
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical exploration of the forces governing the interaction between gold–phthalocyanine and gold surface clusters†
Pablo Castro-Latorre*a,
Sebastián Miranda-Rojas*b and
Fernando Mendizabal *a
*a
aDepartamento de Química, Facultad de Ciencias, Universidad de Chile, Casilla 653, Santiago, Chile. E-mail: hagua@uchile.cl
bDepartamento de Ciencias Químicas, Facultad de Ciencias Exactas, Universidad Andres Bello, Avenida República 275, Santiago, Chile. E-mail: sebastian.miranda@unab.cl
First published on 23rd January 2020
Abstract
Here we aim to explore the nature of the forces governing the adsorption of gold–phthalocyanine on gold substrates. For this, we designed computational models of metal-free phthalocyanine and gold–phthalocyanine deposited over a gold metallic surface represented by cluster models of different sizes and geometries. Thereby, we were able to determine the role of the metal center and of the size of the substrate in the interaction process. For this purpose, we worked within the framework provided by density functional theory, were the inclusion of the semi-empirical correction of the dispersion forces of Grimme's group was indispensable. It has been shown that the interaction between molecules and surfaces is ruled by van der Waals attractive forces, which determine the stabilization of the studied systems and their geometric properties. Their contribution was characterized by energy decomposition analysis and through the visualization of the dispersion interactions by means of the NCI methodology. Moreover, calculations of Density of States (DOS) showed that the molecule-surface system displays a metal–organic interface evidenced by changes in their electronic structure, in agreement with a charge transfer process found to take place between the interacting parts.
Introduction
Nowadays, nanotechnology is a field that attracts a lot of attention from different branches of the scientific community, given that it allows modification of the properties of materials at the molecular level.1 From this perspective, supramolecular chemistry is playing a fundamental role in the development of new applications in nanoscience, since this area of chemistry is more concerned with the intermolecular interactions between different molecular species. These interactions are labelled as non-covalent and are described by electrostatic interactions, hydrogen bonding and van der Waals interactions. The latter holds a special relevance in the description of intermolecular interactions in supramolecular chemistry as it requires precise methods of quantum chemistry to be properly studied.2 A particular system that highlights these properties are phthalocyanines (Pcs).3 They are a well-known class of organic aromatic macrocycles that present good thermal and chemical stability with rich catalytic, electrocatalytic, and coordination properties. Pcs can be modified through the addition of a metal atom at the center of the macrocycle, giving place to a metal phthalocyanine (MPc) as shown in Fig. 1. These are important compounds since they allow the modification of the properties of supramolecular complexes formed by these molecules through self-assembled monolayers.4–8 In this type of interaction, the molecules can interact with the surface of solids forming covalent bonds or deposited over the surface where the most relevant interactions are of non-covalent in nature.9,10 One of the most studied systems is the modification of gold surface with various types of MPcs through direct adsorption on the surface9,11–14 explored by a variety of surface techniques, mainly scanning tunnelling microscopy (STM).4,14–16 Also, there are extensive experimental and theoretical studies regarding first row transition metals atoms modifying Pcs, such as Fe, Co, Cu,10,13–26 but little work has been done using an Au atom coordinating the Pc.27–29 Also, there are theoretical works about the AuPc using DFT in which it has been shown that the spin-density pattern is fully delocalized with an oscillating behaviour.30,31 This is an interesting system since it allows us to study the stabilization of the uncommon +2 oxidation state of Au and to evaluate the aurophilic interaction32,33 between the Au center of the AuPc and the Au(111) surface. The proposed model seeks to offer relevant information about self-assembled monolayers, regarding the interaction between a single molecule and a metallic surface, through ab initio modelling and thus obtaining information of their properties at the molecular level and compare with the experimental system CuPc which holds the same d9 electronic configuration for the metal center.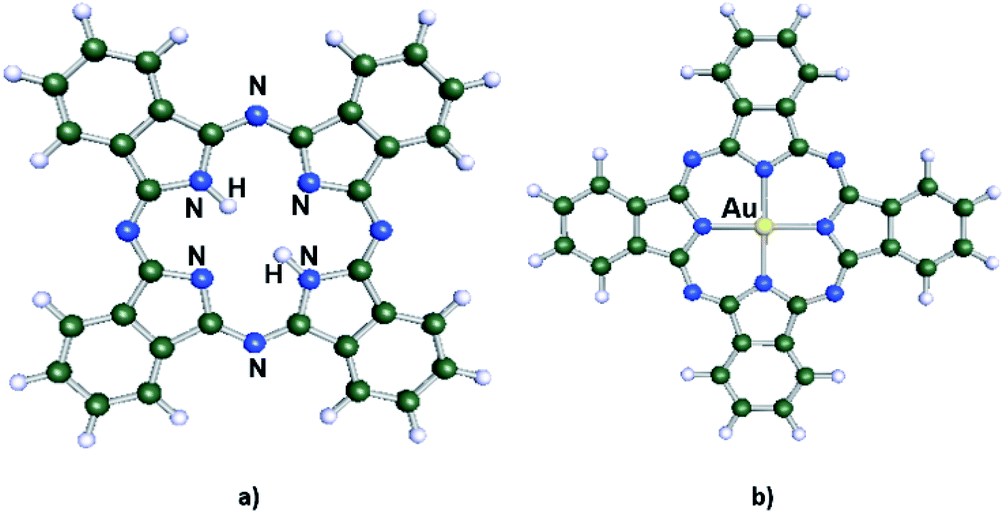 | ||
