous aggregation of molecular networks that results in the deactivation of the molecular components even in solution, which shortens the lifetime of the solutions as coating agents and, concomitantly, limits their practical usability. Therefore, the development of methods and materials that prevent the deactivation of component molecules in the coating agents is an important task.30–32
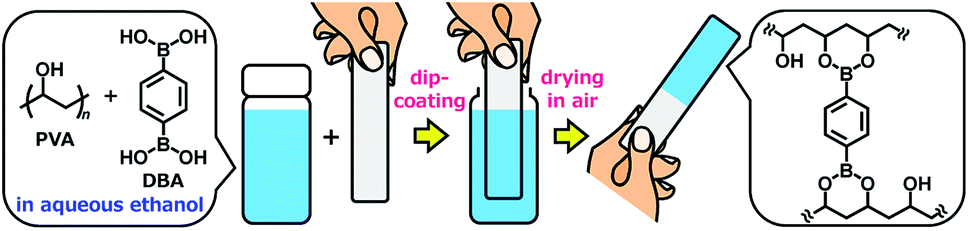
Herein, we report the controlled self-assembly of boronate networks from polyvinyl alcohol (PVA) and benzene-1,4-diboronic acid (DBA) as a sol–gel coating method (Fig. 1). A mixture of PVA and DBA in aqueous ethanol shows negligible changes in viscosity and no precipitation after being stored for more than one month in a sealed bottle, and solvent evaporation of the mixture in open air leads to the formation of a clear and colorless film. Importantly, the resultant PVA/DBA film displays excellent durability in water as well as in organic solvents due to the formation of boronate networks by crosslinking of PVA with DBA through boronate esterification. The films have improved hydrophilicity with antifogging property and resistance to protein and cell fouling owing to the intrinsic properties of PVA. The long pot life of the mixture as a coating agent allows for the coating of a variety of materials including plates of plastics, metals, glass, and silicon, polymer sponges, and glass fiber filter papers by simple cast- and dip-coating methods. The controlled self-assembling process shows adaptive inclusion properties toward a variety of nanoscale materials such as metal–organic coordination polymers and inorganic nanoparticles, affording nanocomposite films. Furthermore, the PVA/DBA film exhibits a unique secondary functionalization reactivity toward boronic acid-appended dyes that converts nonfluorescent materials into fluorescent materials through a simple soaking method under ambient conditions.
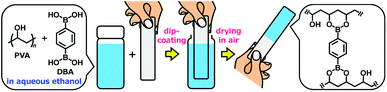 |
| | Fig. 1 A schematic representation of a sol–gel coating method based on the self-assembly of polyvinyl alcohol (PVA) and benzene-1,4-diboronic acid (DBA) through boronate esterification in aqueous ethanol. Solvent evaporation of the mixture in open air leads to the formation of boronate networks self-assembled from PVA and DBA as a coating film. | |
Results and discussion
Controlled boronate self-assembly of polyvinyl alcohol and benzene-1,4-diboronic acid
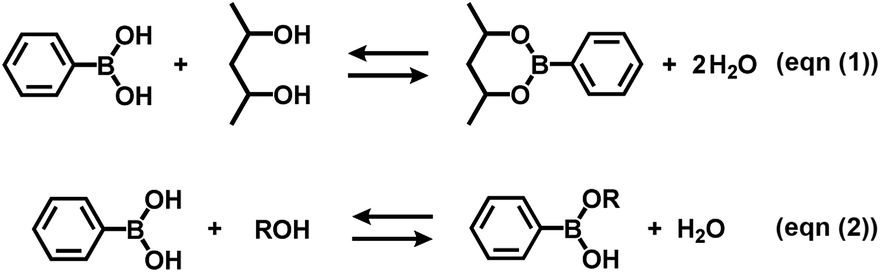
Boronate self-assembly of PVA and DBA was employed as a key approach for the development of a coating method. Molecules having more than two boronic acid groups such as DBA, benzene-1,3-diboronic acid, and biphenyl-4,4′-diboronic acid are known to crosslink PVA in solutions through boronate esterification to form gels with three dimensional network structures at room temperature.33–35 Interestingly, the resultant gels exhibit excellent durability in aqueous solutions over a wide pH range due to the strong binding of the boronic acid groups to the 2,4-pentanediol motifs,36 while boronic acid inherently shows reversible covalent bonding with diols based on the relatively small free energy barriers of the reverse reactions.37–41 Therefore, such boronate gels have been utilized as platforms for adsorbing materials,42,43 microcapsules,44 and solid-based chemosensors45 functioning in aqueous solutions even under basic conditions.42 The excellent durability of the boronate networks prompted us to employ boronate crosslinking of PVA with DBA for the development of a coating agent. However, as in the case of mussel-inspired methods and metal–phenolic assembly methods, PVA and DBA show spontaneous crosslinking reactions in solution that results in the deactivation of the mixtures as coating agents. In fact, the viscosity of the mixture significantly increases through the spontaneous crosslinking reactions, forming gels in less than one minute in dimethylsulfoxide, dimethylformamide, and 2-ethoxyethanol,34 which is a significant drawback for the practical use of the mixture as a coating agent. To overcome this issue, we attempted to prevent the viscosity increase of the mixture by controlling the self-assembling process. After screening several additives such as acids or bases, we found that the addition of water prevents the increase in viscosity in ethanol, and the solution state was thereby significantly prolonged (ESI Fig. S1†). Thus, a mixture at a water fraction of 40% (v/v) showed negligible changes in viscosity even after one month (4.0 ± 0.1 cSt) in a sealed bottle, whereas a mixture at a water fraction of 5% (v/v) underwent gel formation within a few minutes (ESI Fig. S1†). The prolonged pot life with relatively low viscosity allowed us to use the mixture of PVA and DBA as a coating agent. Fig. 2 shows a film prepared on a polystyrene (PS) Petri dish by a cast-coating method using a mixture of PVA and DBA in aqueous ethanol at a 42% (v/v) water fraction ([PVA] unit = 2.3 × 10−1 M and [DBA] = 1.0 × 10−2 M). The resultant clear and colorless film had a thickness of 3.0 ± 0.3 μm (Fig. 2) and the smooth surface was observed by means of field-emission scanning electron microscopy (FE-SEM) (ESI Fig. S2†). Its optical transparency was evinced by a high visible light transmittance (ESI Fig. S3†). Importantly, the film exhibited high durability in water as well as in organic solvents (ESI Fig. S4†). In fact, negligible changes in the weight, thickness, and optical transparency of the film were observed upon immersion in water over a wide pH range even after one week (ESI Tables S1–S3†). To further validate the durability of the film, its structural features were studied by attenuated total reflection Fourier transform infrared (ATR-FT-IR) spectroscopy (ESI Fig. S5†). The spectrum showed a characteristic peak of boronate ester linkages at 660 cm−1 in addition to a peak at 1294 cm−1 (B–O stretching) and peaks arising from PVA at 2926 cm−1 (C–H stretching), 2943 cm−1 (C–H stretching) and 3374 cm−1 (O–H stretching).46–49 Thus, the durability of the film can be attributed to the crosslinking of PVA with DBA through boronate esterification. These results indicate that the addition of water plays a crucial role in preventing boronate esterification between PVA and DBA in the solution state (Fig. 3). In the self-assembling process, water could govern the thermodynamic equilibrium of the dehydrative condensation reaction between the diol moieties of PVA and the boronic acid group of DBA (Fig. 3, (eqn (1))), and also drive thermodynamically and kinetically the reaction toward the solvolysis of the boronic acid groups (Fig. 3, (eqn (2))). Accordingly, in 1H NMR experiments using (2R,4R)-(−)-2,4-pentanediol and phenylboronic acid as model compounds, the addition of water to the mixture in methanol-d4 led to an equilibrium shift toward the reactants (ESI Fig. S6†).
