
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModelling of interactions between Aβ(25–35) peptide and phospholipid bilayers: effects of cholesterol and lipid saturation†
Inna Ermilova* and
Alexander P. Lyubartsev
Department of Materials and Environmental Chemistry, Stockholm University, Stockholm, Sweden. E-mail: ina.ermilova@gmail.com; alexander.lyubartsev@mmk.su.se; Tel: +46 8161193
First published on 23rd January 2020
Abstract
Aggregation of amyloid beta (Aβ) peptides in neuronal membranes is a known promoter of Alzheimer’s disease. To gain insight into the molecular details of Aβ peptide aggregation and its effect on model neuronal membranes, we carried out molecular dynamics simulations of the Aβ(25–35) fragment of the amyloid precursor protein in phospholipid bilayers composed of either fully saturated or highly unsaturated lipids, in the presence or absence of cholesterol. It was found that the peptide does not penetrate through any of the considered membranes, but can reside in the headgroup region and upper part of the lipid tails showing a clear preference to a polyunsaturated cholesterol-free membrane. Due to the ordering and condensing effect upon addition of cholesterol, membranes become more rigid facilitating peptide aggregation on the surface. Except for the case of the cholesterol-free saturated lipid bilayer, the peptides have a small effect on the membrane structure and ordering. It was also found that the most “active” amino-acid for peptide–lipid and peptide–cholesterol interaction is methionine-35, followed by asparagine-27 and serine-26, which form hydrogen bonds between peptides and polar atoms of lipid headgroups. These amino acids are also primarily responsible for peptide aggregation. This work will be relevant for designing strategies to develop drugs to combat Alzheimer’s disease.
1 Introduction
Accumulation of amyloid precursor proteins is a known problem in biochemistry, biophysics and medicine.1–3 This phenomena is related to many diseases, for example, aggregation of such proteins in particular tissues is associated with the development of diabetes, cancer, Alzheimer’s (AD) and Parkinson’s (PD) etc.4–7 Other compounds, such as lipids and cholesterol, in neuronal membranes are also affected by the accumulation of amyloids and become involved in the processes associated with the above listed diseases.8–10 People with high levels of cholesterol in their blood are seen as a potential risk group for heart-failure, cancer and even AD.11–14 However, in human brain tissue, a high level of cholesterol is not necessarily related to bad health; there are regions, called lipid rafts, which need to be rich in cholesterol in order for living organisms to function properly.15–17 The molar ratio between phospholipids and cholesterol in the lipid rafts of a healthy human brain is around 1.15,18 However, in certain neurodegenerative diseases one can observe variations in this composition, which can be helpful in their diagnosis. For instance, in the case of AD the cholesterol–lipid ratio does not differ too much and the change can be considered negligible,15–17,19 while in the case of PD, by looking at the neuronal cell composition, one can define two different stages which are called “incidental PD” and “PD”.18,20Another component of brain tissue membranes which is associated with the development of neurodegenerative diseases, is polyunsaturated lipids, and particularly lipids with docosahexaenoic (22:6(cis), called also ω-3) fatty acid chains. A low amount of phosphatidylethanolamines with tails consisting of 22:6(cis) fatty acids in some areas of a human brain are related to the development of AD as well as PD.15,18,19 The relationship between the content of polyunsaturated phospholipids (which constitutes about 40% of a healthy human brain), cholesterol and amyloid peptides, which is relevant for understanding the molecular mechanisms behind these diseases, has not received much attention experimentally or by modellers. Phospholipids which are used in most studies of interactions of Aβ-peptides with membranes, are typically mono-unsaturated (such as POPC), but the fatty acid composition in different brain tissues does not show any remarkable changes for mono-unsaturated fatty acids in the case of neurodegenerative deceases.19
In this work we investigate the behaviour of Aβ(25–35) peptide, which is a part of the amyloid precursor protein. The reason behind the selection of this particular peptide is its high cytotoxicity in neuronal cells,21,22 as well as experimentally proven negative effects on the memory of animals.23,24 Behaviour of this peptide in different environments, from water to neuronal cells, has been studied in a number of experimental work,25–33 however, information on the molecular interactions of this peptide with different components of neuronal membranes, as well as the effect of the peptide on the membrane structure is still lacking, or inconclusive. For instance, the location of Aβ(25–35) relative to the membrane has still not been clearly determined. Many experiments have considered the peptide being inserted into the fluid phase membrane,28,30,34 but Lau et al.29 have argued that this might be a consequence of the sample preparation during which Aβ(25–35) was added while vesicles were forming. It was also discussed that Aβ(25–35) peptides aggregating in the membrane can form barrel-like pores leading to membrane permeability for Ca2+ ions.35–37 The high content of cholesterol in membranes is related to inhibition of the insertion of the peptide, while cholesterol itself has been seen as a candidate for associations with peptides.28,33,38,39 Low amounts of cholesterol in a membrane was related to the Aβ insertions by Dies et al.,40 which is not coherent with results from Esposito et al.34 and Di Scala et al.35 Unsaturated phospholipids have been seen as promoters of interactions between membranes and peptides, favouring the insertion of the latter.26,32 Partially, controversy between experimental results can be caused by different protocols of sample preparation and the use of membranes composed of different lipids. Another source of uncertainty can be that average experimental data are collected from the whole system including the bulk solvent.
In the absence of unambiguous experimental data, molecular simulations can be an alternative route to gain insight into molecular behaviour of systems of interest. For Aβ(25–35) peptide, a number of computational studies on the atomistic level regarding the structure of the peptide in different media have been carried out.27,41–45 Simulations of Aβ peptides in the lipid bilayer environment are less common. We are aware of only about one work on the simulation of Aβ(25–35) in a lipid bilayer environment which contains 11-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) lipids.46 In that work the peptides were already inserted into the bilayer in a specific arrangement. A number of molecular dynamics (MD) simulations of other Aβ peptides can also be referenced.47–51
In this work, we perform MD simulations of Aβ(25–35) in four different lipid bilayers, two of which are composed of fully saturated 14:0–14:0 PC (DMPC) lipids, and two others of strongly unsaturated 22:6–22:6 PC (DDPC), and within each pair one bilayer is without cholesterol while the other has 50% cholesterol content, according to the ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) provided by Martin et al.15 for cholesterol content in rafts of neuronal membranes. The aim is to investigate both the effect of cholesterol and lipid saturation on the interaction of Aβ(25–35) peptides with the lipid membrane, its partitioning and aggregation. Note that DMPC is the longest fully saturated lipid which forms a liquid phase bilayer at physiological temperature. Another question which we address in this work is which amino-acids of Aβ(25–35) might play a key role in its toxicity, particularly by considering binding to membranes by the strongest hydrogen bonds between peptide and lipid. In previous investigations the toxicity of Aβ(25–35) was related to the methionine-35 residue (MET35) and its position in the C-terminal,25,29,44 while some other studies have considered asparagine-27 (ASN27)31,52 or glycine-25 (GLY25) as being responsible for the accumulation of Aβ(25–35).44 From all-atomistic MD simulations full information regarding the atomic positions can be obtained and one can find out which atoms can build hydrogen bonds, or whether the peptides’ behaviour is determined by the hydrophobic parts of the species.
:1) provided by Martin et al.15 for cholesterol content in rafts of neuronal membranes. The aim is to investigate both the effect of cholesterol and lipid saturation on the interaction of Aβ(25–35) peptides with the lipid membrane, its partitioning and aggregation. Note that DMPC is the longest fully saturated lipid which forms a liquid phase bilayer at physiological temperature. Another question which we address in this work is which amino-acids of Aβ(25–35) might play a key role in its toxicity, particularly by considering binding to membranes by the strongest hydrogen bonds between peptide and lipid. In previous investigations the toxicity of Aβ(25–35) was related to the methionine-35 residue (MET35) and its position in the C-terminal,25,29,44 while some other studies have considered asparagine-27 (ASN27)31,52 or glycine-25 (GLY25) as being responsible for the accumulation of Aβ(25–35).44 From all-atomistic MD simulations full information regarding the atomic positions can be obtained and one can find out which atoms can build hydrogen bonds, or whether the peptides’ behaviour is determined by the hydrophobic parts of the species.
2 Models and simulations setup
The compositions of the simulated systems are given in Table 1. The Aβ(25–35) peptide has the sequence GSNKGAIIGLM. Residue LYS28 was taken as neutral in our simulations, which can be justified as in the membrane environment, and the peptide aggregates, the lysine pKa value drops below 7.53,54 Lipid and cholesterol are described by the fully atomistic Slipids force field,55–57 while for the peptide the General Amber force field (GAFF)58 is used. According to the GAFF definition, the partial atomic charges were determined within the Restricted Electrostatic Potential (RESP) approach59 as implemented in the acpype utility,60 which was also used to generate molecular topology files. Recently, several comparative studies of different force fields to describe peptide structure have been carried out61–64 but their results on the force field performance were ambiguous. GAFF, derived on the basis of the Amber99 parameter set, was designed to provide better transferability of parameters over a wide range of organic molecules, which can be an advantage for the description of intrinsic disordered peptides such as Aβ. Previously, GAFF parameters for solutes have shown good compatibility with the Slipids force field demonstrating consistency with experimental partitioning of the molecules between aqueous and membrane phases.65–67 GAFF parameters have also been used for simulations of leucine zipper peptides68 and for peptide-based drug amphiphile filaments.69,70 Butterfoss et al.71 have used GAFF for blind structure prediction of peptoid molecules. Though the latter study was carried out within the implicit solvation Born model, it showed that GAFF torsional potentials reproduce the experimentally observed conformational properties of these molecules well.| Lipid, PC | Number of lipids | Number of cholesterol molecules | Number of water molecules | Number of peptides |
|---|---|---|---|---|
| 14:0–14:0 | 128 | 0 | 6400 | 14 |
| 22:6–22:6 | 128 | 0 | 6400 | 14 |
| 14:0–14:0 | 128 | 128 | 7680 | 8 |
| 22:6–22:6 | 128 | 128 | 7680 | 8 |
The simulated systems were constructed in the following order: First, four membranes were created. Two of them were pure lipid bilayers (14:0–14:0 PC and 22:6–22:6 PC), the two others consisted of the same lipids but loaded with 50% cholesterol. In the latter case cholesterol molecules were distributed uniformly between the lipids. The bilayers were further hydrated by water. Each system was equilibrated for 100 ns under 1 atm pressure (in the semi-isotropic NPT ensemble) at 303 K. Then the water molecules were taken away, and peptides, preliminarily equilibrated in water for 50 ns simulation, were added in the space outside the bilayer in random positions and orientations, so that no intersections between molecules could occur. Water molecules were inserted again in the remaining space, followed by 100 ns equilibration with restrained peptides under the same conditions. The reason behind this setup was to start from a pre-equilibrated configuration of both membrane and peptides with peptides dispersed outside the bilayer, in order to investigate whether Aβ(25–35) could penetrate the membrane during the simulation or if it would reside outside the bilayer, as well as to investigate the aggregation behaviour of the peptides.