| Fig. 1 Schematic representation of the phthalocyanine macrocycle. (a) Free phthalocyanine (H2Pc) and (b) phthalocyanine with gold as metal center (AuPc) (color code for the atoms type: C = gray, N = blue, H = white, Au = yellow). | ||
Results and discussion
The optimized structures of the Au(II)Pc…Au-surface complexes shown in Fig. 2 adopted a bridge-like conformation, where the metal center was located above a Au–Au bond, interacting with both gold atoms from the surface. The interaction distances between the metal center and the closest gold atoms from the surface along with other selected structural parameters are listed in Table 1. As detailed in the Theoretical and computational details section, we designed four models of different sizes and structures, model1 and model2 which contain 3 layers, with 26 and 58 gold atoms in total, respectively. Then, we have model3 and model4, which are planar models of 58 and 61 gold atoms, respectively. The results for model1 and model2 showed only a slight shortening in one of the interaction distances between the metal center and a gold atom from the surface after the increase of the cluster size, which in some cases is compensated with the increase of the other distance. On the other hand, the smaller model1 cluster causes a major geometrical distortion on both H2Pc and AuPc, as can be seen in Fig. 2, mainly due to an artificial interaction with the cluster border that causes a shortening in the distance between the molecule and the surface's plane that is appreciated in the systems without the dispersion correction. The incorporation of model1, the smaller cluster in this study, was motivated by previous studies,34–36 which have shown the usefulness of this cluster size in SAM's based electrocatalysis and that will allow to evidence the edge effects for this cluster size. Further results will be presented in the ESI.†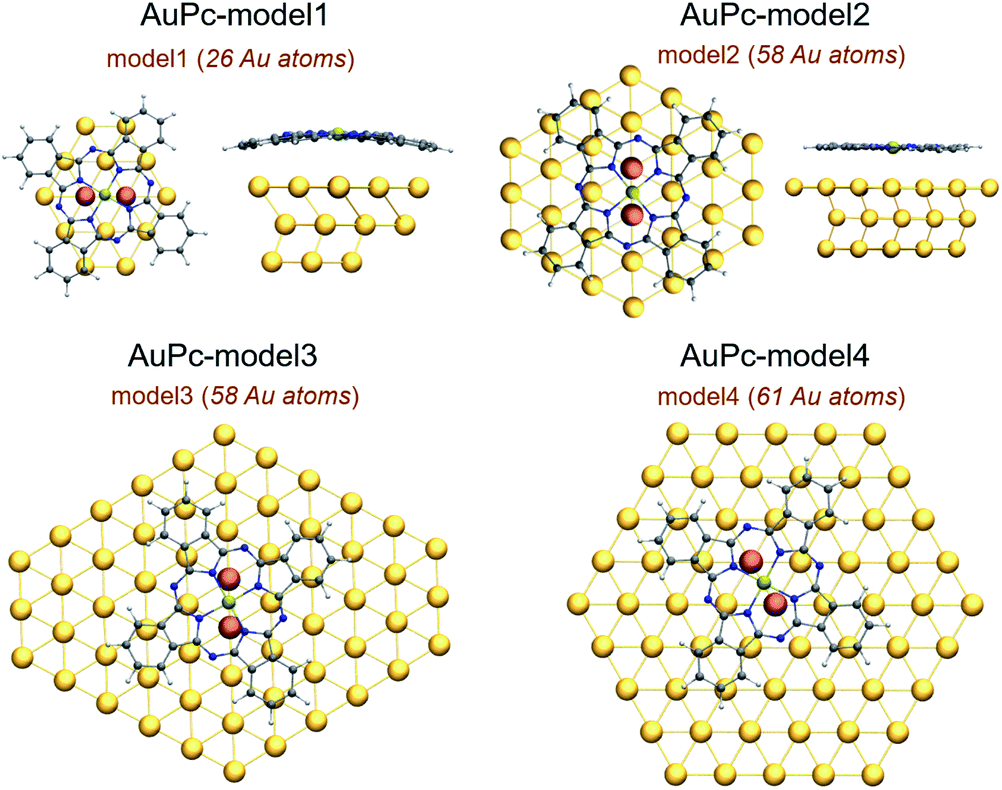 | ||
| Fig. 2 Structure of the interaction mode of AuPc with model1 to model4. In orange are the Au atoms with which the metal center interacts. | ||
| System | Method | Au–Au(1)a | Au–Au(2)b | Au–M–Au | Mol.–surf.c |
|---|---|---|---|---|---|
| a Closest Au–Ausurf. distance.b Second closest Au–Ausurf. distance. In the case of H2Pc, this magnitude is the distance between the H atoms in the center of the molecule and the closest Au atoms in the surface.c Distance from the surface's plane to the molecule's plane. | |||||
| AuPc-model1 | PBE | 3.86 | 3.90 | 43.7° | 3.34 |
| PBE-D3 | 3.54 | 3.60 | 46.8° | 3.18 | |
| H2Pc-model1 | PBE | 3.74 | 3.80 | 3.46 | |
| PBE-D3 | 3.45 | 3.53 | 3.32 | ||
| AuPc-model2 | PBE | 3.81 | 4.04 | 43.1° | 3.60 |
| PBE-D3 | 3.52 | 3.77 | 45.8° | 3.30 | |
| H2Pc-model2 | PBE | 3.71 | 3.97 | 3.53 | |
| PBE-D3 | 3.32 | 3.56 | 3.27 | ||
| AuPc-model3 | PBE-D3 | 3.61 | 3.63 | 47.0° | 3.34 |
| H2Pc-model3 | PBE-D3 | 3.53 | 3.54 | 3.27 | |
| AuPc-model4 | PBE-D3 | 3.54 | 3.73 | 46.6° | 3.37 |
| H2Pc-model4 | PBE-D3 | 3.38 | 3.43 | 3.30 | |
The effect of the inclusion of the dispersion correction was assessed using model1 and model2. According to the results, after its incorporation there is a decrease in the interaction distances between the molecule and the surface, evidencing the relevance of dispersion forces in this interaction. The results obtained for model2 with PBE-D3 showed good agreement with the experimental STM images and theoretical results using a periodic approach available for CuPc, with distances between 3 and 3.25 Å.14–16,19 The shortening in the metal-gold distance has been observed in MPc–gold systems (M = Fe, Cu, Co) previously studied with gold clusters of the same size. The only difference is that MPc–Au distances are shorter than systems with AuPc–Au.16