 |
| | Fig. 2 (a) Photograph of the coating film prepared on a polystyrene Petri dish (9 cm in diameter) through a cast-coating method using a mixture of PVA and DBA in aqueous ethanol as a coating agent. The photograph was taken under ambient light. (b) FE-SEM image of the cross section of the PVA/DBA coating film. | |
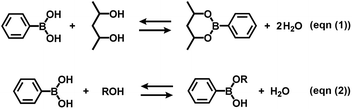 |
| | Fig. 3 Equilibrium equation of the dehydrative condensation between phenylboronic acid and 2,4-pentanediol (eqn (1)), and typical solvolysis reactions of phenylboronic acid with water (R = H) and ethanol (R = CH2CH3) (eqn (2)). Phenylboronic acid and 2,4-pentanediol are represented instead of DBA and PVA, and the formation of sp3-hybridized boronate species in the equilibrium equations are omitted for clarity. | |
Intrinsic properties of the PVA/DBA film
The hydrophilic property of the PVA/DBA film was first evaluated because the hydrophilic modification of material surfaces provides a myriad of functional properties such as improved wettability,50,51 antifogging property,52,53 self-cleaning property,54–56 antifouling property,57–59 and biocompatibility.60–64 These functional properties would be expected for the present PVA/DBA film due to the intrinsic high hydrophilicity of PVA. Thus, the antifogging properties and protein- and cell fouling resistance of the PVA/DBA film were also investigated.
Hydrophilicity. The hydrophilic property of the PVA/DBA film was evaluated by measuring the contact angle of a water droplet on the film (Fig. 4). Water droplets on a uncoated PS Petri dish and a PVA/DBA-coated PS Petri dish showed contact angles of 79° ± 2° and 54° ± 2°, respectively. This significant decrease of the contact angle for the PVA/DBA-coated PS Petri dish indicates that the film maintains the intrinsic hydrophilicity of PVA even after the crosslinking with DBA.
 |
| | Fig. 4 Photographs of water droplets on (a) an uncoated PS Petri dish and (b) a PVA/DBA-coated PS Petri dish. | |
Antifogging property. In this experiment, the inner surface of a PS Petri dish cover was coated with a PVA/DBA film through a cast-coating method. Then, the PVA/DBA-coated PS Petri dish cover and an uncoated PS Petri dish cover were put on respective PS Petri dishes filled with warm water (Fig. 5). From visual inspection, the dish cover without coating treatment showed an obvious fogging (Fig. 5a). On the other hand, the transparency of the PVA/DBA-coated dish cover remained unaltered under the same conditions (Fig. 5b). The optical transmittance at 660 nm of the coated sample showed negligible changes from 91.4% ± 0.2% to 91.8% ± 0.1%, whereas that of the uncoated parent PS Petri dish cover decreased significantly from 90.8% ± 0.3% to 55.3% ± 2.2% after the exposure (ESI Fig. S7†). The antifogging property can be attributed to the hydrophilicity of the PVA/DBA coating film.
 |
| | Fig. 5 Photographs of (a) a PS Petri dish and (b) a PVA/DBA-coated PS Petri dish (4 cm in diameter) after exposure to water vapor from warm water (2 mL, 40 °C). | |
Resistance to protein fouling. The improvement of the resistance to protein and cell fouling of coating films is also significant for applications in various industrial fields including biomedical science.56–64 In the present study, we evaluated the resistance of the PVA/DBA coating film to bovine serum albumin (BSA). Uncoated and PVA/DBA-coated PS samples were immersed in aqueous solutions of BSA (5 mg mL−1 in PBS buffer at pH 7.4, 5 mL) for 24 h at 25 °C, and the adsorption of BSA by the samples were analyzed by means of X-ray photoelectron spectroscopy (XPS). The spectrum of the control PS sample showed an intense peak of N 1s at 400 eV, clearly indicating the adsorption of the BSA to the PS surface (Fig. 6a). In contrast, the coated PS sample gave rise to a minor peak attributable to N 1s at 399 eV (Fig. 6b), which suggests that the PVA/DBA coating film prevents the absorption of the BSA to the surface of the coated PS sample.
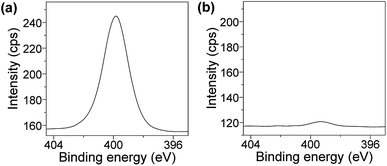 |
| | Fig. 6 XPS spectra at N 1s regions of (a) an uncoated PS Petri dish and (b) a PVA/DBA-coated PS Petri dish treated with BSA in PBS buffer solutions (pH 7.4) for 24 h. | |
Resistance to cell fouling. HeLa cells were cultured for 24 h using PS 24-well culture plates with and without coating treatment (ESI Fig. S8†). The uncoated culture plate showed adhesion of HeLa cells on the surface (Fig. 7a and ESI Fig. S9a†). On the other hand, the formation of aggregated HeLa cells in suspended states was observed in the coated culture plate (ESI Fig. S9b†). In fact, after replacing the culture solution with a fresh solution, negligible adhesion of HeLa cells was detected on the PVA/DBA-coated culture plate (Fig. 7b) and the resistance to cell fouling was also observed toward HaCaT skin keratinocytes (ESI Fig. S10b†). It is noteworthy that HeLa cells incubated using the coated culture plate showed a high survival rate of 86% ± 5% with an abundant cell proliferation from 0.67 × 106 to 2.00 × 106 cells per mL after 24 h, as indicated by trypan blue exclusion assays. The high survival rate of HeLa cells and their robust proliferation are indicative of the low cytotoxicity of the PVA/DBA coating film, which is probably due to the excellent biocompatibility of PVA.65,66
 |
| | Fig. 7 Photographs of (a) an uncoated 24-well PS tissue culture plate and (b) a PVA/DBA-coated 24-well PS tissue culture plate after replacing the HeLa cell-culture solutions with fresh culture media. | |
Potential use of the PVA/DBA film as universal coating
Since the formation of the stable PVA/DBA film is based on the network structures of PVA and DBA and does not require specific interactions with substrates, we envisioned that the present method could be used for the preparation of universal coatings.28,29 To prove our hypothesis, we employed plates of plastics, metals, glass, and silicon, polymer sponges, and glass fiber filter papers as substrates.