After the above procedure, MD simulations were run for 1.05 μs, of which the last 500 ns were used for computations of averages. In all computations, settings were the following: temperature, T = 303 K and pressure, P = 1 atm, maintained by the semi-isotropic pressure coupling scheme using Berendsen barostat72 and by the velocity rescaling thermostate.73 Bonds have been constrained using the LINCS algorithm with 12 iterations.74 The leap-frog algorithm75 with time step 2 fs and a Verlet cut-off scheme76 with cutoff radius 1 nm were used for integration of equations of motion. Final dimensions of the simulation boxes for production runs were about 68 × 68 × 82 Å3 for systems without cholesterol and 72 × 72 × 92 Å3 for systems with cholesterol. Simulation software was GROMACS-4.6.7.77 Analysis of the trajectories was done using routines from Gromacs and from MDynaMix software.78
3 Results
3.1 Density profiles
The final snapshots of the four simulated systems are shown in Fig. 1. One can see that in all cases the peptides reside mostly at the bilayer surface, entering only to the headgroup region of the bilayer. More detailed quantitative analysis of the peptide partitioning in the membrane can be gained from the mass density profiles. Besides showing the preferential positioning of peptides in the membrane, mass density distributions are useful for comparison with the results of X-ray or neutron scattering data which can be used for validation of simulations on the one hand, and for interpretation of experimental data on the other hand. The contribution of peptides to the mass density profiles are shown in Fig. 2, together with the contribution from the lipid phosphate groups, and the hydroxyl and terminal methyl groups of cholesterol in the case of cholesterol-containing membranes.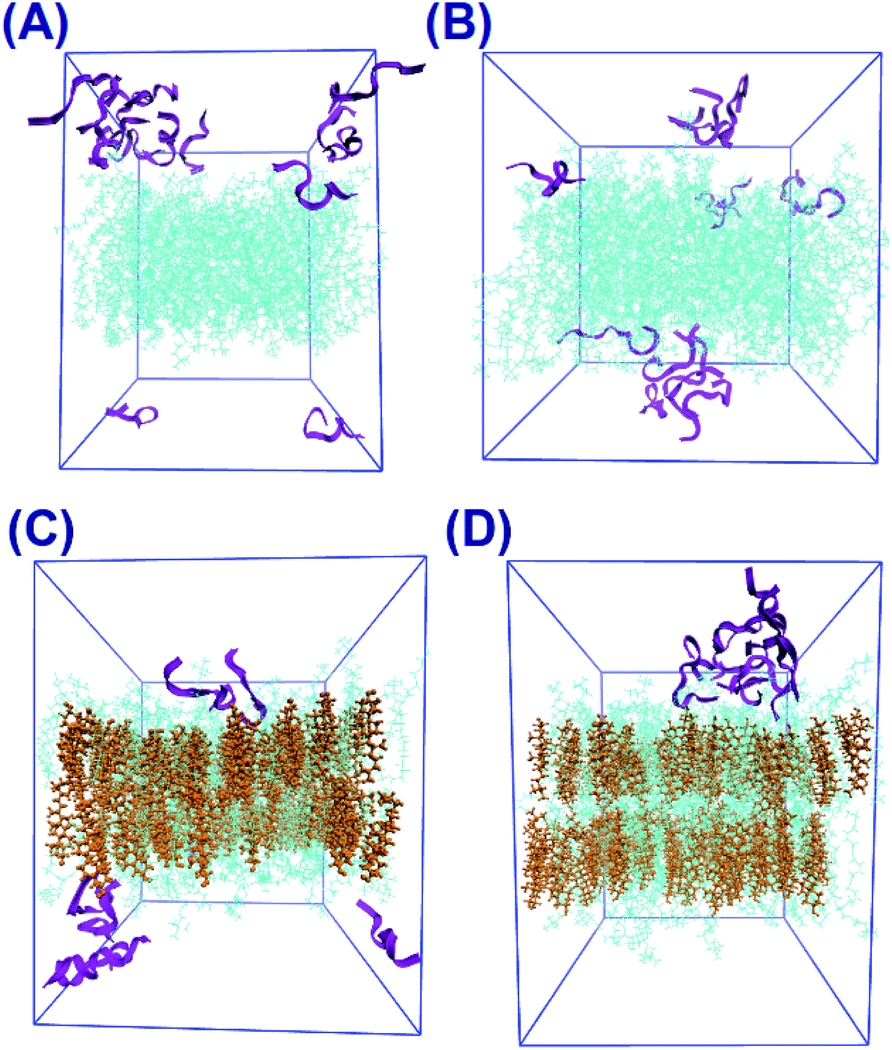 | ||
| Fig. 1 Screenshots from simulations: (A) 14:0–14:0 PC (no cholesterol), (B) 22:6–22:6 PC (no cholesterol), (C) 14:0–14:0 PC (with cholesterol), (D) 22:6–22:6 PC (with cholesterol). Colours on images are: “purple” – peptides, “lime, gray, purple and blue” – atoms on lipids, “yellow” – cholesterol. | ||
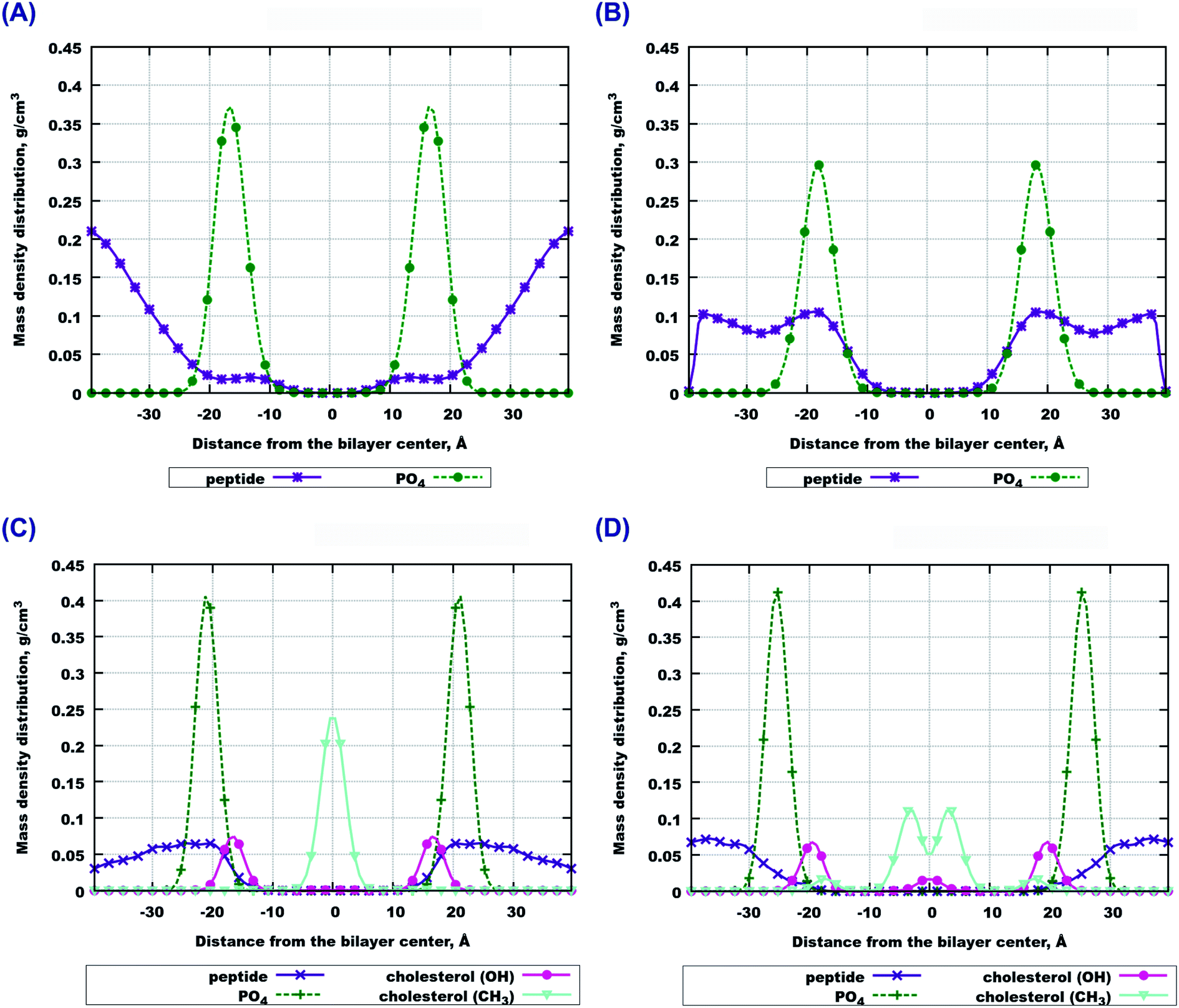 | ||
| Fig. 2 Contributions to the mass density profiles. (A) 14:0–14:0 PC without cholesterol; contributions from the peptides and PO4-groups from lipid heads. (B) 22:6–22:6 PC without cholesterol; contributions from peptides and PO4-groups from the lipid heads. (C) 14:0–14:0 PC with cholesterol; contributions from the peptides, PO4-groups from lipid heads, CH3-terminal and OH-groups from cholesterol. (D) 22:6–22:6 PC with cholesterol; contributions from peptides, PO4-groups from lipid heads, CH3-terminal and OH-groups from cholesterol. | ||
Fig. 2(A and B) demonstrate that peptides in pure lipid bilayers without cholesterol reside deeper in the headgroup region of the membrane in the case of 22:6–22:6 PC compared to 14:0–14:0 PC, for which the mass density distribution of the peptides is concentrated mostly outside the membrane. For membranes loaded with cholesterol (Fig. 2(C and D)) the opposite trend is found: the peptides move further away from the polyunsaturated bilayer while in simulations with 14:0–14:0 PC the peptides are somewhat closer to the membrane. Another observation is that in the presence of cholesterol, the distance between the phosphate groups of the two monolayers increases, making the bilayer thicker. Furthermore, the curve from the hydroxyl groups of cholesterol overlap with the curve from the peptides only in the system containing 14:0–14:0 PC (Fig. 2(C)), while in the system with 22:6–22:6 PC peptides come into contact with the cholesterol hydroxyl groups less frequently. One can also note the density maxima of the cholesterol hydroxyl groups in the bilayer center in the case of 22:6–22:6 PC lipids, which corresponds to the presence of “flipped” orientations of cholesterol in such bilayers, which was discussed in detail in our previous paper.79
In order to investigate which amino-acids make the peptides move locate closer to the bilayer center, we calculated the contributions to the mass density from three amino-acids in the beginning, middle and end of the sequence, that is from GLY25, ALA30 and MET35 residues. Among them, methionine (MET35) seems to be the one which goes the deepest into bilayers, especially in the case of 22:6–22:6 PC (Fig. 3(B and D)). However in the case of pure 14:0–14:0 PC bilayer, methionine is preferably found outside the membrane (Fig. 3(A)). When cholesterol is added, the behaviour trend changes: methionine is more likely to locate closer to the center of a saturated membrane than to the polyunsaturated one (Fig. 3(C and D)). Regarding two other amino-acids (alanine and glycine) one can note that glycine seems to appear closer to the bilayer in cholesterol loaded 22:6–22:6 PC membrane (Fig. 3(C)), while in other cases the behaviour of these species is similar. Density distributions of other amino acids of Aβ(25–35) in the simulated membranes are shown in Fig. S1 of the ESI.†
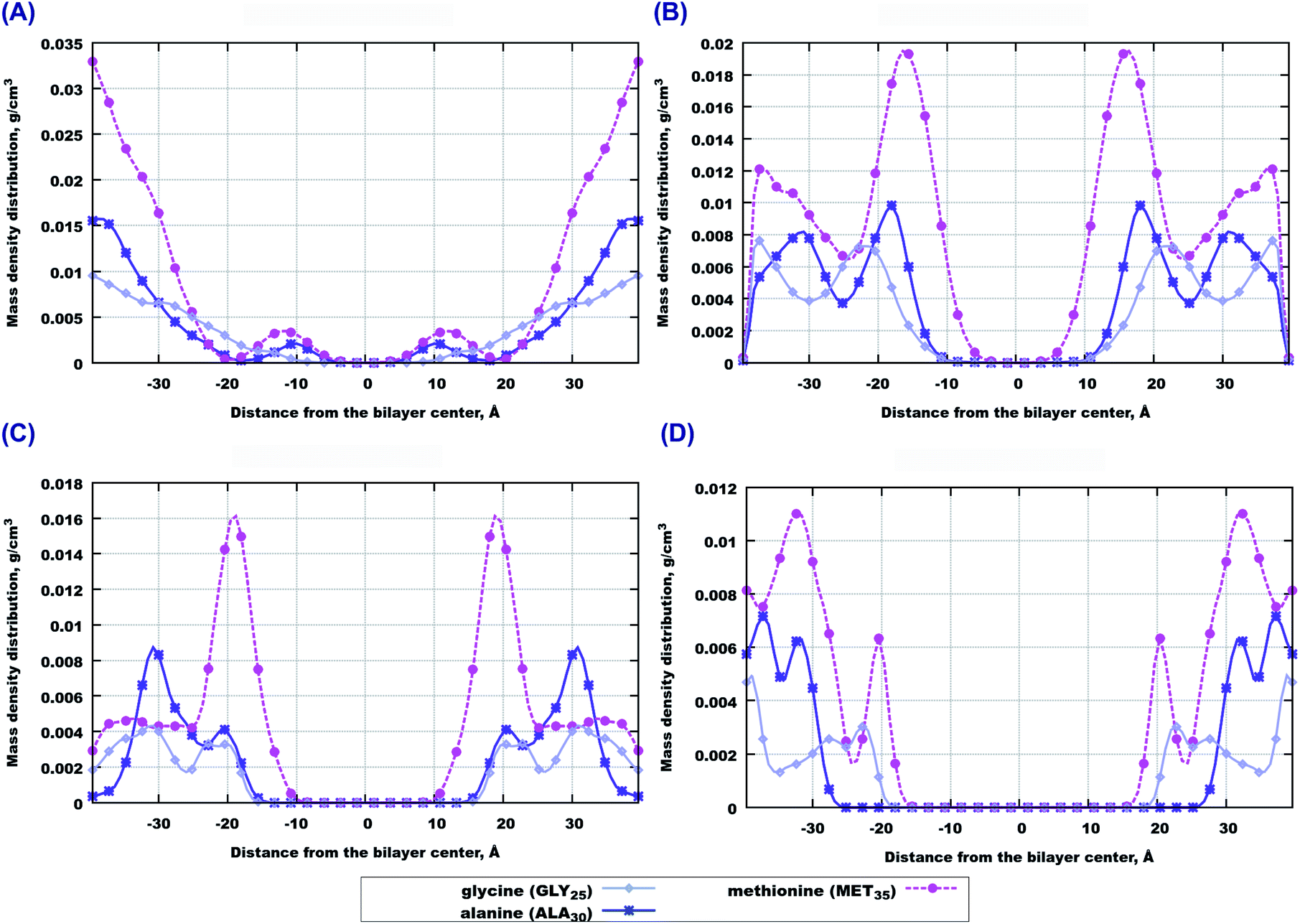 | ||