The interaction energies for the systems optimized without dispersion contribution are listed in Table S1 in ESI,† results that reveal a repulsive character for this interaction. The only system with a minor stabilizing contribution was AuPc–Au58 using PBE with an interaction energy of −6.3 kcal mol−1. These results point out the relevance of the proper description of the dispersion contribution in order to properly represent the stabilizing forces that lead to the formation of these complexes. The interaction energies calculated after the incorporation of the dispersion correction are listed in Table 2 together with the respective EDA results (model1 results are in Table S2†). The interaction energies between AuPc and model2 was of −89.0 kcal mol−1. This is in line with results from periodic calculations that quantify the interaction energy for CuPc in approximately 80 kcal mol−1 using PBE functional with vdWsurf corrections.19 The fact that the dispersion term is higher than the interaction energies with both levels of theory, points out that the dispersion forces are the main stabilizing contribution to the formation of these complexes. This also explains the decrease in the interaction distances after the incorporation of the dispersion correction as detailed above. Regarding the role of the metal center in the interaction strength, the comparison between AuPc-model2 and H2Pc-model2 showed an increase between 17.3 kcal mol−1. According to these results, we calculated a contribution of ca. 19% of the interaction energy provided by the inclusion of Au as the metal center. These results are comparable with the MPc complexes (M = Fe, Co, Cu) on Aun (n = 26, 58). The results showed that dispersion forces rule the MPc–gold interaction, with binding strengths ranging between 61 and 153 kcal mol−1.16 For complexes with MPc (M = Fe, Co and Cu) the interaction energy is higher compared to the AuPc on the same gold cluster. See the Table S3† in ESI.† Such a difference occurs because the MPc complexes have shorter distances over the gold surface. Comparing the resulting interaction energies of AuPc with model2 and model3, an increase of 12.3 kcal mol−1 is observed in the interaction energies. A minor part of that increase comes from the dispersion energy that is related to the effective area of interaction, while the rest of the difference in the interaction comes from a great reduction in the electrostatic and polarization terms of the interaction.
| System | ΔETOT | ΔEELECT | ΔEORB–RELAX | ΔEDISP | ΔEXC | ΔEEXCH–REPUL |
|---|---|---|---|---|---|---|
| AuPc-model2 | −89.0 | 50.8 | 0.0 | −86.2 | −76.4 | 22.8 |
| H2Pc-model2 | −71.7 | 114.1 | 0.0 | −83.3 | −79.4 | −23.2 |
| AuPc-model3 | −101.3 | 26.4 | −0.1 | −88.5 | −75.0 | 35.9 |
| H2Pc-model3 | −76.8 | 98.9 | −6.3 | −86.7 | −76.7 | −6.0 |
| AuPc-model4 | −100.1 | 24.0 | −0.1 | −89.4 | −74.9 | 40.3 |
| H2Pc-model4 | −76.6 | 104.6 | −0.1 | −87.8 | −76.2 | −17.1 |
The results obtained from the EDA calculations corroborate the importance of weak interactions, as can be seen from the dispersion term showed in Table 2. The ex-rep and OICT terms from eqn (1) presented in the Theoretical and computational details section are not included since the interaction does not involve covalent bonds and therefore, the value of such terms according to this EDA approach is 0. Our results reinforce the principle in which the stabilization of systems involving physisorption over a metallic surface is a process driven by weak van der Waals interactions. As a consequence, most of the stabilization is additive in nature and depends on the size of the extended area of contact between the interacting parts. Thereby, the size of the model representing a Au(111) system (model2) plays an important role regarding metal–π interactions, since model2 capture to its full extent the interaction between the molecule and the surface. This can be evidenced from the analysis of the increase in interaction strength after increasing the cluster size from model1 to model2 (results for model1 are shown in Table S4†). Within the context of the used EDA scheme, the electrostatic term represents the classical electronic repulsion, in line with the expected electronic repulsion added after the inclusion of the metal center that causes a decrease both in the electrostatic repulsion and the polarization stabilization. The model3 exhibit only a minor increase for the dispersion term, while the difference in the interaction energy with the layered model comes mainly from the drastic decrease in the electrostatic and polarization terms, thus revealing that edge effects have no influence on the interaction energies.