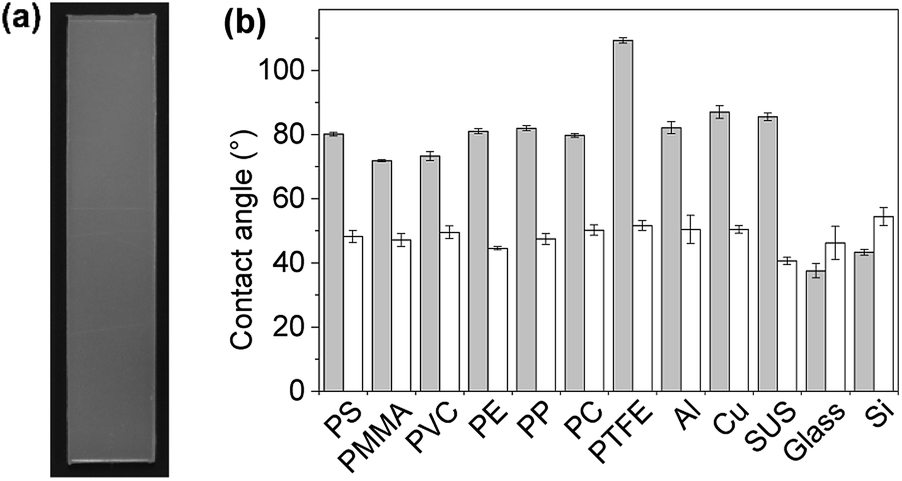
First, plate substrates (50 mm × 10 mm × 1 mm) of polystyrene (PS), polymethylmethacrylate (PMMA), polyvinyl chloride (PVC), polyethylene (PE), polypropylene (PP), polycarbonate (PC), polytetrafluoroethylene (PTFE), aluminum (Al), copper (Cu), stainless steel (SUS), glass (Glass), and silicon (Si) were evaluated (Fig. 8). In this experiment, a dipping method was employed for the coating of these materials with PVA/DBA films, as shown in Fig. 1. Typically, a PE plate was dipped in the coating agent for a few seconds. The resultant plate was dried in air and then rinsed with water several times to afford a colorless and transparent film with a thickness of 0.50 ± 0.13 μm on the PE substrate (Fig. 8a and ESI Fig. S11†). Fig. 8b shows the contact angles of water droplets on the whole series of plate substrates before and after performing the coating treatment. As can be seen, constant values of 48.6° ± 2.6° were obtained for the coated substrates regardless of their chemical composition (ESI Fig. S12–S14†), indicating the formation of PVA/DBA films on these materials. It is noteworthy that this method is applicable to PTFE substrates without requiring any pretreatment such as plasma treatment, most likely due to the amphiphilic nature of PVA. Moreover, the dipping method is applicable not only to plate-shaped substrates but also to materials with complicated structures, such as porous and fibrous materials due to the relatively low viscosity of the coating agent, as was evidenced by the use of polyurethane sponges and glass fiber filter papers as porous and fibrous materials, respectively (Fig. 9). The coating of polyurethane sponges and glass fiber filters was also performed through the dip-coating method. The ATR-FT-IR spectra (ESI Fig. S15 and S16†) and FE-SEM images of the resultant materials (Fig. 9) confirmed the formation of the PVA/DBA coating, while the porous and fibrous structures were preserved.
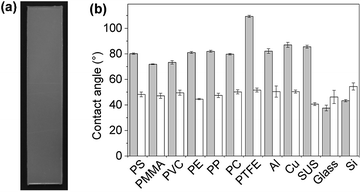 |
| | Fig. 8 (a) Photograph of a PVA/DBA-coated polyethylene (PE) substrate (50 mm × 10 mm × 1 mm) prepared by a dip-coating method. (b) Water contact angles on the uncoated substrates (gray bars) and PVA/DBA-coated substrates (white bars) of PS, polymethyl methacrylate (PMMA), polyvinyl chloride (PVC), PE, polypropylene (PP), polycarbonate (PC), polytetrafluoroethylene (PTFE), aluminum (Al), copper (Cu), stainless steel (SUS), glass (Glass), and silicon (Si). | |
 |
| | Fig. 9 Photographs of (a) a coated polyurethane sponge and (b) a coated glass fiber filter paper. (c) FE-SEM image of a coated glass fiber filter paper. | |
Adaptive inclusion of guest materials in the self-assembling process
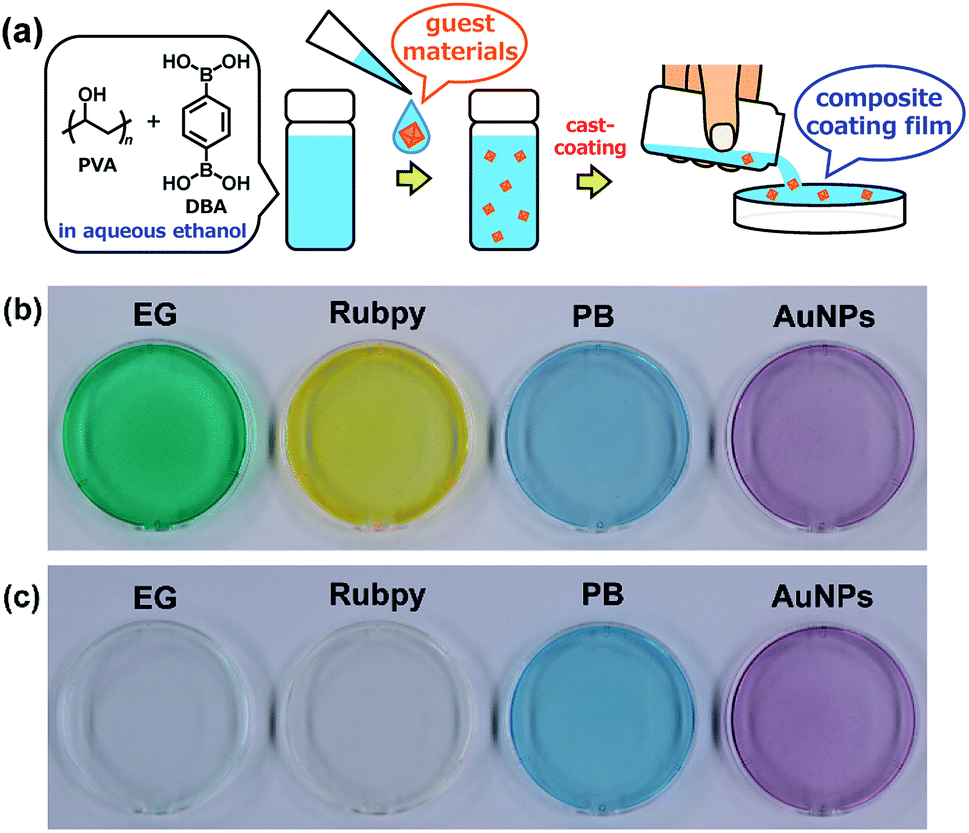