| Fig. 3 Contributions to mass density profiles from specific amino acids of peptides: (A) 14:0–14:0 PC without cholesterol. (B) 22:6–22:6 PC without cholesterol: contributions from amino acids. (C) 14:0–14:0 PC with cholesterol: contributions from amino acids. (D) 22:6–22:6 PC with cholesterol: contributions from amino acids. | ||
Convergence and statistical significance of the density profiles is illustrated in Fig. S2–S5 of the ESI.† In Fig. S2† we show density profiles of the peptides obtained during six sequential 167 ns fragments of the MD trajectories. Fig. S3 and S4† show similar information for three selected amino acids in the peptide. One can see that in most cases the curves generated after 400–600 ns of the simulation are similar to the final one which confirms that results averaged after the equilibration period of 500 ns (which we show in the figures of the main text) correspond to equilibrated systems. The exception is the case of polyunsaturated lipids with cholesterol (Fig. S2D and S4D†) where some slow redistribution of peptides towards the bilayer can be seen. We prescribe this process to the rearrangement of cholesterol and lipids in this system. It was demonstrated previously that cholesterol has poor affinity to polyunsaturated lipids and forms micro-clusters being inserted into the polyunsaturated membrane, the process occurs on a microsecond time scale.57 The observed changes of the peptide distribution are however small, and we believe that even in the case of polyunsaturated lipids with cholesterol our results are qualitatively correct. As an additional argument supporting convergence of the simulations with respect to positioning of the peptides relative to the membrane, we refer to another study where we simulated two other peptides, Aβ(1–28) and Aβ(26–40) in a number of lipid bilayers.80 It was demonstrated that with the same setup as in the present work, with several peptides located initially outside the membrane in random positions, Aβ(26–40) was entering the membrane interior within a few hundred nanoseconds of the simulation and residing there. Aβ(26–40) differs from Aβ(25–35) by the presence of hydrophobic residues in positions 36–40, which provide favourable free energy contribution to be inserted into the membrane. Overall, our data shows that 1 μs simulation time is sufficient to get a grasp on the distribution of the peptides in the simulated systems.
Fig. S5† shows the non-symmetrized density profiles of individual peptides. One can see that while some of the peptides show strong density maxima close to one of the membrane surfaces, others appear on the both sides of the bilayer during the simulation, travelling through the water layer of the periodic box during the production part of the simulations.
3.2 Area per lipid and deuterium order parameters
The increase of bilayer thickness upon addition of cholesterol, illustrated by the above described density profiles, is a well known phenomena related to the condensing and ordering effect of cholesterol,79,81,82 which is expressed in a reduction of the partial area per lipid, and in an increase of the ordering of lipid acyl chains. In order to investigate whether the presence of peptides can change the structure of the membrane, we have computed average areas and deuterium order parameters and compared them with our previous simulations of the same bilayers without peptides.79Area per lipid is an important characteristic of a membrane affecting many properties of the membrane organization.83,84 Table 2 shows the calculated average area per molecule (lipid and cholesterol) computed in this work (in simulations with peptides) and taken from our previous work79 (without peptides). One can see that in the case of cholesterol-free bilayers, addition of peptides leads to the increase of the average area per lipid, which is especially pronounced for the polyunsaturated 22:6–22:6 PC bilayer. This is consistent with observations of the density profiles in Fig. 2(A and B) showing higher presence of peptides in the headgroup region of the 22:6–22:6 PC membrane. The result shows that the presence of peptides increases the area and perhaps deforms the membrane, coherent with the simulation results of Lemkul et al.85
| Lipid (PC) + chol. | No peptides | With peptides |
|---|---|---|
| 14:0–14:0 + 0% | 62.7 ± 0.6 | 65.0 ± 0.6 |
| 14:0–14:0 + 50% | 40.3 ± 0.6 | 40.25 ± 0.2 |
| 22:6–22:6 + 0% | 70.0 ± 0.6 | 79.0 ± 0.7 |
| 22:6–22:6 + 50% | 40.5 ± 0.6 | 40.5 ± 0.2 |
Considering simulations with cholesterol loaded bilayers, one can see that due to the presence of cholesterol the average area per molecule is significantly decreased. The reduction of area per lipid in membranes with a high amount of cholesterol has been seen before experimentally and in models,28,79,82,86–90 and is related not only to the fact that cholesterol is a smaller molecule than a PC lipid, but also that lipids are becoming more ordered and leave less free space in the membrane interior which other molecules can take. This can explain why peptides do not penetrate into cholesterol loaded membranes (as discussed in the previous section) and do not affect average area per lipid.