We analyzed the differences in the charge transfer process by using the natural population analysis (NPA), results listed in Table 3. Since the interacting parts are neutral, this analysis gives information about the charge transfer between the subunits after forming the complex. Interestingly, this only occurred when the Au was incorporated at the center of the Pc scaffold. These results reveal the role of Au as the metal center in the binding nature as an indispensable chemical block able to assist the charge transfer process with direction from the molecule to the gold substrate and are in agreement with experimental evidence of CuPc37 and previous theoretical results21 regarding the direction of the process. For other metal centers such as Fe or Ni, the charge transfer process goes from the gold substrate towards the molecule, showing that AuPc with its metal center with d9 electronic configuration follows the trend of CuPc in the charge transfer process.
| System | Method | Au-cluster | Au | Pc |
|---|---|---|---|---|
| AuPc | PBE-D3 | 1.21 | −1.21 | |
| AuPc-model2 | PBE-D3 | −0.71 | 1.27 | −0.57 |
| H2Pc-model2 | PBE-D3 | −0.01 | 0.01 | |
| AuPc-model3 | PBE-D3 | −0.88 | 1.27 | −0.38 |
| H2Pc-model3 | PBE-D3 | −0.10 | 0.10 | |
| AuPc-model4 | PBE-D3 | −0.87 | 1.27 | −0.40 |
| H2Pc-model4 | PBE-D3 | −0.04 | 0.04 |
In general, the interaction between AuPc and the four models is characterized by a donor–acceptor behavior, with the substrate (Au cluster) as the electron acceptor. According to this, the decrease in the electrostatic repulsion observed for AuPc-model2 is a consequence of the transfer of electronic charge between the fragments that allows to decrease the electronic overlapping between the subunits, which also leads to a decrease in the polarization of the fragments. To understand the nature of the charge transfer process that seems to be the second main source of stabilization besides the dispersion contribution, we calculated the fragmental electronic chemical potential as described in the methodology section. This chemical descriptor provides a measure of the electron escape tendency from the system, being a suitable property to understand the directionality of charge transfer process. Being the μ defined as the partial derivative of the energy with respect to the change in the number of electrons at constant external potential, the more negative the value of μ, the larger the change-decrease (negative slope) in energy under the addition of an electron; and therefore, the higher the stabilization towards receiving an electron. Thus, the differences between the μ of the subunits of the complex not only provides the tendency towards charge transfer, but its sign provide information about the direction of the process. We arbitrarily defined Δμ as the difference between the electronic chemical potential from the substrate (μSUBS) and the ligands (μAuPc and μH2Pc). A simple comparison between AuPc and H2Pc from the data listed in Table 4 (results for model1 are in Table S5†) shows that the former has a higher tendency towards donating electronic charge. Then, after comparing the μ of AuPc and H2Pc with the μSUBS of the substrate, the largest value of Δμ was obtained with AuPc, indicating that the AuPc–gold cluster systems will carry out more charge transfer than the H2Pc–gold cluster complex. The negative sign of Δμ in both types of complexes indicates that the gold substrate has a higher μSUBS than both ligands, thus being the most suitable electron acceptor of the complex. Actually, for the complexes with H2Pc the charge transfer was close to zero indicating that probably there is a threshold in which the difference between the electronic chemical potentials may trigger the electron transfer process. According to our results, PBE-D3 level of theory is able to provide a proper description of the electronic phenomenon involved in the AuPc–gold interaction.
| System | Method | μAu-substrate | μAuPc | Δμ |
|---|---|---|---|---|
| AuPc-model2 | PBE-D3 | −5.11 | −3.71 | −1.40 |
| H2Pc-model2 | PBE-D3 | −5.10 | −4.30 | −0.80 |
| AuPc-model3 | PBE-D3 | −5.41 | −3.70 | −1.71 |
| H2Pc-model3 | PBE-D3 | −5.41 | −4.29 | −1.12 |
| AuPc-model4 | PBE-D3 | −5.29 | −3.70 | −1.59 |
| H2Pc-model4 | PBE-D3 | −5.29 | −4.30 | −0.99 |
To characterize the nature of the non-covalent interactions arising at the interface between AuPc/H2Pc and the surface of the gold substrate, we calculated the non-covalent interaction index (NCI), results shown in Fig. 3. This index allows visualizing different types of interactions in real space through a color code. As identified by the green areas from the NCI surface, the main stabilizing contribution to the non-covalent part is located at the interatomic space between the AuPc/H2Pc and the gold surface atoms. While most of the stabilizing interactions are close to the center region of the interface; at the outer region of the macrocycle, where the electron delocalization takes part, there is a slightly destabilizing interaction located between the delocalized π-bonds from the phenyl rings and the gold surface. The addition of the gold atom at the center of the Pc incorporates a stabilizing interaction with the gold surface that accounts for the increase in the dispersion contribution after the inclusion of the gold center. Another important aspect is that the contribution of the gold atom of AuPc is related to the dispersion term, a characteristic feature of aurophilic interaction, although in this case is between a gold center of oxidation state +2 and a neutral metallic Au surface, which is 2.9 kcal mol−1 with PBE-D3 by model 2. As stated above, the van der Waals force dominates the interaction between these molecules and the surface, and it is related to the area that the molecules occupy over the surface.