Adaptive encapsulation of guest materials is a unique characteristic of molecular self-assembly.67–70 The adaptability allows for the encapsulation of various materials such as organic dyes, inorganic nanocrystals, and biomacromolecules, regardless of their size, shape, and chemical composition, into the network structures. Thus, the encapsulation ability of the present boronate networks was evaluated. Erio green B (EG), tris(2,2′-bipyridyl)ruthenium(II) chloride (Rubpy), Prussian blue (PB), and gold nanoparticles (AuNPs) were selected as guest materials (Fig. 10). These guest materials were dissolved in water, and the resultant solutions were simply added to the coating agents, respectively. Casting the resultant coating agents on top of PS Petri dish covers afforded transparent films, indicating that the encapsulation of these guest materials proceeded without aggregation in the resultant films. Interestingly, significant leakages of the anionic organic dye EG and the cationic dye Rubpy were observed within a few minutes when the films were immersed in water, whereas negligible leakages of metal–organic coordination polymer nanoparticles (PB) and inorganic nanoparticles (AuNPs) were detected even after immersion in water for 24 h (Fig. 10 and ESI Fig. S17–S21†). This selective leakage of guest materials clearly indicates the guest-dependent diffusive mass transfer in the boronate networks. It is also noteworthy that nanoscale materials such as metal–organic coordination polymer nanoparticles and inorganic nanoparticles are entrapped stably in the films, which could provide a platform for the fabrication of nanocomposite materials in many applications.71–80
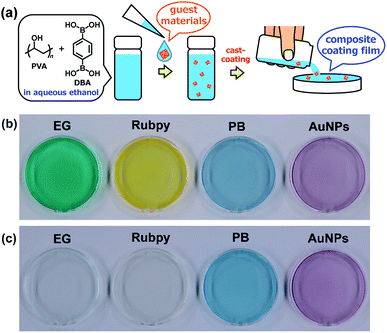 |
| | Fig. 10 (a) Schematic representation of the preparation of composite PVA/DBA coating films. Guest materials in the coating agents are encapsulated into the boronate networks of PVA and DBA upon solvent evaporation of the mixtures. Photographs of composite coating films before (b) and after (c) immersion in water for 2 h. These coating films were prepared on PS Petri dishes (4 cm in diameter) using coating agents containing erio green B (EG), tris(2,2′-bipyridyl)ruthenium(II) chloride (Rubpy), Prussian blue (PB), and gold nanoparticles (AuNPs) as guest materials, respectively. | |
Secondary functionalization reactivity of the PVA/DBA film
Secondary functionalization of coating films is also important for the practical usability of coating agents because it provides the materials with desired properties by attaching chemical modifiers to the coating films.21,29
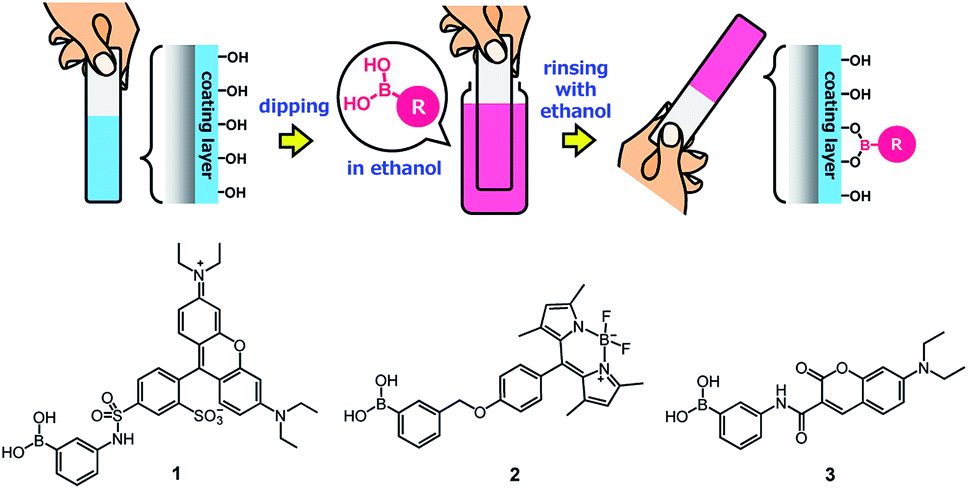
Owing to the presence of abundant unreacted diol moieties in PVA in the films, the PVA/DBA coating films can be modified with boronic acids because we have previously reported the modification of microparticles, films, fibers, and sponges of solid PVA with boronic acid-appended dyes by a simple soaking technique.81–83 Therefore, we investigated the applicability of this functionalization technique to the present PVA/DBA coating films (Fig. 11).