The ordering effects of cholesterol can be illustrated by deuterium order parameters. Order parameters for CH bonds of both lipid tails in each of the four systems are displayed in Fig. 4, and compared with those obtained in previous work79 in simulations without peptides. It can be seen that in the case of pure 14:0–14:0 PC bilayer the presence of peptides leads to a substantial decrease of the order parameters, while in other cases the presence of peptides does not show any noticeable influence. Such behaviour can be understood by considering the following: cholesterol-containing bilayers are more condensed and ordered and they do not let Aβ(25–35) peptides penetrate inside the bilayer, which is consistent with the observations of the density profiles and area per lipid. Order parameters in lipid tails do not change upon addition of peptides (blue and green lines in Fig. 4). Pure bilayers are more fluid than cholesterol-containing ones and have a larger area per lipid with more space between lipid headgroups, so that peptides can penetrate into the headgroup region. In the case of 14:0–14:0 PC this leads to further increase of the area per lipid (since some peptides are inserted between the headgroups) and a decrease in ordering. The highly unsaturated 22:6–22:6 PC bilayers are even more flexible and fluid, and have already low (about 0.05) order parameters. One can note that while the area per lipid strongly increases upon addition of peptides to the 22:6–22:6 PC bilayer, the order parameters are effected only marginally. It is likely that highly unsaturated fatty acid chains favour even larger areas per lipid as the one observed in the pure 22:6–22:6 PC bilayer, but this is counterweighted by the attraction between dipole moments of the headgroups. When peptides are inserted between the headgroups, this increases the average distance between the lipid heads and area per lipid, without affecting the structure of the hydrocarbon tails. Still, a small decrease of the order parameter of pure 22:6–22:6 bilayers upon addition of peptides can be observed.
 | ||
| Fig. 4 Deuterium order parameters for lipid tails (A) 14:0–14:0 PC (sn − 1), (B) 14:0–14:0 PC (sn − 2), (C) 22:6–22:6 PC (sn − 1), (D) 22:6–22:6 PC (sn − 2), order parameters for systems without any peptide (red and green curves labeled as “no cholesterol” and “with cholesterol”, respectively). Reproduced from Ref. 79 with permission from the Royal Society of Chemistry. | ||
3.3 Radial distribution functions
To obtain detailed insight into differences in the affect of peptides on the considered bilayers we computed a number of radial distribution functions (RDF) between peptides and lipids or cholesterol. First we consider RDF between molecular centers of mass which illustrate general features of molecular association and clustering. In Fig. 5(A), peptide–peptide RDF show that these RDF are higher in cholesterol-containing systems, which indicates that peptides have a stronger tendency to cluster in such systems. Saturation of lipid tails seems to have an affect as well, which can be seen from the higher values of RDF at shorter distances for 22:6–22:6 PC. | ||
| Fig. 5 RDF between molecular centers of mass. (A) Peptide–peptide, (B) peptide–lipid, (C) peptide–cholesterol. | ||
Going further with peptide–lipid RDF (Fig. 5(B)) and peptide–cholesterol RDF (Fig. 5(C)), one can see that they do not have significant maxima at short distances, except the system with cholesterol loaded 14:0–14:0 PC which shows values of about 1 at 10 Å distance. The reason for such behavior of peptide–lipid RDF is that peptides reside mostly outside the membrane or in the membrane surface area at a certain distance from the lipid centers of mass. Cholesterol does not attract peptides either, which can be observed from Fig. 5(C). Peptide–cholesterol RDF start growing at shorter distances in the case of 14:0–14:0 PC bilayer which is explained because this bilayer is thinner than the 22:6–22:6 PC bilayer.
Among the polar atoms of Aβ(25–35) peptide and lipid headgroups, the highest RDF maxima were found for RDF between oxygen’s of the phosphate groups of lipids, and hydrogens in the hydroxyl groups in methionine MET35 and serine SER26 residues of the peptide (Fig. 6). In the bilayer consisting of 14:0–14:0 PC (Fig. 6(A)) one can see approximately equally high peaks for both amino acids, while for the other three bilayers, methionine shows stronger hydrogen bonding to phosphate groups than serine.
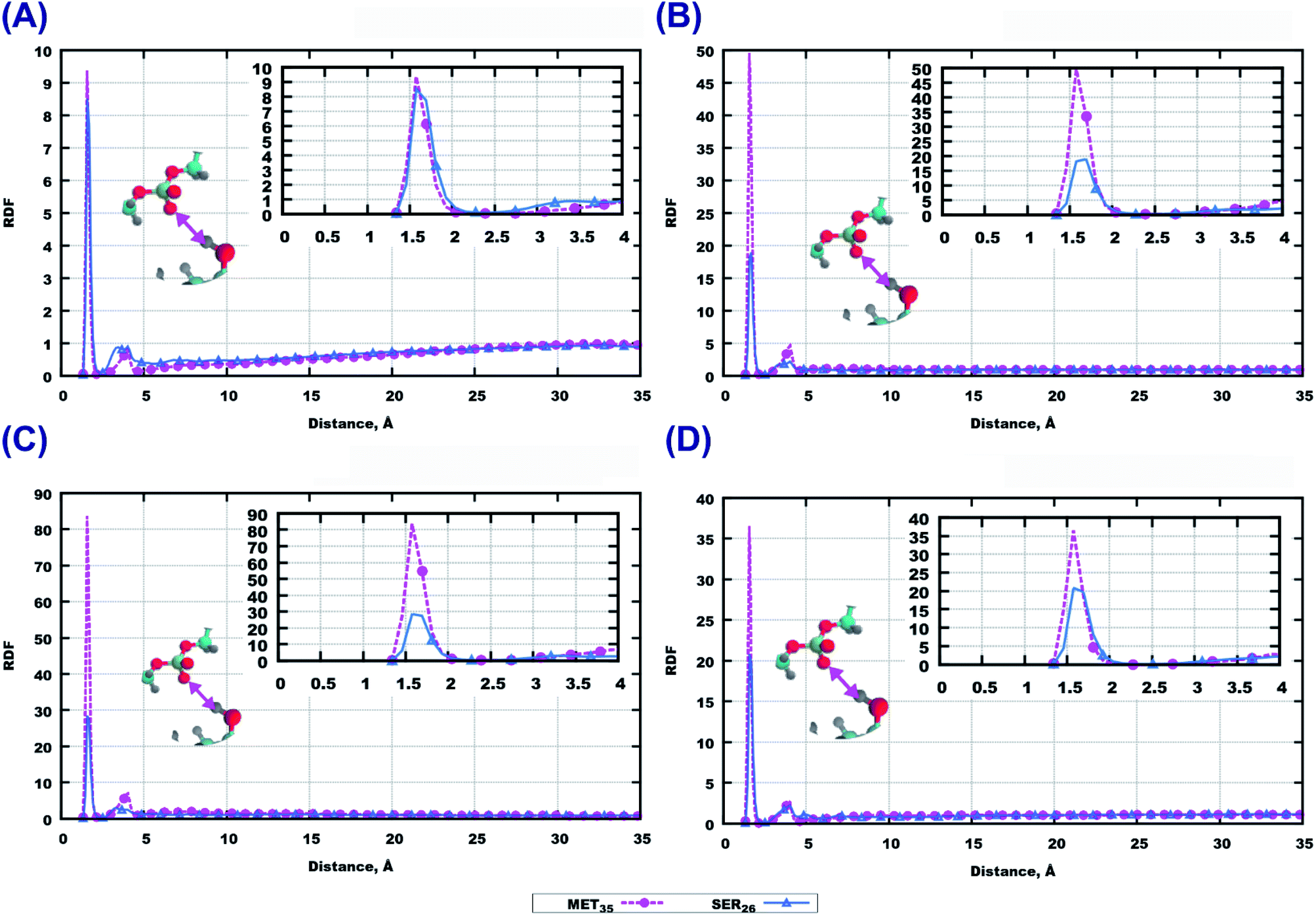 | ||
| Fig. 6 RDF between oxygens from phosphatic groups in lipids and hydrogens from hydroxyl groups from methionine MET35 and serine SER26 from the peptide. (A) 14:0–14:0 PC with Aβ(25–35) in water. (B) 22:6–22:6 PC with Aβ(25–35) in water. (C) 14:0–14:0 PC with Aβ(25–35) and 50% cholesterol in water. (D) 22:6–22:6 PC with Aβ(25–35) and 50% cholesterol in water. Colours on atoms: red – oxygen, cyan – carbon, yellow – phosphorus, gray – hydrogen, blue – nitrogen. | ||
For other residues, we show RDF between hydrogen’s of the amino group of the peptide backbone and phosphate oxygen’s in Fig. S6 and S7 of the ESI.† In the system with pure 14:0–14:0 PC the most pronounced peak is for LYS28, while in the 14:0–14:0 PC bilayer loaded with cholesterol the peak for LEU34 is prominent (Fig. S6(A and C)†). For a polyunsaturated bilayer without cholesterol, the highest peaks were observed for MET35 and LEU34, while for the cholesterol loaded membrane the maximum values of RDF appeared for ASN27 and SER26. Next, taking into account that peptides can penetrate up to the upper parts of lipid tails (as follows from the mass density profiles, Fig. 2 and 3), we plotted RDF between oxygen’s in the ester groups of lipid tails, and hydrogen’s in the hydroxyl groups in peptides (Fig. S8 of the ESI†). The most significant peaks can be seen for methionine and serine for the membrane built of 22:6–22:6 PC without cholesterol (Fig. S8(B)†), but comparing these with respective RDF at the phosphate group (Fig. 6), it is clear that hydrogen bonding is of less importance here.