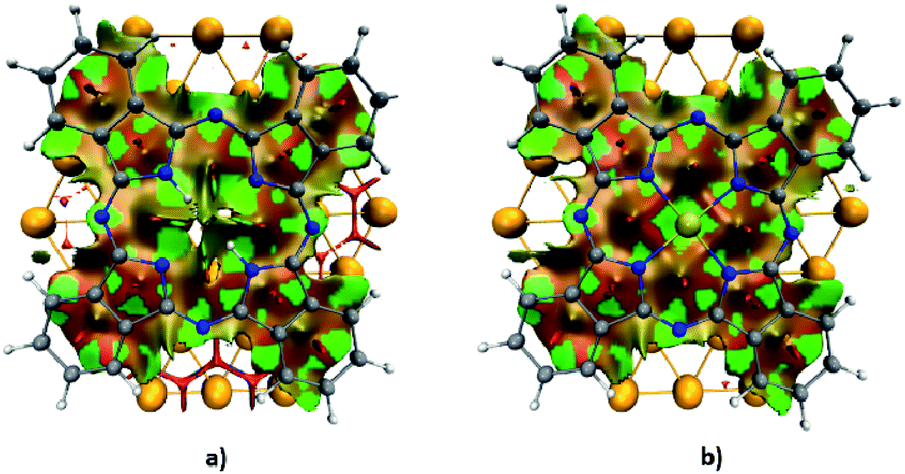 | ||
| Fig. 3 NCI index surface representation (isovalue = 0.5) of the (a) H2Pc–Au58 and (b) AuPc–Au58 complexes. | ||
In order to study the electronic structure of the metal–organic interface,2,18,37–43 density of states (DOS) calculations were carried out to determine the effects of the surface–molecule interaction. In Fig. 4 is displayed a DOS diagram of the interacting system AuPc–Au58 (green line) along with a diagram of sub-systems AuPc (blue line) and Au(111) surface (red line). Also is displayed the sum of the diagrams of the separate sub-systems (yellow line) to compare with the DOS of the interacting system. The zero at the diagram is set to be the Fermi level (EF) of the Au(111) surface cluster model, indicated by the vertical black line. The broad peak of the Fermi level of AuPc is due to the degeneracy of a d9 electronic configuration in a square–planar coordination environment. It is observed that the peak corresponding to the Fermi level of the AuPc (indicated by the arrow) is shifted by ∼0.9 eV towards the Fermi level of the Au(111) in the interacting system. This is appreciated by a decrease in the DOS around the EF of AuPc when comparing the sum of the isolated sub-systems with the interacting system, and an increase in the DOS at the EF of the Au(111) substrate in the interacting system. The Energy Level Alignment43 is a characteristic feature of an organic molecule or polymer interacting with a metallic substrate, in which the electronic structure of the interface and it's principal properties such as charge transfer character is determined by electrostatic and polarization components of the interaction.44 This is useful to rationalize the magnitudes of these terms provided by the EDA calculation discussed above, since they provide a wave-function based description of the electronic structure of the interacting systems. It is an interesting feature that this system exhibit such a displacement on the density of states given that is a weakly interacting one, on a physisorption regime based on π-molecules on a clean metal surface where its stability is driven mainly from van der Waals forces.45 However, this interaction is not easily compassed on basic interface models such as the integer charge-transfer model (ICT) or induced density of interfacial states (IDIS)46 because of the presence of d orbitals from the metal center. Nevertheless, the Fermi level shift in the diagram is in accordance with the calculated difference of electronic chemical potential as discussed in Table 4, related to the direction of charge transfer, thus showing that the energy level alignment is related to the equilibration of the chemical potential, as has been suggested by previous work on the field.47
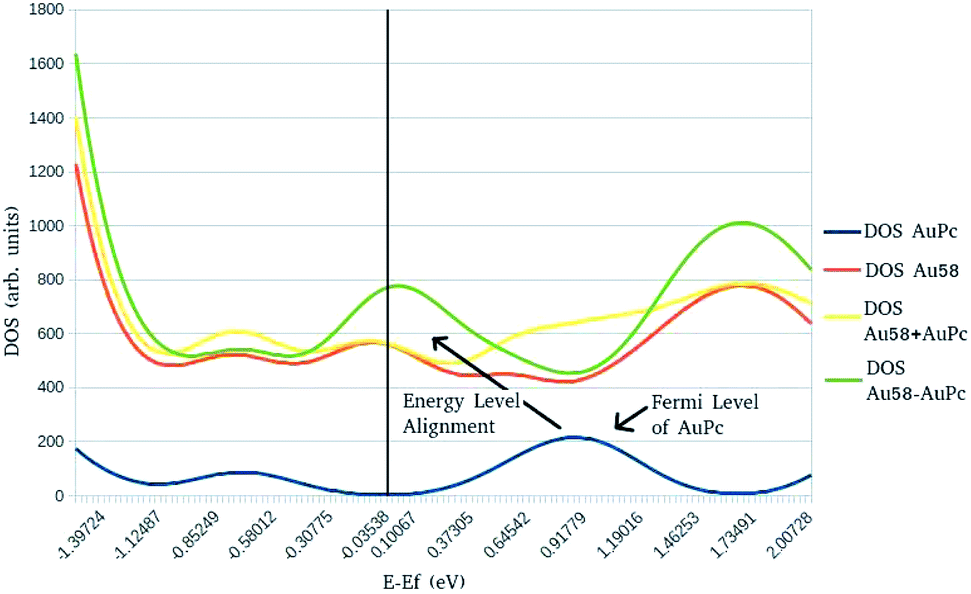 | ||
| Fig. 4 Density of States (DOS) diagram for the AuPc–Au58 complex. | ||
A DOS diagram of AuPc–Au58 using the flat cluster was also drafted (Fig. S1, ESI†) and it highlights the importance of a layered cluster model to reproduce the electronic structure of the interface. This can be explained by the fact that the cluster model fails to account for the electrostatic and polarization terms (Table 2) that define the metal–organic interface and the energy level alignment. The importance of those terms in the interface are also described by charge rearrangements and cumulative charge transfer in periodic calculations,19 and is supportive of the need of a layered cluster to accurately describe the interface electronic structure.