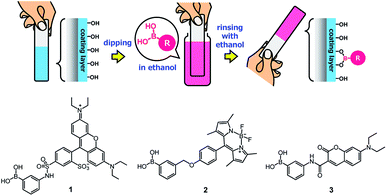 |
| | Fig. 11 Secondary functionalization of a PVA/DBA-coated substrate with a boronic acid as a chemical modifier and chemical structures of boronic acid-appended rhodamine (1), borondipyrromethene (2), and diethylaminocoumarin (3) dyes employed for the evaluation of the secondary functionalization of the PVA/DBA coating films. | |
A series of fluorescent dyes having boronic acid groups were employed as chemical modifiers to evaluate the secondary functionalization reactivity of the PVA/DBA coating films (Fig. 11). Fig. 12 shows the photographs of an uncoated PE plate and a PVA/DBA-coated PE plate after soaking in ethanol solutions of a boronic acid-appended rhodamine dye (1) at room temperature. The PVA/DBA-coated PE plate clearly developed a red color due to the attachment of 1, whereas the uncoated PE plate remained colorless. The attachment of 1 was also evinced by the observation of red fluorescence from the PVA/DBA-coated PE plate under UV light (λem = 365 nm), and a fluorescence peak was detected at 592 nm (Fig. 12e) with a fluorescent quantum yield of 0.40. The selective binding of 1 to the PVA/DBA-coated PE plate indicates the occurrence of boronate esterification of 1 with the PVA/DBA coating film on the PE plate. In fact, the use of a boronic acid-free rhodamine dye as a control dye resulted in negligible adsorption of this dye to the PVA/DBA-coated PE plate (ESI Fig. S22 and S23†). The PVA/DBA-coated PE substrates were also converted into fluorescent materials when a boronic acid-appended borondipyrromethene dye (2) (Fig. 13a) and a boronic acid-appended diethylaminocoumarin dye (3) were employed (ESI Fig. S24–S28†). In addition to the PE plates, PVA/DBA-coated polyurethane sponges (Fig. 13b) and glass fiber filter papers (Fig. 13c) were functionalized with these boronic acid-appended dyes, affording fluorescent materials with relatively high quantum yields (ESI Fig. S29–S40†). The digital microscope image of the fluorescence glass fiber filter papers revealed that the fluorescent boronic acids were uniformly attached to the fibrous structures (ESI Fig. S41†). The durability of the series of functionalized materials in water (ESI Fig. S42–S44†) illustrates the potential utility of the secondary functionalization method for the creation of functional hydrogel films on a wide range of materials via the boronate sol–gel method (ESI Fig. S45†).
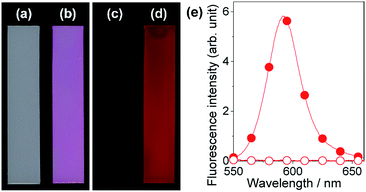 |
| | Fig. 12 Photographs of an uncoated PE substrate (a and c) and a PVA/DBA-coated PE substrate (b and d) after immersion in ethanol in the presence of a boronic acid-appended rhodamine dye (1) as a chemical modifier. The photographs were taken under ambient light (a and b) and UV light at 365 nm (c and d), respectively. (e) Fluorescence spectra of the uncoated PE substrate (open circle) and the PVA/DBA-coated PE substrate (solid circle) after the treatments. | |
 |
| | Fig. 13 Photographs of secondary functionalized PVA/DBA-coated PE substrate with 2 (a), PVA/DBA-coated polyurethane sponge with 3 (b), and PVA/DBA-coated glass fiber filter paper with 1 (c). The photograph was taken under UV light (365 nm). | |
Conclusions
We have demonstrated the controlled self-assembly of polyvinyl alcohol (PVA) and benzene-1,4-diboronic acid (DBA) as a sol–gel coating method. A mixture of PVA and DBA in aqueous ethanol showed negligible changes in viscosity and no precipitation after being stored for more than one month in a sealed bottle, and the solvent evaporation of the mixture in open air afforded a clear and colorless film. The resultant film exhibited excellent durability in water and in organic solvents, as well as improved antifogging property and resistance to protein and cell fouling. The prolonged pot life of the mixture allowed preparing coating films by using cast- and dip-coating methods. In addition, due to the formation of boronate network structures, the coating method could be applied to a variety of materials regardless of their shapes and chemical characteristics. The adaptive encapsulation of nanoscale guest materials such as metal–organic coordination polymer nanoparticles and inorganic nanoparticles to form nanocomposite materials was observed during the self-assembling process. Furthermore, the PVA/DBA film exhibited a unique secondary functionalization reactivity, binding a series of boronic acid-appended dyes, which allowed converting nonfluorescent materials into fluorescent materials by a simple immersion method under ambient conditions. Antimicrobial evaluation of the present coating films such as experiments using Gram-negative bacteria, Gram-positive bacteria and fungi is the subject of future investigation to evaluate their practical utility.
The present boronate self-assembly constitutes a supramolecular sol–gel coating method that offers rapid and easy access to various functional coating films with desired properties for a wide range of materials used in the laboratory and those in our daily life.
Experiment
Materials and methods
Unless otherwise described, reagents and solvents used for this study were commercially available and used as supplied. Water-soluble polyvinyl alcohol (PVA) was purchased from Sigma-Aldrich (MW = 9000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80% hydrolyzed, product number: 360627) and used as supplied. PB was purchased from Sigma-Aldrich (product number: 03899) and used as supplied. Gold nanoparticles were prepared according to a reported method using N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) as a reducing agent.69 Compounds 1 and 2 were synthesized following reported procedures.81,84 HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (heat inactivated at 56 °C before use) and 1% penicillin/streptomycin/amphotericin B at 37 °C in a humidified atmosphere with 5% CO2.
000, 80% hydrolyzed, product number: 360627) and used as supplied. PB was purchased from Sigma-Aldrich (product number: 03899) and used as supplied. Gold nanoparticles were prepared according to a reported method using N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) as a reducing agent.69 Compounds 1 and 2 were synthesized following reported procedures.81,84 HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (heat inactivated at 56 °C before use) and 1% penicillin/streptomycin/amphotericin B at 37 °C in a humidified atmosphere with 5% CO2.
FE-SEM was conducted by a JSM-7500F (JEOL, acceleration voltage: 5 kV). For FE-SEM measurements, specimens were coated with osmium by using a Meiwafosis Neoc-Pro osmium coater. FT-IR spectra were recorded on a JASCO FT/IR-4100 spectrometer equipped with a diamond ATR crystal (JASCO PRO 450S). Contact angle measurements were performed using a Drop Master DM300 (Kyowa Interface Science). XPS spectra were acquired using a JPS-9010MX (JEOL). Digital photographs were taken with a digital camera (Canon, EOS Kiss X8i). UV-vis absorption spectra were recorded on a UV-3600 spectrometer (Shimadzu). Fluorescence measurements were performed using a spectrofluorometer (JASCO, FP-8500) equipped with a substandard light source (ESC-842), and the fluorescence quantum yields were determined by using an integrating sphere (IL835). The digital microscope images of the coated glass fiber filter paper functionalized with 1 were recorded by a HIROX RH-8800 digital microscope.