The RDF computed between the same type of atoms of peptides and lipid headgroups are noticeably different in some considered cases, while lipid head-groups are the same in all simulations. This fact raises the question regarding possible interactions between atoms on lipid tails and peptides, for example, due to hydrophobic associations where the hydrocarbon parts of the Aβ(25–35) could be involved. For this purpose RDF between peptide residues and the first four groups of atoms in lipid tails were computed. Some of these results are presented in Fig. 7(A and B). While these RDF do not show high maxima, one can infer that there are more often contacts between peptide atoms and atoms in the upper part of the lipid tails in the case of pure 22:6–22:6 PC bilayer, compared to 14:0–14:0 PC. The 22:6–22:6 PC bilayer has a larger area per lipid, which facilitates peptide penetration into the bilayer headgroup area.
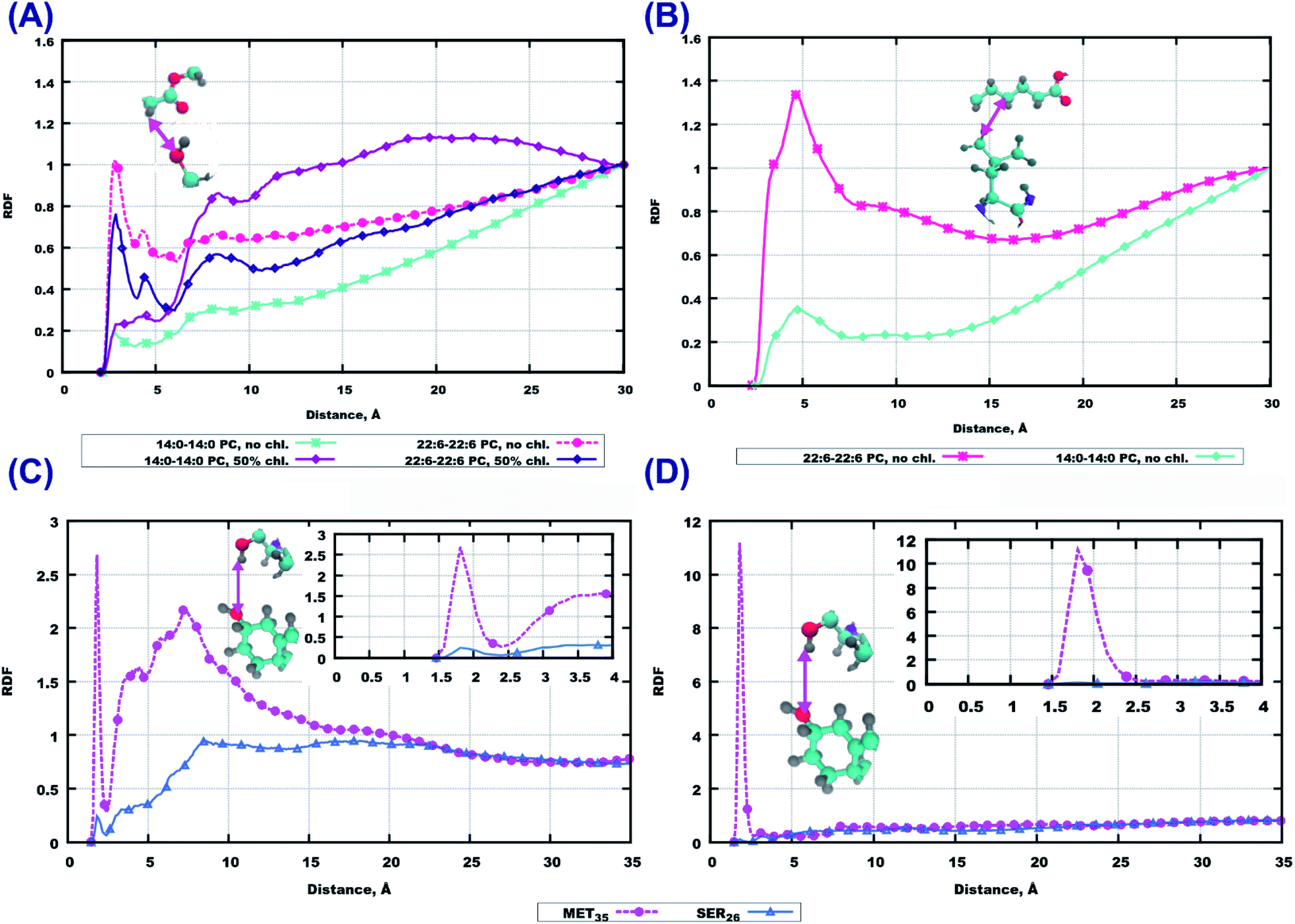 | ||
| Fig. 7 RDF of selected atoms in peptides and atoms on other molecules. (A) oxygen from the OH-group in SER26 and the hydrogen on the second carbon atom of a lipid tail, (B) hydrogen atoms from CH3-groups in LEU34 and the fourth carbon atom on a lipid tail, (C) oxygen from the hydroxyl group in cholesterol and the hydrogen atom from hydroxyl groups in MET35 and SER26 in systems with 14:0–14:0 PC, (D) oxygen from the hydroxyl group in cholesterol and the hydrogen atom from hydroxyl groups in MET35 and SER26 in systems with 22:6–22:6 PC. Colours on atoms: red – oxygen, cyan – carbon, yellow – phosphorus, gray – hydrogen, blue – nitrogen. | ||
RDF for the interaction of the GLY25 residue with others are shown in Fig. S9 and S10 of the ESI.† Remarkably, this part of the peptide sequence which did not seem to play a role in interactions with the lipids of membranes, has shown a strong involvement in interactions with other peptides. For instance, the saturated lipid bilayer GLY25 associates strongest with GLY29, then with the side chain of ASN27 and then with other amino-acids (ALA30, LYS28, GLY25, SER26). In the case of polyunsaturated membrane, ASN27; ILE31, MET35 and LEU34 can be seen as important actors while the other amino-acids do not show any remarkable peaks. Moving towards cholesterol loaded systems, again saturation of lipid tails seems to be an important factor: for systems with 14:0–14:0 PC, ILE31, ALA30, GLY29, GLY25 show strong interactions; while for 22:6–22:6 PC SER26 gives the highest peak followed by ALA30, LYS28, and ASN27. Other amino acids show lower peaks or did not show anything at all (Fig. S9 and S10 of the ESI†).
Moving further to the middle of the sequence we discuss RDF for ALA30 which are given in Fig. S11 and S12 of the ESI.† Here the oxygen atoms show even stronger hydrogen bonds compared to GLY25. It is remarkable, that for all systems containing 14:0–14:0 PC, the strongest associations are observed with hydrogen’s from amino-groups in ALA30. For simulations with 22:6–22:6 PC the situation differs: the highest peaks in the system without cholesterol belong to SER26, ASN27 and GLY25; while for the membrane loaded with cholesterol LYS28, ASN27 and GLY25 associate most strongly with alanine.
The highest RDF peaks for interactions between peptide atoms were found for the methionine MET35 residue, which is the last residue in the peptide. The corresponding RDF is shown in Fig. 8. In the pure 14:0–14:0 membrane one can see that the highest peaks of RDF belong to ASN27 (backbone) and SER26, followed by LEU34 and MET35 (Fig. 8 (A)). Furthermore, there is a high probability for association with other amino-acids of the peptide (Fig. S13(A) of the ESI†). A similar trend can be observed for methionine in pure 22:6–22:6 PC as rather high peaks on RDF are seen for all residues of Aβ(25–35) (Fig. 8(B) and S13(B) of the ESI†). The situation is somewhat different for bilayers loaded with cholesterol. Thus, in the case of 14:0–14:0 PC bilayer, methionine shows strong binding to 5 amino-acids which are located at the ends of the peptide (Fig. 8(C) and S13(C) of the ESI†), while in the case of 22:6–22:6 PC one can see again high peaks on RDF for all residues of the sequence, with the highest one for ILE31 and ILE32, followed by LEU34 and ASN27 (Fig. 8 and S13 in ESI†).