The DOS diagram of the layered model2 (Fig. S2†) resembles the one obtained with periodic methods of calculation.48,49 Another important aspect is that the dispersion correction does not modify the DOS diagram. This is an expected result since the dispersion correction is semi-empirical and it does not modify the wave function of the systems being considered and thus, the energy level alignment displayed by this system may be regarded as a feature that arise from the geometry of the Au(111) cluster rather than the consideration of van der Waals forces, thus showing that the cluster model is able to reproduce some important properties regarding molecule–surface interactions.
Conclusions
The results here presented allowed to define the nature of the interaction between AuPc and a gold-based substrate, revealing two main components responsible of the binding strength: the dispersion forces and the charge transfer process characterized by a donor–acceptor type of interaction. The EDA calculations allowed describing the relevance of the charge transfer in the decrease of the electrostatic repulsion between the interacting systems and the effect of the Au atom in the interaction can be quantified to ∼12.3 kcal mol−1, in line with aurophilic interactions of dispersion origin; together with the importance of Au as the metal center to favor the flow of electronic charge towards the gold substrate, showing the same trend of CuPc which holds the same electronic configuration for the metal center. From a methodological point of view, to properly describe this new framework of chemical assemblies it is mandatory to incorporate the dispersion correction within the scope of DFT calculations, as the lack of dispersion leads to a repulsive interaction. The electrostatic and polarization terms also provide a basis for rationalizing the electronic structure of the interface and the energy level alignment displayed in DOS diagrams. EDA and DOS results jointly indicate the importance of a layered cluster model to reliably reproduce the metal–organic interface. This result indicates the feasibility of the cluster model as an approximation to surface related phenomena and prove that the Au58 cluster size is adequate, but still further work is required to provide a systematic approach to surface science within the cluster model approximation.Theoretical and computational details
The interaction of AuPc with the Au(111) surface was simulated using four different gold clusters depicted in Fig. 2. The first consists of 26 atoms distributed in three layers of 14, 8 and 4 gold atoms, and is defined as model1. The second is formed by 58 atoms also distributed in three layers composed of 30, 14 and 14 gold atoms, and defined as model2. The third and fourth models are planar clusters, one of 58 and the second of 61 gold atoms, defined as model3 and model4, respectively. This selection of clusters allowed to assess any edge effects and their influence on geometric parameters, interaction energies and electronic structure. The Au–Au distance in the cluster is 2.89 Å, taken from the bulk structure of the surface.50,51 The use of clusters of different size allowed us to define the relevance of the cluster size in the proper description of the interaction between these systems and how it can affect the metal center–substrate interaction. In this study we chose the Au(II)Pc system to avoid excessive contribution from the electrostatic term and because it represents an understudied state of this type of systems. Also, the results could be related to experimental evidence obtained with CuPc, which holds the same electronic configuration for the metal center. To evaluate the interaction of the gold atom in the structure of the phthalocyanine with Au(111) represented by model1 and model2, the results will be compared with a reference system of metal-free phthalocyanine H2Pc over the Au(111) surface. The modelling of these systems was performed by placing the AuPc and H2Pc over the Au(111) cluster, aligning the center of the macrocycle with the center of the surface cluster, as seen in Fig. 2. The geometry of the macrocycle system was freely optimized on top of the gold surface. Meanwhile, for the gold clusters we adopted a mixed scheme, where in the first layer we only fixed the outer Au atoms allowing to relax all the inner atoms from the first layer. For those models consisting of multiple layers, the second and third layer was also fixed. With this scheme we avoided the loss of the expected structure coming from its solid surface nature.All calculations were carried out within the Density Functional Theory framework, using GGA PBE(Perdew–Burke–Ernzenhof)52 exchange–correlation functionals as implemented in the Turbomole package.54,55 This functional was chosen to be consistent with previous publications regarding surface chemistry and related phenomena. The meta-GGA TPSS (Tao, Perdew, Staroverov, Scuseria)53 functional was also used to obtain a comparison in the description of surface chemistry with the more broadly used and more validated PBE functional.21,56 The comparison is provided in the ESI.† Since we are interested in the supramolecular assemblies that this molecules form over a surface, the inclusion of dispersion interactions is fundamental to accurately describe the non-covalent interaction between the macrocycle and the Au(111) surface. This is incorporated using the “DFT-D3” methodology.57 For Au, Stuttgart pseudo relativistic effective core potentials (ECP) were used, with 19 valence electrons.58 Two f-type polarization functions were added Au (αf = 0.20, 1.19).21 All atoms were treated with Ahlrichs type def2-TZVP basis set,59 adding one d-type polarization function for the optimizations, and subsequently the energies were corrected by single point calculations using the def2-TZVP basis set.