NMR spectra were recorded on an AVANCE-500 spectrometer (Bruker). Tetramethylsilane (TMS) was used as an internal standard (δ = 0 ppm) for 1H NMR (500 MHz) and 13C NMR (126 MHz) measurements. For 11B NMR (160 MHz) measurements, BF3·OEt2 (δ = 0 ppm) was used as an external standard. All NMR spectra were recorded at 298 K. High-resolution mass spectrometry (HRMS) was performed by a micrOTOF mass spectrometer (Bruker) in an electrospray ionization (ESI) mode, and Tuning-Mix was used as a calibration standard.
Synthesis
7-(Diethylamino)-N-(3-dihydroxyborylphenyl)-2-oxo-2H-chromene-3-carboxamide (3): 3-aminophenyboronic acid (0.16 g, 1.17 mmol) and 7-(diethylamino)coumarin-3-carboxylic acid (0.30 g, 1.15 mmol) were dissolved in dry dichloromethane (20 mL) under a nitrogen atmosphere, and the resultant solution was cooled using an ice bath. Then, 1-[bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide hexafluorophosphate (HBTU, 0.46 g, 1.20 mmol) and N,N-diisopropylethylamine (0.19 g, 1.5 mmol) were added to the solution under a nitrogen atmosphere at 0 °C, and the mixture was stirred at room temperature for 12 h. Dichloromethane (200 mL) was then added to the reaction mixture. The organic layer was washed with saturated aqueous ammonium chloride solution (200 mL) and then dried over anhydrous sodium sulfate. The resultant solution was concentrated using a rotary evaporator to afford compound 3 as a yellow precipitate (0.38 g) in 57% yield. 1H NMR (500 MHz, DMSO-d6): δ (ppm) = 1.16 (t, 6H, J = 7.0 Hz), 3.51 (q, 4H, J = 7.0 Hz), 6.69 (d, 1H, J = 2.2 Hz), 6.86 (dd, 1H, J = 2.4 and 9.1 Hz), 7.34 (dd, 1H, J = 7.7 and 7.7 Hz), 7.53 (d, 1H, J = 7.4 Hz), 7.75 (d, 1H, J = 9.1 Hz), 7.83 (s, 1H), 7.99 (d, 1H, J = 9.3 Hz), 8.10 (s, 2H), 8.78 (s, 1H), 10.76 (s, 1H); 13C NMR (126 MHz, DMSO-d6): δ (ppm) = 12.2, 44.3, 95.8, 107.8, 109.0, 110.3, 119.6, 123.7, 128.9, 131.8, 138.2, 148.1, 152.7, 157.3, 160.6, 162.1; 11B NMR (160 MHz, DMSO-d6): δ (ppm) = 28.1; HRMS (ESI): m/z [3 + H]+ calcd. for C20H22BN2O5, 381.1620; found, 381.1621.
Preparation of coating agents
PVA (1.21 g) was dissolved in a mixture of water (42 mL) and ethanol (33 mL). The suspension was kept at 60 °C until complete dissolution, and the resultant PVA solution was cooled down to room temperature. Separately, DBA (0.17 g) was dissolved in ethanol (25 mL). The ethanol solution was then added to the PVA solution to obtain a colorless solution with 1.2% (w/v) of PVA (2.3 × 10−1 unit M) and 0.17% (w/v) of DBA (1.0 × 10−2 M) in a 58% (v/v) ethanol solution with 42% (v/v) water.
Coating procedures
To prepare PVA/DBA films in Petri dishes (9 cm in diameter), the mixture of PVA and DBA in aqueous ethanol (2.4 mL) was poured into the Petri dish directly through a cast-coating method. The sample was dried under ambient conditions and the resultant film was rinsed with water several times. Plate samples of polystyrene, polymethyl methacrylate, polyvinyl chloride, polyethylene, polypropylene, polycarbonate, polytetrafluoroethylene, aluminum, copper, stainless steel, glass, and silicon plates were employed without pretreatment. These materials were coated by a dipping method. Polyurethane sponges and glass fiber filter papers were also coated as described for the plate samples.
Antifogging property and resistance to protein and cell fouling
The inner surface of a PS Petri dish cover (4 cm in diameter) was coated with the PVA/DBA film through the cast-coating method by using 0.45 mL of mixture. The coated dish cover was put on a PS Petri dish bottom with warm water (2 mL, 40 °C), and the optical transmittance of the exposure cover was recorded at 660 nm by UV-vis absorption spectroscopy.
To evaluate the resistance of the PVA/DBA film toward protein fouling, the uncoated PS sample and the coated PS sample were immersed in aqueous solutions of BSA (5 mg mL−1, PBS buffer, pH 7.4, 5 mL) for 24 h at 25 °C, and the resultant samples were analyzed by means of XPS. In the experiments for evaluating the antifouling property of the PVA/DBA film toward HeLa cells, the inner surface of a PS 24-well culture plate was coated with the PVA/DBA films by casting 20 μL of mixture into each well (1.5 cm in diameter) to obtain films with a thickness of 0.52 ± 0.14 μm. HeLa cells (0.67 × 106 cells per mL) in the culture media (500 μL) were cultured in each cell of the coated culture plate for 24 h. The resultant cells were collected by centrifugation, and 250 μL of a 0.25 w/v% trypsin solution was added to the collected cells. After 1 min incubation, the culture media were added to the mixtures, and the cell viability was evaluated by a trypan blue exclusion assay using a Countess II automated cell counter. HaCaT skin keratinocytes (1 × 106 cells per mL) in the culture media (400 μL) were also employed.
Preparation of composite PVA/DBA coating films
EG, Rubpy and PB were dissolved in water (4 mM). The particle diameters of PB (4 mM) and AuNPs (5 mM) in the aqueous solutions were determined to be 85 ± 23 nm and 6.2 ± 1.4 nm, respectively. The aqueous solutions were added to mixtures of PVA and DBA in aqueous ethanol to obtain solutions with 1.2% (w/v) of PVA and 0.17% (w/v) of DBA in a 58% (v/v) ethanol solution with 42% (v/v) water containing the guest materials (0.8 mM for EG, Rubpy and PB, and 0.5 mM for AuNPs, respectively). The top surfaces of PS Petri dish covers (4 cm) were coated with the coating agents (0.48 mL) by a cast-coating method. The samples were dried under ambient conditions, and then the UV-vis absorption spectra of the resultant samples were measured before and after immersion in water.