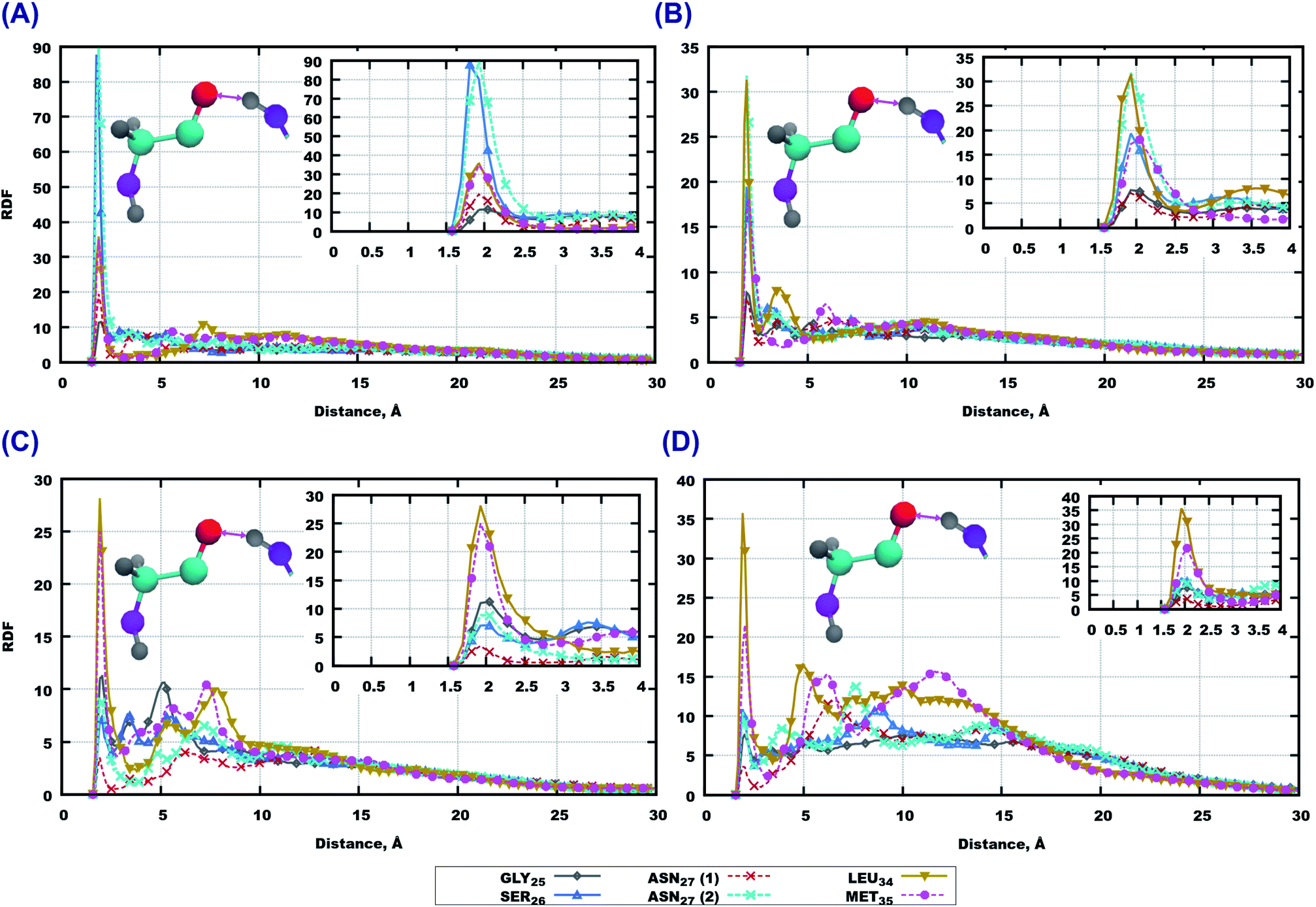 | ||
| Fig. 8 RDF between oxygen atoms on MET35 and hydrogen atoms in amino-groups on other amino-acids. (A) 14:0–14:0 PC and Aβ(25–35) in water, (B) 22:6–22:6 PC and Aβ(25–35) in water, (C) 14:0–14:0 PC, cholesterol and Aβ(25–35) in water, (D) 22:6–22:6 PC, cholesterol and Aβ(25–35) in water. Colours on atoms: red – oxygen, cyan – carbon, yellow – phosphorus, gray – hydrogen, blue – nitrogen. | ||
As further evidence of peptide association and clustering, we display contact maps of the peptides in Fig. S14–S17 of the ESI.† In each case, the MD trajectory was divided into 5 fragments of 200 ns, and the results obtained within each fragment are shown on separate panels. Off-diagonal features of the density maps show relatively frequent inter-molecular contact amongst the peptides, which are not persistent during the simulations. Thus, the peptides do not form stable aggregated structures, but change their intermolecular contacts on a hundred-nanosecond time scale.
3.4 Peptide conformations
Peptide three-dimensional conformations are determined mainly by the ϕ and ψ torsional angles of the backbone. These conformations may depend on the environment (polar vs. hydrophobic) as well as whether peptides are dissolved in a solvent or aggregated in clusters. We have compared simulated conformations of Aβ(25–35) with those found in the Protein Data Bank for three kinds of environments: hexafluoroisopropanol (HFIP)/water (80%/20%), hexafluoroisopropanol/water (20%/80%), and a 100 mM aqueous solution of SDS.27 In all cases the authors provided around 20 frames obtained by the best fit to NMR spectroscopy data, which were generated using SANDER module with DYANA derived restraints from AMBER 5.0 software.91,92In Fig. S18 and S19 of the ESI† we show the most populated ranges of ϕ and ψ torsional angles for each amino acid in the peptide sequence, together with experimental ranges from Ref. 27 for two (HFIP)/water ratios (80%/20% and 20%/80%), and for peptides in an aqueous solution of SDS. For our simulation results we show the range of angles for which the distribution histogram exceeds unite value (corresponding to the uniform distribution), while the experimental ranges are shown as average values with variance. The average values and variances of the torsional angles are also given in Table S1 and S2 of the ESI.† One can see that while for some torsional angles there is an overlap, for other angles the results are different. For a number of torsion angles our results show multimodal distribution, with the average value between the maxima and closer to the experimental average. We should note that results from the three experiments carried out in different solvents are different from each other, and experimental conditions were also different from our simulations where peptides were aggregated in smaller clusters at the membrane surface. Furthermore, the structures obtained experimentally have been initially generated by simulations using the AMBER1991 force field with DYANA restraints,91 and those providing best fit to NMR data were used to determine the structures, thus they can still be biased to the used force field.
We have also determined secondary structures of the peptides and plotted them as a function of the simulation time in Fig. S21–S24 of the ESI.† The secondary structure maps show that peptides have predominantly either “turn” or free chain conformation, which changes over simulation time and from one peptide molecule to another. This observation is consistent with conclusions from experimental studies27,37 that Aβ(25–35) in aqueous media shows irregular, polymorphic structural behaviour with prevailing β-turn conformations.
4 Discussion
Our results show that the location of peptides relative to membranes, as well as their association behaviour, depend on the membrane composition. Particularly, such factors as saturation of lipids and the presence of cholesterol affect fluidity and ordering of the membrane, which in turn influences the penetration of molecular species into the bilayers. Previously, various experimental and simulation techniques93–98 showed that bilayers composed of saturated lipids are generally more ordered systems characterized by lower area per lipid, higher temperature of gel phase transition and higher order parameters, while the presence of double bonds in acyl chains was related to disorder and higher fluidity.99–101 Higher cholesterol content in membranes has been related to an increase of order and rigidity.79,82,102–106 Our simulations confirm this picture showing by the deuterium order parameters and areas per lipid, that the most disordered membrane was the pure 22:6–22:6 PC bilayer, while addition of 50% of cholesterol increased the order in both 14:0–14:0 PC and 22:6–22:6 PC bilayers (Fig. 4). It seems reasonable that peptides penetrate more and deeper into a highly disordered membrane (pure 22:6–22:6 PC), and less into ordered membranes in the presence of cholesterol, with pure saturated 14:0–14:0 PC membrane taking intermediate place. As a consequence, the pure 22:6–22:6 PC allows the peptide to reside in the upper part of lipid tails while cholesterol promoted the aggregation of Aβ(25–35) at the membrane surface. Similar behaviour of Aβ peptides was observed earlier in experimental work26,29,32,39,40,107 and in simulations.45,47,108 We can further note that observations made for Aβ(25–35) might be valuable for other amyloid peptides. For example, it was shown by Owen et al.109 and others28,47 that insertion of the full Aβ(1–42) peptide into the membrane can be hampered by membrane rigidity, that is, by a similar mechanism as observed for Aβ(25–35) in this work.However, behaviour of the peptides relative to the membrane is determined not only by ordering and packing effects. Specific interactions of amino acids with various lipid moieties and formation of hydrogen bonds affect these processes too. Thus, methionine (MET35), located in the C-terminal of Aβ(25–35), seems to have the highest potency to bind to the membrane. This was also observed earlier in experiments and simulations of other lipid bilayers.34,110 Two other amino-acids, SER26 and ASN27, also consistently appear to be good promoters for hydrogen bonding between peptide and lipids. Hydrophobic interactions do not seem to be important actors here, since the highest peaks were not significant to the relevant RDF (Fig. 7(A and B)). For the cholesterol-containing bilayers one can also observe strong binding of MET35 to the hydroxyl group of cholesterol. Furthermore, since development of AD is related to the aggregation of Aβ peptides and particularly Aβ(25–35), a remarkable finding is that in addition to activity with lipids, MET35 shows frequent formation of hydrogen bonds with NH groups of the backbone for all amino-acids in the peptide. ASN27 also shows often high RDF peaks for peptide–peptide binding. Thus, it appears from our simulations that MET35 and ASN27 residues play a key role in the aggregation of Aβ(25–35). These findings are coherent with conclusions from previous work where possible toxicity of Aβ(25–35), as well as other Aβ peptides is related to MET35 and ASN27 residues.25,29,45,111–113 Thus, Varadarajan et al.25 associated MET35 with free radicals oxidative stress, by observing that the substitution of MET35 by structurally similar norleucine removed the toxic effect. Friedemann et al.113 found that oxidation of MET35 residues have a dumping effect on the aggregation of Aβ(25–35). Decrease in toxicity of Aβ(25–35) was also observed upon substitution of the ASN27 residue.31,52
Concluding this discussion, two amino-acids can be highlighted since they have shown significant interactions with different compounds in the four considered systems. MET35 appears to be the most pronounced promoter for binding between peptides and lipids in the bilayer as well as between peptides themselves. Perhaps, the reason for “activity” of this amino-acid in Aβ(25–35) is the fact that it is situated in the C-terminal and thus is able to bind by the carboxyl oxygen, as Tsai et al.45 showed that the reversed sequence has no toxic effects.
Still, the processes in living cells are much more complex than the ones which were studied here. There is still no consensus about the exact toxicity pathways of Aβ peptides, where various mechanisms such as oxidative stress, lipid peroxidation, cytotoxicity are discussed. Our simulations show that cytotoxicity via direct membrane damage due to the presence of Aβ(25–35) is unlikely since the peptide does not penetrate into the membrane interior, and exerts a rather limited effect on the membrane properties such as average lipid area and the order parameters. Pore formation by Aβ peptides was considered as a possible cytotoxicity mechanism in a number of works,35,37,46 for example the study by Kandel et al.37 shows that addition of Aβ(25–35) peptides to membrane vesicles leads to the appearance of Ca2+-conducting pores. That study was performed with anionic membranes with 30 mol% of POPG lipids, which have a strong affinity to the LYS-residue of Aβ(25–35). In another study of the same group36 it was demonstrated that a decrease in membrane anionic charge significantly suppresses the peptide binding to membrane and pore formation, which is consistent with our simulations performed with uncharged membranes, and illustrates also that membrane lipid composition is an important factor to consider while discussing the interaction of amyloid peptides with neuronal membranes.