The counterpoise correction has been used to avoid the basis set superposition error (BSSE) in the calculation of interaction energies, through single point calculations, based on the optimized geometry obtained.60 To gain more insight on the interaction between the macrocycle and the surface, energy decomposition analysis (EDA) calculations were performed.61,62 This methodology expresses the interaction energy in different physical components, comprising an electrostatic term (ES), the exchange–repulsion term (EXR), a term labelled orbital interaction and charge transfer (OICT), polarization contribution (POL) and the dispersion term (DISP), as expressed in eqn (1).
| ΔE = EES + EEXR + EOICT + EPOL + EDISP | (1) |
Also, density of states (DOS) calculations were performed to obtain information regarding the electronic structure of the systems. DOS has been built via a Gaussian distribution of the one-electron eigenstates obtained by the DFT methods described above, and considering a bandwidth of 0.27 eV.
To characterize the intermolecular interactions, the non-covalent interaction index (NCI) was used.63 This is a qualitative tool for describing attractive and repulsive interactions in real space, based on the reduced density gradient (RDG) and can be interpreted by color coded isosurfaces that represent those interactions. Hydrogen bonds are labelled as strong interactions and are represented by blue color. Weak van der Waals interactions are shown in green color, while repulsive steric interactions are displayed in red color. Natural Population Analysis (NPA)64 calculations were used to study the charge transfer processes that may be involved in these systems. Finally, the fragmental electronic chemical potential was calculated according to the following equation:
| μ = (I + A)/2 = (εLUMO + εHOMO)/2 | (2) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financed by FONDECYT under Project 1180158 (Conicyt-Chile).References
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS.
- J. Hermann, R. A. DiStasio and A. Tkatchenko, Chem. Rev., 2017, 117, 4714–4758 CrossRef CAS PubMed.
- J. H. Zagal, F. Bedioui and J.-P. Dodelet, N4-macrocyclic metal complexes, Springer Science & Business Media, 2007 Search PubMed.
- K. Nilson, J. Åhlund, B. Brena, E. Göthelid, J. Schiessling, N. Mårtensson and C. Puglia, J. Chem. Phys., 2007, 127, 114702 CrossRef PubMed.
- T. Nyokong and F. Bedioui, J. Porphyr. Phthalocyanines, 2006, 10, 1101–1115 CrossRef CAS.
- Z. Li, B. Li, J. Yang and J. G. Hou, Acc. Chem. Res., 2010, 43, 954–962 CrossRef CAS PubMed.
- J. D. Baran and J. A. Larsson, Phys. Chem. Chem. Phys., 2010, 12, 6179–6186 RSC.
- J. D. Baran and J. A. Larsson, J. Phys. Chem. C, 2012, 116, 9487–9497 CrossRef CAS.
- F. Schreiber, J. Phys.: Condens. Matter, 2004, 16, R881–R900 CrossRef CAS.
- Y. H. Jiang, W. D. Xiao, L. W. Liu, L. Z. Zhang, J. C. Lian, K. Yang, S. X. Du and H.-J. Gao, J. Phys. Chem. C, 2011, 115, 21750–21754 CrossRef CAS.
- T. Komeda, H. Isshiki and J. Liu, Sci. Technol. Adv. Mater., 2010, 11, 54602 CrossRef PubMed.
- S. Ahmadi, M. N. Shariati, S. Yu and M. Göthelid, J. Chem. Phys., 2012, 137, 84705 CrossRef PubMed.
- K. Nilson, J. Åhlund, M.-N. Shariati, E. Göthelid, P. Palmgren, J. Schiessling, S. Berner, N. Mårtensson and C. Puglia, J. Phys. Chem. C, 2010, 114, 12166–12172 CrossRef CAS.
- M. Takada and H. Tada, Jpn. J. Appl. Phys., 2005, 44, 5332–5335 CrossRef CAS.
- X. Lu, K. W. Hipps, X. D. Wang and U. Mazur, J. Am. Chem. Soc., 1996, 118, 7197–7202 CrossRef CAS.
- X. Lu and K. W. Hipps, J. Phys. Chem. B, 1997, 101, 5391–5396 CrossRef CAS.
- L. Massimi, M. Angelucci, P. Gargiani, M. G. Betti, S. Montoro and C. Mariani, J. Chem. Phys., 2014, 140, 244704 CrossRef PubMed.
- H. Yamane and N. Kosugi, J. Phys. Chem. C, 2016, 120, 24307–24313 CrossRef CAS.
- L. Y. Huang, E. Wruss, A. D. Egger, S. Kera, N. Ueno, A. W. Saidi, T. Bucko, T. S. A. Wee and E. Zojer, Molecules, 2014, 19, 3145 Search PubMed.
- Z. H. Cheng, L. Gao, Z. T. Deng, N. Jiang, Q. Liu, D. X. Shi, S. X. Du, H. M. Guo and H.-J. Gao, J. Phys. Chem. C, 2007, 111, 9240–9244 CrossRef CAS.
- S. Miranda-Rojas, P. Sierra-Rosales, A. Munoz-Castro, R. Arratia-Perez, J. H. Zagal and F. Mendizabal, Phys. Chem. Chem. Phys., 2016, 18, 29516–29525 RSC.
- A. Mugarza, R. Robles, C. Krull, R. Korytár, N. Lorente and P. Gambardella, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155437 CrossRef.