Secondary functionalization of PVA/DBA coating films
PE substrates, polyurethane sponges, and glass fiber filters were coated with PVA/DBA films through a dip-coating method, and the resultant samples were dried under ambient conditions. The coated PE substrates were then immersed in ethanol solutions of dyes (50 mL, 2.0 × 10−5 M) for 10 min, and the resultant samples were rinsed with ethanol several times. The coated polyurethane sponges and glass fiber filter papers were immersed in the ethanol solutions for 1 min and 30 s, respectively, and the resultant samples were rinsed with ethanol several times.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP18K05088 and JP19H02704.
References
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- M. Fialkowski, K. J. M. Bishop, R. Klajn, S. K. Smoukov, C. J. Campbell and B. A. Grzybowski, J. Phys. Chem. B, 2006, 110, 2482–2496 CrossRef CAS PubMed.
- J. A. Zasadzinski, R. Viswanathan, L. Madsen, J. Garnaes and D. K. Schwartz, Science, 1994, 263, 1726–1733 CrossRef CAS PubMed.
- G. Decher, J. D. Hong and J. Schmitt, Thin Solid Films, 1992, 210, 831–835 CrossRef.
- K. Ariga and T. Kunitake, Acc. Chem. Res., 1998, 31, 371–378 CrossRef CAS.
- D. H. McCullough and S. L. Regen, Chem. Commun., 2004, 2787–2791 RSC.
- K. Ariga, T. Nakanishi and J. P. Hill, Soft Matter, 2006, 2, 465–477 RSC.
- K. Ariga, Y. Yamauchi, T. Mori and J. P. Hill, Adv. Mater., 2013, 25, 6477–6512 CrossRef CAS PubMed.
- L. H. Dubois and R. G. Nuzzo, Annu. Rev. Phys. Chem., 1992, 43, 437–463 CrossRef CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- R. Maoz, E. Frydman, S. R. Cohen and J. Sagiv, Adv. Mater., 2000, 12, 424–429 CrossRef CAS.
- D. S. Ginger, H. Zhang and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 30–45 CrossRef PubMed.
- S. Onclin, B. J. Ravoo and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2005, 44, 6282–6304 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- P. Bertrand, A. Jonas, A. Laschewsky and R. Legras, Macromol. Rapid Commun., 2000, 21, 319–348 CrossRef CAS.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271–1293 CrossRef CAS.
- K. Ariga, J. P. Hill and Q. M. Ji, Phys. Chem. Chem. Phys., 2007, 9, 2319–2340 RSC.
- J. Borges and J. F. Mano, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- Q. Wei, K. Achazi, H. Liebe, A. Schulz, P. L. M. Noeske, I. Grunwald and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 11650–11655 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS PubMed.
- J. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J. J. Richardson, Y. Yan, K. Peter, D. von Elverfeldt, C. E. Hagemeyer and F. Caruso, Angew. Chem., Int. Ed. Engl., 2014, 53, 5546–5551 CrossRef CAS PubMed.
- M. A. Rahim, H. Ejima, K. L. Cho, K. Kempe, M. Müllner, J. P. Best and F. Caruso, Chem. Mater., 2014, 26, 1645–1653 CrossRef CAS.
- H. A. Lee, Y. Ma, F. Zhou, S. Hong and H. Lee, Acc. Chem. Res., 2019, 52, 704–713 CrossRef CAS PubMed.
- Q. Wei and R. Haag, Mater. Horiz., 2015, 2, 567–577 RSC.
- S. H. Hong, S. Hong, M.-H. Ryou, J. W. Choi, S. M. Kang and H. Lee, Adv. Mater. Interfaces, 2016, 3, 1500857 CrossRef.
- C. Schlaich, M. Li, C. Cheng, I. S. Donskyi, L. Yu, G. Song, E. Osorio, Q. Wei and R. Haag, Adv. Mater. Interfaces, 2018, 5, 1701254 CrossRef.
- X. Yao, J. Liu, C. Yang, X. Yang, J. Wei, Y. Xia, X. Gong and Z. Suo, Adv. Mater., 2019, 31, 1903062 CrossRef PubMed.
- R. Nishiyabu, H. Kobayashi and Y. Kubo, RSC Adv., 2012, 2, 6555–6561 RSC.
- T. T. Duncan and R. G. Weiss, Colloid Polym. Sci., 2018, 296, 1047–1056 CrossRef CAS.
- T. T. Duncan, B. H. Berrie and R. G. Weiss, ACS Appl. Mater. Interfaces, 2017, 9, 28069–28078 CrossRef CAS PubMed.
- C. D. Roy and H. C. Brown, J. Organomet. Chem., 2007, 692, 784–790 CrossRef CAS.
- N. Fujita, S. Shinkai and T. D. James, Chem.–Asian J., 2008, 3, 1076–1091 CrossRef CAS PubMed.
- K. Severin, Dalton Trans., 2009, 5254–5264 RSC.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124–1150 RSC.
- S. D. Bull, M. G. Davidson, J. M. H. van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS PubMed.
- M. Arzt, C. Seidler, D. Y. W. Ng and T. Weil, Chem.–Asian J., 2014, 9, 1994–2003 CrossRef CAS PubMed.
- G. M. Peters, X. Chi, C. Brockman and J. L. Sessler, Chem. Commun., 2018, 54, 5407–5409 RSC.
- S. Ren, P. Sun, A. Wu, N. Sun, L. Sun, B. Dong and L. Zheng, New J. Chem., 2019, 43, 7701–7707 RSC.
- M. A. P. Nunes, P. M. P. Gois, M. E. Rosa, S. Martins, P. C. B. Fernandes and M. H. L. Ribeiro, Tetrahedron, 2016, 72, 7293–7305 CrossRef CAS.
- R. Nishiyabu, S. Ushikubo, Y. Kamiya and Y. Kubo, J. Mater. Chem. A, 2014, 2, 15846–15852 RSC.
- R. W. Tilford, W. R. Gemmill, H.-C. zur Loye and J. J. Lavigne, Chem. Mater., 2006, 18, 5296–5301 CrossRef CAS.
- B. M. Rambo and J. J. Lavigne, Chem. Mater., 2007, 19, 3732–3739 CrossRef CAS.