5 Conclusions
Aβ(25–35) peptide, which is believed to be the most toxic among other fragments of amyloid precursor proteins, was simulated in the presence of lipid bilayers in order to understand which details of the peptide structure are responsible for its aggregation and how this process is affected by the composition of the membrane. Four MD simulations of lipid bilayers with fully saturated and strongly unsaturated lipids in the presence of Aβ(25–35) peptide have been carried out for this purpose.The simulations show that in the case of bilayers without cholesterol, Aβ(25–35) penetrates deeper into the bilayer built of polyunsaturated 22:6–22:6 PC lipids, while it stays on the surface of the membrane composed of 14:0–14:0 PC. When both membranes were loaded with cholesterol, the peptide did not enter the membrane, with slightly stronger preference to associate with lipid head-groups of 14:0–14:0 PC rather than of 22:6–22:6 PC. Such behaviour can be related to the ordering and packing effects in the cholesterol-containing bilayers: denser packing and higher ordering prevent peptides to enter the membrane. Another important conclusion from the simulations is the role of methionine (MET35) residue which was found to form the most strong hydrogen bonds with the polar groups of lipid and cholesterol as well as with other peptides, inducing peptide aggregation. The results regarding possible activity of methionine are coherent with experimental findings which relate toxicity of Aβ peptides to the presence of MET35 in the sequences.25,29,113 Other important residues involved in strong hydrogen bonding are ASN27 and SER26.
The molecular insight into the behaviour of Aβ(25–35) in lipid membranes can be relevant for designing strategies for drug design against AD. One possibility is decreasing the ordering (or increasing fluidity) of the membranes. This idea is supported by experimental data regarding the fatty acid composition of neuronal membranes in a human brain,15,19 which shows a high index of lipid unsaturation for the group of “healthy” people while the index is strongly reduced in an AD brain. Cholesterol lowering medicines can be considered for this purpose, taking into account that cholesterol was seen as a promoter of Aβ accumulation even in this work. Effecting membrane saturation is another option: peptides aggregate less in polyunsaturated membranes. Finally, another direction could be to develop medicines inhibiting the aggregation of peptides by blocking the most active residues which are found to be promoters of amyloid aggregation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Swedish Research Council (Vetenskapsrådet), grant no. 2017-03950. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) through the Center for Parallel Computing (PDC), National Supercomputer Center (NSC), and High Performance Computing Center North (HPC2N).References
- A. Ramamoorthy, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1601–1602 CrossRef CAS PubMed.
- E. H. Koo, P. T. Lansbury and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9989–9990 CrossRef CAS PubMed.
- M. Hashimoto, E. Rockenstein, L. Crews and E. Masliah, NeuroMol. Med., 2003, 4, 21–35 CrossRef CAS.
- L. M. Dember, J. Am. Soc. Nephrol., 2006, 17, 3458–3471 CrossRef CAS PubMed.
- A. Abedini and A. M. Schmidt, FEBS Lett., 2013, 587, 1119–1127 CrossRef CAS PubMed.
- E. Masliah, E. Rockenstein, I. Veinbergs, Y. Sagara, M. Mallory, M. Hashimoto and L. Mucke, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12245–12250 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- P. D. Schley, D. N. Brindley and C. J. Field, J. Nutr., 2007, 137, 548–553 CrossRef CAS PubMed.
- C. R. Santos and A. Schulze, FEBS J., 2012, 279, 2610–2623 CrossRef CAS PubMed.
- S. A. Shobab and G.-Y. H. H. Feldman, Lancet Neurol., 2005, 4, 841–852 CrossRef PubMed.
- N. C. Serrano, E. Guio-Mahecha, D. C. Quintero-Lesmes, S. Becerra-Bayona, M. C. Paez, M. Beltran, V. M. Herrera, L. J. Leon, D. Williams and J. P. Casas, Atherosclerosis, 2018, 189–194 CrossRef CAS PubMed.
- H.-Y. Tang, C.-H. Wang, H.-Y. Ho, P.-T. Wu, C.-L. Hung, C.-Y. Huang, P.-R. Wu, Y.-H. Yeh and M.-L. Cheng, Redox Biol., 2018, 14, 499–508 CrossRef CAS PubMed.
- A. Leiter, N. A. Bickell, D. LeRoith, A. Nayak, S. M. Feldman, N. B. Friedman, A. Estabrook, T. A. King, K. Fei and R. Franco, et al., Horm. Cancer, 2018, 9, 55–61 CrossRef CAS PubMed.
- M. R. Freeman and K. R. Solomon, J. Cell. Biochem., 2004, 91, 54–69 CrossRef CAS PubMed.
- V. Martin, N. Fabelo, G. Santpere, B. Puig, R. Martin, I. Ferrer and M. Diaz, J. Alzheimer’s Dis., 2010, 19, 489–502 CAS.
- N. Fabelo, V. Martín, R. Marín, D. Moreno, I. Ferrer and M. Díaz, Neurobiol. Aging, 2014, 35, 1801–1812 CrossRef CAS PubMed.
- N. Fabelo, V. Martín, R. Marín, G. Santpere, E. Aso, I. Ferrer and M. Díaz, J. Neuropathol. Exp. Neurol., 2012, 71, 868–881 CrossRef CAS PubMed.
- N. Fabelo, V. Martín, G. Santpere, R. Marín, L. Torrent, I. Ferrer and M. Díaz, Mol. Med., 2011, 17, 1107 CAS.
- M. Söderberg, C. Edlund, K. Kristenssin and G. Dallner, Lipids, 1991, 26, 421–425 CrossRef PubMed.
- R. Marin, N. Fabelo, V. Martin, P. Garcia-Esparcia, I. Ferrer, D. Quinto-Alemany and M. Diaz, Neurobiol. Aging, 2017, 49, 52–59 CrossRef CAS PubMed.
- D. J. Selkoe, J. Alzheimer’s Dis., 2001, 3, 75–80 CAS.
- F. Fang and G. Liu, Acta Pharmacol. Sin., 2006, 27, 651–658 CrossRef CAS PubMed.
- S. Delobette, A. Privat and T. Maurice, Eur. J. Pharmacol., 1997, 319, 1–4 CrossRef CAS PubMed.
- S.-Y. Chen, J. W. Wright and C. D. Barnes, Brain Res., 1996, 720, 54–60 CrossRef CAS PubMed.
- S. Varadarajan, S. Yatin, J. Kanski, F. Jahanshani and D. A. Butterfield, Brain Res. Bull., 1999, 50, 133–141 CrossRef CAS PubMed.
- S. Dante, T. Hauss and N. A. Dencher, Biochemistry, 2003, 42, 13667–13672 CrossRef CAS PubMed.
- A. M. D’Ursi, M. R. Armenante, R. Guerrini, S. Salvadori, G. Sorrentino and D. Picone, J. Med. Chem., 2004, 47, 4231–4238 CrossRef PubMed.
- S. Dante, T. Hauß and N. A. Dencher, Eur. Biophys. J., 2006, 35, 523–531 CrossRef CAS PubMed.
- T.-L. Lau, J. D. Gehman, J. D. Wade, K. Perez, C. L. Masters, K. J. Barnham and F. Separovic, Biochim. Biophys. Acta, 2007, 1768, 2400–2408 CrossRef CAS PubMed.
- G. D’Errico, G. Vitiello, O. Ortona, A. Tedeschi, A. Ramunno and A. M. D’Ursi, Biochim. Biophys. Acta, 2008, 1778, 2710–2716 CrossRef PubMed.
- M. Naldi, J. Fiori, M. Pistolozzi, A. F. Drake, C. Bertucci, R. Wu, K. Mlynarczyk, S. Filipek, A. D. Simone and V. Adrisano, ACS Chem. Neurosci., 2012, 3, 952–962 CrossRef CAS PubMed.
- G. Vitiello, S. Marino, A. M. D’Ursi and G. D’Errico, Langmuir, 2013, 29, 14239–14245 CrossRef CAS PubMed.
- A. Cuco, A. P. Serro, J. P. Farinha, B. Saramago and A. da Silva, Colloids Surf., B, 2016, 141, 10–18 CrossRef CAS PubMed.
- C. Esposito, A. Tedeschi, M. Scrima, G. D’Errico, M. F. Ottaviani, P. Rovero and A. M. D’Ursi, J. Pept. Sci., 2006, 12, 766–774 CrossRef CAS PubMed.
- C. Di Scala, H. Chahinian, N. Yahi, N. Garmy and J. Fantini, Biochemistry, 2014, 53, 4489–4502 CrossRef CAS PubMed.
- N. Kandel, T. Zheng, Q. Huo and S. A. Tatulian, J. Phys. Chem. B, 2017, 121, 10293–10305 CrossRef CAS PubMed.
- N. Kandel, J. O. Matos and S. A. Tatulian, Sci. Rep., 2019, 9, 2689 CrossRef PubMed.
- J. Singh and M. Peric, Chem. Phys. Lipids, 2018, 216, 39–47 CrossRef CAS PubMed.
- J.-F. Labbé, T. Lefèvre, A.-A. Guay-Bègin and M. Auger, Phys. Chem. Chem. Phys., 2013, 15, 7228–7239 RSC.
- H. Dies, L. Toppozini and M. C. Rheinstädter, PLoS One, 2014, 9, 1–17 CrossRef PubMed.
- G. Wei and J.-E. Shea, Biophys. J., 2006, 91, 1638–1647 CrossRef CAS PubMed.
- B. Ma and R. Nussinov, Biophys. J., 2006, 90, 3365–3374 CrossRef CAS PubMed.
- S.-W. Lee and Y.-M. Kim, Bull. Korean Chem. Soc., 2004, 25, 838–842 CrossRef CAS.
- M. Kittner and V. Knecht, J. Phys. Chem. B, 2010, 114, 15288–15295 CrossRef CAS PubMed.
- H.-H. G. Tsai, J.-B. Lee, Y.-C. Shih, L. Wan, F.-K. Shieh and C.-Y. Chen, ChemMedChem, 2014, 2014, 1002–1011 CrossRef PubMed.