- F. Evangelista, A. Ruocco, R. Gotter, A. Cossaro, L. Floreano, A. Morgante, F. Crispoldi, M. G. Betti and C. Mariani, J. Chem. Phys., 2009, 131, 174710 CrossRef CAS PubMed.
- H. Peisert, M. Knupfer, T. Schwieger, J. M. Auerhammer, M. S. Golden and J. Fink, J. Appl. Phys., 2002, 91, 4872–4878 CrossRef CAS.
- S. Li, J. Hao, F. Li, Z. Niu, Z. Hu and L. Zhang, J. Phys. Chem. C, 2014, 118, 27843–27849 CrossRef CAS.
- P. Gargiani, M. Angelucci, C. Mariani and M. G. Betti, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 85412 CrossRef.
- A. Laguna and M. Laguna, Coord. Chem. Rev., 1999, 193–195, 837–856 CrossRef CAS.
- E. W. Y. Wong, A. Miura, M. D. Wright, Q. He, C. J. Walsby, S. Shimizu, N. Kobayashi and D. B. Leznoff, Chem.–Eur. J., 2012, 18, 12404–12410 CrossRef CAS PubMed.
- A. MacCragh and W. S. Koski, J. Am. Chem. Soc., 1965, 87, 2496–2497 CrossRef CAS.
- D. Carrascal, L. Fernández-Seivane and J. Ferrer, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 184415 CrossRef.
- M. E. Azim-Araghi, L. M. Goodarzi and J. Baedi, Mater. Sci. Semicond. Process., 2013, 16, 1736–1741 CrossRef CAS.
- H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS.
- H. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Chem. Phys., 2005, 311, 151–161 CrossRef CAS.
- P. Castro-Latorre, S. Miranda-Rojas, L. Barrientos and F. Mendizabal, Mol. Simul., 2019, 45, 1447–1453 CrossRef CAS.
- I. Ponce, J. F. Silva, R. Oñate, S. Miranda-Rojas, A. Muñoz-Castro, R. Arratia-Pérez, F. Mendizabal and J. H. Zagal, J. Phys. Chem. C, 2011, 115, 23512–23518 CrossRef CAS.
- I. Ponce, J. F. Silva, R. Oñate, M. C. Rezende, M. A. Paez, J. H. Zagal, J. Pavez, F. Mendizabal, S. Miranda-Rojas, A. Muñoz-Castro and R. Arratia-Pérez, J. Phys. Chem. C, 2012, 116, 15329–15341 CrossRef CAS.
- J. Xiao and P. A. Dowben, J. Phys.: Condens. Matter, 2008, 21, 52001 CrossRef.
- X. Crispin, V. Geskin, A. Crispin, J. Cornil, R. Lazzaroni, W. R. Salaneck and J.-L. Brédas, J. Am. Chem. Soc., 2002, 124, 8131–8141 CrossRef CAS.
- K. Toyoda, I. Hamada, K. Lee, S. Yanagisawa and Y. Morikawa, J. Chem. Phys., 2010, 132, 134703 CrossRef PubMed.
- K. W. Hipps and U. Mazur, J. Porphyr. Phthalocyanines, 2012, 16, 273–281 CrossRef CAS.
- D. Cahen and A. Kahn, Adv. Mater., 2003, 15, 271–277 CrossRef CAS.
- R. Otero, A. L. Vázquez de Parga and J. M. Gallego, Surf. Sci. Rep., 2017, 72, 105–145 CrossRef CAS.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11, 605–625 CrossRef CAS.
- O. L. A. Monti, J. Phys. Chem. Lett., 2012, 3, 2342–2351 CrossRef CAS PubMed.
- S. P. Jarvis, S. Taylor, J. D. Baran, D. Thompson, A. Saywell, B. Mangham, N. R. Champness, J. A. Larsson and P. Moriarty, J. Phys. Chem. C, 2015, 119, 27982–27994 CrossRef CAS.
- S. Braun, W. R. Salaneck and M. Fahlman, Adv. Mater., 2009, 21, 1450–1472 CrossRef CAS.
- S. Braun, M. P. de Jong, W. Osikowicz and W. R. Salaneck, Appl. Phys. Lett., 2007, 91, 202108 CrossRef.
- A. Singhal, S. Kancharlapalli and S. K. Ghosh, J. Mol. Model., 2018, 24, 217 CrossRef PubMed.
- P. F. Cafe, A. G. Larsen, W. Yang, A. Bilic, I. M. Blake, M. J. Crossley, J. Zhang, H. Wackerbarth, J. Ulstrup and J. R. Reimers, J. Phys. Chem. C, 2007, 111, 17285–17296 CrossRef CAS.
- A. Ricca and C. W. Bauschlicher, Chem. Phys. Lett., 2003, 372, 873–877 CrossRef CAS.
- C. W. Bauschlicher and A. Ricca, Chem. Phys. Lett., 2003, 367, 90–94 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, J. Chem. Theory Comput., 2018, 14, 3083–3090 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- B. G. Janesko, ed. E. R. Johnson, Density Functional Theory Beyond the Generalized Gradient Approximation for Surface Chemistry, Springer International Publishing, Cham, 2015, pp. 25–51 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 14102 CrossRef.
- H. L. Yu, Phys. Rev. B: Solid State, 1977, 15, 3609–3616 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07959a |
| This journal is © The Royal Society of Chemistry 2020 |