- R. Nishiyabu, S. Teraoka, Y. Matsushima and Y. Kubo, ChemPlusChem, 2012, 77, 201–209 CrossRef CAS.
- M. K. Smith and B. H. Northrop, Chem. Mater., 2014, 26, 3781–3795 CrossRef CAS.
- S. Li, J. Huang, Z. Chen, G. Chen and Y. Lai, J. Mater. Chem. A, 2017, 5, 31–55 RSC.
- R. K. Gupta, G. J. Dunderdale, M. W. England and A. Hozumi, J. Mater. Chem. A, 2017, 5, 16025–16058 RSC.
- I. R. Durán and G. Laroche, Adv. Colloid Interface Sci., 2019, 263, 68–94 CrossRef PubMed.
- I. R. Durán and G. Laroche, Prog. Mater. Sci., 2019, 99, 106–186 CrossRef.
- P. Ragesh, V. Anand Ganesh, S. V. Nair and A. S. Nair, J. Mater. Chem. A, 2014, 2, 14773–14797 RSC.
- V. A. Ganesh, H. K. Raut, A. S. Nair and S. Ramakrishna, J. Mater. Chem., 2011, 21, 16304–16322 RSC.
- I. P. Parkin and R. G. Palgrave, J. Mater. Chem., 2005, 15, 1689–1695 RSC.
- R. Zhang, Y. Liu, M. He, Y. Su, X. Zhao, M. Elimelech and Z. Jiang, Chem. Soc. Rev., 2016, 45, 5888–5924 RSC.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- D. Rana and T. Matsuura, Chem. Rev., 2010, 110, 2448–2471 CrossRef CAS.
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS.
- C. Blaszykowski, S. Sheikh and M. Thompson, Chem. Soc. Rev., 2012, 41, 5599–5612 RSC.
- S. R. Meyers and M. W. Grinstaff, Chem. Rev., 2012, 112, 1615–1632 CrossRef CAS PubMed.
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed.
- N. Hadjesfandiari, K. Yu, Y. Mei and J. N. Kizhakkedathu, J. Mater. Chem. B, 2014, 2, 4968–4978 RSC.
- A. Miyamoto, S. Lee, N. F. Cooray, S. Lee, M. Mori, N. Matsuhisa, H. Jin, L. Yoda, T. Yokota, A. Itoh, M. Sekino, H. Kawasaki, T. Ebihara, M. Amagai and T. Someya, Nat. Nanotechnol., 2017, 12, 907–913 CrossRef CAS PubMed.
- A. C. Wilkinson, R. Ishida, M. Kikuchi, K. Sudo, M. Morita, R. V. Crisostomo, R. Yamamoto, K. M. Loh, Y. Nakamura, M. Watanabe, H. Nakauchi and S. Yamazaki, Nature, 2019, 571, 117–121 CrossRef CAS PubMed.
- I. Imaz, J. Hernando, D. Ruiz-Molina and D. Maspoch, Angew. Chem., Int. Ed., 2009, 48, 2325–2329 CrossRef CAS PubMed.
- R. Nishiyabu, N. Hashimoto, T. Cho, K. Watanabe, T. Yasunaga, A. Endo, K. Kaneko, T. Niidome, M. Murata, C. Adachi, Y. Katayama, M. Hashizume and N. Kimizuka, J. Am. Chem. Soc., 2009, 131, 2151–2158 CrossRef CAS PubMed.
- R. Nishiyabu, C. Aimé, R. Gondo, T. Noguchi and N. Kimizuka, Angew. Chem., Int. Ed., 2009, 48, 9465–9468 CrossRef CAS PubMed.
- X. Yan, P. Zhu, J. Fei and J. Li, Adv. Mater., 2010, 22, 1283–1287 CrossRef CAS PubMed.
- Q. Wu, X. J. Wang, S. A. Rasaki, T. Thomas, C. X. Wang, C. Zhang and M. H. Yang, J. Mater. Chem. C, 2018, 6, 4508–4515 RSC.
- W. Zhou, J. Zhuang, W. Li, C. Hu, B. Lei and Y. Liu, J. Mater. Chem. C, 2017, 5, 8014–8021 RSC.
- A. Cosgun, R. Fu, W. Jiang, J. Li, J. Song, X. Song and H. Zeng, J. Mater. Chem. C, 2015, 3, 257–264 RSC.
- R. Z. Liang, D. P. Yan, R. Tian, X. J. Yu, W. Y. Shi, C. Y. Li, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2014, 26, 2595–2600 CrossRef CAS.
- Z. Q. Cheng, F. L. Zhang, W. Liu, L. Y. Cui and L. J. Kang, RSC Adv., 2015, 5, 54182–54187 RSC.
- E. F. de Melo, N. D. C. Santana, K. G. B. Alves, G. F. de Sá, C. P. de Melo, M. O. Rodrigues and S. A. Júnior, J. Mater. Chem. C, 2013, 1, 7574–7581 RSC.
- Y. Deng, D. Zhao, X. Chen, F. Wang, H. Song and D. Shen, Chem. Commun., 2013, 49, 5751–5753 RSC.
- V. K. Rao and T. P. Radhakrishnan, J. Mater. Chem. A, 2013, 1, 13612–13618 RSC.
- X. Fang, H. Ma, S. L. Xiao, M. W. Shen, R. Guo, X. Y. Cao and X. Y. Shi, J. Mater. Chem., 2011, 21, 4493–4501 RSC.
- H. M. Yang, J. R. Hwang, D. Y. Lee, K. B. Kim, C. W. Park, H. R. Kim and K. W. Lee, Sci. Rep., 2018, 8, 11476 CrossRef PubMed.
- R. Nishiyabu and A. Shimizu, Chem. Commun., 2016, 52, 9765–9768 RSC.
- R. Nishiyabu, S. Iizuka, S. Minegishi, H. Kitagishi and Y. Kubo, Chem. Commun., 2017, 53, 3563–3566 RSC.
- R. Nishiyabu, M. Tomura, T. Okade and Y. Kubo, New J. Chem., 2018, 42, 7392–7398 RSC.
- R. Nishiyabu, Y. Sugino and Y. Kubo, Chem. Commun., 2013, 49, 9869–9871 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, spectroscopic and analytical data. See DOI: 10.1039/c9ra08208e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yuki Takahashia,
Taro Yabukia,
Shoji Gommoria,
Yuki Yamamotoa,
Hiroaki Kitagishi
*a,
Yuki Takahashia,
Taro Yabukia,
Shoji Gommoria,
Yuki Yamamotoa,
Hiroaki Kitagishi