- Z. Chang, Y. Luo, Y. Zhang and G. Wei, J. Phys. Chem. B, 2011, 115, 1165–1174 CrossRef CAS PubMed.
- C. H. Davis and M. L. Berkowitz, Biophys. J., 2009, 96, 785–797 CrossRef CAS PubMed.
- C. H. Davis and M. L. Berkowitz, J. Phys. Chem. B, 2009, 113, 14480–14486 CrossRef CAS PubMed.
- C. H. Davis and M. L. Berkowitz, Proteins: Struct., Funct., Bioinf., 2010, 78, 2533–2545 CAS.
- L. N. Zhao, S. W. Chiu, J. Benoit, L. Y. Chew and Y. G. Mu, J. Phys. Chem. B, 2011, 115, 12247–12256 CrossRef CAS PubMed.
- J. A. Lemkul and D. R. Bevan, Biochemistry, 2013, 52, 4971–4980 CrossRef CAS PubMed.
- T. Kohno, K. Kobayashi, T. Maeda, K. Sato and A. Takashima, Biochemistry, 1996, 35, 16094–16104 CrossRef CAS PubMed.
- D. G. Isom, C. A. Castaneda, B. R. Cannon and B. Garcia-Moreno, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260–5265 CrossRef CAS PubMed.
- N. J. Gleason, V. V. Vostrikov, D. V. Greathouse and R. E. Koeppe II, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1692–1695 CrossRef CAS PubMed.
- J. P. M. Jämbeck and A. P. Lyubartsev, J. Phys. Chem. B, 2012, 116, 3164–3179 CrossRef PubMed.
- J. P. M. Jämbeck and A. P. Lyubartsev, J. Chem. Theory Comput., 2013, 9, 774–784 CrossRef PubMed.
- I. Ermilova and A. P. Lyubartsev, J. Phys. Chem. B, 2016, 120, 12826–12842 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- A. Sousa da Silva and W. Vranken, BMC Res. Notes, 2012, 5, 367 CrossRef PubMed.
- S. R. Gerben, J. A. Lemkul, A. M. Brown and D. R. Bevan, J. Biomol. Struct. Dyn., 2014, 32, 1817–1832 CrossRef CAS PubMed.
- A. K. Somavarapu and K. P. Kepp, ChemPhysChem, 2016, 16, 3278–3289 CrossRef PubMed.
- M. Carballo-Pacheco and B. Strodel, Protein Sci., 2017, 26, 174–185 CrossRef CAS PubMed.
- P. Robustelli, S. Piana and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4758–E4766 CrossRef CAS PubMed.
- J. P. M. Jämbeck and A. P. Lyubartsev, J. Phys. Chem. Lett., 2013, 4, 1781–1787 CrossRef PubMed.
- M. Paloncýová, G. Fabre, R. H. DeVane, P. Trouillas, K. Berka and M. Otyepka, J. Chem. Theory Comput., 2014, 10, 4143–4151 CrossRef PubMed.
- I. Ermilova and A. P. Lyubartsev, Phys. Chem. Chem. Phys., 2017, 19, 28263–28274 RSC.
- X. Xu, X. Xiao, S. Xu and H. Liu, Phys. Chem. Chem. Phys., 2016, 18, 25465–25473 RSC.
- M. Kang, P. Zhang, H. Cui and S. M. Loverde, Macromolecules, 2016, 49, 994–1001 CrossRef CAS PubMed.
- M. Kang, H. Cui and S. M. Loverde, Soft Matter, 2017, 13, 7721–7730 RSC.
- G. L. Butterfoss, B. Yoo, J. N. Jaworski, I. Chorny, K. A. Dill, R. N. Zuckermann, R. Bonneau, K. Kirshenbaum and V. A. Voetz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14320–14325 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- H. J. C. Berendsen, D. van der Spoel and R. Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- M. F. M. Sciacca, F. Lolicato, G. D. Mauro, D. Milardi, L. D’Urso, C. Satriano, A. Ramamoorthy and C. La Rosa, Biophys. J., 2016, 111, 140–151 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- A. P. Lyubartsev and A. Laaksonen, Comput. Phys. Commun., 2000, 128, 565–589 CrossRef CAS.
- I. Ermilova and A. P. Lyubartsev, Soft Matter, 2019, 15, 78–93 RSC.
- N. Ntarakas, I. Ermilova and A. P. Lyubartsev, Eur. Biophys. J., 2019, 48, 813–824 CrossRef CAS PubMed.
- W.-C. Hung, M.-T. Lee, F.-Y. Chen and H. W. Huang, Biophys. J., 2007, 92, 3960–3967 CrossRef CAS PubMed.
- T. Róg, M. Pasenkiewicz-Gierula, I. Vattulainen and M. Karttunen, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 97–121 CrossRef PubMed.
- J. F. Nagle, Annu. Rev. Phys. Chem., 1980, 31, 157–195 CrossRef CAS.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, 2000, 1469, 159–195 CrossRef CAS.
- J. A. Lemkul and D. R. Bevan, FEBS J., 2009, 276, 3060–3075 CrossRef CAS PubMed.
- A. M. Smondyrev and M. L. Berkowitz, Biophys. J., 1999, 77, 2075–2089 CrossRef CAS PubMed.
- O. Edholm and J. F. Nagle, Biophys. J., 2005, 89, 1827–1832 CrossRef CAS PubMed.
- C. L. Armstrong, D. Marquardt, H. Dies, N. Kučerka, Z. Yamani, T. A. Harroun, J. Katsaras, A.-C. Shi and M. C. Rheinstädter, PLoS One, 2013, 8, e66162 CrossRef CAS PubMed.
- N. Kučerka, J. D. Perimutter, J. Pan, S. Tristram-Nagle and J. Katsaras, Biophys. J., 2008, 95, 2792–2805 CrossRef PubMed.
- P. S. Niemelä, S. Ollila, M. T. Hyvönen, M. Karttunen and I. Vattulainen, PLoS Comput. Biol., 2007, 3, e34 CrossRef PubMed.
- P. Güntert, C. Mumenthaler and K. Wüthrich, J. Mol. Biol., 1997, 273, 283–298 CrossRef PubMed.
- S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, S. Profeta and P. Weiner, J. Am. Chem. Soc., 1984, 106, 765–784 CrossRef CAS.
- S. R. Shaikh, V. Cherezov, M. Caffrey, S. P. Soni, D. LoCascio, W. Stillwell and S. R. Wassall, J. Am. Chem. Soc., 2006, 128, 5375–5383 CrossRef CAS PubMed.
- F. W. Stetter, L. Cwiklik, P. Jungwirth and T. Hugel, Biophys. J., 2014, 107, 1167–1175 CrossRef CAS PubMed.
- T. A. Harroun and J. K. S. R. Wassal, Biochemistry, 2006, 45, 1227–1233 CrossRef CAS PubMed.
- D. Marquardt, N. Kučerka, S. R. Wassall, T. A. Harroun and K. Katsaras, Chem. Phys. Lipids, 2016, 199, 17–25 CrossRef CAS PubMed.
- W. D. Bennett, J. L. MacCallum, M. J. Hinner, S. J. Marrink and D. P. Tieleman, J. Am. Chem. Soc., 2009, 131, 12714–12720 CrossRef CAS PubMed.
- W. D. Bennett, J. L. MacCallum and D. P. Tieleman, J. Am. Chem. Soc., 2009, 131, 1972–1978 CrossRef CAS PubMed.
- N. Kučerka, S. Tristram-Nagle and J. F. Nagle, J. Membr. Biol., 2006, 208, 193–202 CrossRef PubMed.
- M. Bachar, P. Brunelle, D. P. Tieleman and A. Rauk, J. Phys. Chem. B, 2004, 108, 7170–7179 CrossRef CAS.
- J. B. Klauda, V. Monje, T. Kim and W. Im, J. Phys. Chem. B, 2012, 116, 9424–9431 CrossRef CAS PubMed.
- O. G. Mouritsen and K. Jørgensen, Chem. Phys. Lipids, 1994, 73, 3–25 CrossRef CAS PubMed.
- S. A. Pandit, V. S. W. Chiu, R. J. Mashl, E. Jakobsson and H. L. Scott, Biophys. J., 2004, 87, 1092–1100 CrossRef CAS PubMed.
- W. K. Subczynski, M. Pasenkiewicz-Gierula, J. Widomska, L. Mainali and M. Raguz, Cell Biochem. Biophys., 2017, 75, 369–385 CrossRef CAS PubMed.
- H. I. Petrache, D. Harries and V. A. Parsegian, Macromol. Symp., 2005, 39–50 CrossRef.
- S. R. Wassall, M. R. Brzustovicz, S. R. Shaikh, V. Cherezov, M. Caffrey and W. Stillwell, Chem. Phys. Lipids, 2004, 132, 79–88 CAS.
- A. Choucair, M. Chakrapani, B. Chakravarthy, J. Katsaras and L. Johnston, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 146–154 CrossRef CAS PubMed.
- A. K. Smith and D. K. Klimov, J. Chem. Inf. Model., 2018, 58, 1053–1065 CrossRef CAS PubMed.
- M. C. Owen, W. Kulig, C. Poojari, T. Rog and B. Strodel, Biochim. Biophys. Acta, Biomembr., 2018, 1709–1720 CrossRef CAS PubMed.
- F. Tofoleanu and N.-V. Buchete, Prion, 2012, 6, 339–345 CrossRef CAS PubMed.
- D. A. Butterfield and A. I. Bush, Neurobiol. Aging, 2004, 25, 563–568 CrossRef CAS PubMed.
- M. E. Clementi, S. Marini, M. Coletta, F. Orsini, B. Giardina and F. Misiti, FEBS Lett., 2005, 579, 2913–2918 CrossRef CAS PubMed.
- M. Friedemann, E. Helk, A. Tiiman, K. Zovo, P. Palumaa and V. Tõugu, Biochemistry and Biophysics Reports, 2015, 3, 94–99 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and tables showing density profiles, radial distribution functions, contact maps, average values of torsional angles, secondary structures. See DOI: 10.1039/c9ra06424a |
| This journal is © The Royal Society of Chemistry 2020 |